
Identification of miRNAs and Their Targets in Cunninghamia lanceolata Under Low Phosphorus Stress Based on Small RNA and Degradome Sequencing


Abstract
:1. Introduction
2. Results
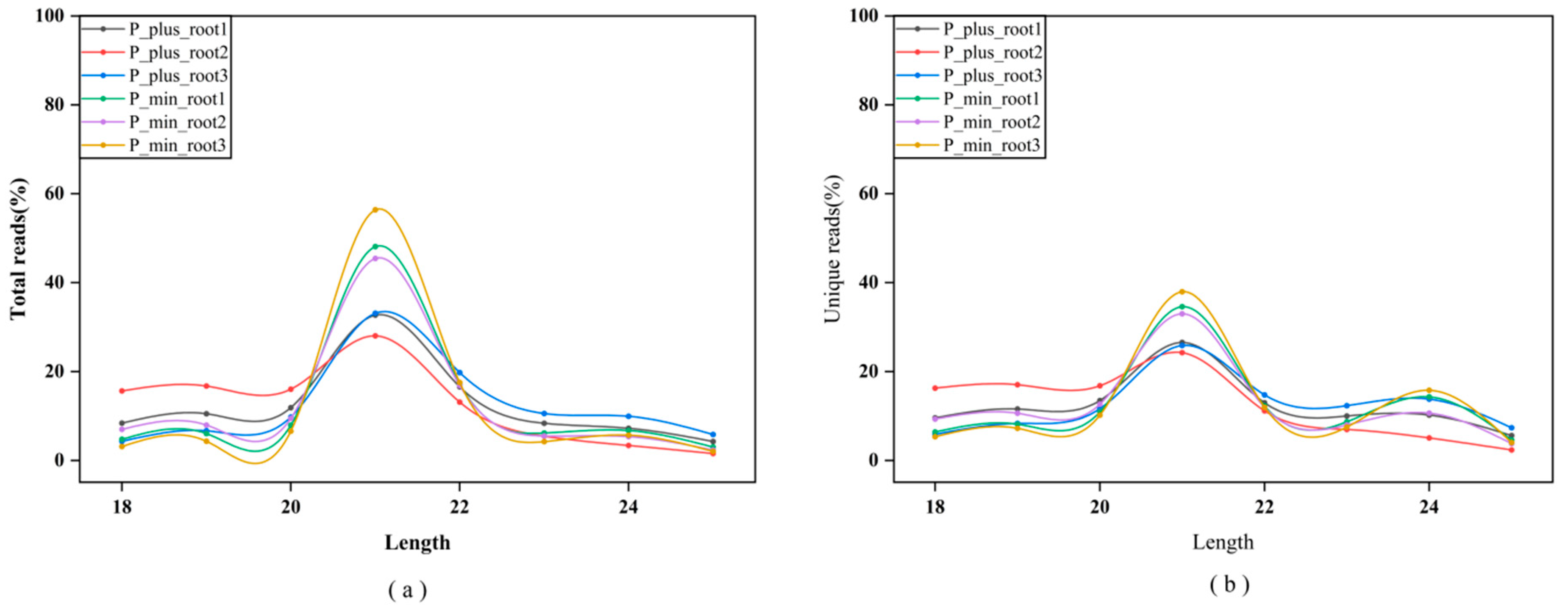
2.1. Small RNA Sequencing and Analysis
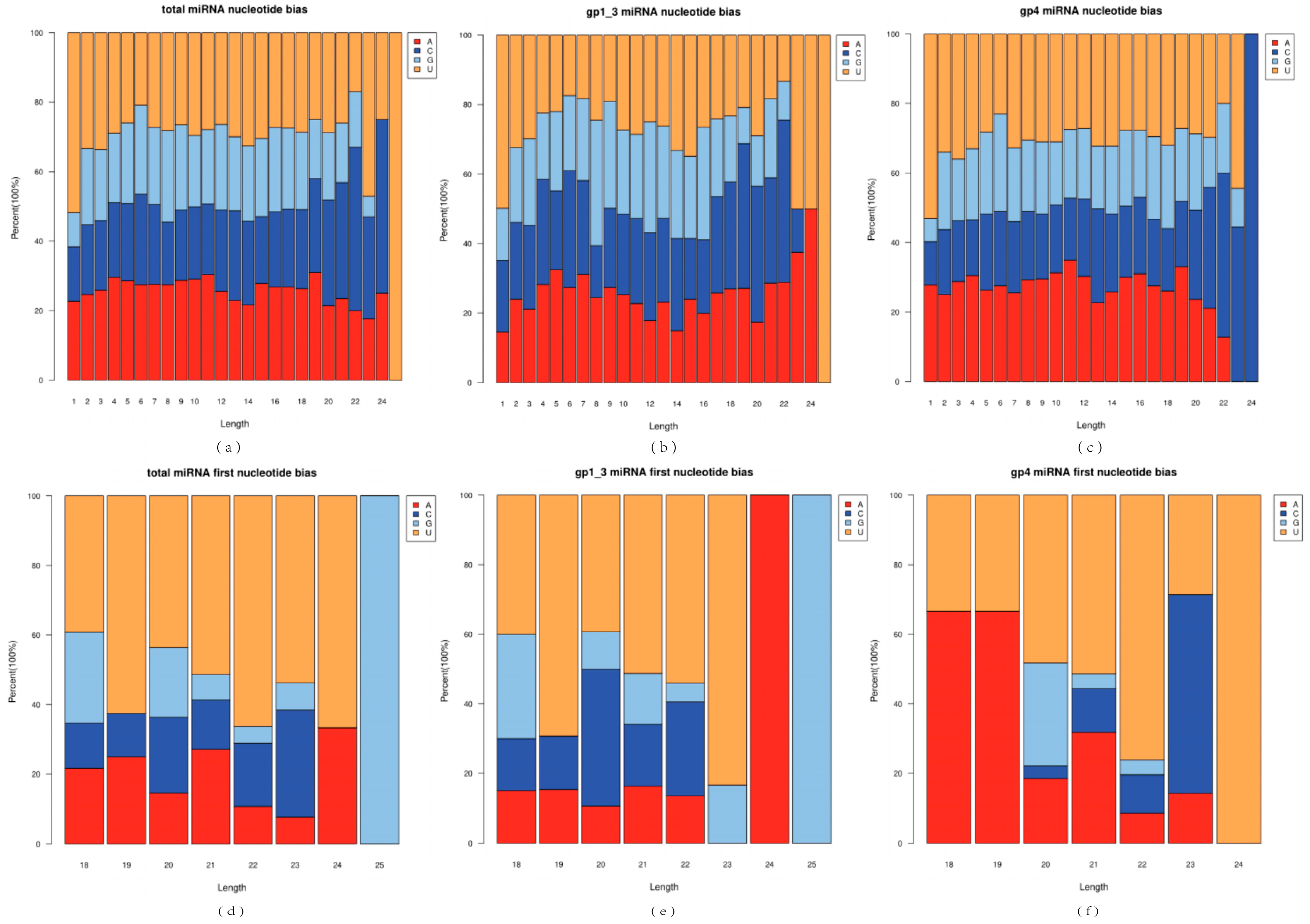
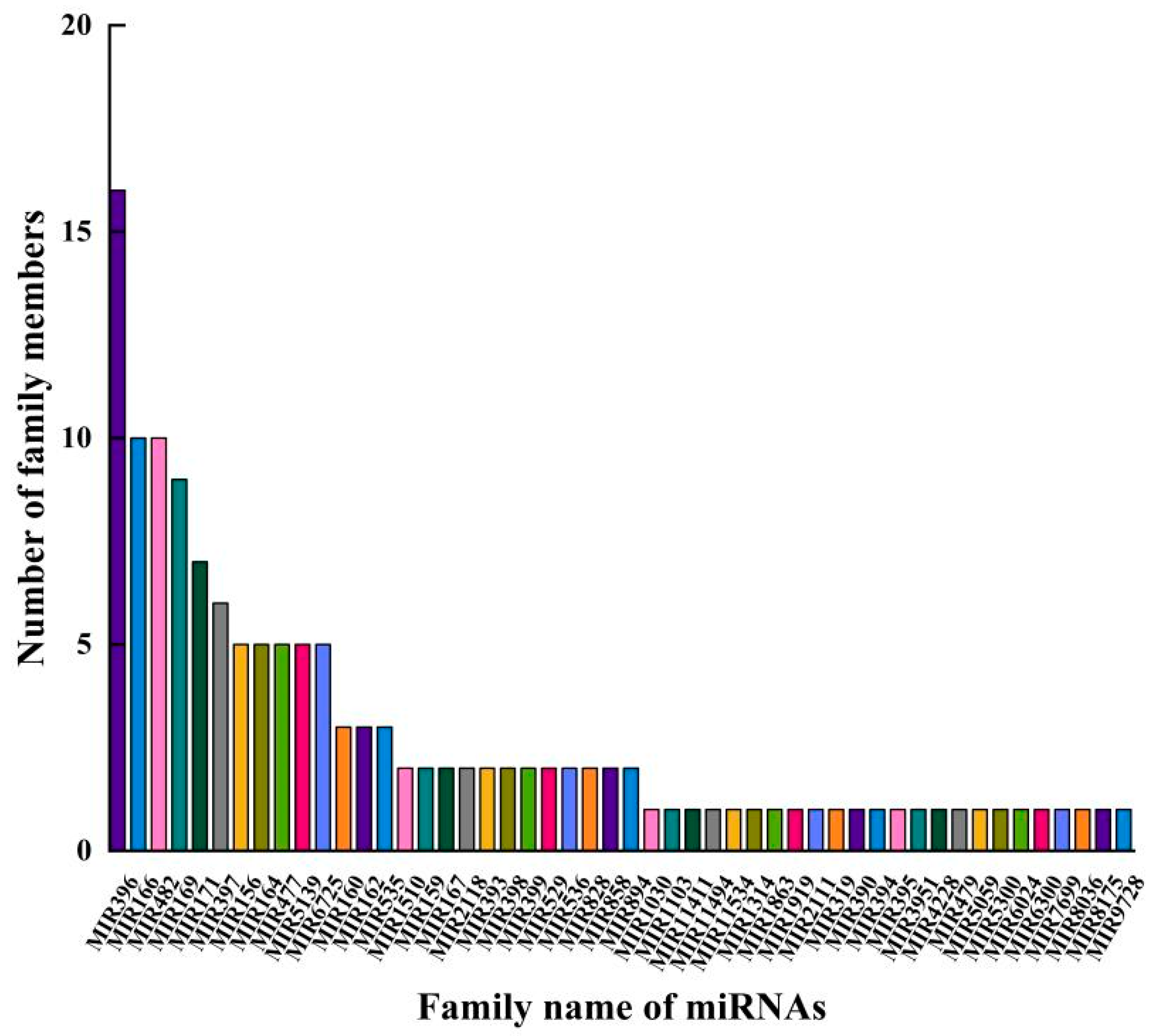
2.2. Identification of Known and Novel miRNA
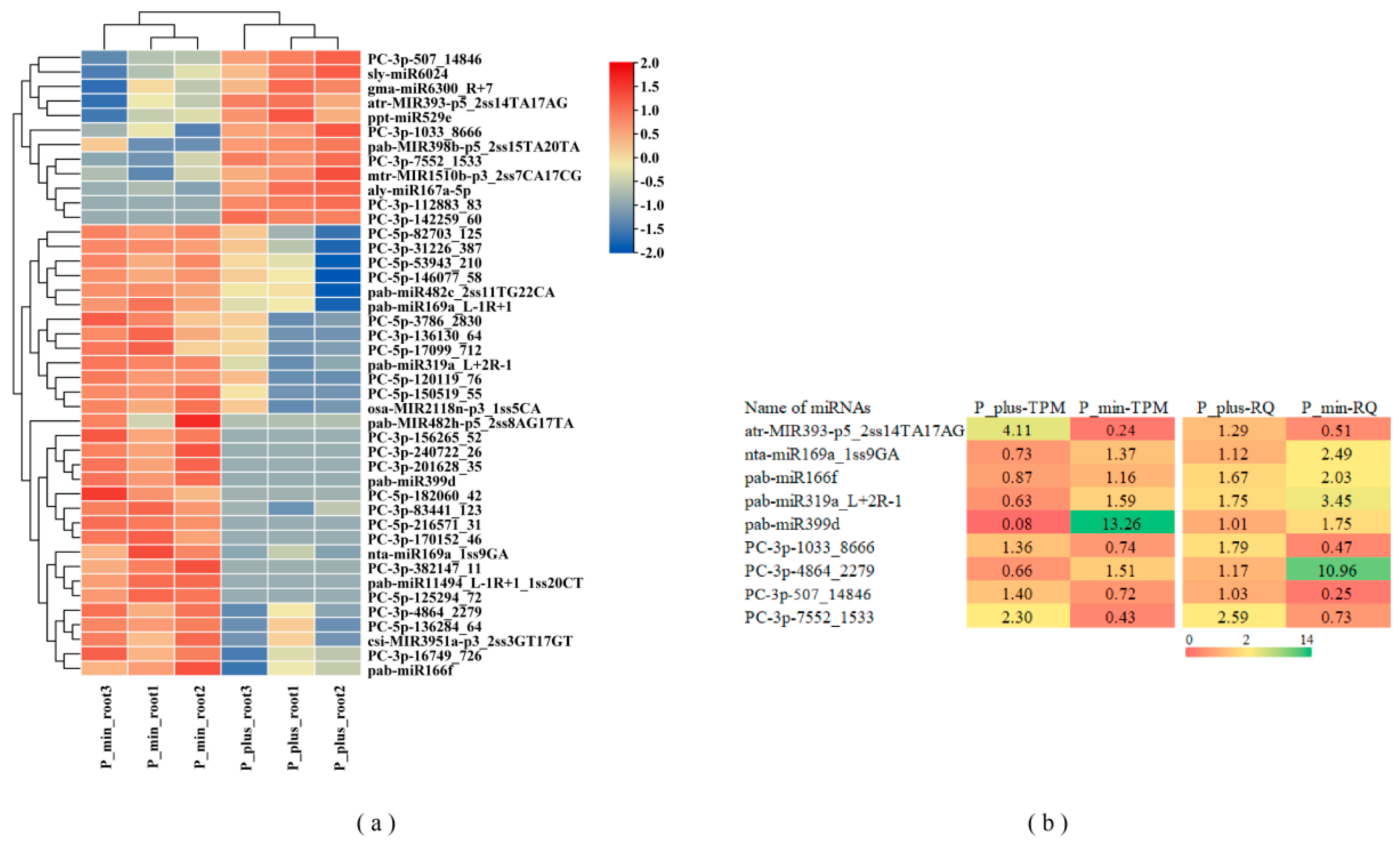
2.3. Differentially Expressed miRNA
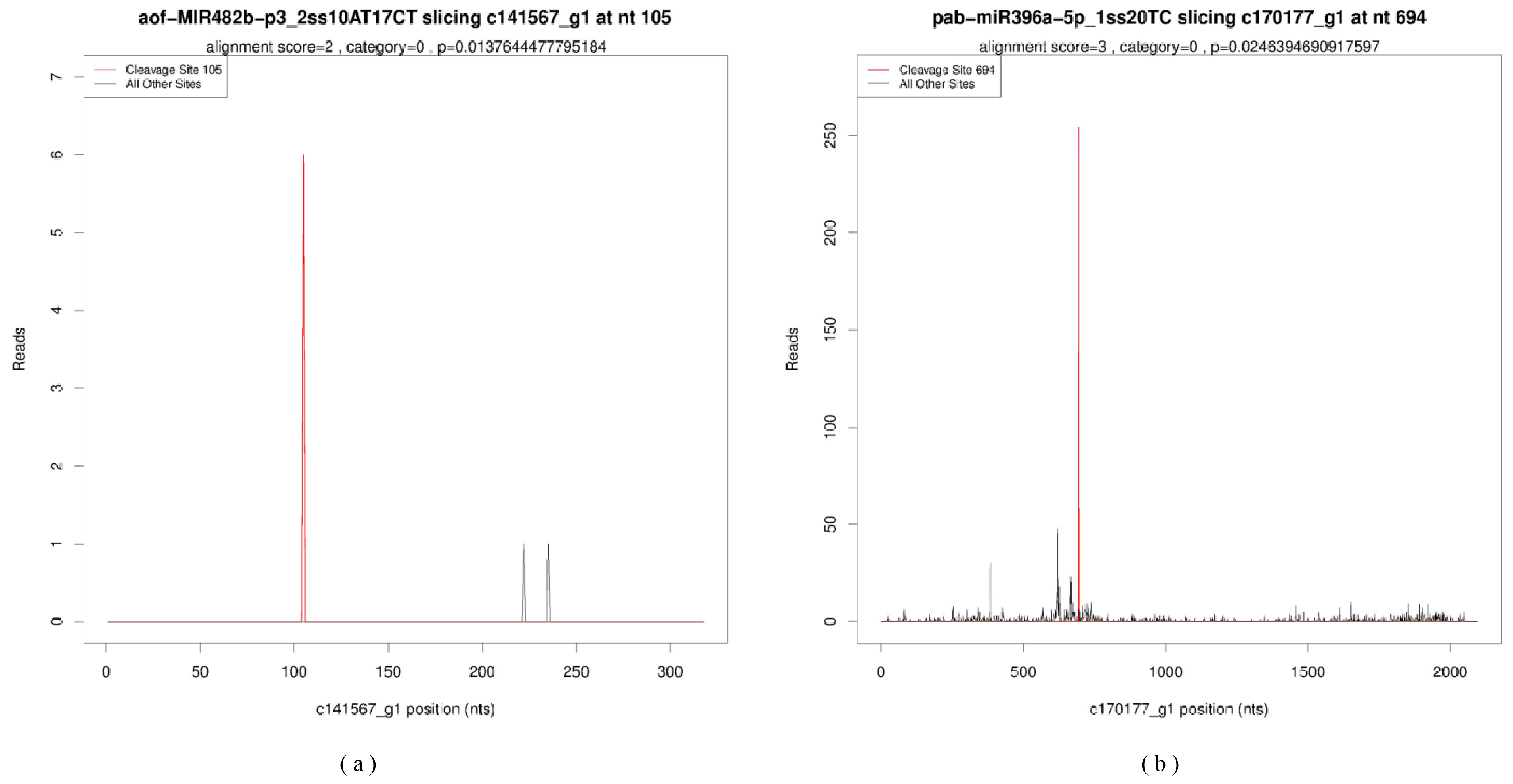
2.4. Target Genes Identification and Functional Enrichment Based on Degradome Sequencing
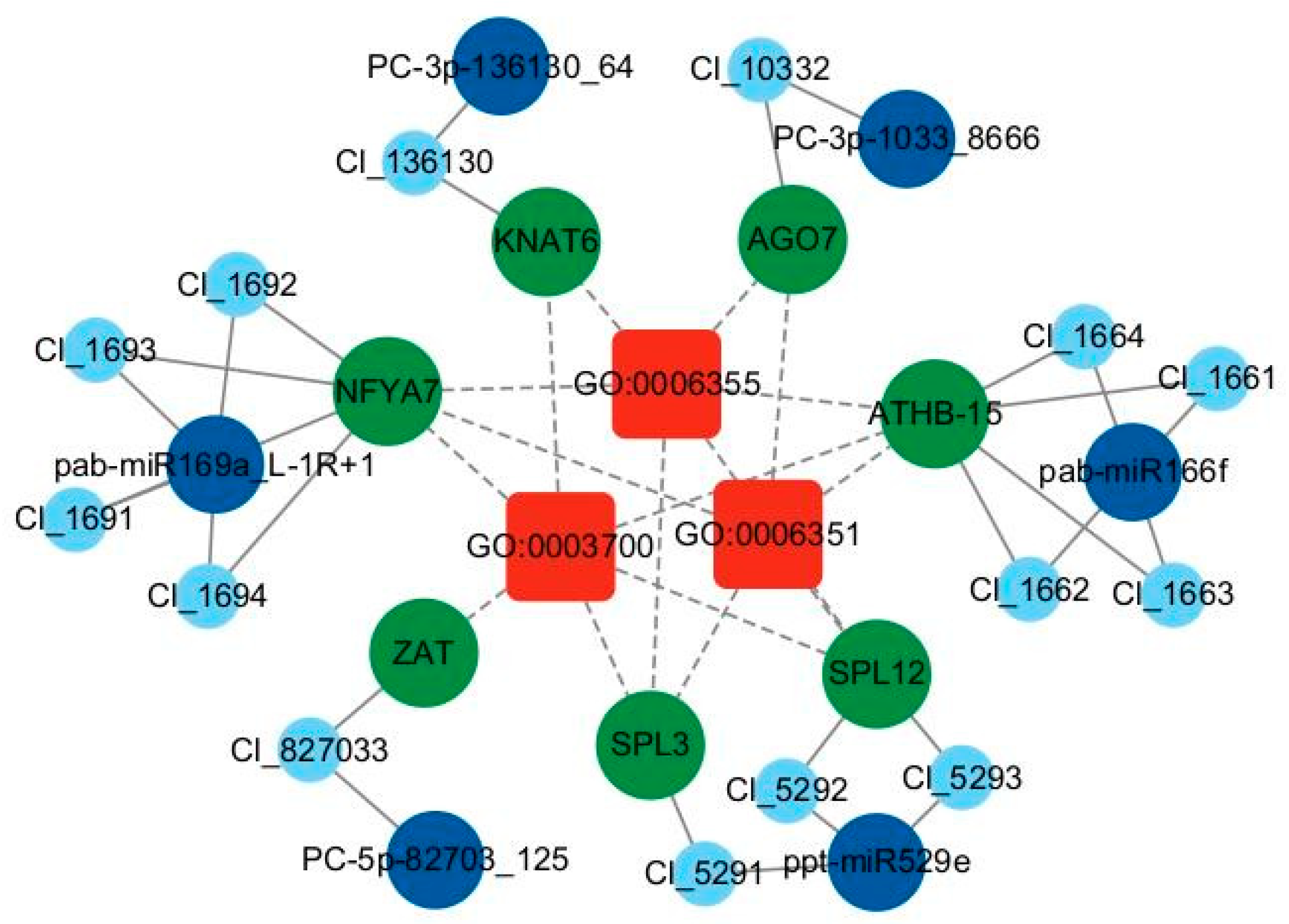
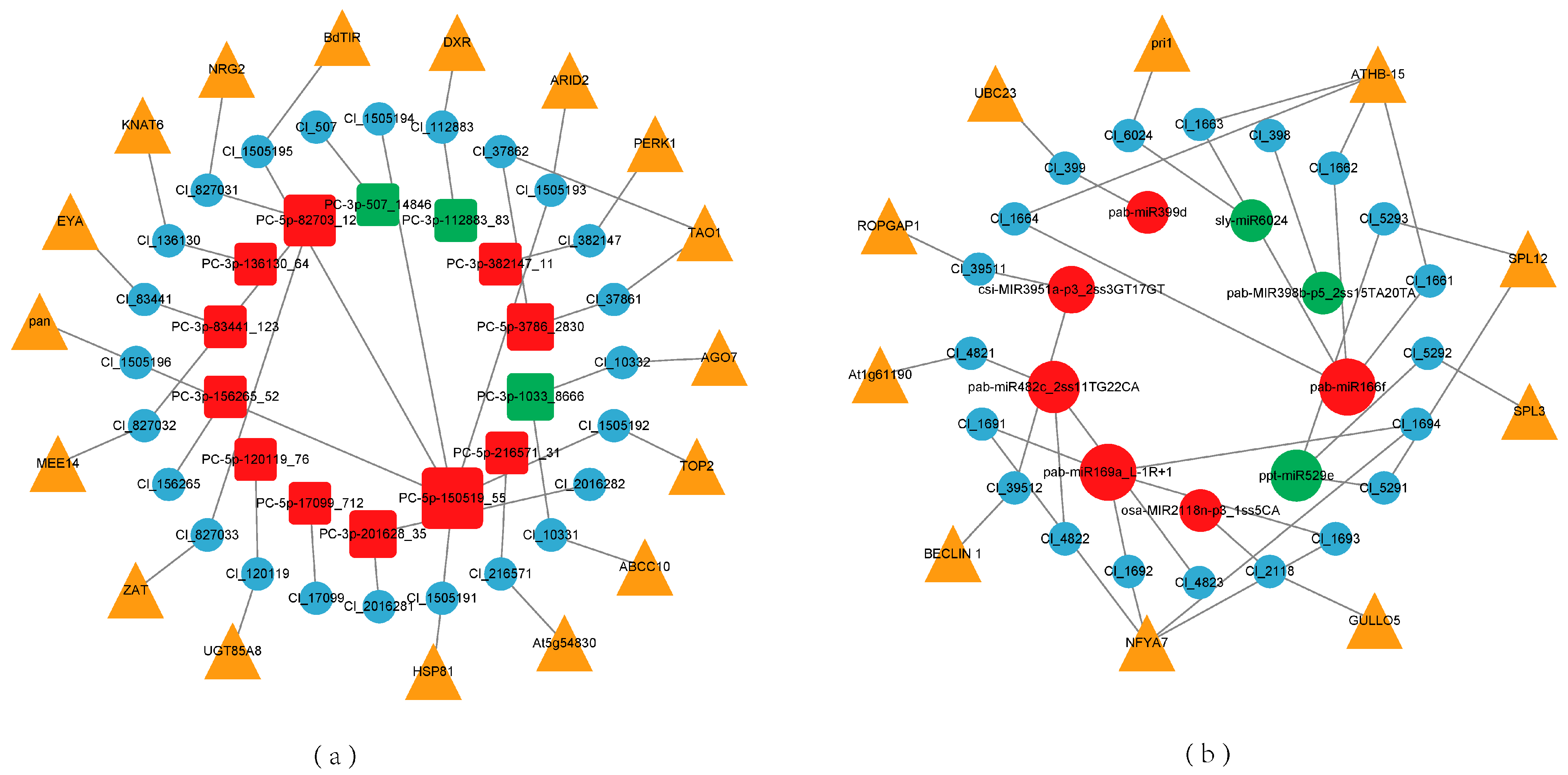
2.5. miRNA Regulatory Network and Key Modules in Response to Low Phosphorus Stress
3. Discussion
4. Materials and Methods
4.1. Plant Materials and RNA Extraction
4.2. Small RNA Library Construction and Sequencing
4.3. Filtering of Clean Reads and Identification of miRNAs
4.4. Differential Expression Analysis of miRNA
4.5. Degradome Sequencing, Target Identification and Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Liang, X.; Liu, A.; Ma, X.; Feng, L.; Chen, Y. The effect of phosphorus stress on the activity of acid phosphatase in different Chinese fir clones. J. Plant Ecol. 2005, 1, 54–59. [Google Scholar]
- Jiang, Y.; Hu, Z.; Han, Z.; Zhang, J.; Han, S.; Hao, L. Growth characteristics of Cunninghamia lanceolata in China. Sci. Rep. 2022, 12, 18179. [Google Scholar] [CrossRef] [PubMed]
- Sun, K.; Sun, H.; Lu, G.; Fang, L.; Wan, Z.; Tan, Z. Study on the effects of stand density management of Chinese fir plantation in Northern China. Front. Plant Sci. 2023, 14, 1130299. [Google Scholar] [CrossRef]
- Fan, S.H.; Sheng, W.T.; Ma, X.Q. The Impact of Continuous Planting on the Productivity of Cunninghamia Lanceolata Plantations at Different Developmental Stages. J. For. Res. 2003, 5, 560–567. [Google Scholar]
- Shi, J.; Zhao, B.; Zheng, S.; Zhang, X.; Wang, X.; Dong, W.; Xie, Q.; Wang, G.; Xiao, Y.; Chen, F.; et al. A phosphate starvation response-centered network regulates mycorrhizal symbiosis. Cell 2021, 184, 5527–5540. [Google Scholar] [CrossRef]
- Vance, C.P. Symbiotic Nitrogen Fixation and Phosphorus Acquisition, Plant Nutrition in a World of Declining Renewable Resources. Plant Physiol. 2001, 127, 390–397. [Google Scholar] [CrossRef]
- Bieleski, R.L. Phosphate pools, phosphate transport, and phosphate availability. Annu. Rev. Plant Physiol. 1973, 24, 225–252. [Google Scholar] [CrossRef]
- Ham, B.K.; Chen, J.; Yan, Y.; Lucas, W. Insights into plant phosphate sensing and signaling. Curr. Opin. Biotechnol. 2018, 49, 1–9. [Google Scholar] [CrossRef]
- He, M.; Xu, Q.; Xia, Y.; Yang, M.; Fan, Y.; Yang, Y. Mechanisms of plant phosphorus acquisition and their responses to global changes. J. Plant Ecol. 2023, 47, 291–305. [Google Scholar] [CrossRef]
- Liu, C.; Bai, Z.; Luo, Y.; Zhang, Y.F.; Wang, Y.F. Multiomics dissection of Brassica napus L. lateral roots and endophytes interactions under phosphorus starvation. Nat. Commun. 2024, 15, 9732. [Google Scholar] [CrossRef]
- Vives-Peris, V.; Ollas, C. Root exudates: From plant to rhizosphere and beyond. Plant Cell Rep. 2020, 39, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Hong, W.T.; Han, Y.; Xu, Z.K.; Cheng, L.Y. Targeting internal phosphorus re-utilization to improve plant phosphorus use efficiency. Plant Nutr. Fertil. 2021, 27, 2216–2228. [Google Scholar]
- Hu, B.; Wang, W.; Deng, K.; Li, H.; Zhang, Z. MicroRNA399 is involved in multiple nutrient starvation responses in rice. Front. Plant Sci. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Fujii, H.; Chiou, T.J.; Lin, S.I.; Aung, K.; Zhu, J.K. A miRNA Involved in Phosphate-Starvation Response in Arabidopsis. Curr. Biol. 2005, 15, 2038–2043. [Google Scholar] [CrossRef]
- Ji, J.Y.; Li, Q.; Zeng, Y.L. Research Progress in miR169/NFYA Module Responding to Abiotic Stress in Plants. Biotechnol. Inf. 2022, 38, 27–34. [Google Scholar]
- Kant, S.; Peng, M.S.; Rothstein, S.J. Genetic Regulation by NLA and MicroRNA827 for Maintaining Nitrate-Dependent Phosphate Homeostasis in Arabidopsis. PLoS Genet. 2011, 7, e1002021. [Google Scholar] [CrossRef]
- Barrera-Rojas, C.H.; Rocha, G.H.; Polverari, L. miR156-targeted SPL10 controls Arabidopsis root meristem activity and root-derived de novo shoot regeneration via cytokinin responses. J. Exp. Bot. 2020, 71, 934–950. [Google Scholar] [CrossRef]
- Liang, C.Y.; Liao, H. Molecular mechanisms underlying the responses of plant roots to low P stress. Chin. Bull. Life Sci. 2015, 3, 389–397. [Google Scholar]
- Qing, Z.H.; Yong, Z.Y. Expression of miR399 and P responsive genes and their relationship in white lupin under P stress. Acta Pedol. Sin. 2010, 47, 971–978. [Google Scholar]
- Deng, H.Y.; Huang, R.; Hu, D.H.; Wang, R.H.; Wei, R.P.; Yan, S. Further Mining and Characterization of miRNA Resource in Chinese Fir (Cunninghamia lanceolata). Genes 2022, 13, 2137. [Google Scholar] [CrossRef]
- Cao, D.C.; Xu, H.M.; Zhao, Y.Y.; Deng, X.; Liu, Y.X. Transcriptome and Degradome Sequencing Reveals Dormancy Mechanisms of Cunninghamia lanceolata Seeds. Plant Physiol. 2016, 172, 2347–2362. [Google Scholar] [CrossRef] [PubMed]
- Wan, L.C.; Wang, F.; Guo, X.Q.; Lu, S.F.; Qiu, Z.B.; Zhao, Y.Y.; Zhang, H.Y.; Lin, J.X. Identification and characterization of small non-coding RNAs from Chinese fir by high throughput sequencing. BMC Plant Biol. 2012, 12, 146. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Z.B.; Li, X.J.; Zhao, Y.Y.; Zhang, M.M.; Wan, Y.L.; Cao, D.C.; Lu, S.F.; Lin, J.X. Genome-wide analysis reveals dynamic changes in expression of microRNAs during vascular cambium development in Chinese fir, Cunninghamia lanceolata. J. Exp. Bot. 2015, 66, 3041–3054. [Google Scholar] [CrossRef]
- Whiteford, N.; Skelly, T.; Curtis, C.; Ritchie, M.E.; Löhr, A.; Zaranek, A.W.; Abnizova, I.; Brown, C. Swift: Primary data analysis for the Illumina Solexa sequencing platform. Bioinformatics 2009, 25, 2194–2199. [Google Scholar] [CrossRef]
- Han, X.J.; Yin, H.F.; Song, X.X.; Zhang, Y.X.; Liu, M.Y.; Sang, J.; Jiang, J.; Li, J.H.; Zhuo, R.Y. Integration of small RNAs, degradome and transcriptome sequencing in hyperaccumulator Sedum alfredii uncovers a complex regulatory network and provides insights into cadmium phytoremediation. Plant Biotechnol. J. 2016, 14, 1470–1483. [Google Scholar] [CrossRef]
- Aung, K.; Lin, S.I.; Wu, C.C.; Huang, Y.T.; Su, C.L.; Chiou, T.J. pho2, a Phosphate Overaccumulator, Is Caused by a Nonsense Mutation in a MicroRNA399 Target Gene. Plant Physiol. 2006, 141, 1000–1011. [Google Scholar] [CrossRef]
- Kuo, H.F.; Chiou, T.J. The Role of MicroRNAs in Phosphorus Deficiency Signaling. Plant Physiol. 2011, 156, 1016–1024. [Google Scholar] [CrossRef]
- Guo, T.; Li, G.L.; Wei, Q.; Liang, Y.H. Research progress on the functions of plant MicroRNA. Acta Bot. Boreali-Occident. Sin. 2011, 31, 2347–2354. [Google Scholar]
- Ding, Y.H. High-Throughput Sequencing Analysis of Small RNAs in Cotton Male Sterility Caused By High Temperature Stress and Functional Validation of MicroRNAs. Ph.D. Thesis, Huazhong Agricultural University, Wuhan, China, 2018. [Google Scholar]
- Zhang, K.W. Analysis of the High Photosynthetic Efficiency Mechanism of the Maize Mutant Qi319-96 Under Low Phosphorus Stress and the Creation of Transgenic Salt- and Drought-Tolerant Cotton New Germplasm. Ph.D. Thesis, Shandong University, Jinan, China, 2017. [Google Scholar]
- Li, J.S. Differential Expression of Maize MicroRNAs Under Drought Stress. Ph.D. Thesis, Sichuan Agricultural University, Ya’an, China, 2014. [Google Scholar]
- Zhang, L.; Wang, L.H.; Gui, H.R.; Wang, H.S. Research progress on soil heavy metal stress and plant-related miRNAs. North. Hortic. 2017, 18, 180–185. [Google Scholar]
- Kozomara, A.; Birgaoanu, M.; Griffiths-Jones, S. MiRBase: From microRNA sequences to function. Nucleic Acids Res. 2019, 47, D155–D162. [Google Scholar] [CrossRef]
- Li, J.; Guo, G.H.; Guo, W.W.; Guo, G.G.; Tong, D.; Ni, Z.F.; Sun, Q.X.; Yao, Y.Y. miRNA164-directed cleavage ofZmNAC1confers lateral rootdevelopment in maize (Zea mays L.). BMC Plant Biol. 2012, 12, 220. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.Y.; Guo, G.G.; Ni, Z.F.; Sunkar, R.; Du, J.K.; Zhu, J.K.; Sun, Q.X. Cloning and characterization of microRNAs from wheat (Triticum aestivum L.). Genome Biol. 2007, 8, 96–109. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Tan, T.M.; Xu, C.C.; Huang, S.P.; Tan, J.; Zhang, M.; Wang, C.L.; Xie, C.H. Genome-wide transcriptome profiling reveals novel insights into Luffa cylindrica browning. Biochem. Biophys. Res. Commun. 2015, 463, 1243–1249. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.J.; Wang, J.W.; Ni, Z.X.; Meng, X.; Feng, Y.H.; Yang, Z.Q.; Xu, L.A. Small RNA and degradome sequencing reveal roles of miRNAs in strobilus development in masson pine (Pinus massoniana). Ind. Crops Prod. 2020, 154, 112724. [Google Scholar] [CrossRef]
- Yakovlev, I.A.; Fossdal, C.G. In silico analysis of small RNAs suggest roles for noveland conserved miRNAs in the formation of epigenetic memory in somatic embryos of Norway spruce. Front. Physiol. 2017, 8, 674. [Google Scholar] [CrossRef]
- Ujino-Ihara, T.; Ueno, S.; Uchiyama, K.; Futamura, N. Comprehensive analysis of small RNAs expressed in developing male strobili of Cryptomeria japonica. PLoS ONE 2018, 13, e0193665. [Google Scholar] [CrossRef]
- Hsieh, L.C.; Lin, S.I.; Shih, A.; Chen, J.W.; Lin, W.Y.; Tseng, C.Y.; Li, W.H.; Chiou, T.J. Uncovering Small RNA-Mediated Responses to Phosphate Deficiency in Arabidopsis by Deep Sequencing. Plant Physiol. 2009, 151, 2120–2132. [Google Scholar] [CrossRef]
- Li, Z.Y.; Xu, H.Y.; Li, Y.; Wan, X.F.; Ma, Z.; Cao, J.; Li, Z.S.; He, F.; Wang, Y.F.; Wan, L.Q. Analysis of physiological and miRNA responses to Pi deficiency in alfalfa (Medicago sativa L.). Plant Mol. Biol. 2018, 96, 473–492. [Google Scholar] [CrossRef]
- Xu, F.; Liu, Q.; Chen, L.Y.; Kuang, J.B.; Walk, T.; Wang, J.X.; Liao, H. Genome-wide identification of soybean microRNAs and their targets reveals their organ-specificity and responses to phosphate starvation. BMC Genom. 2013, 14, 66. [Google Scholar] [CrossRef]
- Bari, R. PHO2, MicroRNA399, and PHR1 Define a Phosphate-Signaling Pathway in Plants. Plant Physiol. 2006, 141, 988–999. [Google Scholar] [CrossRef]
- Wang, K. Molecular Mechanism Study of ZmmiR399 in Maize Adaptation to Low Phosphorus Stress. Master’s Thesis, Chinese Academy of Agricultural Sciences, Beijing, China, 2018. [Google Scholar]
- Tao, P. Preliminary Study on the Regulation of Soybean Phosphorus Balance and Flowering by MiR399. Master’s Thesis, South China Agricultural University, Guangzhou, China, 2017. [Google Scholar]
- Kang, M.M. Identification and Functional Analysis of Small Molecule RNAs and Their Target Genes in Maize. Ph.D. Thesis, China Agricultural University, Beijing, China, 2014. [Google Scholar]
- Pant, B.D.; Musialak-Lange, M.; Nuc, P.; May, P.; Buhtz, A.; Kehr, J.; Walther, D.; Scheible, W.R. Identification of Nutrient-Responsive Arabido- psis and Rapeseed MicroRNAs by Comprehensive Real-Time Polymerase Chain Reaction Profilin -g and Small RNA Sequencing. Plant Physiol. 2009, 150, 1541–1555. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ling, H.Q. Study on the synergistic mechanism of high efficiency of nitrogen and phosphorus and high yield in wheat. Sci. Technol. Innov. Her. 2016, 13, 177. [Google Scholar]
- Sha, A.; Chen, Y.H.; Ba, H.P.; Shan, Z.H.; Zhang, X.J.; Wu, X.J.; Qiu, D.Z.; Chen, S.L.; Zhou, X. Identification of Glycine Max MicroRNAs in response to phosphorus deficiency. J. Plant Biol. 2012, 55, 268–280. [Google Scholar] [CrossRef]
- Li, L.; Wang, Y.; Wang, K.Y.; Zhao, M.Z.; Sun, C.Y.; Lin, Y.P.; Zhang, M.P. The structure, function, and expression patterns of the HD-ZIP gene family. Mol. Plant Breed. 2018, 16, 781–790. [Google Scholar]
- Lei, K.J.; Ren, J.; Zhu, Y.Y.; An, G.Y. The Arabidopsis SPL1 gene is involved in regulating rhizosphere acidification under low phosphorus conditions. Acta Bot. Sin. 2016, 51, 184–193. [Google Scholar]
- Ren, J. The Molecular Mechanism of MiR156 and Its Target Gene SPL9 Regulating MiR399f Under Low Phosphorus Stress. Master’s Thesis, Henan University, Kaifeng, China, 2018. [Google Scholar]
- Shen, Y. Research on the Role of OsRGA1 in Rice Response to Strigolactones and Low Phosphorus Stress. Master’s Thesis, Yangzhou University, Yangzhou, China, 2024. [Google Scholar]
- Wang, Y.; Liu, W.; Shen, H.; Zhu, X.S.; Zhai, L.L.; Xu, L. Identification of Radish (Raphanus sativus L.) miRNAs and Their Target Genes to Explore miRNA-Mediated Regulatory Networks in Lead (Pb) Stress Responses by High-Throughput Sequencing and Degradome Analysis. Plant Mol. Biol. Report. 2015, 33, 358–376. [Google Scholar] [CrossRef]
- Li, M.; Su, S.; Wu, P.F.; Cameron, K.; Zhang, Y.; Chen, W.T.; Ma, X.Q. Transcriptome Characterization of the Chinese fir (Cunninghamia lanceolata (Lamb.) Hook.) and Expression Analysis of Candidate Phosphate Transporter Genes. Forests 2017, 8, 420. [Google Scholar] [CrossRef]
- Addo-Quaye, C.; Miller, W.; Axtell, M.J. CleaveLand: A pipeline for using degradome data to find cleaved small RNA targets. Bioinformatics 2009, 25, 130–131. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Raw Reads | 3ADT&Length Filter | Junk Reads | Rfam Reads | Valid Reads | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Total | Uniq | Total | Uniq | Total | Uniq | Total | Uniq | Total | Uniq | |
| P_plus_root1 | 16,035,908 (100%) | 4,620,097 (100%) | 3,285,048 (20.49%) | 1,748,224 (37.84%) | 59,001 (0.37%) | 24,745 (0.54%) | 1,582,806 (9.87%) | 47,073 (1.02%) | 11,101,097 (69.23%) | 2,799,745 (60.60%) |
| P_plus_root2 | 12,653,242 (100%) | 4,175,035 (100%) | 4,294,605 (33.94%) | 2,026,027 (48.53%) | 41,521 (0.33%) | 18,307 (0.44%) | 1,081,337 (8.55%) | 35,145 (0.84%) | 7,227,792 (57.12%) | 2,095,238 (50.18%) |
| P_plus_root3 | 17,352,907 (100%) | 5,277,691 (100%) | 3,184,645 (18.35%) | 1,928,169 (36.53%) | 80,397 (0.46%) | 34,711 (0.66%) | 1,379,134 (7.95%) | 44,820 (0.85%) | 12,703,527 (73.21%) | 3,269,733 (61.95%) |
| P_min_root1 | 21,267,612 (100%) | 5,185,861 (100%) | 4,349,634 (20.45%) | 2,129,049 (41.05%) | 71,407 (0.34%) | 26,599 (0.51%) | 1,586,045 (7.46%) | 31,782 (0.61%) | 15,245,191 (71.68%) | 2,998,143 (57.81%) |
| P_min_root2 | 15,786,575 (100%) | 4,599,854 (100%) | 4,039,757 (25.59%) | 1,967,958 (42.78%) | 64,517 (0.41%) | 24,490 (0.53%) | 1,057,089 (6.7%) | 26,580 (0.58%) | 10,615,959 (67.25%) | 2,580,559 (56.10%) |
| P_min_root3 | 19,893,744 (100%) | 4,505,317 (100%) | 2,746,217 (13.8%) | 1,392,144 (30.90%) | 83,072 (0.42%) | 29,281 (0.65%) | 872,518 (4.39%) | 21,718 (0.48%) | 16,184,397 (81.35%) | 3,061,902 (67.96%) |
| Sample | DeP_plus (Number) | DeP_plus (Ratio) | DeP_min (Number) | DeP_min (Ratio) | Sum (Number) | Sum (Ratio) |
|---|---|---|---|---|---|---|
| Raw Reads | 39,016,365 | / | 40,529,349 | / | 79,545,714 | / |
| reads < 15 nt after removing 3 adaptor | 213,292 | 0.55% | 234,398 | 0.58% | 447,690 | 0.56% |
| Mappable Reads | 38,803,073 | 99.45% | 40,294,951 | 99.42% | 79,098,024 | 99.44% |
| Unique Raw Reads | 9,111,132 | / | 9,732,496 | / | 16,259,475 | / |
| Unique reads < 15 nt after removing 3 adaptor | 71,443 | 0.78% | 74,632 | 0.77% | 122,517 | 0.75% |
| Unique Mappable Reads | 9,039,689 | 99.22% | 9,657,864 | 99.23% | 16,136,958 | 99.25% |
| Transcript Mapped Reads | 29,310,460 | 75.12% | 30,967,313 | 76.41% | 60,277,773 | 75.78% |
| Unique Transcript Mapped Reads | 5,787,569 | 63.52% | 6,075,096 | 62.42% | 9,648,644 | 59.34% |
| Number of input Transcript | 638,227 | / | 638,227 | / | 638,227 | / |
| Number of Coverd Transcript | 163,561 | 25.63% | 167,571 | 26.26% | 187,329 | 29.35% |
| This Study | Wan et al., 2012 [22] | Qiu et al., 2015 [23] | Cao et al., 2016 [21] | Deng et al., 2022 [20] | MiRNA Sequence |
|---|---|---|---|---|---|
| aly-miR167a-5p | cln-miR167a | - | - | - | UGAAGCUGCCAGCAUGAUCUA |
| atr-miR164a | cln-miR164a | aly-miR164a | - | - | UGGAGAAGCAGGGCACGUGCA |
| cln-miR162 | cln-miR162d | - | cln-miR162 | - | UUGAUAAACCUCUGCAUCCAG |
| cln-miR164 | cln-miR164b | aly-miR164c | - | - | UGGAGAAGCAGGGCACGUGCG |
| cln-miR6725 | cln-miRn1 | cln-miR6725 | - | - | UGGCAUCUGUCGAGGUCAUCUA |
| fve-miR397 | - | aly-miR397a | - | - | UCAUUGAGUGCAGCGUUGAUG |
| mdm-miR160a | cln-miR160a | aly-miR160a | - | cln-miR160 | UGCCUGGCUCCCUGUAUGCCA |
| mes-miR167a | cln-miR167f | - | - | - | UGAAGCUGCCAGCAUGAUCUG |
| mes-miR2111a | cln-miR2111a | - | - | - | UAAUCUGCAUCCUGAGGUUUA |
| pab-miR156m | cln-miR156i | mtr-miR156g | - | cln-miR156 | UUGACAGAAGAUAGAGGGCAC |
| pab-miR159a_L+1R+1_1ss22CT | - | - | - | cln-miR159c | UUUGGUUUGAAGGGAGCUCUA |
| pab-miR166f | cln-miR166a | aly-miR166a | - | cln-miR166 | UCGGACCAGGCUUCAUUCCCC |
| pab-miR166f_L+2R-2 | cln-miR166l | bdi-miR166f | - | - | UCUCGGACCAGGCUUCAUUCC |
| pab-miR166f_R-2 | cln-miR166c | vvi-miR166a | - | - | UCGGACCAGGCUUCAUUCC |
| pab-miR390a | cln-miR390a | aly-miR390a | cln-miR390 | - | AAGCUCAGGAGGGAUAGCGCC |
| pab-miR396a-3p | - | - | - | cln-miR396c | CUCAAGAAAGCUGUGGGAAA |
| pab-miR396b | - | pab-miR396b | cln-miR396a | cln-miR396b | UUCCACGGCUUUCUUGAACUU |
| pab-miR396g | cln-miR396b | aly-miR396b | - | - | UUCCACAGCUUUCUUGAACUU |
| pab-miR396g_1ss21TG | cln-miR396a | aau-miR396 | - | - | UUCCACAGCUUUCUUGAACUG |
| pab-miR397a_L+1R-1_1ss15GA | - | - | - | cln-miR23 | UUAUUGAGUGCAGCAUUGACG |
| pab-miR399d | cln-miR399a | aly-miR399b | - | cln-miR399d | UGCCAAAGGAGAGUUGCCCUG |
| pab-miR482c_1ss11TG | - | pab-miR482c | - | - | TCTTTCCTACGCCTCCCATTCC |
| pab-miR535a | - | osa-miR535 | - | - | UGACAACGAGAGAGAGCACGC |
| pab-miR536a_1ss20CA | - | - | - | cln-miR536 | CCGUGCCAAGCUGCGUGCAAC |
| ppe-MIR482e-p3_2ss14TC17CT_1 | - | - | - | cln-miR48 | UCUUGCCUAUUCCCCCUAUGCC |
| ppt-MIR477d-p5_1ss18TC | - | - | - | cln-miR107 | CCUCUCCCUCAAAGGCUCCCA |
| ppt-miR529e | - | ppt-miR529e | cln-miR529 | cln-miR529a | AGAAGAGAGAGAGUACAGCCC |
| pvu-miR482-3p_L-2R+2_1ss5CG | - | - | - | cln-miR68 | UUGCCAAUUCCGCCCAUUCCUA |
| tcc-miR169m_L-1R+1_1ss2GA | - | mtr-miR169d | - | - | AAGCCAAGGAUGACUUGCCGG |
| PC-3p-10_276448 | - | cln-miR01 | - | cln-miR26 | UCUUUCCUUUACCACCGAUACC |
| PC-3p-11729_1020 | - | - | - | cln-miR101 | AUGUAACAAAGUAAAGCUGCC |
| PC-3p-1603_5981 | - | - | - | cln-miR98 | ACGACUGGCAUGUUGAGCACA |
| PC-3p-1692_5734 | - | cln-miR07 | - | - | AAUCUAAUGGAAGCCAGUGUU |
| PC-3p-17_141789 | - | - | - | cln-miR92 | UUUUCCCUGUACCACCCAUUCC |
| PC-3p-18_140528 | - | cln-miR02 | - | - | UACCCAAUGGAUCUUCCCAACU |
| PC-3p-2_787472 | - | - | - | cln-miR08 | UUUUCCCUGAACCACCCAUUCC |
| PC-3p-30807_392 | - | - | - | cln-miR72 | UCAAUGCUGUACUCAAUAACG |
| PC-3p-4437_2467 | - | - | - | cln-miR75 | CCACAUUGAUGAAUUGAUUUC |
| PC-3p-5_354638 | - | - | - | cln-miR50 | UAAUGGCUAGUGGUAACUUACC |
| PC-5p-1647_5857 | - | - | - | cln-miR24 | UGUUUCUGUUUGUUGACAAUG |
| PC-5p-17099_712 | - | - | - | cln-miR39 | AAUCAAUUCAUCAAUGUGGCA |
| PC-5p-216_29280 | - | - | - | cln-miR59 | UUUGAGUGAAUCCAGAGUCUCU |
| PC-5p-25017_487 | - | - | - | cln-miR65 | UGAAGAGAGAGAGCAUAGCCA |
| PC-5p-3222_3238 | - | - | - | cln-miR46 | UCCUCCUAACUGGUGUGAGCUUU |
| PC-5p-9664_1213 | - | - | - | cln-miR51 | UUCUUUGCUCUGUUAUGCUCC |
| PC-5p-970_9048 | - | - | - | cln-miR14 | UAUAGGGGUAAUGGACAAACU |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, M.; Ye, X.; Zhao, Z.; Zeng, Y.; Huang, C.; Ma, X.; Shuai, P. Identification of miRNAs and Their Targets in Cunninghamia lanceolata Under Low Phosphorus Stress Based on Small RNA and Degradome Sequencing. Int. J. Mol. Sci. 2025, 26, 3655. https://doi.org/10.3390/ijms26083655
Li M, Ye X, Zhao Z, Zeng Y, Huang C, Ma X, Shuai P. Identification of miRNAs and Their Targets in Cunninghamia lanceolata Under Low Phosphorus Stress Based on Small RNA and Degradome Sequencing. International Journal of Molecular Sciences. 2025; 26(8):3655. https://doi.org/10.3390/ijms26083655
Chicago/Turabian StyleLi, Meng, Xiaopeng Ye, Ziyu Zhao, Yifan Zeng, Chaozhang Huang, Xiangqing Ma, and Peng Shuai. 2025. "Identification of miRNAs and Their Targets in Cunninghamia lanceolata Under Low Phosphorus Stress Based on Small RNA and Degradome Sequencing" International Journal of Molecular Sciences 26, no. 8: 3655. https://doi.org/10.3390/ijms26083655
APA StyleLi, M., Ye, X., Zhao, Z., Zeng, Y., Huang, C., Ma, X., & Shuai, P. (2025). Identification of miRNAs and Their Targets in Cunninghamia lanceolata Under Low Phosphorus Stress Based on Small RNA and Degradome Sequencing. International Journal of Molecular Sciences, 26(8), 3655. https://doi.org/10.3390/ijms26083655