
Ultrasound-Assisted Synthesis of Substituted Chalcone-Linked 1,2,3-Triazole Derivatives as Antiproliferative Agents: In Vitro Antitumor Activity and Molecular Docking Studies

 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Results and Discussion
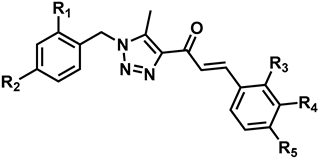
2.1. Synthesis of Chalcone Derivatives
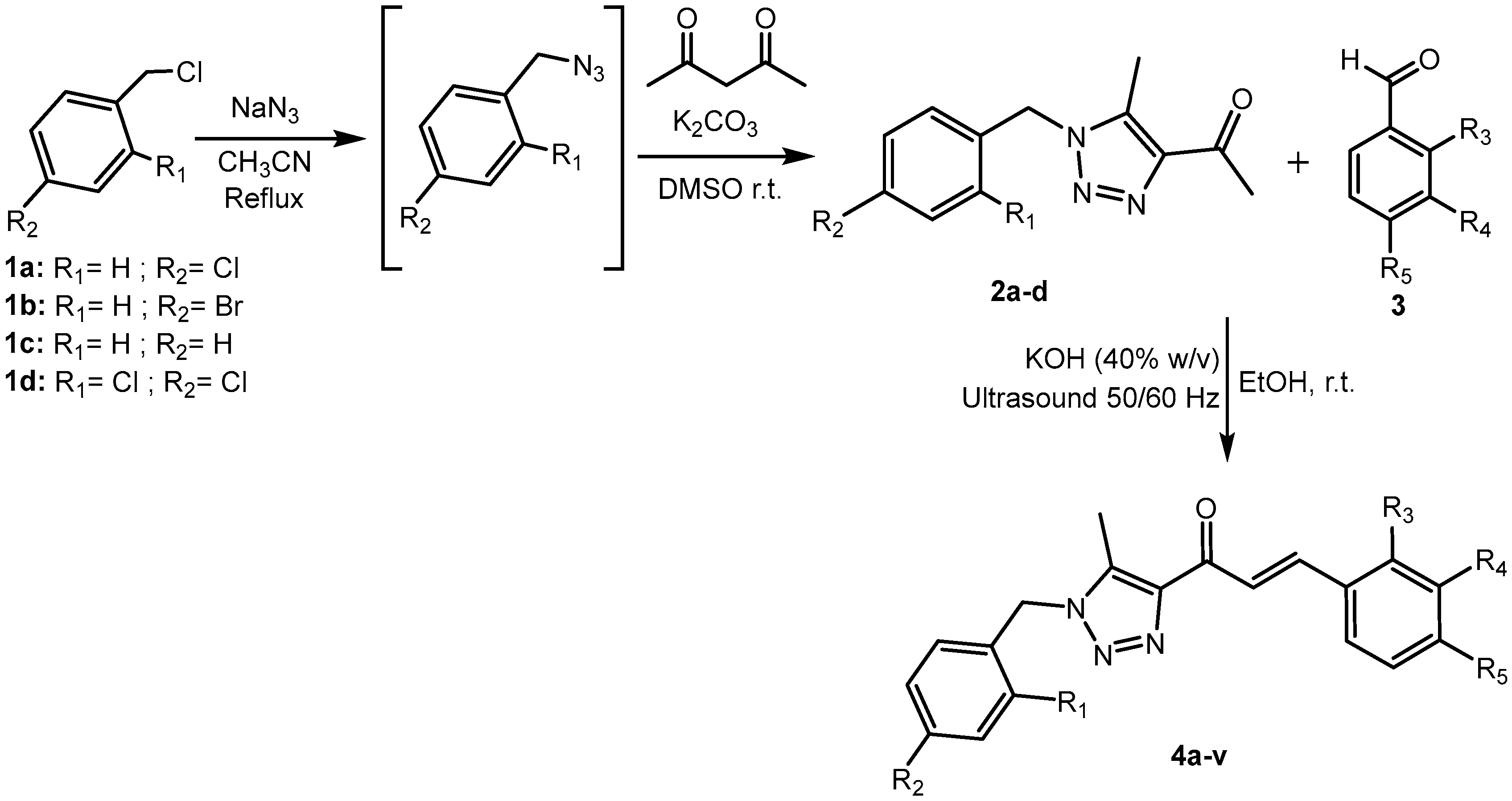
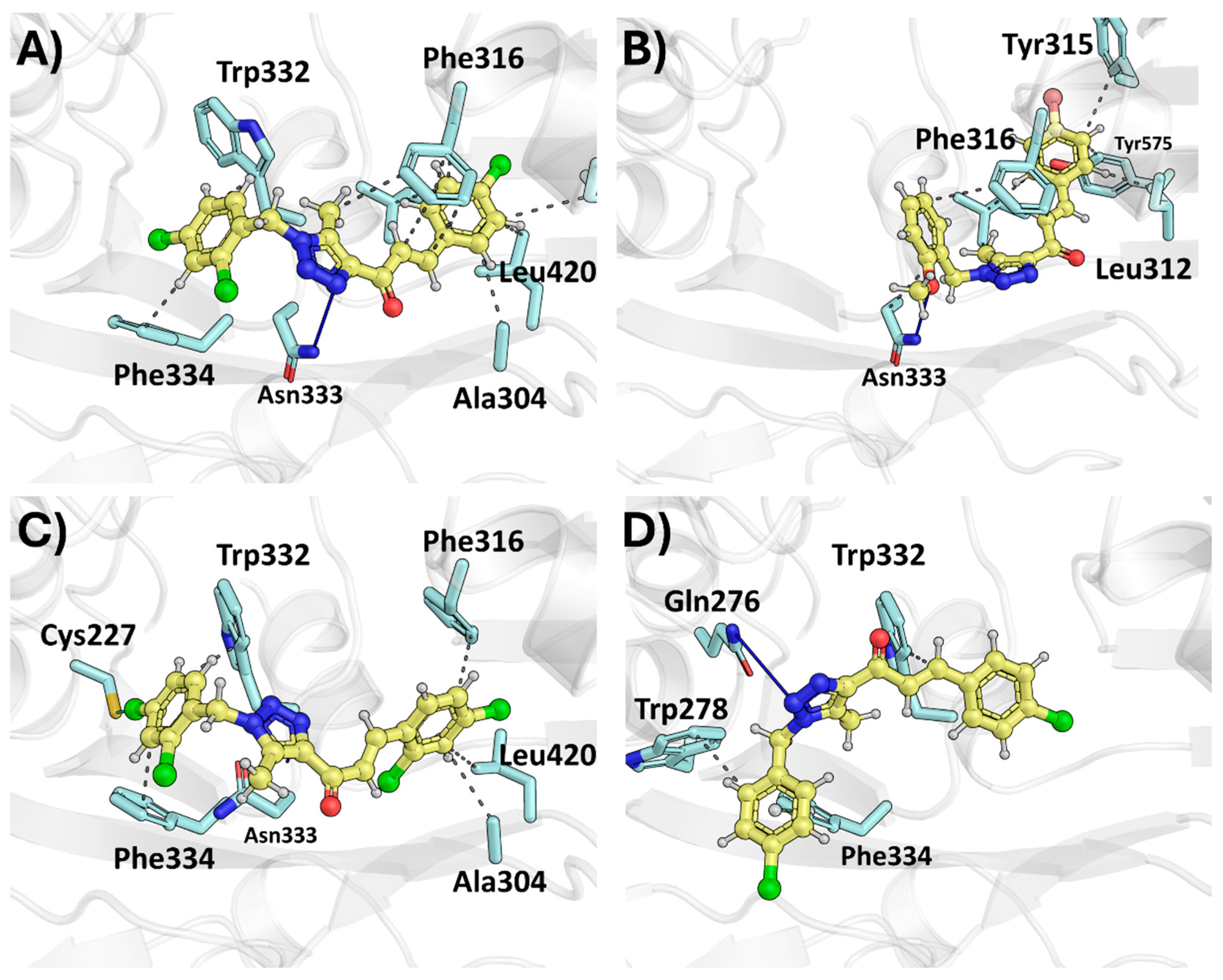
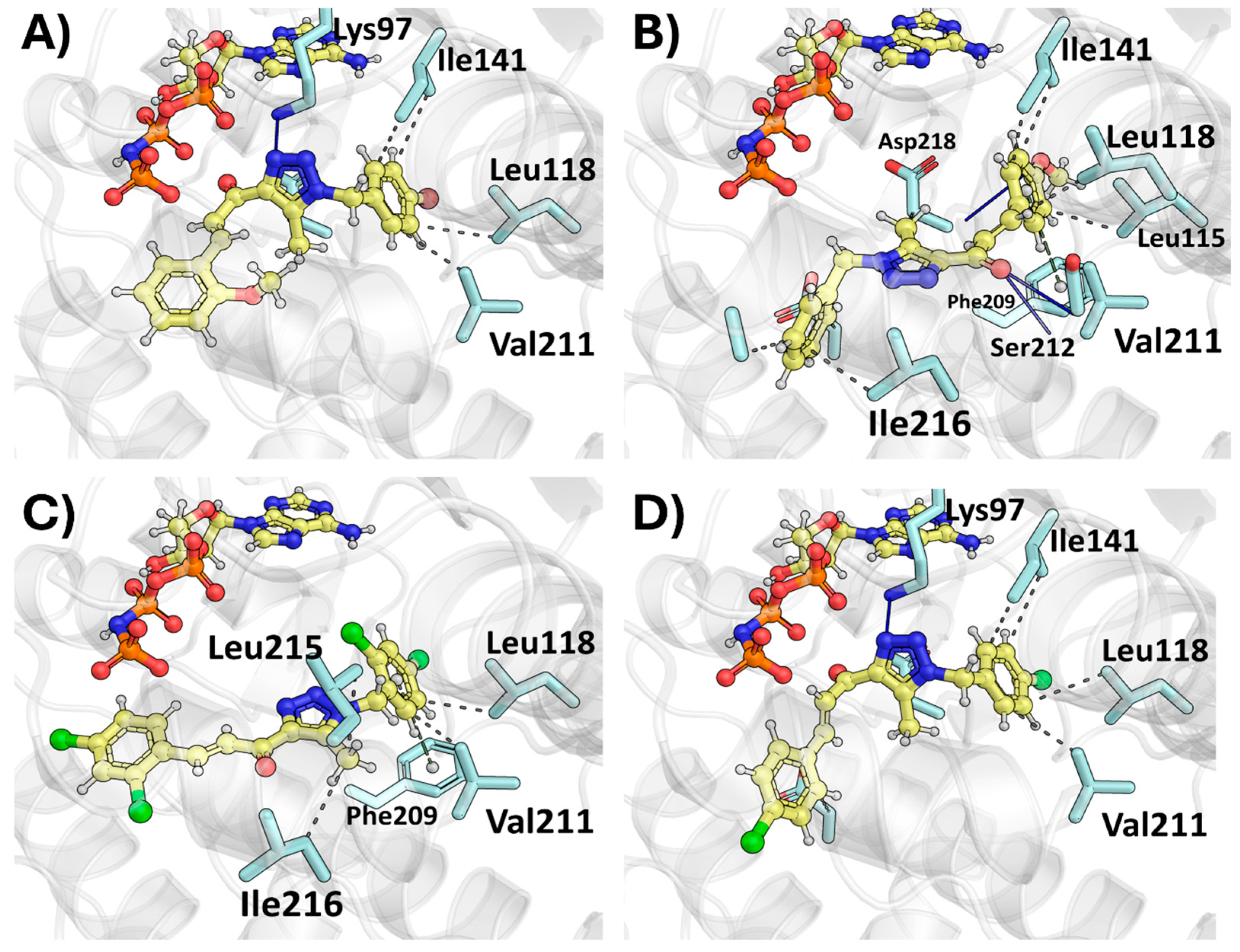
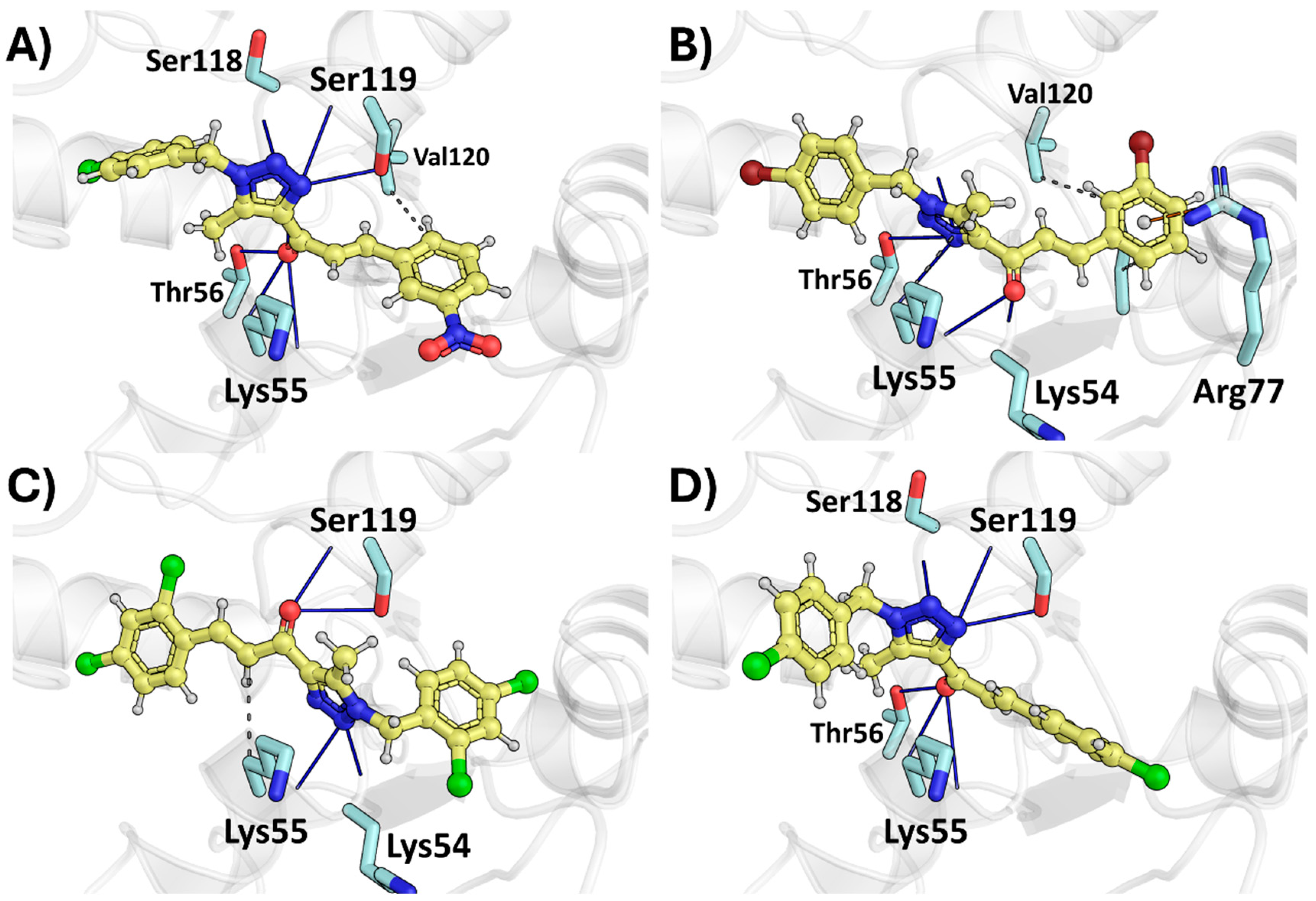
2.2. Molecular Docking
2.2.1. Therapeutic Target Involved in U937—VEGFR Kinase
2.2.2. Therapeutic Target Involved in T98G—Tissue Transglutaminase, TG2
2.2.3. Therapeutic Target Involved in Gb-d1—MEK
2.2.4. Therapeutic Target Involved in HeLa—Dihydrofolate Reductase, DHFR
2.3. Cytotoxic Activities
3. Materials and Methods
3.1. General Section
3.2. General Procedure for Synthesis of Triazolic Chalcones (4a–4v) (Exemplified for Synthesis of 4a)

- (E)-3-(4-bromophenyl)-1-(1-(4-chlorobenzyl)-5-methyl-1H-1,2,3-triazol-4-yl)-2-propen-1-one (4a).
- (E)-3-(4-chlorophenyl)-1-(1-(4-chlorobenzyl)-5-methyl-1H-1,2,3-triazol-4-yl)-2-propen-1-one (4b).
- (E)-1-((4-chlorobenzyl)-5-methyl-1H-1,2,3-triazol-4-yl)-3-(4-methoxyphenyl)-2-propen-1-one (4c).
- (E)-3-(2,4-dichlorophenyl)-1-(1-(4-chlorobenzyl)-5-methyl-1H-1,2,3-triazol-4-yl)-2-propen-1-ona (4d).
- (E)-3-(2-chlorophenyl)-1-(1-(4-chlorobenzyl)-5-methyl-1H-1,2,3-triazol-4-yl)-2-propen-1-one (4e).
- (E)-1-((4-bromobenzyl)-5-methyl-1H-1,2,3-triazol-4-yl)-3-(3-bromophenyl)-2-propen-1-one (4f).
- (E)-1-((4-chlorobenzyl)-5-methyl-1H-1,2,3-triazol-4-yl)-3-(3-nitrophenyl)-2-propen-1-one (4g).
- (E)-1-((4-bromobenzyl)-5-methyl-1H-1,2,3-triazol-4-yl)-3-(2-chlorophenyl)-2-propen-1-one (4h).
- (E)-1-((1-(4-bromobenzyl)-5-methyl-1H-1,2,3-triazol-4-yl)-3-(2,4-dichlorophenyl)-2-propen-1-one (4i).
- (E)-1-((4-bromobenzyl)-5-methyl-1H-1,2,3-triazol-4-yl)-3-(4-methoxyphenyl)-2-propen-1-one (4j).
- (E)-1-((4-bromobenzyl)-5-methyl-1H-1,2,3-triazol-4-yl)-3-(2-methoxyphenyl)-2-propen-1-one (4k).
- (E)-1-((4-bromobenzyl)-5-methyl-1H-1,2,3-triazol-4-yl)-3-(3,4-dimethoxyphenyl)-2-propen-1-one (4l).
- (E)-1-(benzyl)-5-methyl-1H-1,2,3-triazol-4-yl)-3-phenyl-2-propen-1-one (4m).
- (E)-1-(benzyl)-5-methyl-1H-1,2,3-triazol-4-yl)-3-(4-chlorophenyl)-2-propen-1-one (4n).
- (E)-1-(bencil)-5-metil-1H-1,2,3-triazol-4-il)-3-(2,4-diclorofenil)-2-propen-1-ona (4o).
- (E)-1-(benzyl)-5-methyl-1H-1,2,3-triazol-4-yl)-3-(4-methoxyphenyl)-2-propen-1-one (4p).
- (E)-1-(2,4-dichlorobenzyl)-5-methyl-1H-1,2,3-triazol-4-yl)-3-(4-chlorophenyl)-2-propen-1-one (4q).
- (E)-1-(2,4-dichlorobenzyl)-5-methyl-1H-1,2,3-triazol-4-yl)-3-(2-chlorophenyl)-2-propen-1-one (4r).
- (E)-1-(2,4-dichlorobenzyl)-5-methyl-1H-1,2,3-triazol-4-yl)-3-(2,4-dichlorophenyl)-2-propen-1-one (4s).
- (E)-1-(2,4-dichlorobenzyl)-5-methyl-1H-1,2,3-triazol-4-yl)-3-(3,4-dimethoxyphenyl)-2-propen-1-one (4t).
- (E)-1-(2-methoxybenzyl)-5-methyl-1H-1,2,3-triazol-4-yl)-3-(4-bromophenyl)-2-propen-1-one (4v).
3.3. Computational Method
3.4. Cytotoxixity Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Khan, S.A.; Akhtar, M.J.; Gogoi, U.; Meenakshi, D.U.; Das, A. An Overview of 1,2,3-Triazole-Containing Hybrids and Their Potential Anticholinesterase Activities. Pharmaceuticals 2023, 16, 179. [Google Scholar] [CrossRef] [PubMed]
- Lengerli, D.; Ibis, K.; Nural, Y.; Banoglu, E. The 1,2,3-Triazole All-in-One Ring System in Drug Discovery: A Good Bioisostere, a Good Pharmacophore, a Good Linker, and a Versatile Synthetic Tool. Expert. Opin. Drug. Discov. 2022, 17, 1209–1236. [Google Scholar] [CrossRef] [PubMed]
- Matin, M.M.; Matin, P.; Rahman, M.R.; Ben Hadda, T.; Almalki, F.A.; Mahmud, S.; Ghoneim, M.M.; Alruwaily, M.; Alshehri, S. Triazoles and Their Derivatives: Chemistry, Synthesis, and Therapeutic Applications. Front. Mol. Biosci. 2022, 9, 864286. [Google Scholar] [CrossRef]
- Bozorov, K.; Zhao, J.; Aisa, H.A. 1,2,3-Triazole-Containing Hybrids as Leads in Medicinal Chemistry: A Recent Overview. Bioorg. Med. Chem. 2019, 27, 3511–3531. [Google Scholar] [CrossRef]
- Cunha, A.C.; Figueiredo, J.M.; Tributino, J.L.M.; Miranda, A.L.P.; Castro, H.C.; Zingali, R.B.; Fraga, C.A.M.; De Souza, M.C.B.V.; Ferreira, V.F.; Barreiro, E.J. Antiplatelet Properties of Novel N-Substituted-Phenyl-1,2,3-Triazole-4-Acylhydrazone Derivatives. Bioorg. Med. Chem. 2003, 11, 2051–2059. [Google Scholar] [CrossRef] [PubMed]
- Jordão, A.K.; Ferreira, V.F.; Lima, E.S.; de Souza, M.C.B.V.; Carlos, E.C.L.; Castro, H.C.; Geraldo, R.B.; Rodrigues, C.R.; Almeida, M.C.B.; Cunha, A.C. Synthesis, Antiplatelet and in Silico Evaluations of Novel N-Substituted-Phenylamino-5-Methyl-1H-1,2,3-Triazole-4-Carbohydrazides. Bioorg. Med. Chem. 2009, 17, 3713–3719. [Google Scholar] [CrossRef]
- Cogan, D.A.; Aungst, R.; Breinlinger, E.C.; Fadra, T.; Goldberg, D.R.; Hao, M.H.; Kroe, R.; Moss, N.; Pargellis, C.; Qian, K.C.; et al. Structure-Based Design and Subsequent Optimization of 2-tolyl-(1,2,3-Triazol-1-yl-4-carboxamide) Inhibitors of P38 MAP Kinase. Bioorg. Med. Chem. Lett. 2008, 18, 3251–3255. [Google Scholar] [CrossRef]
- Chen, P.C.; Patil, V.; Guerrant, W.; Green, P.; Oyelere, A.K. Synthesis and Structure-Activity Relationship of Histone Deacetylase (HDAC) Inhibitors with Triazole-Linked Cap Group. Bioorg. Med. Chem. 2008, 16, 4839–4853. [Google Scholar] [CrossRef]
- Bhat, B.A.; Reddy, P.B.; Agrawal, S.K.; Saxena, A.K.; Kumar, H.M.S.; Qazi, G.N. Studies on Novel 4β-[(4-substituted)-1,2,3-Triazol-1-yl] Podophyllotoxins as Potential Anticancer Agents. Eur. J. Med. Chem. 2008, 43, 2067–2072. [Google Scholar] [CrossRef]
- Kelley, J.L.; Koble, C.S.; Davis, R.G.; McLean, E.W.; Soroko, F.E.; Barrett, R. Cooper 1-(fluorobenzyl)-4-Amino-1H-1,2,3-Triazolo [4,5-c]Pyridines: Synthesis and Anticonvulsant Activity. J. Med. Chem. 1995, 38, 4131–4134. [Google Scholar] [CrossRef]
- Mahmood, S.; Khan, S.G.; Rasul, A.; Christensen, J.B.; Abourehab, M.A.S. Ultrasound Assisted Synthesis and In Silico Modelling of 1,2,4-Triazole Coupled Acetamide Derivatives of 2-(4-Isobutyl Phenyl)Propanoic Acid as Potential Anticancer Agents. Molecules 2022, 27, 7984. [Google Scholar] [CrossRef] [PubMed]
- Calderone, V.; Fiamingo, F.L.; Amato, G.; Giorgi, I.; Livi, O.; Martelli, A.; Martinotti, E. 1,2,3-Triazol-Carboxanilides and 1,2,3-Triazol-(N-Benzyl)-Carboxamides as BK-Potassium Channel Activators. XII. Eur. J. Med. Chem. 2008, 43, 2618–2626. [Google Scholar] [CrossRef]
- Holla, B.S.; Mahalinga, M.; Karthikeyan, M.S.; Poojary, B.; Akberali, P.M.; Kumari, N.S. Synthesis, Characterization and Antimicrobial Activity of Some Substituted 1,2,3-Triazoles. Eur. J. Med. Chem. 2005, 40, 1173–1178. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.Y.; Li, C.S.; Cao, L.T.; Bai, X.Q.; Zhao, D.H.; Sun, S.M. New Ursolic Acid Derivatives Bearing 1,2,3-Triazole Moieties: Design, Synthesis and Anti-Inflammatory Activity in Vitro and in Vivo. Mol. Divers. 2022, 26, 1129–1139. [Google Scholar] [CrossRef]
- Kuntala, N.; Mareddy, J.; Telu, J.R.; Banothu, V.; Pal, S.; Anireddy, J.S. Nonsteroidal Anti-Inflammatory Drugs Based New 1,2,3-Triazole Derivatives: Their Design, One-Pot Synthesis and in Vitro Evaluation. J. Heterocycl. Chem. 2021, 58, 2018–2032. [Google Scholar] [CrossRef]
- Alvarez, R.; Velázquez, S.; San-félix, A.; Aquaro, S.; De Clercq, E.; Pemo, C.; Karlsson, A.; Balzarini, J.; Camarasa, M.J. 1,2,3-Triazole-[2,5-Bis-O-(Tert-Butyldimethylsilyl)-Beta.-D-Ribofuranosyl]-3′-Spiro-5″-(4″-Amino-1″,2″-Oxathiole 2″,2″-Dioxide) (TSAO) Analogs: Synthesis and Anti-HIV-1 Activity. J. Med. Chem. 1994, 37, 4185–4194. [Google Scholar] [CrossRef] [PubMed]
- Silva, F.d.C.d.; de Souza, M.C.B.V.; Frugulhetti, I.I.P.; Castro, H.C.; Souza, S.L.d.O.; de Souza, T.M.L.; Rodrigues, D.Q.; Souza, A.M.T.; Abreu, P.A.; Passamani, F.; et al. Synthesis, HIV-RT Inhibitory Activity and SAR of 1-Benzyl-1H-1,2,3-Triazole Derivatives of Carbohydrates. Eur. J. Med. Chem. 2009, 44, 373–383. [Google Scholar] [CrossRef]
- Jordão, A.K.; Afonso, P.P.; Ferreira, V.F.; de Souza, M.C.B.V.; Almeida, M.C.B.; Beltrame, C.O.; Paiva, D.P.; Wardell, S.M.S.V.; Wardell, J.L.; Tiekink, E.R.T.; et al. Antiviral Evaluation of N-Amino-1,2,3-Triazoles against Cantagalo Virus Replication in Cell Culture. Eur. J. Med. Chem. 2009, 44, 3777–3783. [Google Scholar] [CrossRef]
- da Silva, E.N.; Menna-Barreto, R.F.S.; Pinto, M.d.C.F.R.; Silva, R.S.F.; Teixeira, D.V.; de Souza, M.C.B.V.; De Simone, C.A.; De Castro, S.L.; Ferreira, V.F.; Pinto, A.V. Naphthoquinoidal [1,2,3]-Triazole, a New Structural Moiety Active against Trypanosoma Cruzi. Eur. J. Med. Chem. 2008, 43, 1774–1780. [Google Scholar] [CrossRef]
- da Silva Júnior, E.N.; de Souza, M.C.B.V.; Fernandes, M.C.; Menna-Barreto, R.F.S.; Pinto, M.d.C.F.R.; de Assis Lopes, F.; de Simone, C.A.; Andrade, C.K.Z.; Pinto, A.V.; Ferreira, V.F.; et al. Synthesis and Anti-Trypanosoma Cruzi Activity of Derivatives from nor-Lapachones and Lapachones. Bioorg. Med. Chem. 2008, 16, 5030–5038. [Google Scholar] [CrossRef]
- Gill, C.; Jadhav, G.; Shaikh, M.; Kale, R.; Ghawalkar, A.; Nagargoje, D.; Shiradkar, M. Clubbed [1,2,3] Triazoles by Fluorine Benzimidazole: A Novel Approach to H37Rv Inhibitors as a Potential Treatment for Tuberculosis. Bioorg. Med. Chem. Lett. 2008, 18, 6244–6247. [Google Scholar] [CrossRef] [PubMed]
- Upadhayaya, R.S.; Kulkarni, G.M.; Vasireddy, N.R.; Vandavasi, J.K.; Dixit, S.S.; Sharma, V.; Chattopadhyaya, J. Design, Synthesis and Biological Evaluation of Novel Triazole, Urea and Thiourea Derivatives of Quinoline against Mycobacterium Tuberculosis. Bioorg. Med. Chem. 2009, 17, 4681–4692. [Google Scholar] [CrossRef]
- Wilkinson, B.L.; Bornaghi, L.F.; Wright, A.D.; Houston, T.A.; Poulsen, S.A. Anti-Mycobacterial Activity of a Bis-Sulfonamide. Bioorg. Med. Chem. Lett. 2007, 17, 1355–1357. [Google Scholar] [CrossRef] [PubMed]
- Castagnolo, D.; Radi, M.; Dessì, F.; Manetti, F.; Saddi, M.; Meleddu, R.; De Logu, A.; Botta, M. Synthesis and Biological Evaluation of New Enantiomerically Pure Azole Derivatives as Inhibitors of Mycobacterium Tuberculosis. Bioorg. Med. Chem. Lett. 2009, 19, 2203–2205. [Google Scholar] [CrossRef] [PubMed]
- Dabak, K.; Sezer, Ö.; Akar, A.; Anaç, O. Synthesis and Investigation of Tuberculosis Inhibition Activities of Some 1,2,3-Triazole Derivatives. Eur. J. Med. Chem. 2003, 38, 215–218. [Google Scholar] [CrossRef]
- Costa, M.S.; Boechat, N.; Rangel, É.A.; da Silva, F.d.C.; de Souza, A.M.T.; Rodrigues, C.R.; Castro, H.C.; Junior, I.N.; Lourenço, M.C.S.; Wardell, S.M.S.V.; et al. Synthesis, Tuberculosis Inhibitory Activity, and SAR Study of N-Substituted-Phenyl-1,2,3-Triazole Derivatives. Bioorg. Med. Chem. 2006, 14, 8644–8653. [Google Scholar] [CrossRef]
- Ferreira, S.B.; Costa, M.S.; Boechat, N.; Bezerra, R.J.S.; Genestra, M.S.; Canto-Cavalheiro, M.M.; Kover, W.B.; Ferreira, V.F. Synthesis and Evaluation of New Difluoromethyl Azoles as Antileishmanial Agents. Eur. J. Med. Chem. 2007, 42, 1388–1395. [Google Scholar] [CrossRef]
- Beena; Kumar, N.; Rohilla, R.K.; Roy, N.; Rawat, D.S. Synthesis and Antibacterial Activity Evaluation of Metronidazole-Triazole Conjugates. Bioorg. Med. Chem. Lett. 2009, 19, 1396–1398. [Google Scholar] [CrossRef]
- Poonia, N.; Kumar, A.; Kumar, V.; Yadav, M.; Lal, K. Recent Progress in 1H-1,2,3-Triazoles as Potential Antifungal Agents. Curr. Top Med. Chem. 2021, 21, 2109–2133. [Google Scholar] [CrossRef]
- Kong, Y.; Wang, K.; Edler, M.C.; Hamel, E.; Mooberry, S.L.; Paige, M.A.; Brown, M.L. A Boronic Acid Chalcone Analog of Combretastatin A-4 as a Potent Anti-Proliferation Agent. Bioorg. Med. Chem. 2010, 18, 971–977. [Google Scholar] [CrossRef]
- Dhaliwal, J.S.; Moshawih, S.; Goh, K.W.; Loy, M.J.; Hossain, S.; Hermansyah, A.; Kotra, V.; Kifli, N.; Goh, H.P.; Kaur, S.; et al. Pharmacotherapeutics Applications and Chemistry of Chalcone Derivatives. Molecules 2022, 27, 7062. [Google Scholar] [CrossRef]
- Zhuang, C.; Zhang, W.; Sheng, C.; Zhang, W.; Xing, C.; Miao, Z. Chalcone: A Privileged Structure in Medicinal Chemistry. Chem. Rev. 2017, 117, 7762–7810. [Google Scholar] [CrossRef] [PubMed]
- Mezgebe, K.; Melaku, Y.; Mulugeta, E. Synthesis and Pharmacological Activities of Chalcone and Its Derivatives Bearing N-Heterocyclic Scaffolds: A Review. ACS Omega 2023, 8, 19194–19211. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, H.H.H.; Abd El-Hafeez, A.A.; Ebeid, K.; Mekkawy, A.I.; Abourehab, M.A.S.; Wafa, E.I.; Alhaj-Suliman, S.O.; Salem, A.K.; Ghosh, P.; Abuo-Rahma, G.E.D.A.; et al. New 1,2,3-Triazole Linked Ciprofloxacin-Chalcones Induce DNA Damage by Inhibiting Human Topoisomerase I& II and Tubulin Polymerization. J. Enzyme Inhib. Med. Chem. 2022, 37, 1346–1363. [Google Scholar] [CrossRef]
- Zhang, S.Y.; Fu, D.J.; Yue, X.X.; Liu, Y.C.; Song, J.; Sun, H.H.; Liu, H.M.; Zhang, Y.B. Design, Synthesis and Structure-Activity Relationships of Novel Chalcone-1,2,3-Triazole-Azole Derivates as Antiproliferative Agents. Molecules 2016, 21, 653. [Google Scholar] [CrossRef] [PubMed]
- Kınalı, M.; Çol, S.; Çoban, C.Ç.; Türk, M.; Aydın, G.; Emirik, M.; Baran, A. Chalcone-Based Dipolar Cycloaddition of Novel Heteroaromatic Compounds: Their Anticancer Examination. J. Mol. Struct. 2023, 1293, 136244. [Google Scholar] [CrossRef]
- Yadav, P.; Lal, K.; Kumar, A.; Guru, S.K.; Jaglan, S.; Bhushan, S. Green Synthesis and Anticancer Potential of Chalcone Linked-1,2,3-Triazoles. Eur. J. Med. Chem. 2017, 126, 944–953. [Google Scholar] [CrossRef]
- Zhao, L.; Mao, L.; Hong, G.; Yang, X.; Liu, T. Design, Synthesis and Anticancer Activity of Matrine-1H-1,2,3-Triazole-Chalcone Conjugates. Bioorg. Med. Chem. Lett. 2015, 25, 2540–2544. [Google Scholar] [CrossRef]
- Constantinescu, T.; Lungu, C.N. Anticancer Activity of Natural and Synthetic Chalcones. Int. J. Mol. Sci. 2021, 22, 11306. [Google Scholar] [CrossRef]
- Ouyang, Y.; Li, J.; Chen, X.; Fu, X.; Sun, S.; Wu, Q. Chalcone Derivatives: Role in Anticancer Therapy. Biomolecules 2021, 11, 894. [Google Scholar] [CrossRef]
- Yadav, M.; Lal, K.; Kumar, A.; Kumar, A.; Kumar, D. Indole-Chalcone Linked 1,2,3-Triazole Hybrids: Facile Synthesis, Antimicrobial Evaluation and Docking Studies as Potential Antimicrobial Agents. J. Mol. Struct. 2022, 1261, 132867. [Google Scholar] [CrossRef]
- Singh, G.; Arora, A.; Kalra, P.; Maurya, I.K.; Ruizc, C.E.; Estebanc, M.A.; Sinha, S.; Goyal, K.; Sehgal, R. A Strategic Approach to the Synthesis of Ferrocene Appended Chalcone Linked Triazole Allied Organosilatranes: Antibacterial, Antifungal, Antiparasitic and Antioxidant Studies. Bioorg. Med. Chem. 2019, 27, 188–195. [Google Scholar] [CrossRef]
- Mahapatra, D.K.; Bharti, S.K.; Asati, V. Chalcone Scaffolds as Anti-Infective Agents: Structural and Molecular Target Perspectives. Eur. J. Med. Chem. 2015, 101, 496–524. [Google Scholar] [CrossRef]
- Singh, P.; Anand, A.; Kumar, V. Recent Developments in Biological Activities of Chalcones: A Mini Review. Eur. J. Med. Chem. 2014, 85, 758–777. [Google Scholar] [CrossRef]
- Sooknual, P.; Pingaew, R.; Phopin, K.; Ruankham, W.; Prachayasittikul, S.; Ruchirawat, S.; Prachayasittikul, V. Synthesis and Neuroprotective Effects of Novel Chalcone-Triazole Hybrids. Bioorg. Chem. 2020, 105, 104384. [Google Scholar] [CrossRef]
- Chen, X.B.; Shi, D.Q. Synthesis and Biological Activity of Novel Phosphonate Derivatives Containing of Pyridyl and 1,2,3-Triazole Rings. Phosphorus Sulfur Silicon Relat. Elem. 2008, 183, 1134–1144. [Google Scholar] [CrossRef]
- Nelson, R.; Kesternich, V.; Pérez-Fehrmann, M.; Jaldin, S.; Marcourt, L.; Christen, P. Regiospecific Synthesis of 1,4,5-Trisubstituted 1,2,3-Triazoles via Enolate-Azide Cycloaddition between 1,3-Dicarbonyl Compounds and Aryl Azides. J. Chem. Res. 2016, 40, 453–457. [Google Scholar] [CrossRef]
- Jarag, K.J.; Pinjari, D.V.; Pandit, A.B.; Shankarling, G.S. Synthesis of Chalcone (3-(4-Fluorophenyl)-1-(4-Methoxyphenyl)Prop-2-En-1-One): Advantage of Sonochemical Method over Conventional Method. Ultrason. Sonochem. 2011, 18, 617–623. [Google Scholar] [CrossRef] [PubMed]
- Haghi, A.; Salami, M.; Kian, M.M.; Nikbakht, M.; Mohammadi, S.; Chahardouli, B.; Rostami, S.; Malekzadeh, K. Effects of Sorafenib and Arsenic Trioxide on U937 and KG-1 Cell Lines: Apoptosis or Autophagy? Cell J. 2020, 22, 253–262. [Google Scholar] [CrossRef]
- Inaba, H.; Rubnitz, J.E.; Coustan-Smith, E.; Li, L.; Furmanski, B.D.; Mascara, G.P.; Heym, K.M.; Christensen, R.; Onciu, M.; Shurtleff, S.A.; et al. Phase I Pharmacokinetic and Pharmacodynamic Study of the Multikinase Inhibitor Sorafenib in Combination with Clofarabine and Cytarabine in Pediatric Relapsed/Refractory Leukemia. J. Clin. Oncol. 2011, 29, 3293–3300. [Google Scholar] [CrossRef]
- McTigue, M.; Murray, B.W.; Chen, J.H.; Deng, Y.L.; Solowiej, J.; Kania, R.S. Molecular Conformations, Interactions, and Properties Associated with Drug Efficiency and Clinical Performance among VEGFR TK Inhibitors. Proc. Natl. Acad. Sci. USA 2012, 109, 18281–18289. [Google Scholar] [CrossRef]
- Gundemir, S.; Monteagudo, A.; Akbar, A.; Keillor, J.W.; Johnson, G.V.W. The Complex Role of Transglutaminase 2 in Glioblastoma Proliferation. Neuro. Oncol. 2017, 19, 208–218. [Google Scholar] [CrossRef] [PubMed]
- Lindemann, I.; Boettcher, J.; Oertel, K.; Weber, J.; Hils, M.; Pasternack, R.; Heine, A.; Klebe, G. Inhibitors of Transglutaminase 2: A Therapeutic Option in Celiac Disease. RCSB Protein Data Bank 2011, 3S3J. [Google Scholar] [CrossRef]
- Canale, M.; Monti, M.; Rapposelli, I.G.; Ulivi, P.; Sullo, F.G.; Bartolini, G.; Tiberi, E.; Frassineti, G.L. Gallbladder Cancer. Cancers 2021, 12, 5671. [Google Scholar] [CrossRef]
- Gonzalez-Del Pino, G.L.; Li, K.; Park, E.; Schmoker, A.M.; Ha, B.H.; Eck, M.J. Allosteric MEK Inhibitors Act on BRAF/MEK Complexes to Block MEK Activation. Proc. Natl. Acad. Sci. USA 2021, 118, e2107207118. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Fehrmann, M.; Kesternich, V.; Puelles, A.; Quezada, V.; Salazar, F.; Christen, P.; Castillo, J.; Cárcamo, J.G.; Castro-Alvarez, A.; Nelson, R. Synthesis, Antitumor Activity, 3D-QSAR and Molecular Docking Studies of New Iodinated 4-(3H)-Quinazolinones 3N-Substituted. RSC Adv. 2022, 12, 21340–21352. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid Colorimetric Assay for Cellular Growth and Survival: Application to Proliferation and Cytotoxicity Assays. J. Lmmunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- González-Calderón, D.; Santillán-Iniesta, I.; González-González, C.A.; Fuentes-Benítes, A.; González-Romero, C. A Novel and Facile Synthesis of 1,4,5-Trisubstituted 1,2,3-Triazoles from Benzylic Alcohols through a One-Pot, Three-Component System. Tetrahedron Lett. 2015, 56, 514–516. [Google Scholar] [CrossRef]
- Shafran, Y.M.; Beryozkina, T.V.; Efimov, I.V.; Bakulev, V.A. Synthesis of β-Azolyl- and β-Azolylcarbonylenamines and Their Reactions with Aromatic Azides. Chem. Heterocycl. Compd. 2019, 55, 704–715. [Google Scholar] [CrossRef]
- Jin, G.; Zhang, J.; Fu, D.; Wu, J.; Cao, S. One-Pot, Three-Component Synthesis of 1,4,5-Trisubstituted 1,2,3-Triazoles Starting from Primary Alcohols. Eur. J. Org. Chem. 2012, 2012, 5446–5449. [Google Scholar] [CrossRef]
- Pérez-Ferhmann, M.; Kesternich, V.; Cáceres, M.; Salazar, F.; Cárdenas, A.; Cataldo, F.; Brito, I. Crystal Structure of 1-(1-(2,4-Dichlorobenzyl)-5-Methyl-1H-1,2,3-Triazol-4-yl)Ethanone, C12H11Cl2N3O. Z. Fur. Krist.—New Cryst. Struct. 2014, 229, 367–368. [Google Scholar] [CrossRef]
- Shanmugavelan, P.; Sathishkumar, M.; Nagarajan, S.; Ponnuswamy, A. A Facile Synthesis of 1,2,3-Triazolyl Indole Hybrids via SbCl3-Catalysed Michael Addition of Indoles to 1,2,3-Triazolyl Chalcones. J. Chem. Sci. 2012, 124, 941–950. [Google Scholar] [CrossRef]
- Bhabha, G.; Ekiert, D.C.; Jennewein, M.; Zmasek, C.M.; Tuttle, L.M.; Kroon, G.; Dyson, H.J.; Godzik, A.; Wilson, I.A.; Wright, P.E. Divergent Evolution of Protein Conformational Dynamics in Dihydrofolate Reductase. Nat. Struct. Mol. Biol. 2013, 20, 1243–1249. [Google Scholar] [CrossRef]
- Schrödinger Release 2022-1: LigPrep; Schrödinger LLC: New York, NY, USA, 2022.
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein-Ligand Complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 2. Enrichment Factors in Database Screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comp. | R1 | R2 | R3 | R4 | R5 | Yield (%) |
|---|---|---|---|---|---|---|
| 4a | H | Cl | H | H | Br | 90 |
| 4b | H | Cl | H | H | Cl | 85 |
| 4c | H | Cl | H | H | OMe | 91 |
| 4d | H | Cl | Cl | H | Cl | 90 |
| 4e | H | Cl | Cl | H | H | 75 |
| 4f | H | Br | H | Br | H | 82 |
| 4g | H | Cl | H | NO2 | H | 60 |
| 4h | H | Br | Cl | H | H | 85 |
| 4i | H | Br | Cl | H | Cl | 71 |
| 4j | H | Br | H | H | OMe | 56 |
| 4k | H | Br | OMe | H | H | 85 |
| 4l | H | Br | H | OMe | OMe | 79 |
| 4m | H | H | H | H | H | 84 |
| 4n | H | H | H | H | Cl | 90 |
| 4o | H | H | Cl | H | Cl | 98 |
| 4p | H | H | H | H | OMe | 50 |
| 4q | Cl | Cl | H | H | Cl | 95 |
| 4r | Cl | Cl | Cl | H | H | 86 |
| 4s | Cl | Cl | Cl | H | Cl | 98 |
| 4t | Cl | Cl | H | OMe | OMe | 80 |
| 4v | OMe | H | H | H | Br | 80 |
| 1H-NMR (δ/ppm) | 13C-NMR (δ/ppm) | IR (cm−1) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Comp. | R1 | R2 | R3 | R4 | R5 | Hα | Hβ | J (Hz) | C=O | C=O |
| 4a | H | Cl | H | H | Br | 7.80 | 8.02 | 16.0 | 184.2 | 1684 |
| 4b | H | Cl | H | H | Cl | 7.82 | 8.00 | 16.0 | 184.2 | 1661 |
| 4c | H | Cl | H | H | OMe | 7.82 | 7.90 | 15.9 | 184.3 | 1656 |
| 4d | H | Cl | Cl | H | Cl | 8.00 | 8.22 | 16.0 | 183.8 | 1669 |
| 4e | H | Cl | Cl | H | H | 8.03 | 8.31 | 15.9 | 184.1 | 1667 |
| 4f | H | Br | H | Br | H | 7.78 | 8.02 | 16.0 | 183.9 | 1667 |
| 4g | H | Cl | H | NO2 | H | 7.89 | 8.13 | 16.0 | 183.4 | 1661 |
| 4h | H | Br | Cl | H | H | 8.03 | 8.31 | 16.0 | 184.1 | 1667 |
| 4i | H | Br | Cl | H | Cl | 8.00 | 8.22 | 16.0 | 183.8 | 1667 |
| 4j | H | Br | H | H | OMe | 7.85 | 7.92 | 15.9 | 184.5 | 1658 |
| 4k | H | Br | OMe | H | H | 8.07 | 8.26 | 16.1 | 184.8 | 1657 |
| 4l | H | Br | H | OMe | OMe | 7.85 | 7.91 | 15.9 | 184.3 | 1653 |
| 4m | H | H | H | H | H | 7.88 | 8.06 | 16.0 | 184.4 | 1663 |
| 4n | H | H | H | H | Cl | 7.82 | 8.02 | 16.0 | 184.0 | 1662 |
| 4o | H | H | Cl | H | Cl | 7.82 | 8.02 | 15.9 | 183.6 | 1667 |
| 4p | H | H | H | H | OMe | 7.85 | 7.93 | 15.9 | 184.4 | 1657 |
| 4q | Cl | Cl | H | H | Cl | 7.84 | 8.01 | 16.0 | 183.9 | 1666 |
| 4r | Cl | Cl | Cl | H | H | 8.04 | 8.33 | 15.9 | 183.7 | 1666 |
| 4s | Cl | Cl | Cl | H | Cl | 8.02 | 8.24 | 16.0 | 183.6 | 1669 |
| 4t | Cl | Cl | H | OMe | OMe | 7.87 | 7.92 | 16.0 | 184.2 | 1656 |
| 4v | OMe | H | H | H | Br | 7.80 | 7.04 | 16.0 | 184.1 | 1661 |
| VEGFR (U937) | TG2 (T98G) | MEK (Gb-d1) | DHFR (HeLa) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Comp | Activity | Score | Comp | Activity | Score | Comp | Activity | Score | Comp | Activity | Score |
| 4a | 4.60 | −8.54 | 4q | 12.30 | −7.56 | 4k | 20.00 | −8.55 | 4h | 42.50 | −4.84 |
| 4c | 4.40 | −8.00 | 4v | 11.00 | −8.35 | 4o | 16.00 | −8.84 | 4f | 44.50 | −5.59 |
| 4b | 200.00 | −6.32 | 4b | 200.00 | −4.33 | 4b | 200.00 | −7.24 | 4b | 200.00 | −4.10 |
| 4s | 200.00 | −6.14 | 4s | 200.00 | −4.02 | 4s | 200.00 | −7.14 | 4s | 200.00 | −4.45 |
 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| IC50 (µM) | ||||||||||
| Comp. | R1 | R2 | R3 | R4 | R5 | U937 | T98G | Gb-d1 | Hela | G-415 |
| 4a | H | Cl | H | H | Br | 4.6 ± 0.28 | >200 | >200 | >200 | >200 |
| 4b | H | Cl | H | H | Cl | >200 | >200 | >200 | >200 | >200 |
| 4c | H | Cl | H | H | OMe | 4.4 ± 0.2 | >200 | >200 | >200 | >200 |
| 4d | H | Cl | Cl | H | Cl | 10.8 ± 0.6 | 24.6 ± 0.1 | >200 | >200 | >200 |
| 4e | H | Cl | Cl | H | H | 12.3 ± 0.3 | >200 | >200 | >200 | >200 |
| 4f | H | Br | H | Br | H | 11 | 21.8 ± 0.3 | 22.3 ± 1.8 | 42.5 ± 0.7 | 27.6 ± 0.2 |
| 4g | H | Cl | H | NO2 | H | 9.6 ± 0.14 | 16 | 24 | 44.5 ± 2.1 | 51 |
| 4h | H | Br | Cl | H | H | 11.3 ± 0.3 | 22 | 17.2 ± 1.2 | >200 | >200 |
| 4i | H | Br | Cl | H | Cl | 17.5 | 20.3 ± 1.1 | 58.8 ± 2.5 | >200 | >200 |
| 4j | H | Br | H | H | OMe | 9.5 ± 0.7 | 66.5 ± 1.6 | 39.5 ± 0.3 | >200 | >200 |
| 4k | H | Br | OMe | H | H | 17.8 ± 0.4 | 44.3 ± 1.8 | 20 | >200 | >200 |
| 4l | H | Br | H | OMe | OMe | 12.3 ± 0.3 | 43.1 | 23 ± 2.8 | >200 | >200 |
| 4m | H | H | H | H | H | 8 | 52 ± 0.7 | 32.2 ± 1.1 | 86 | 26 ± 4.2 |
| 4n | H | H | H | H | Cl | 11 | 45.6 | >200 | >200 | >200 |
| 4o | H | H | Cl | H | Cl | 10 | 29 | 16 ± 3.5 | 95.5 ± 0.7 | >200 |
| 4p | H | H | H | H | OMe | 83 ± 1.4 | >200 | >200 | >200 | >200 |
| 4q | Cl | Cl | H | H | Cl | >200 | 12.3 ± 0.3 | >200 | >200 | >200 |
| 4r | Cl | Cl | Cl | H | H | >200 | 14 | >200 | >200 | >200 |
| 4s | Cl | Cl | Cl | H | Cl | >200 | >200 | >200 | >200 | >200 |
| 4t | Cl | Cl | H | OMe | OMe | 16 | >200 | 20 ± 0.7 | >200 | >200 |
| 4v | OMe | H | H | H | Br | 8.2 ± 0.3 | 11 ± 0.9 | >200 | >200 | >200 |
| Paclitaxel | 6 | 21 | 41 | 6.2 | 10 | |||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cáceres, M.; Kesternich, V.; Pérez-Fehrmann, M.; Castroagudin, M.; Nelson, R.; Quezada, V.; Christen, P.; Castro-Alvarez, A.; Cárcamo, J.G. Ultrasound-Assisted Synthesis of Substituted Chalcone-Linked 1,2,3-Triazole Derivatives as Antiproliferative Agents: In Vitro Antitumor Activity and Molecular Docking Studies. Int. J. Mol. Sci. 2025, 26, 3389. https://doi.org/10.3390/ijms26073389
Cáceres M, Kesternich V, Pérez-Fehrmann M, Castroagudin M, Nelson R, Quezada V, Christen P, Castro-Alvarez A, Cárcamo JG. Ultrasound-Assisted Synthesis of Substituted Chalcone-Linked 1,2,3-Triazole Derivatives as Antiproliferative Agents: In Vitro Antitumor Activity and Molecular Docking Studies. International Journal of Molecular Sciences. 2025; 26(7):3389. https://doi.org/10.3390/ijms26073389
Chicago/Turabian StyleCáceres, Manuel, Víctor Kesternich, Marcia Pérez-Fehrmann, Mariña Castroagudin, Ronald Nelson, Víctor Quezada, Philippe Christen, Alejandro Castro-Alvarez, and Juan G. Cárcamo. 2025. "Ultrasound-Assisted Synthesis of Substituted Chalcone-Linked 1,2,3-Triazole Derivatives as Antiproliferative Agents: In Vitro Antitumor Activity and Molecular Docking Studies" International Journal of Molecular Sciences 26, no. 7: 3389. https://doi.org/10.3390/ijms26073389
APA StyleCáceres, M., Kesternich, V., Pérez-Fehrmann, M., Castroagudin, M., Nelson, R., Quezada, V., Christen, P., Castro-Alvarez, A., & Cárcamo, J. G. (2025). Ultrasound-Assisted Synthesis of Substituted Chalcone-Linked 1,2,3-Triazole Derivatives as Antiproliferative Agents: In Vitro Antitumor Activity and Molecular Docking Studies. International Journal of Molecular Sciences, 26(7), 3389. https://doi.org/10.3390/ijms26073389