

Disruption of P2Y2 Signaling Promotes Breast Tumor Cell Dissemination by Reducing ATP-Dependent Calcium Elevation and Actin Localization to Cell Junctions

 , , , , , ,
, , , , , ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
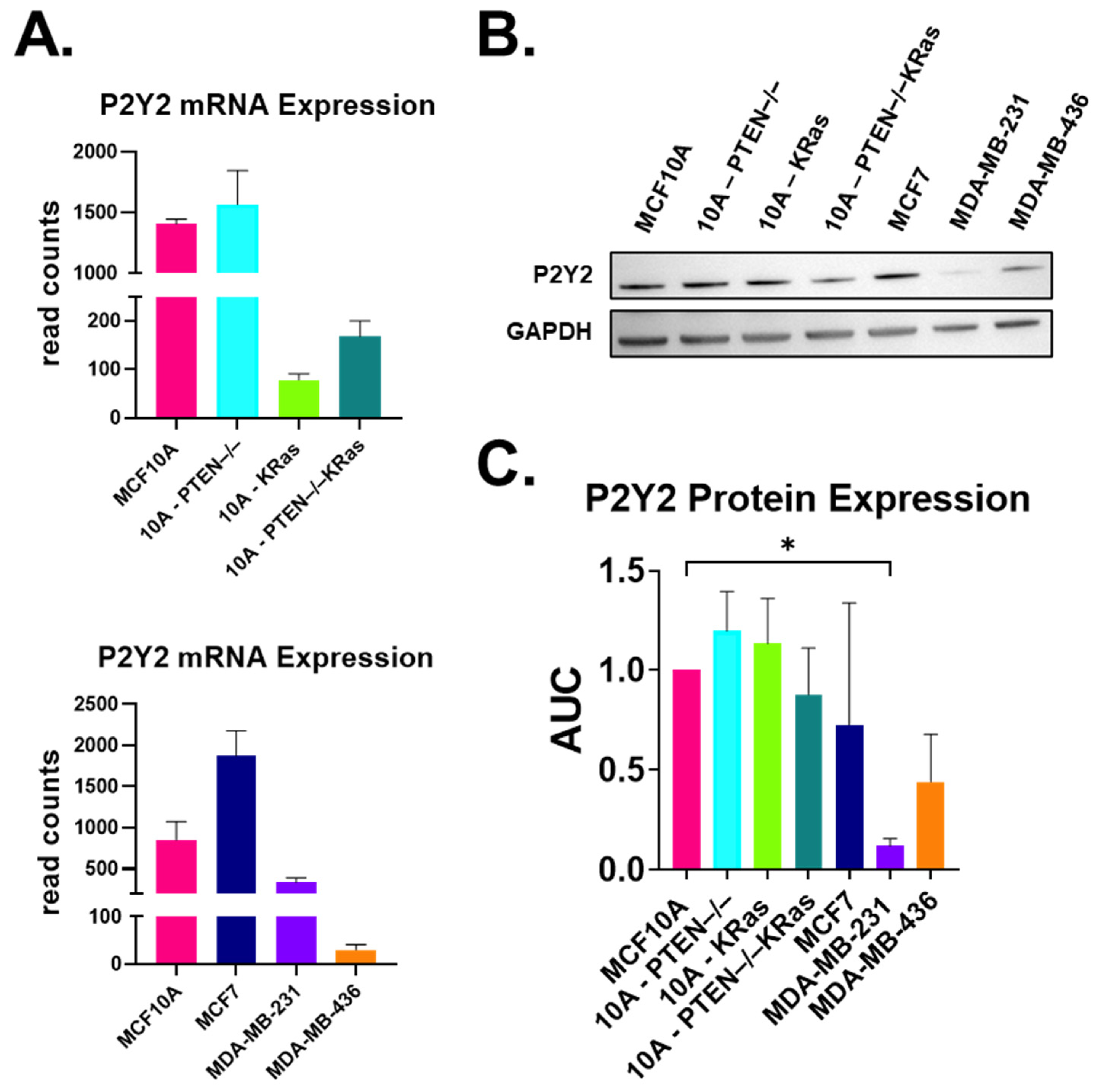
2.1. P2Y2 Receptor Expression Is Lower at the mRNA and Protein Levels in Metastatic MDA-MB-231 Cells Compared to Non-Tumorigenic MCF10As
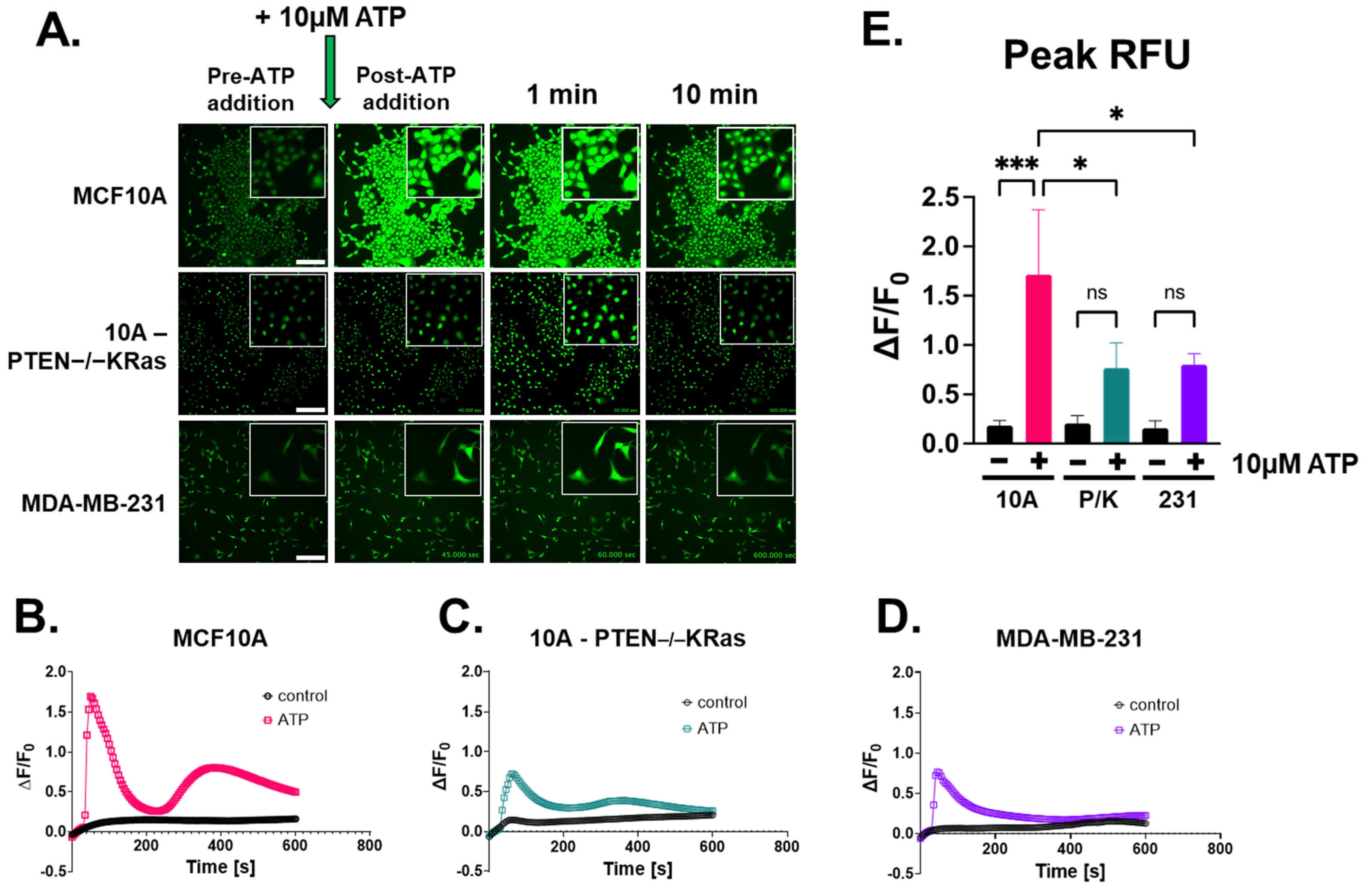
2.2. Extracellular ATP Stimulates Calcium Signaling Across Breast Epithelial and Cancer Cell Lines
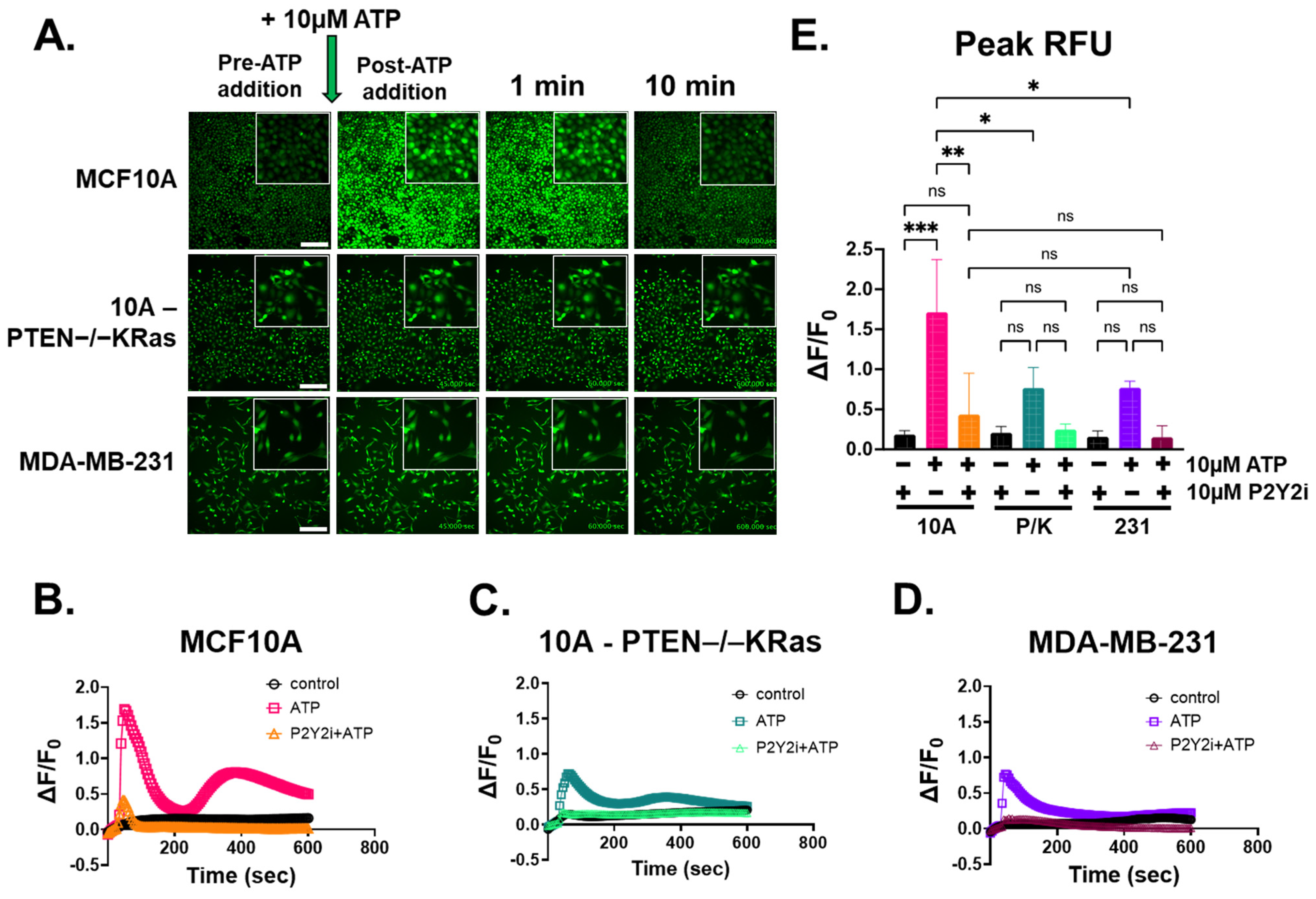
2.3. ATP-Induced Intracellular Ca2+ Response Is Inhibited by P2Y2 Antagonist in Breast Epithelial Cells
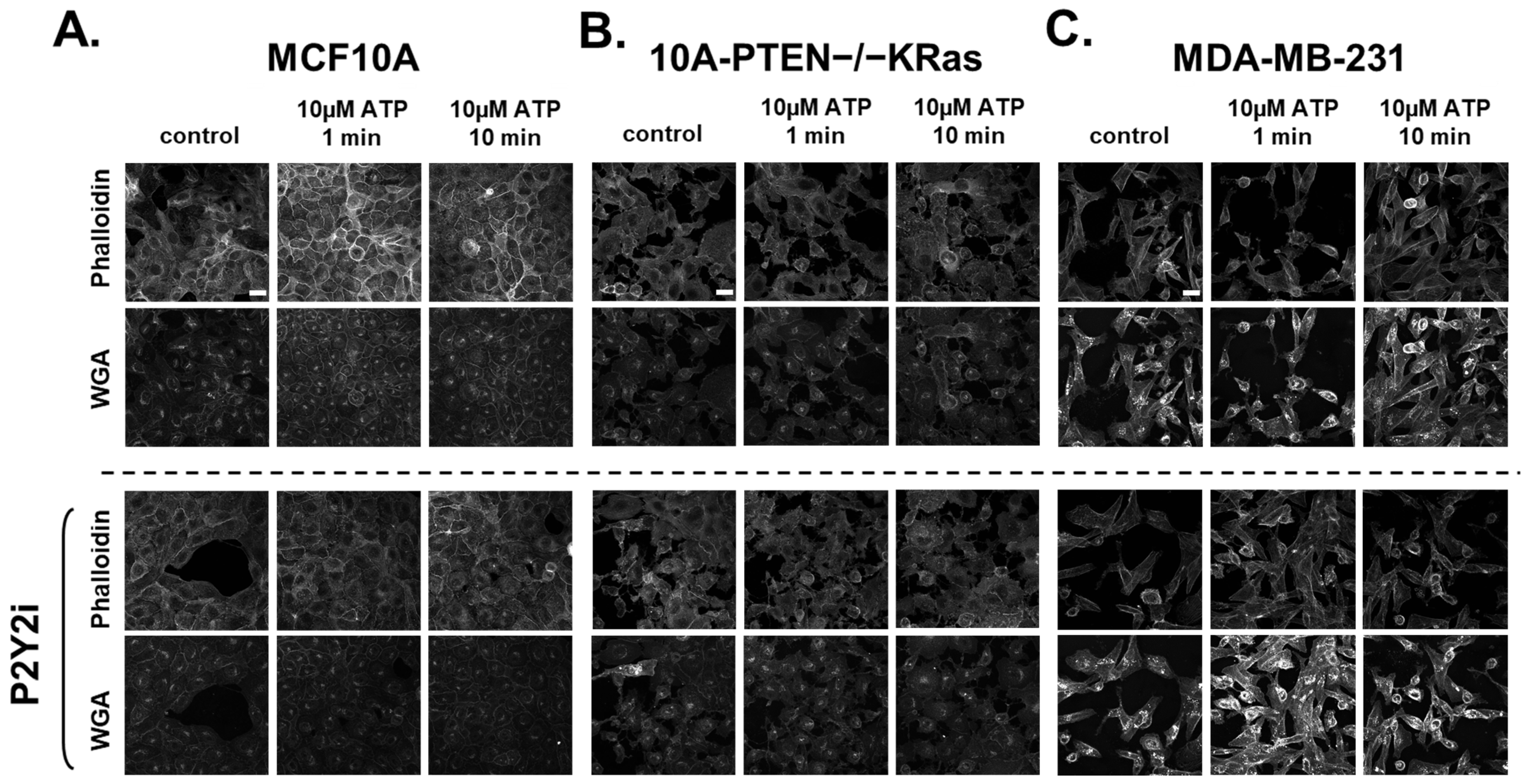
2.4. Rapid Changes in the Actin Cortex Occur After ATP-Induced Ca2+ Increases in Breast Epithelial Cells
2.5. P2Y2i Treatment Increases Cell Dissemination and Spheroid Size in Non-Tumorigenic Breast Epithelial Cells
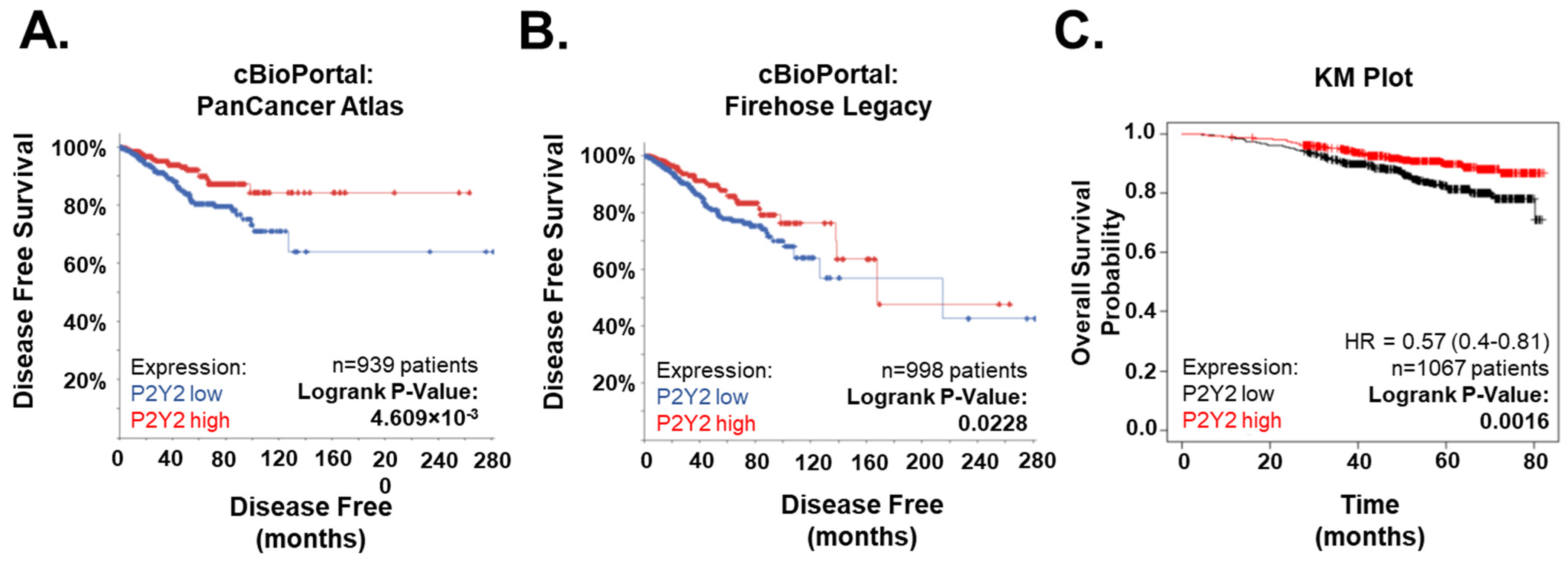
2.6. Low P2Y2 mRNA Expression Is Correlated with Decreased Survival and Disease-Free Progression in Breast Cancer Patients
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Culture
4.3. Live Imaging
4.4. Quantitative Plate Reader
4.5. RNA Seq
4.6. Western Blotting
4.7. Immunofluorescence
4.8. 3-Dimensional Cell Dissemination Assay
4.9. Bioinformatics
4.10. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ATP | Adenosine triphosphate |
| ADP | Adenosine diphosphate |
| UTP | Uridine triphosphate |
| P2Y2 | Purinergic receptor P2Y2 |
| TME | Tumor microenvironment |
| EGF | Epidermal growth factor |
| EMT | Epithelial to mesenchymal transition |
| TNBC | Triple negative breast cancer |
| Ca2+ | Calcium |
| CaAR | Calcium-mediated actin reset |
| HBSS | Hanks’ Balanced Salt Solution |
| DMSO | Dimethyl sulfoxide |
| ddH2O | Double-distilled water |
| P2Y2i | Antagonist for P2Y2, AR-C 118925XX |
| WGA | Wheat germ agglutinin |
References
- Schäfer, M.; Werner, S. Cancer as an overhealing wound: An old hypothesis revisited. Nat. Rev. Mol. Cell Biol. 2008, 9, 628–638. [Google Scholar] [CrossRef] [PubMed]
- Foster, D.S.; Jones, R.E.; Ransom, R.C.; Longaker, M.T.; Norton, J.A. The evolving relationship of wound healing and tumor stroma. JCI Insight 2018, 3, e99911. [Google Scholar] [CrossRef] [PubMed]
- Cordeiro, J.V.; Jacinto, A. The role of transcription-independent damage signals in the initiation of epithelial wound healing. Nat. Rev. Mol. Cell Biol. 2013, 14, 249–262. [Google Scholar] [CrossRef] [PubMed]
- Arwert, E.N.; Hoste, E.; Watt, F.M. Epithelial stem cells, wound healing and cancer. Nat. Rev. Cancer 2012, 12, 170–180. [Google Scholar] [CrossRef]
- Balkwill, F.R.; Capasso, M.; Hagemann, T. The tumor microenvironment at a glance. J. Cell Sci. 2012, 125, 5591–5596. [Google Scholar] [CrossRef]
- Hanahan, D.; Coussens, L.M. Accessories to the Crime: Functions of Cells Recruited to the Tumor Microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef]
- Di Virgilio, F.; Sarti, A.C.; Falzoni, S.; De Marchi, E.; Adinolfi, E. Extracellular ATP and P2 purinergic signalling in the tumour microenvironment. Nat. Rev. Cancer 2018, 18, 601–618. [Google Scholar] [CrossRef]
- Pellegatti, P.; Raffaghello, L.; Bianchi, G.; Piccardi, F.; Pistoia, V.; Di Virgilio, F. Increased Level of Extracellular ATP at Tumor Sites: In Vivo Imaging with Plasma Membrane Luciferase. PLoS ONE 2008, 3, e2599. [Google Scholar] [CrossRef]
- Yang, H.; Geng, Y.; Wang, P.; Zhou, Y.; Yang, H.; Huo, Y.; Zhang, H.; Li, Y.; He, H.; Tian, X.; et al. Extracellular ATP promotes breast cancer invasion and epithelial-mesenchymal transition via hypoxia-inducible factor 2α signaling. Cancer Sci. 2019, 110, 2456–2470. [Google Scholar] [CrossRef]
- Vultaggio-Poma, V.; Sarti, A.C.; Di Virgilio, F. Extracellular ATP: A Feasible Target for Cancer Therapy. Cells 2020, 9, 2496. [Google Scholar] [CrossRef]
- Burnstock, G.; Williams, M. P2 Purinergic Receptors: Modulation of Cell Function and Therapeutic Potential. J. Pharmacol. Exp. Ther. 2000, 295, 862–869. [Google Scholar] [CrossRef] [PubMed]
- Li, W.-H.; Qiu, Y.; Zhang, H.-Q.; Liu, Y.; You, J.-F.; Tian, X.-X.; Fang, W.-G. P2Y2 receptor promotes cell invasion and metastasis in prostate cancer cells. Br. J. Cancer 2013, 109, 1666–1675. [Google Scholar] [CrossRef] [PubMed]
- Li, W.-H.; Qiu, Y.; Zhang, H.-Q.; Tian, X.-X.; Fang, W.-G. P2Y2 Receptor and EGFR Cooperate to Promote Prostate Cancer Cell Invasion via ERK1/2 Pathway. PLoS ONE 2015, 10, e0133165. [Google Scholar] [CrossRef] [PubMed]
- Schulien, I.; Hockenjos, B.; van Marck, V.; Ayata, C.K.; Follo, M.; Thimme, R.; Hasselblatt, P. Extracellular ATP and Purinergic P2Y2 Receptor Signaling Promote Liver Tumorigenesis in Mice by Exacerbating DNA Damage. Cancer Res. 2020, 80, 699–708. [Google Scholar] [CrossRef]
- Gendron, F.-P.; Placet, M.; Arguin, G. P2Y2 Receptor Functions in Cancer: A Perspective in the Context of Colorectal Cancer. In Advances in Experimental Medicine and Biology; Atassi, M.Z., Ed.; In Protein Reviews; Springer: Singapore, 2017; Volume 19, pp. 91–106. [Google Scholar] [CrossRef]
- Hu, L.-P.; Zhang, X.-X.; Jiang, S.-H.; Tao, L.-Y.; Li, Q.; Zhu, L.-L.; Yang, M.-W.; Huo, Y.-M.; Jiang, Y.-S.; Tian, G.-A.; et al. Targeting Purinergic Receptor P2Y2 Prevents the Growth of Pancreatic Ductal Adenocarcinoma by Inhibiting Cancer Cell Glycolysis. Clin. Cancer Res. 2019, 25, 1318–1330. [Google Scholar] [CrossRef]
- Yang, G.; Zhang, S.; Zhang, Y.; Zhou, Q.; Peng, S.; Zhang, T.; Yang, C.; Zhu, Z.; Zhang, F. The inhibitory effects of extracellular ATP on the growth of nasopharyngeal carcinoma cells via P2Y2 receptor and osteopontin. J. Exp. Clin. Cancer Res. 2014, 33, 53. [Google Scholar] [CrossRef]
- Davis, F.M.; Kenny, P.A.; Soo, E.T.-L.; van Denderen, B.J.W.; Thompson, E.W.; Cabot, P.J.; Parat, M.-O.; Roberts-Thomson, S.J.; Monteith, G.R. Remodeling of Purinergic Receptor-Mediated Ca2+ Signaling as a Consequence of EGF-Induced Epithelial-Mesenchymal Transition in Breast Cancer Cells. PLoS ONE 2011, 6, e23464. [Google Scholar] [CrossRef]
- Azimi, I.; Beilby, H.; Davis, F.M.; Marcial, D.L.; Kenny, P.A.; Thompson, E.W.; Roberts-Thomson, S.J.; Monteith, G.R. Altered purinergic receptor-Ca2+ signaling associated with hypoxia-induced epithelial-mesenchymal transition in breast cancer cells. Mol. Oncol. 2016, 10, 166–178. [Google Scholar] [CrossRef]
- Qiu, Y.; Liu, Y.; Li, W.-H.; Zhang, H.-Q.; Tian, X.-X.; Fang, W.-G. P2Y2 receptor promotes the migration and invasion of breast cancer cells via EMT-related genes Snail and E-cadherin. Oncol. Rep. 2018, 39, 138–150. [Google Scholar] [CrossRef]
- Boucher, I.; Rich, C.; Lee, A.; Marcincin, M.; Trinkaus-Randall, V. The P2Y2 receptor mediates the epithelial injury response and cell migration. Am. J. Physiol. Cell Physiol. 2010, 299, C411–C421. [Google Scholar] [CrossRef]
- de Araújo, J.B.; Kerkhoff, V.V.; de Oliveira Maciel, S.F.V.; de Resende e Silva, D.T. Targeting the purinergic pathway in breast cancer and its therapeutic applications. Purinergic Signal. 2021, 17, 179–200. [Google Scholar] [CrossRef] [PubMed]
- Waks, A.G.; Winer, E.P. Breast Cancer Treatment: A Review. JAMA 2019, 321, 288–300. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA. Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Prior, I.A.; Lewis, P.D.; Mattos, C. A Comprehensive Survey of Ras Mutations in Cancer. Cancer Res. 2012, 72, 2457–2467. [Google Scholar] [CrossRef]
- Hwang, K.-T.; Kim, B.H.; Oh, S.; Park, S.Y.; Jung, J.; Kim, J.; Choi, I.S.; Jeon, S.Y.; Kim, W.-Y. Prognostic Role of KRAS mRNA Expression in Breast Cancer. J. Breast Cancer 2019, 22, 548–561. [Google Scholar] [CrossRef]
- Tokumaru, Y.; Oshi, M.; Katsuta, E.; Yan, L.; Satyananda, V.; Matsuhashi, N.; Futamura, M.; Akao, Y.; Yoshida, K.; Takabe, K. KRAS signaling enriched triple negative breast cancer is associated with favorable tumor immune microenvironment and better survival. Am. J. Cancer Res. 2020, 10, 897–907. [Google Scholar]
- Thompson, K.N.; Whipple, R.A.; Yoon, J.R.; Lipsky, M.; Charpentier, M.S.; Boggs, A.E.; Chakrabarti, K.R.; Bhandary, L.; Hessler, L.K.; Martin, S.S.; et al. The combinatorial activation of the PI3K and Ras/MAPK pathways is sufficient for aggressive tumor formation, while individual pathway activation supports cell persistence. Oncotarget 2015, 6, 35231–35246. [Google Scholar] [CrossRef]
- Chadet, S.; Jelassi, B.; Wannous, R.; Angoulvant, D.; Chevalier, S.; Besson, P.; Roger, S. The activation of P2Y2 receptors increases MCF-7 breast cancer cells migration through the MEK-ERK1/2 signalling pathway. Carcinogenesis 2014, 35, 1238–1247. [Google Scholar] [CrossRef]
- Pratt, S.J.; Hernández-Ochoa, E.O.; Lee, R.M.; Ory, E.C.; Lyons, J.S.; Joca, H.C.; Johnson, A.; Thompson, K.; Bailey, P.; Lee, C.J.; et al. Real-time scratch assay reveals mechanisms of early calcium signaling in breast cancer cells in response to wounding. Oncotarget 2018, 9, 25008–25024. [Google Scholar] [CrossRef]
- Wales, P.; E Schuberth, C.; Aufschnaiter, R.; Fels, J.; García-Aguilar, I.; Janning, A.; Dlugos, C.P.; Schäfer-Herte, M.; Klingner, C.; Wälte, M.; et al. Calcium-mediated actin reset (CaAR) mediates acute cell adaptations. eLife 2016, 5, e19850. [Google Scholar] [CrossRef]
- Dushek, O.; Mueller, S.; Soubies, S.; Depoil, D.; Caramalho, I.; Coombs, D.; Valitutti, S. Effects of Intracellular Calcium and Actin Cytoskeleton on TCR Mobility Measured by Fluorescence Recovery. PLoS ONE 2008, 3, e3913. [Google Scholar] [CrossRef] [PubMed]
- Lehne, F.; Pokrant, T.; Parbin, S.; Salinas, G.; Großhans, J.; Rust, K.; Faix, J.; Bogdan, S. Calcium bursts allow rapid reorganization of EFhD2/Swip-1 cross-linked actin networks in epithelial wound closure. Nat. Commun. 2022, 13, 2492. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.E.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef]
- Lánczky, A.; Győrffy, B. Web-Based Survival Analysis Tool Tailored for Medical Research (KMplot): Development and Implementation. J. Med. Internet Res. 2021, 23, e27633. [Google Scholar] [CrossRef]
- Gilbert, S.; Oliphant, C.; Hassan, S.; Peille, A.; Bronsert, P.; Falzoni, S.; Di Virgilio, F.; McNulty, S.; Lara, R. ATP in the tumour microenvironment drives expression of nfP2X7, a key mediator of cancer cell survival. Oncogene 2019, 38, 194–208. [Google Scholar] [CrossRef]
- McEwan, T.B.-D.; Sophocleous, R.A.; Cuthbertson, P.; Mansfield, K.J.; Sanderson-Smith, M.L.; Sluyter, R. Autocrine regulation of wound healing by ATP release and P2Y2 receptor activation. Life Sci. 2021, 283, 119850. [Google Scholar] [CrossRef]
- Schumacher, D.; Strilic, B.; Sivaraj, K.K.; Wettschureck, N.; Offermanns, S. Platelet-Derived Nucleotides Promote Tumor-Cell Transendothelial Migration and Metastasis via P2Y2 Receptor. Cancer Cell 2013, 24, 130–137. [Google Scholar] [CrossRef]
- Eun, S.Y.; Ko, Y.S.; Park, S.W.; Chang, K.C.; Kim, H.J. P2Y2 nucleotide receptor-mediated extracellular signal-regulated kinases and protein kinase C activation induces the invasion of highly metastatic breast cancer cells. Oncol. Rep. 2015, 34, 195–202. [Google Scholar] [CrossRef]
- Jin, H.; Eun, S.Y.; Lee, J.S.; Park, S.W.; Lee, J.H.; Chang, K.C.; Kim, H.J. P2Y2receptor activation by nucleotides released from highly metastatic breast cancer cells increases tumor growth and invasion via crosstalk with endothelial cells. Breast Cancer Res. 2014, 16, R77. [Google Scholar] [CrossRef]
- Zhang, J.-L.; Liu, Y.; Yang, H.; Zhang, H.-Q.; Tian, X.-X.; Fang, W.-G. ATP-P2Y2-β-catenin axis promotes cell invasion in breast cancer cells. Cancer Sci. 2017, 108, 1318–1327. [Google Scholar] [CrossRef]
- Stewart, T.A.; Yapa, K.T.D.S.; Monteith, G.R. Altered calcium signaling in cancer cells. Biochim. Biophys. Acta BBA Biomembr. 2015, 1848, 2502–2511. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.S. Myosins in Osteoclast Formation and Function. Biomolecules 2018, 8, 157. [Google Scholar] [CrossRef] [PubMed]
- Rüegg, J.C. Cardiac contractility: How calcium activates the myofilaments. Naturwissenschaften 1998, 85, 575–582. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; King, N.C.; Sinoway, L.I. ATP concentrations and muscle tension increase linearly with muscle contraction. J. Appl. Physiol. 2003, 95, 577–583. [Google Scholar] [CrossRef]
- Ahangar, P.; Strudwick, X.L.; Cowin, A.J. Wound Healing from an Actin Cytoskeletal Perspective. Cold Spring Harb. Perspect. Biol. 2022, 14, a041235. [Google Scholar] [CrossRef]
- Kindon, N.; Davis, A.; Dougall, I.; Dixon, J.; Johnson, T.; Walters, I.; Thom, S.; McKechnie, K.; Meghani, P.; Stocks, M.J. From UTP to AR-C118925, the discovery of a potent non nucleotide antagonist of the P2Y2 receptor. Bioorg. Med. Chem. Lett. 2017, 27, 4849–4853. [Google Scholar] [CrossRef]
- Amano, S.; Inoue, K. Effect of topical 3% diquafosol sodium on eyes with dry eye disease and meibomian gland dysfunction. Clin. Ophthalmol. 2017, 11, 1677–1682. [Google Scholar] [CrossRef]
- Wiedemar, N.; Hauser, D.A.; Mäser, P. 100 Years of Suramin. Antimicrob. Agents Chemother. 2020, 64, e01168–19. [Google Scholar] [CrossRef]
- Lustberg, M.B.; Pant, S.; Ruppert, A.S.; Shen, T.; Wei, Y.; Chen, L.; Brenner, L.; Shiels, D.; Jensen, R.R.; Berger, M.; et al. Phase I/II trial of non-cytotoxic suramin in combination with weekly paclitaxel in metastatic breast cancer treated with prior taxanes. Cancer Chemother. Pharmacol. 2012, 70, 49–56. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]
- Doyle, A.D. Generation of 3D collagen gels with controlled, diverse architectures. Curr. Protoc. Cell Biol. Editor. Board. Juan Bonifacino Al 2016, 72, 10.20.1–10.20.16. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mull, M.L.; Pratt, S.J.P.; Thompson, K.N.; Annis, D.A.; Lee, R.M.; Ju, J.A.; Gilchrist, D.E.; Stemberger, M.B.; Boyman, L.; Lederer, W.J.; et al. Disruption of P2Y2 Signaling Promotes Breast Tumor Cell Dissemination by Reducing ATP-Dependent Calcium Elevation and Actin Localization to Cell Junctions. Int. J. Mol. Sci. 2025, 26, 4286. https://doi.org/10.3390/ijms26094286
Mull ML, Pratt SJP, Thompson KN, Annis DA, Lee RM, Ju JA, Gilchrist DE, Stemberger MB, Boyman L, Lederer WJ, et al. Disruption of P2Y2 Signaling Promotes Breast Tumor Cell Dissemination by Reducing ATP-Dependent Calcium Elevation and Actin Localization to Cell Junctions. International Journal of Molecular Sciences. 2025; 26(9):4286. https://doi.org/10.3390/ijms26094286
Chicago/Turabian StyleMull, Makenzy L., Stephen J. P. Pratt, Keyata N. Thompson, David A. Annis, Rachel M. Lee, Julia A. Ju, Darin E. Gilchrist, Megan B. Stemberger, Liron Boyman, William J. Lederer, and et al. 2025. "Disruption of P2Y2 Signaling Promotes Breast Tumor Cell Dissemination by Reducing ATP-Dependent Calcium Elevation and Actin Localization to Cell Junctions" International Journal of Molecular Sciences 26, no. 9: 4286. https://doi.org/10.3390/ijms26094286
APA StyleMull, M. L., Pratt, S. J. P., Thompson, K. N., Annis, D. A., Lee, R. M., Ju, J. A., Gilchrist, D. E., Stemberger, M. B., Boyman, L., Lederer, W. J., Vitolo, M. I., & Martin, S. S. (2025). Disruption of P2Y2 Signaling Promotes Breast Tumor Cell Dissemination by Reducing ATP-Dependent Calcium Elevation and Actin Localization to Cell Junctions. International Journal of Molecular Sciences, 26(9), 4286. https://doi.org/10.3390/ijms26094286