
Lung Microbiome in Lung Cancer: A Systematic Review

 , , ,
, , ,  ,
,  , ,
, , 
Abstract
:1. Introduction
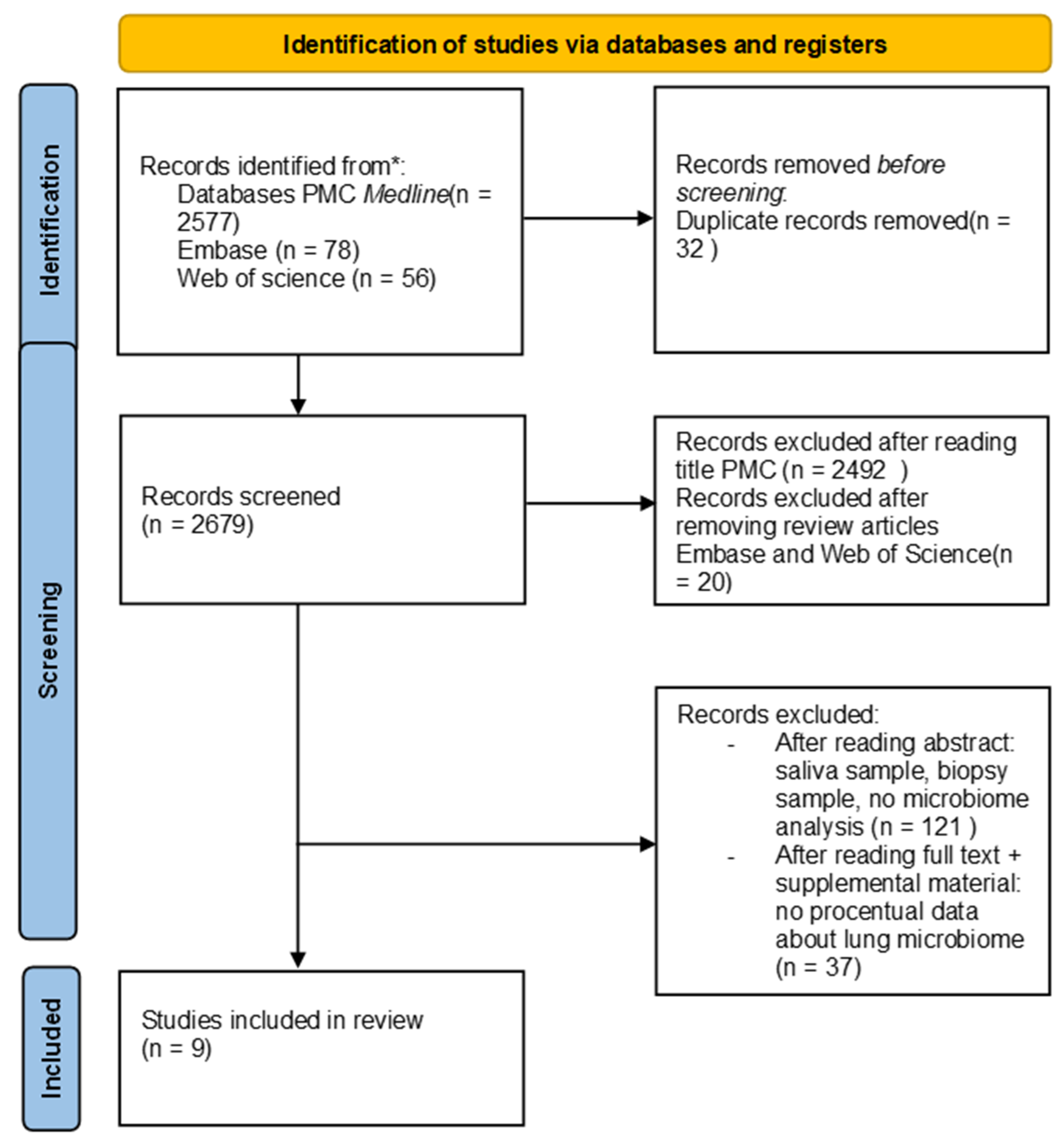
2. Material and Methods
2.1. Search Strategy
2.2. Studies Selection and Eligibility Criteria
2.3. Study Objectives
2.4. Data Extraction and Synthesis
3. Results
3.1. Literature Search
3.2. Characteristics of the Included Studies
3.2.1. Studies Objective
3.2.2. Inclusion/Exclusion Criteria
3.2.3. Bronchoalveolar Lavage Sample Collection
3.2.4. Insights from Reviewed Studies
3.3. Comparative Analysis of Microbial Phyla and Genera Distribution in Lung Cancer
3.4. Patient Demographics and Tumor Histology in Selected Studies
3.5. Alpha Diversity
4. Discussion
Funding
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020 GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Ursell, L.K.; Metcalf, J.L.; Parfrey, L.W.; Knight, R. Defining the human microbiome. Nutr. Rev. 2013, 70, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Luo, J.L.; Ali, M.K.; Spiekerkoetter, E.; Nicolls, M.R. The Human Respiratory Microbiome: Current Understandings and Future Directions. Am. J. Respir. Cell Mol. Biol. 2023, 68, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Natalini, J.G.; Singh, S.; Segal, L.N. The dynamic lung microbiome in health and disease. Nat. Rev. Microbiol. 2023, 21, 222–235. [Google Scholar] [CrossRef] [PubMed]
- de Steenhuijsen Piters, W.A.A.; Binkowska, J.; Bogaert, D. Early Life Microbiota and Respiratory Tract Infections. Cell Host Microbe 2020, 28, 223–232. [Google Scholar] [CrossRef]
- Cox, M.; Ege, M.J. The Lung Microbiome; Cox, M., Ege, M.J., von Mutius, E., Eds.; European Respiratory Society: Brussels, Belgium, 2019. [Google Scholar]
- Mathieu, E.; Escribano-Vazquez, U.; Descamps, D.; Cherbuy, C.; Langella, P.; Riffault, S.; Remot, A.; Thomas, M. Paradigms of lung microbiota functions in health and disease, particularly, in asthma. Front. Physiol. 2018, 9, 1168. [Google Scholar] [CrossRef]
- Guarner, F.; Malagelada, J.R. Gut flora in health and disease. Lancet 2003, 361, 512–519. [Google Scholar] [CrossRef]
- Yagi, K.; Huffnagle, G.B.; Lukacs, N.W.; Asai, N. The Lung Microbiome during Health and Disease. Int. J. Mol. Sci. 2021, 22, 10872. [Google Scholar] [CrossRef]
- Zheng, L.; Sun, R.; Zhu, Y.; Li, Z.; She, X.; Jian, X.; Yu, F.; Deng, X.; Sai, B.; Wang, L.; et al. Lung microbiome alterations in NSCLC patients. Sci. Rep. 2021, 11, 11736. [Google Scholar] [CrossRef]
- Kuczynski, J.; Liu, Z.; Lozupone, C.; McDonald, D.; Fierer Knight, R. Microbial community resemblance methods differ in their ability to detect biologically relevant patterns. Nat. Methods 2010, 7, 813–819. [Google Scholar] [CrossRef]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [PubMed]
- Hartman, A.L.; Riddle, S.; McPhillips, T.; Ludäscher, B.; Eisen, J.A. Introducing W.A.T.E.R.S.: A Workflow for the Alignment, Taxonomy, and Ecology of Ribosomal Sequences. BMC Bioinform. 2010, 11, 317. [Google Scholar] [CrossRef] [PubMed]
- Cole, J.R.; Wang, Q.; Cardenas, E.; Fish, J.; Chai, B.; Farris, R.J.; Kulam-Syed-Mohideen, A.S.; McGarrell, D.M.; Marsh, T.; Garrity, G.M.; et al. The Ribosomal Database Project: Improved alignments and new tools for rRNA analysis. Nucleic Acids Res. 2009, 37, D141–D145. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef]
- Huse, S.M.; Ye, Y.; Zhou, Y.; Fodor, A.A. A Core Human Microbiome as Viewed through 16S rRNA Sequence Clusters. PLoS ONE 2012, 7, e34242. [Google Scholar] [CrossRef]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef]
- Page, M.J.; McKenzie, J.E.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D.; Shamseer, L.; Tetzlaff, J.M.; Akl, E.A.; Brennan, S.E.; et al. The PRISMA 2020 statement: An updated guideline for reporting systematic reviews. BMJ 2021, 372, n71. [Google Scholar] [CrossRef]
- Bingula, R.; Filaire, E.; Molnar, I.; Delmas, E.; Berthon, J.Y.; Vasson, M.P.; Bernalier-Donadille, A.; Filaire, M. Characterisation of microbiota in saliva, bronchoalveolar lavage fluid, non-malignant, peritumoural and tumour tissue in non-small cell lung cancer patients: A cross-sectional clinical trial. Respir. Res. 2020, 21, 129. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Huang, Y.; Zhang, Z.; Liao, J.; Ding, Y.; Fang, X.; Liu, L.; Luo, J.; Kong, J. A Preliminary Study of Microbiota Diversity in Saliva and Bronchoalveolar Lavage Fluid from Patients with Primary Bronchogenic Carcinoma. Med. Sci. Monit. 2019, 25, 2819–2834. [Google Scholar] [CrossRef]
- Jang, H.J.; Choi, J.Y.; Kim, K.; Yong, S.H.; Kim, Y.W.; Kim, S.Y.; Kim, E.Y.; Jung, J.Y.; Kang, Y.A.; Park, M.S.; et al. Relationship of the lung microbiome with PD-L1 expression and immunotherapy response in lung cancer. Respir. Res. 2021, 22, 322. [Google Scholar] [CrossRef]
- Zhuo, M.; An, T.; Zhang, C.; Wang, Z. Characterization of Microbiota in Cancerous Lung and the Contralateral Non-Cancerous Lung Within Lung Cancer Patients. Front. Oncol. 2020, 10, 1584. [Google Scholar] [CrossRef]
- Gomes, S.; Cavadas, B.; Ferreira, J.C.; Marques, P.I.; Monteiro, C.; Sucena, M.; Sousa, C.; Vaz Rodrigues, L.; Teixeira, G.; Pinto, P.; et al. Profiling of lung microbiota discloses differences in adenocarcinoma and squamous cell carcinoma. Sci. Rep. 2019, 9, 12838. [Google Scholar] [CrossRef] [PubMed]
- Seixas, S.; Kolbe, A.R.; Gomes, S.; Sucena, M.; Sousa, C.; Rodrigues, L.V.; Teixeira, G.; Pinto, P.; Tavares de Abreu, T.; Bárbara, C.; et al. Comparative analysis of the bronchoalveolar microbiome in Portuguese patients with different chronic lung disorders. Sci. Rep. 2021, 11, 15042. [Google Scholar] [CrossRef]
- Lee, S.H.; Sung, J.Y.; Yong, D.; Chun, J.; Kim, S.Y.; Song, J.H.; Chung, K.S.; Kim, E.Y.; Jung, J.Y.; Kang, Y.A.; et al. Characterization of microbiome in bronchoalveolar lavage fluid of patients with lung cancer comparing with benign mass like lesions. Lung Cancer 2016, 102, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Li, Y.; Suo, L.; Zhang, W.; Cao, H.; Wang, R.; Luan, J.; Yu, X.; Dong, L.; Wang, W.; et al. Characterizing microbiota and metabolomics analysis to identify candidate biomarkers in lung cancer. Front. Oncol. 2022, 12, 1058436. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.J.; Lee, E.; Cho, Y.J.; Lee, S.H. Subtype-Based Microbial Analysis in Non-small Cell Lung Cancer. Tuberc. Respir. Dis. 2023, 86, 294–303. [Google Scholar] [CrossRef] [PubMed]
- Zeng, W.; Zhao, C.; Yu, M.; Chen, H.; Pan, Y.; Wang, Y.; Bao, H.; Ma, H.; Ma, S. Alterations of lung microbiota in patients with non-small cell lung cancer. Bioengineered 2022, 3, 6665–6677. [Google Scholar] [CrossRef]
- Liu, X.; Sun, W.; Ma, W.; Wang, H.; Xu, K.; Zhao, L.; He, Y. Smoking related environmental microbes affecting the pulmonary microbiome in Chinese population. Sci. Total Environ. 2022, 829, 154652. [Google Scholar] [CrossRef]
- Dickson, R.P.; Erb-Downward, J.R.; Freeman, C.M.; Lisa McCloskey, L.; Beck, J.M.; Huffnagle, G.B.; Curtis, J.L. Spatial Variation in the Healthy Human Lung Microbiome and the Adapted Island Model of Lung Biogeography. Ann. Am. Thorac. Soc. 2015, 12, 821–830. [Google Scholar] [CrossRef]
- Karakasidis, E.; Kotsiou, O.S.; Gourgoulianis, K.I. Lung and Gut Microbiome in COPD. J. Pers. Med. 2023, 13, 804. [Google Scholar] [CrossRef]
- Dickson, R.P.; Erb-Downward, J.R.; Martinez, F.J.; Huffnagle, G.B. The Microbiome and the Respiratory Tract. Annu. Rev. Physiol. 2016, 78, 481–504. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Shariff, M.; Chaturvedi, G.; Chaturvedi, G.; Sharma, A.; Goel, N.; Yadav, M.; Mortensen, M.S.; Sørensen, S.J.; Mukerji, M.; et al. Comparative analysis of the alveolar microbiome in COPD, ECOPD, Sarcoidosis, and ILD patients to identify respiratory illnesses specific microbial signatures. Sci. Rep. 2021, 11, 3963. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.J.; Nelson, C.E.; Brodie, E.L.; DeSantis, T.Z.; Baek, M.S.; Liu, J.; Woyke, T.; Allgaier, M.; Bristow, J.; Wiener-Kronish, J.P.; et al. Airway microbiota and bronchial hyperresponsiveness in patients with suboptimally controlled asthma. J. Allergy Clin. Immunol. 2011, 127, 372–381. [Google Scholar] [CrossRef] [PubMed]
- Tsay, J.-C.T.; Wu, B.G.; Sulaiman, I.; Gershner, K.; Schluger, R.; Li, Y.; Yie, T.A.; Meyn, P.; Olsen, E.; Perez, L.; et al. Lower Airway Dysbiosis Affects Lung Cancer Progression. Cancer Discov. 2021, 11, 293–307. [Google Scholar] [CrossRef]
- Einarsson, G.G.; Comer, D.M.; McIlreavey, L.; Parkhill, J.; Ennis, M.; Tunney, M.M.; Elborn, J.S. Community dynamics and the lower airway microbiota in stable chronic obstructive pulmonary disease, smokers and healthy non-smokers. Thorax 2016, 71, 795–803. [Google Scholar] [CrossRef]
- Loverdos, K.; Bellos, G.; Kokolatou, L.; Vasileiadis, I.; Giamarellos, E.; Pecchiari, M.; Koulouris, N.; Koutsoukou, A.; Rovina, N. Lung Microbiome in Asthma: Current Perspectives. J. Clin. Med. 2019, 8, 1967. [Google Scholar] [CrossRef] [PubMed]
- Morris, A.; Beck, J.M.; Schloss, P.D.; Campbell, T.B.; Crotherset, K.; Curtis, J.L.; Flores, S.C.; Fontenot, A.P.; Ghedin, E.; Huang, L.; et al. Comparison of the respiratory microbiome in healthy nonsmokers and smokers. Am. J. Respir. Crit. Care Med. 2013, 187, 1067–1075. [Google Scholar] [CrossRef]
- Becker, A.; Vella, G.; Galata, V.; Rentz, K.; Beisswenger, C.; Herr, C.; Walter, J.; Tierling, S.; Slevogt, H.; Keller, A.; et al. The composition of the pulmonary microbiota in sarcoidosis—An observational study. Respir. Res. 2019, 20, 46. [Google Scholar] [CrossRef]
- Molyneaux, P.L.; Cox, M.J.; Willis-Owen, S.A.; Mallia, P.; Russellet, K.E.; Russell, A.M.; Murphy, E.; Johnston, S.L.; Schwartz, D.A.; Wells, A.U.; et al. The role of bacteria in the pathogenesis and progression of idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2014, 190, 906–913. [Google Scholar] [CrossRef]
- Kwok, B.; Wu, B.G.; Kocak, I.F.; Sulaiman, I.; Schluger, R.; Li, Y.; Anwer, R.; Goparaju, C.; Ryan, D.J.; Sagatelian, M.; et al. Pleural fluid microbiota as a biomarker for malignancy and prognosis. Sci. Rep. 2023, 13, 2229. [Google Scholar] [CrossRef]
- Budden, K.F.; Shukla, S.D.; Rehman, S.F.; Bowerman, K.L.; Keely, S.; Hugenholtz, P.; Armstrong-James, D.P.H.; Adcock, I.M.; Chotirmall, S.H.; Chung, K.F.; et al. Functional effects of the microbiota in chronic respiratory disease. Lancet Respir. Med. 2019, 7, 907–920. [Google Scholar] [CrossRef] [PubMed]
- Dickson, R.P.; Erb-Downward, J.R.; Huffnagle, G.B. Towards an ecology of the lung: New conceptual models of pulmonary microbiology and pneumonia pathogenesis. Lancet Respir. Med. 2014, 2, 238–246. [Google Scholar] [CrossRef]
- Scher, J.U.; Joshua, V.; Artacho, A.; Abdollahi-Roodsaz, S.; Öckinger, J.; Kullberg, S.; Sköld, M.; Eklund, A.; Grunewald, J.; Clemente, J.C.; et al. The lung microbiota in early rheumatoid arthritis and autoimmunity. Microbiome 2016, 4, 60. [Google Scholar] [CrossRef] [PubMed]
- Wiscovitch-Russo, R.; Singh, H.; Oldfield, L.M.; Fedulov, A.V.; Gonzalez-Juarbe, N. An optimized approach for processing of frozen lung and lavage samples for microbiome studies. PLoS ONE 2022, 17, e0265891. [Google Scholar] [CrossRef]
- Oren, A.; Garrity, G.M. Valid publication of the names of forty-two phyla of prokaryotes. Int. J. Syst. Evol. Microbiol. 2021, 71, 005056. [Google Scholar] [CrossRef] [PubMed]
- Masuhiro, K.; Tamiya, M.; Fujimoto, K.; Koyama, S.; Naito, Y.; Osa, A.; Hirai, T.; Suzuki, H.; Okamoto, N.; Shiroyama, T.; et al. Bronchoalveolar lavage fluid reveals factors contributing to the efficacy of PD-1 blockade in lung cancer. JCI Insight 2022, 7, e157915. [Google Scholar] [CrossRef]
- Doocey, C.M.; Finn, K.; Murphy, C.; Guinane, C.M. The impact of the human microbiome in tumorigenesis, cancer progression, and biotherapeutic development. BMC Microbiol. 2022, 22, 53. [Google Scholar] [CrossRef]

{kind=link}
| Authors | Country | Inclusion Criteria | No * | What Was Compared | Sample | Method of Analysis | Alpha Diversity | Main Results |
|---|---|---|---|---|---|---|---|---|
| Bingula R. et al. (2020) [19] | France |
| 15 | Microbiota analysis in saliva, BAL (collected directly from the excised lobe), as well as in non-malignant, peritumoral, and tumoral tissues. |
| Illumina MiSeq technology (San Diego, CA, USA), performed 16S ribosomal rRNA targeted region V3–V4. | The Shannon diversity index and Faith’s phylogenetic diversity showed no significant differences in alpha diversity metrics across the four lung samples. | At phylum level: Firmicutes 45.7%; Bacteriodes 13.3%; Actinobacteria 11.9%; Proteobacteria 28%; Fusobacteria 0.23%; Cyanobacteria 0.16%; Acidobacteria 0.11%; Other 0.07% At genus level: Pseudomonas 10.3%; Blautia 5.9%; Streptococcus 5.1%; Capnocytophaga 4.8%; Acinetobacter 2.9%; Prevotella 2.3% Propionibacterium 2.3%; Lactobacillus 2.1%; Sphingomonas 1.8%; Bacteroides 1.5%; Veillonella 1.4%; other each <1%. |
| Wang K. et al. (2019) [20] | China |
| 47 | The variation in microbiota diversity between the oral cavity and bronchoalveolar lavage fluid (BALF) of lung cancer patients compared to healthy controls. |
| Illumina MiSeq technology, performed 16S ribosomal rRNA targeted region V4. QIAamp DNA Microbiome Kit. | Shannon and Simpson indexes. Lung cancer patients exhibited lower microbiota diversity in both the lungs and oral cavity compared to healthy controls. | At phylum level: Firmicutes 38.42%; Fusobacteria 5.12%; Spirochaetes 0.11%; Tenericutes 0.11%; Synergistetes 0.03%; |
| Jang, H.J. et al. (2021) [21] | South Korea | Pathologically diagnosed with non-small cell lung cancer (NSCLC). | 84 | Variations in the lung microbiomes of patients with lung cancer. |
| Illumina HiSeq technology, performed 16S ribosomal rRNA targeted region V3–V4. FastDNA® SPIN Kit for Soil CleanPCR kit. | Shannon and Simpson. The difference was not statistically significant (Shannon index: p = 0.307; Simpson index: p = 0.540). | At phylum level: PD-L1 > 10%: Bacteroidetes 39.4%; Firmicutes 30.5%; Proteobacteria 19.1%; Fusobacteria 6.4%; Acinetobacter 3.2%; PD-L1 < 10% Bacteroidetes 39.4%; Proteobacteria 28.2%; Firmicutes 23.2%; Fusobacteria 5.1%; Acinetobacter 2.8% At genus level: PD-L1 > 10%: Prevotella; Streptococcus; Veillonella; Haemophilus; Neisseria; Porphyromonas; Fusobacterium; Megasphaera; Leptotrichia; Rothia; Escheichia; PD-L1 < 10%: Prevotella; Neisseria; Haemophilus; Veillonella; Streptococcus; Porphyromonas; Fusobacterium; Megasphaera; Leptotrichia; Rothia; Pseudomonas. |
| Zhuo M. et al. (2020) [22] | China | Lung cancer—no one with cancer treatment. | 50 | Association of the microbiota with lung cancer. |
| Illumina MiSeq technology, performed 16S ribosomal rRNA targeted region V3–V4 PowerSoil DNA Isolation Kit. | Shannon diversity index and Simpson diversity index. There was no significant difference in alpha diversity between the cancerous and normal lung samples. | At phylum level: Affected lung: Proteobacteria: 34.2%; Firmicutes: 27.96%; Bacteroides: 21.46%; Actinobacteria: 5.79%; Fusobacteria: 5.39%; Cyanobacteria: 1.23%; Spirochaerae: 1.12%; TM7 (Saccharibacteria): 0.53%; Acidobacteria: 0.53%; Tenericutes: 0.5%; Others: 1.2% Normal lung: Proteobacteria: 32.95%; Bacteroides: 26.65%; Firmicutes: 26.46%; Fusobacteria: 5.02%; Actinobacteria: 4.39%; Spirochaerae: 0.97%; TM7 (Saccharibacteria): 0.65%; Cyanobacteria: 0.56%; Acidobacteria: 0.55%; Tenericutes: 0.32%; Others: 1.43%. At genus level: Affected lung: Streptococcus: 10.78%; Neisseria: 7.54%; Alloprevotella: 5.22%; Prevotella_7: 4.88%; Haemophilus: 4.8%; Veillonella: 4.25%; Fusobacterium: 4.14%; Prevotella: 3.93%; Ochrobactrum: 3.25%; Porphyromonas: 3.25%; Other: 47.95%. Normal lung: Streptococcus: 12.04%; Neisseria: 9.37%; Prevotella_7: 7.1%; Alloprevotella: 6.57%; Haemophilus: 5.65%; Prevotella: 5.28%; Porphyromonas: 4.78%; Veillonella: 4.53%; Fusobacterium: 3.96%; Stenotrophomonas: 3.86%; Other: 47.95%. |
| Gomes S. et al. (2019) [23] | Portugal | Subjects undergoing bronchoscopy for evaluation of lung disease at three hospitals in Portugal. | 49 | Microbiota in LC vs controller. |
| V3–V4, V4–V6 regions of the 16S rRNA gene DNA Mini kit (Qiagen). | Simpson and Shannon. SCC cases were in average more diverse than ADC. | At phylum level: Proteobacteria 38.7%; Firmicutes 25.4%; Actinobacteria 16.5%; Bacteroidetes 13.3%; Spirochaetes 2.2%; Fusobacteria 2.1%; TM7 0.7%; OD1 0.5%; SR1 0.3%; Tenericutes 0.2%; Synergistetes 0.1%; Others 0.0%. At genus level: Haemophilus 29.5%; Streptococcus 10.9%; Corynebacterium 8.2%; Actinomyces 7.4%; Prevotella 5.8%; Veillonella 5.0%; Neisseria 3.6%; Selenomonas 2.8%; Parvimonas 2.4%; Porphyromonas 2.4%; Aggregatibacter 2.1%; Treponema 2.1%; Fusobacterium 2.1%; Propionibacterium 2.0%; Bulleidia 1.9%; Peptostreptococcus 1.2%; Pseudomonas 1.1%; Granulicatella 0.9%; Oribacterium 0.9%; Actinobacillus 0.8%; Bifidobacterium 0.6%; Campylobacter 0.5%; Sphingobacterium 0.5%; Staphylococcus 0.5%; Sphaerochaeta 0.5%; Filifactor 0.4%; Leptotrichia 0.4%; Scardovia 0.3%; Stenotrophomonas 0.3%; Moraxella 0.3%; Capnocytophaga 0.3%; Rothia 0.2%; Lactobacillus 0.2%; Megasphaera 0.2%; Morganella 0.2%; Acholeplasma 0.2%; Flavobacterium 0.1%; Catonella 0.1%; Aerococcus 0.1%; Cupriavidus 0.1%; TG5 0.1%; Sphingomonas 0.1%; Phenylobacterium 0.1%; Pedobacter 0.1%; Dialister 0.1%; Others 0.1%. |
| Seixas S. et al. (2021) [24] | Portugal |
| 49 | LC vs other lung disease. |
| Illumina MiSeq technology, performed 16S ribosomal rRNA targeted region V4 DNA Mini kit (Qiagen). | The Shannon, ACE, Simpson, Fisher, and Phylogenetic (Faith’s) diversity indices showed no significant variation in alpha diversity between the LC and non-LC groups. | At phylum level: Firmicutes 47.11%; Proteobacteria 31.35%; Bacteroidetes 15.52%; Actinobacteria 2.80%; At genus level: Escherichia/Shigella 8.80%; Bacillus 7.66%; Streptococcus 7.45%; Salmonella 7.40%; Staphylococcus 7.27%; Lactobacillus 6.41%; Prevotella 6.09%; Veillonella 6.00%; Pseudomonas 3.56%; Haemophilus 3.21%; Others (each <1%). |
| Lee S.H. et al. (2016) [25] | South Korea |
| 20 | The microbiomes of patients with lung cancer were characterized and compared to those with benign mass-like lesions. |
| Illumina HiSeq technology, performed 16S ribosomal rRNA targeted region V1–V3. | Chao1 estimation and Shannon more complex diversity with higher abundance and α-diversity. | At phylum level: Bacteroidetes: 39.5%; Firmicutes: 29.7%; Proteobacteria: 22.8%; Fusobacteria: 4.5%; Actinobacteria: 2.1%; Spirochaetes: 0.4%; TM7: 0.5%; SR1: 0.3%; Tenericutes: 0.1%. At genus level: Prevotella: 30.8%; Neisseria: 13.8%; Veillonella: 11.4%; Streptococcus: 10.9%; Haemophilus: 7.2%; Alloprevotella: 6.1%; Fusobacterium: 2.2%. Megasphaera: 2.2%; Porphyromonas: 2.0%; Leptotrichia: 1.8%; Campylobacter: 1.1%; Actinomyces: 0.8%. |
| Liu B. et al. (2022) [26] | China |
| 7 | Explore the characteristics of lung microbiota and metabolites in patients, and identify potential biomarkers for lung cancer diagnosis. |
| Illumina MiSeq technology, performed 16S ribosomal rRNA targeted region V3-V4 FastDNA Spin Kit (MP Biomedicals, Shanghai, China). | Shannon, Chao, ace Lower abundance in alpha diversity. | At phylum level: Proteobacteria 45.05%; Firmicutes 28.31%; Bacteroidota 14.89%; Actinobacteriota 7.15%; Fusobacteriota 2.41%; Patescibacteria 1.25%; others 0.94%. At genus level: Pseudomonas 35.14%; Streptococcus 14.34%; Prevotella 9.55%; Neisseria 6.81%; Veillonella 4.85%; Actinomyces 4.6%; Granulicatella 3.53%; Alloprevotella 3.25%; Leptotrichia 1.27 %; Fusobacterium 1.13%; Porphyromonas 1.12%; Haemophilus 1.07%; Rhodococcus 0.91%; Klebsiella 0.05%; Lactobacillus 0.12%; Bacillus 0.11%; others 12.15%. |
| Jang, H.J. et al. (2023) [27] | South Korea | Patients who were pathologically diagnosed with NSCLC. | 84 | Differences in lung microbiomes among lung cancer patients based on histological type. |
| Illumina MiSeq technology, performed 16S ribosomal rRNA targeted region V3–V4. | Shannon and Simpson α -diversity was different between the two types of lung cancer. | At phylum level: ADK Bacteroidetes 40.8%; Proteobacteria 24.9%; Firmicutes 24.1%; Fusobacteria 6.0%; Actinobacteria 2.8% SCC Bacteroidetes 35.0%; Firmicutes 29.3%; Proteobacteria 27.8%; Fusobacteria 3.8%; Actinobacteria 3.3%. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lucaciu, S.-R.; Domokos, B.; Puiu, R.; Ruta, V.; Motoc, S.N.; Rajnoveanu, R.; Todea, D.; Stoia, A.M.; Man, A.M. Lung Microbiome in Lung Cancer: A Systematic Review. Microorganisms 2024, 12, 2439. https://doi.org/10.3390/microorganisms12122439
Lucaciu S-R, Domokos B, Puiu R, Ruta V, Motoc SN, Rajnoveanu R, Todea D, Stoia AM, Man AM. Lung Microbiome in Lung Cancer: A Systematic Review. Microorganisms. 2024; 12(12):2439. https://doi.org/10.3390/microorganisms12122439
Chicago/Turabian StyleLucaciu, Sergiu-Remus, Bianca Domokos, Ruxandra Puiu, Victoria Ruta, Stefania Nicoleta Motoc, Ruxandra Rajnoveanu, Doina Todea, Anca Mirela Stoia, and Adina Milena Man. 2024. "Lung Microbiome in Lung Cancer: A Systematic Review" Microorganisms 12, no. 12: 2439. https://doi.org/10.3390/microorganisms12122439
APA StyleLucaciu, S.-R., Domokos, B., Puiu, R., Ruta, V., Motoc, S. N., Rajnoveanu, R., Todea, D., Stoia, A. M., & Man, A. M. (2024). Lung Microbiome in Lung Cancer: A Systematic Review. Microorganisms, 12(12), 2439. https://doi.org/10.3390/microorganisms12122439