
PNPLA3 Polymorphism Is Inversely Correlated with Aortic Stiffness in Patients with Metabolic Dysfunction-Associated Steatotic Liver Disease Without Fibrosis

 ,
, 
 , ,
, ,
Abstract
:1. Introduction
2. Results
2.1. MASLD Patients’ Characteristics
2.2. Observed and Reported Allele Frequencies of the Minor G Allele in PNPLA3 rs738409
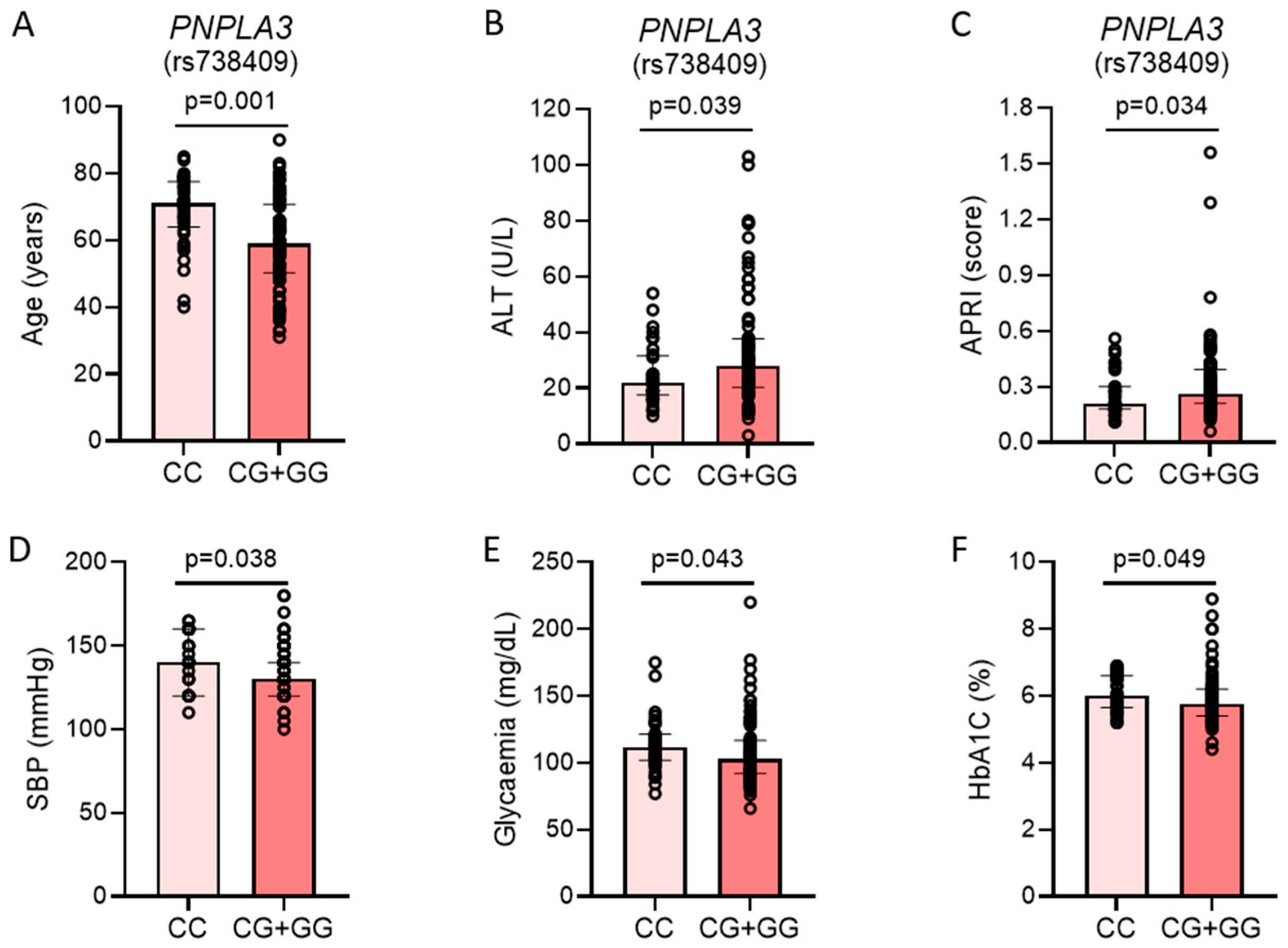
2.3. Metabolic Differences Between Patients with and Without PNPLA3 rs738409[G] Allele
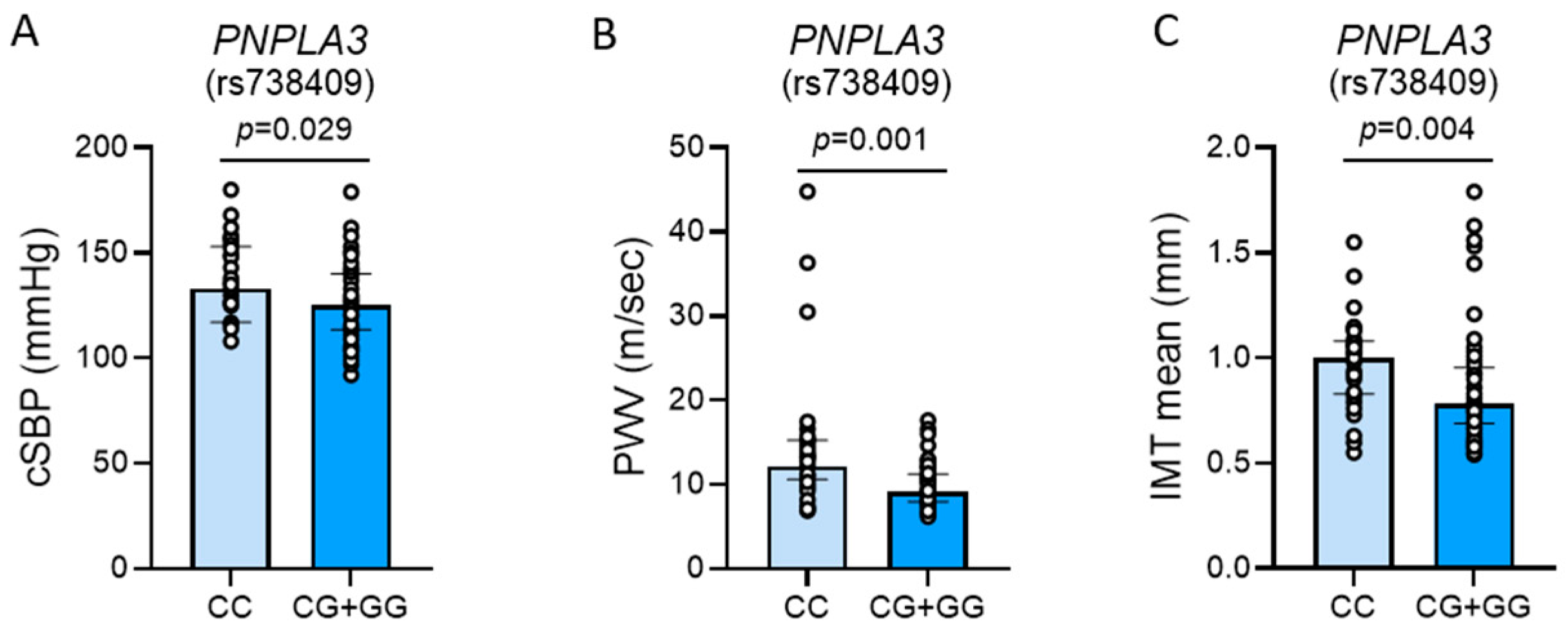
2.4. Vascular Differences Between Patients with and Without PNPLA3 rs738409[G] Allele
2.5. Multivariate Regression Analysis
3. Discussion
4. Materials and Methods
4.1. Study Design
4.2. Clinical Assessment, General Biochemistries, and Noninvasive Assessment of Liver Fibrosis
4.3. PNPLA3 rs738409 Genotyping
4.4. Arterial Tonometry
4.5. Ultrasound Examination of the Carotid Arteries
4.6. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Targher, G.; Byrne, C.D.; Tilg, H. MASLD: A systemic metabolic disorder with cardiovascular and malignant complications. Gut 2024, 73, 691–702. [Google Scholar] [CrossRef] [PubMed]
- Rinella, M.E.; Lazarus, J.V.; Ratziu, V.; Francque, S.M.; Sanyal, A.J.; Kanwal, F.; Romero, D.; Abdelmalek, M.F.; Anstee, Q.M.; Arab, J.P.; et al. A multisociety Delphi consensus statement on new fatty liver disease nomenclature. Hepatology 2023, 78, 1966–1986. [Google Scholar] [CrossRef]
- Romeo, S.; Kozlitina, J.; Xing, C.; Pertsemlidis, A.; Cox, D.; Pennacchio, L.A.; Boerwinkle, E.; Cohen, J.C.; Hobbs, H.H. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2008, 40, 1461–1465. [Google Scholar] [CrossRef] [PubMed]
- Hyysalo, J.; Gopalacharyulu, P.; Bian, H.; Hyotylainen, T.; Leivonen, M.; Jaser, N.; Juuti, A.; Honka, M.J.; Nuutila, P.; Olkkonen, V.M.; et al. Circulating triacylglycerol signatures in nonalcoholic fatty liver disease associated with the I148M variant in PNPLA3 and with obesity. Diabetes 2014, 63, 312–322. [Google Scholar] [CrossRef]
- Salameh, H.; Hanayneh, M.A.; Masadeh, M.; Naseemuddin, M.; Matin, T.; Erwin, A.; Singal, A.K. PNPLA3 as a Genetic Determinant of Risk for and Severity of Non-alcoholic Fatty Liver Disease Spectrum. J. Clin. Transl. Hepatol. 2016, 4, 175–191. [Google Scholar] [CrossRef] [PubMed]
- Unalp-Arida, A.; Ruhl, C.E. Patatin-Like Phospholipase Domain-Containing Protein 3 I148M and Liver Fat and Fibrosis Scores Predict Liver Disease Mortality in the U.S. Population. Hepatology 2020, 71, 820–834. [Google Scholar] [CrossRef]
- Lonardo, A.; Ballestri, S.; Targher, G. “Not all forms of NAFLD were created equal”. Do metabolic syndrome-related NAFLD and PNPLA3-related NAFLD exert a variable impact on the risk of early carotid atherosclerosis? Atherosclerosis 2017, 257, 253–255. [Google Scholar] [CrossRef]
- Pipitone, R.M.; Ciccioli, C.; Infantino, G.; La Mantia, C.; Parisi, S.; Tulone, A.; Pennisi, G.; Grimaudo, S.; Petta, S. MAFLD: A multisystem disease. Ther. Adv. Endocrinol. Metab. 2023, 14, 1–23. [Google Scholar] [CrossRef]
- Petta, S.; Valenti, L.; Marchesini, G.; Di Marco, V.; Licata, A.; Camma, C.; Barcellona, M.R.; Cabibi, D.; Donati, B.; Fracanzani, A.; et al. PNPLA3 GG genotype and carotid atherosclerosis in patients with non-alcoholic fatty liver disease. PLoS ONE 2013, 8, e74089. [Google Scholar] [CrossRef]
- Di Costanzo, A.; D’Erasmo, L.; Polimeni, L.; Baratta, F.; Coletta, P.; Di Martino, M.; Loffredo, L.; Perri, L.; Ceci, F.; Montali, A.; et al. Non-alcoholic fatty liver disease and subclinical atherosclerosis: A comparison of metabolically- versus genetically-driven excess fat hepatic storage. Atherosclerosis 2017, 257, 232–239. [Google Scholar] [CrossRef]
- Castaldo, L.; Laguzzi, F.; Strawbridge, R.J.; Baldassarre, D.; Veglia, F.; Vigo, L.; Tremoli, E.; de Faire, U.; Eriksson, P.; Smit, A.J.; et al. Genetic Variants Associated with Non-Alcoholic Fatty Liver Disease Do Not Associate with Measures of Sub-Clinical Atherosclerosis: Results from the IMPROVE Study. Genes 2020, 11, 1243. [Google Scholar] [CrossRef]
- Kozlitina, J.; Sookoian, S. Global Epidemiological Impact of PNPLA3 I148M on Liver Disease. Liver Int. 2025, 45, e16123. [Google Scholar] [CrossRef]
- Gil-Gomez, A.; Rojas, A.; Garcia-Lozano, M.R.; Munoz-Hernandez, R.; Gallego-Duran, R.; Maya-Miles, D.; Montero-Vallejo, R.; Gato, S.; Gallego, J.; Frances, R.; et al. Impact of a Loss-of-Function Variant in HSD17B13 on Hepatic Decompensation and Mortality in Cirrhotic Patients. Int. J. Mol. Sci. 2022, 23, 11840. [Google Scholar] [CrossRef] [PubMed]
- Vespasiani-Gentilucci, U.; Gallo, P.; Porcari, A.; Carotti, S.; Galati, G.; Piccioni, L.; De Vincentis, A.; Dell’Unto, C.; Vorini, F.; Morini, S.; et al. The PNPLA3 rs738409 C > G polymorphism is associated with the risk of progression to cirrhosis in NAFLD patients. Scand. J. Gastroenterol. 2016, 51, 967–973. [Google Scholar] [CrossRef]
- Chirinos, J.A.; Segers, P.; Hughes, T.; Townsend, R. Large-Artery Stiffness in Health and Diseasse: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2019, 74, 1237–1263. [Google Scholar] [CrossRef]
- Climie, R.E.; Alastruey, J.; Mayer, C.C.; Schwarz, A.; Laucyte-Cibulskiene, A.; Voicehovska, J.; Bianchini, E.; Bruno, R.M.; Charlton, P.H.; Grillo, A.; et al. Vascular ageing: Moving from bench towards bedside. Eur. J. Prev. Cardiol. 2023, 30, 1101–1117. [Google Scholar] [CrossRef] [PubMed]
- Salvi, P.; Valbusa, F.; Kearney-Schwartz, A.; Labat, C.; Grillo, A.; Parati, G.; Benetos, A. Non-Invasive Assessment of Arterial Stiffness: Pulse Wave Velocity, Pulse Wave Analysis and Carotid Cross-Sectional Distensibility: Comparison between Methods. J. Clin. Med. 2022, 11, 2225. [Google Scholar] [CrossRef]
- Van Bortel, L.M.; Laurent, S.; Boutouyrie, P.; Chowienczyk, P.; Cruickshank, J.K.; De Backer, T.; Filipovsky, J.; Huybrechts, S.; Mattace-Raso, F.U.; Protogerou, A.D.; et al. Expert consensus document on the measurement of aortic stiffness in daily practice using carotid-femoral pulse wave velocity. J. Hypertens. 2012, 30, 445–448. [Google Scholar] [CrossRef]
- Townsend, R.R.; Wilkinson, I.B.; Schiffrin, E.L.; Avolio, A.P.; Chirinos, J.A.; Cockcroft, J.R.; Heffernan, K.S.; Lakatta, E.G.; McEniery, C.M.; Mitchell, G.F.; et al. Recommendations for Improving and Standardizing Vascular Research on Arterial Stiffness: A Scientific Statement From the American Heart Association. Hypertension 2015, 66, 698–722. [Google Scholar] [CrossRef]
- Mancia, G.; Kreutz, R.; Brunstrom, M.; Burnier, M.; Grassi, G.; Januszewicz, A.; Muiesan, M.L.; Tsioufis, K.; Agabiti-Rosei, E.; Algharably, E.A.E.; et al. 2023 ESH Guidelines for the management of arterial hypertension The Task Force for the management of arterial hypertension of the European Society of Hypertension: Endorsed by the International Society of Hypertension (ISH) and the European Renal Association (ERA). J. Hypertens. 2023, 41, 1874–2071. [Google Scholar] [CrossRef]
- Pirazzi, C.; Adiels, M.; Burza, M.A.; Mancina, R.M.; Levin, M.; Stahlman, M.; Taskinen, M.R.; Orho-Melander, M.; Perman, J.; Pujia, A.; et al. Patatin-like phospholipase domain-containing 3 (PNPLA3) I148M (rs738409) affects hepatic VLDL secretion in humans and in vitro. J. Hepatol. 2012, 57, 1276–1282. [Google Scholar] [CrossRef] [PubMed]
- Walker, R.W.; Belbin, G.M.; Sorokin, E.P.; Van Vleck, T.; Wojcik, G.L.; Moscati, A.; Gignoux, C.R.; Cho, J.; Abul-Husn, N.S.; Nadkarni, G.; et al. A common variant in PNPLA3 is associated with age at diagnosis of NAFLD in patients from a multi-ethnic biobank. J. Hepatol. 2020, 72, 1070–1081. [Google Scholar] [CrossRef]
- Habib, S. Metabolic dysfunction-associated steatotic liver disease heterogeneity: Need of subtyping. World J. Gastrointest. Pathophysiol. 2024, 15, 92791. [Google Scholar] [CrossRef]
- Pais, R.; Giral, P.; Khan, J.F.; Rosenbaum, D.; Housset, C.; Poynard, T.; Ratziu, V.; Group, L.S. Fatty liver is an independent predictor of early carotid atherosclerosis. J. Hepatol. 2016, 65, 95–102. [Google Scholar] [CrossRef]
- Zhou, Y.Y.; Zhou, X.D.; Wu, S.J.; Fan, D.H.; Van Poucke, S.; Chen, Y.P.; Fu, S.W.; Zheng, M.H. Nonalcoholic fatty liver disease contributes to subclinical atherosclerosis: A systematic review and meta-analysis. Hepatol. Commun. 2018, 2, 376–392. [Google Scholar] [CrossRef] [PubMed]
- Targher, G.; Bertolini, L.; Padovani, R.; Rodella, S.; Tessari, R.; Zenari, L.; Day, C.; Arcaro, G. Prevalence of nonalcoholic fatty liver disease and its association with cardiovascular disease among type 2 diabetic patients. Diabetes Care 2007, 30, 1212–1218. [Google Scholar] [CrossRef]
- Mantovani, A.; Csermely, A.; Petracca, G.; Beatrice, G.; Corey, K.E.; Simon, T.G.; Byrne, C.D.; Targher, G. Non-alcoholic fatty liver disease and risk of fatal and non-fatal cardiovascular events: An updated systematic review and meta-analysis. Lancet Gastroenterol. Hepatol. 2021, 6, 903–913. [Google Scholar] [CrossRef]
- Baratta, F.; D’Erasmo, L.; Bini, S.; Pastori, D.; Angelico, F.; Del Ben, M.; Arca, M.; Di Costanzo, A. Heterogeneity of non-alcoholic fatty liver disease (NAFLD): Implication for cardiovascular risk stratification. Atherosclerosis 2022, 357, 51–59. [Google Scholar] [CrossRef]
- Wu, J.T.; Liu, S.S.; Xie, X.J.; Liu, Q.; Xin, Y.N.; Xuan, S.Y. Independent and joint correlation of PNPLA3 I148M and TM6SF2 E167K variants with the risk of coronary heart disease in patients with non-alcoholic fatty liver disease. Lipids Health Dis. 2020, 19, 29. [Google Scholar] [CrossRef]
- Akuta, N.; Kawamura, Y.; Arase, Y.; Saitoh, S.; Fujiyama, S.; Sezaki, H.; Hosaka, T.; Kobayashi, M.; Kobayashi, M.; Suzuki, Y.; et al. PNPLA3 genotype and fibrosis-4 index predict cardiovascular diseases of Japanese patients with histopathologically-confirmed NAFLD. BMC Gastroenterol. 2021, 21, 434. [Google Scholar] [CrossRef]
- Ruschenbaum, S.; Schwarzkopf, K.; Friedrich-Rust, M.; Seeger, F.; Schoelzel, F.; Martinez, Y.; Zeuzem, S.; Bojunga, J.; Lange, C.M. Patatin-like phospholipase domain containing 3 variants differentially impact metabolic traits in individuals at high risk for cardiovascular events. Hepatol. Commun. 2018, 2, 798–806. [Google Scholar] [CrossRef] [PubMed]
- Ben-Shlomo, Y.; Spears, M.; Boustred, C.; May, M.; Anderson, S.G.; Benjamin, E.J.; Boutouyrie, P.; Cameron, J.; Chen, C.H.; Cruickshank, J.K.; et al. Aortic pulse wave velocity improves cardiovascular event prediction: An individual participant meta-analysis of prospective observational data from 17,635 subjects. J. Am. Coll. Cardiol. 2014, 63, 636–646. [Google Scholar] [CrossRef]
- Laurent, S.; Boutouyrie, P.; Asmar, R.; Gautier, I.; Laloux, B.; Guize, L.; Ducimetiere, P.; Benetos, A. Aortic stiffness is an independent predictor of all-cause and cardiovascular mortality in hypertensive patients. Hypertension 2001, 37, 1236–1241. [Google Scholar] [CrossRef]
- Bernardi, S.; Giudici, F.; Barbato, V.; Zanatta, L.; Grillo, A.; Fabris, B. Meta-analysis on the Effect of Mild Primary Hyperparathyroidism and Parathyroidectomy Upon Arterial Stiffness. J. Clin. Endocrinol. Metab. 2021, 106, 1832–1843. [Google Scholar] [CrossRef]
- Gasparotto, M.; Di Pierro, G.; Toffoli, B.; Grillo, A.; Bressan, M.; Fiorentin, M.; Di Luozzo, L.; Fischetti, F.; Zen, M.; Fabris, B.; et al. Preliminary Study on Pulse Wave Changes in Patients with Inflammatory Arthropathies Treated with bDMARDs. J. Clin. Med. 2024, 13, 2684. [Google Scholar] [CrossRef] [PubMed]
- Cooper, L.L.; Prescott, B.R.; Xanthakis, V.; Benjamin, E.J.; Vasan, R.S.; Hamburg, N.M.; Long, M.T.; Mitchell, G.F. Association of Aortic Stiffness and Pressure Pulsatility With Noninvasive Estimates of Hepatic Steatosis and Fibrosis: The Framingham Heart Study. Arterioscler. Thromb. Vasc. Biol. 2024, 44, 1704–1715. [Google Scholar] [CrossRef] [PubMed]
- Visseren, F.L.J.; Mach, F.; Smulders, Y.M.; Carballo, D.; Koskinas, K.C.; Back, M.; Benetos, A.; Biffi, A.; Boavida, J.M.; Capodanno, D.; et al. 2021 ESC Guidelines on cardiovascular disease prevention in clinical practice. Eur. Heart J. 2021, 42, 3227–3337. [Google Scholar] [CrossRef]
- Touboul, P.-J.; Hennerici, M.G.; Meairs, S.; Adams, H.; Amarenco, P.; Bornstein, N.; Csiba, L.; Desvarieux, M.; Ebrahim, S.; Hernandez Hernandez, R.; et al. Mannheim Carotid Intima-Media Thickness and Plaque Consensus (2004–2006–2011): An Update on Behalf of the Advisory Board of the 3rd, 4th and 5th Watching the Risk Symposia, at the 13th, 15th and 20th European Stroke Conferences, Mannheim, Germany, 2004, Brussels, Belgium, 2006, and Hamburg, Germany, 2011. Cerebrovasc. Dis. 2012, 34, 290–296. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Variable | All Patients (n = 117) | PNPLA3 CC Genotype (n = 37) | PNPLA3 CG or GG Genotype (n = 80) | p-Value |
|---|---|---|---|---|
| Age (years) | 63 (55–73) | 71 (65–77) | 59 (51–70) | 0.001 |
| Sex M F | 67/117 (57.3%) 50/117 (42.7%) | 25/37 (67.6%) 12/37 (32.4%) | 42/80 (52.5%) 38/80 (47.5%) | 0.12 |
| BMI (Kg/m2) | 28.5 (26.2–29.5) | 27.8 (26.2–31.1) | 29.0 (26.3–32.5) | 0.35 |
| SBP (mmHg) | 135 (120–145) | 140 (120–160) | 130 (120–140) | 0.04 |
| DBP (mmHg) | 80 (70–80) | 80 (70–80) | 80 (70–80) | 0.65 |
| Glycaemia (mg/dL) | 106 (94–119) | 112 (103–121) | 102.5 (92–116) | 0.04 |
| HbA1c (%) | 5.8 (5.5–6.3) | 6.0 (5.7–6.6) | 5.75 (5.4–6.2) | 0.049 |
| HOMA Index | 2.58 (2.01–4.13) | 2.99 (2.54–4.21) | 2.27 (1.65–3.86) | 0.08 |
| Diabetes (yes/no) | 42/117 (35.9%) | 17/37 (45.9%) | 25/80 (31.25%) | 0.12 |
| Metformin treatment | 33/117 (28.2%) | 14/37 (37.8%) | 19/80 (23.75%) | 0.11 |
| GLP-1RA treatment | 10/117 (8.55%) | 3/37 (8.1%) | 7/80 (8.75%) | 0.91 |
| SGLT2i treatment | 16/117 (13.7%) | 4/37 (10.8%) | 12/80 (15.0%) | 0.54 |
| Total cholesterol (mg/dL) | 187 (160–213) | 176 (160–207) | 193 (161–218) | 0.06 |
| HDL (mg/dL) | 50 (43–60) | 53 (44–58) | 50 (43–61) | 0.79 |
| LDL (mg/dL) | 114.7 (88.7–135.6) | 104.2 (70.2–129.8) | 118.2 (90.3–137.9) | 0.03 |
| Triglycerides (mg/dL) | 120 (89–148) | 121 (87–137) | 119.5 (90.5–156) | 0.67 |
| Lipid-lowering therapy (yes/no) | 56/101 (55.45%) | 21/30 (70.0%) | 35/71 (49.3%) | 0.06 |
| Creatinine (mg/dL) | 0.88 (0.71–1.03) | 0.91 (0.81–1.07) | 0.83 (0.70–1.01) | 0.06 |
| AST (U/L) | 24 (20–29) | 24 (19–28) | 23.5 (20–32) | 0.19 |
| ALT (U/L) | 25 (19–36) | 22 (18–31) | 28 (21–37) | 0.04 |
| GGT (U/L) | 29 (20–44) | 28 (19–44) | 30 (21–44) | 0.62 |
| Ferritin (ug/L) | 106.0 (37.2–203.2) | 66.6 (34.8–189.0) | 110.0 (53.9–211.4) | 0.18 |
| NFS (score) | −0.91 (−2.04–0.06) | −0.54 (−1.56–−0.25) | −1.14 (−2.28–0.18) | 0.51 |
| FIB-4 (score) | 1.35 (0.85–1.78) | 1.54 (0.96–1.83) | 1.19 (0.81–1.74) | 0.16 |
| APRI (score) | 0.26 (0.20–0.36). | 0.22 (0.18–0.29) | 0.26 (0.22–0.37) | 0.04 |
| Hepatic stiffness (kPa) | 4.71 (4.02–5.73) | 4.96 (4.12–5.59) | 4.63 (3.98–5.91) | 0.99 |
| Gene (SNPs) | n | Observed Genotype Frequency in Our Cohort | Observed Allele Frequency in Our Cohort | Reported Allele Frequency | p | ||||
|---|---|---|---|---|---|---|---|---|---|
| MM | Mm | mm | M | m | M | m | |||
| PNPLA3 (rs738409) | 117 | CC 37 (31.6%) | CG 59 (50.4%) | GG 21 (18.0%) | C 0.57 | G 0.43 | C 0.79 | G 0.21 | <0.0001 a [12] |
| PNPLA3 (rs738409) | 117 | CC 37 (31.6%) | CG 59 (50.4%) | GG 21 (18.0%) | C 0.57 | G 0.43 | C 0.52 | G 0.48 | 0.328 b [14] |
| Variable | PNPLA3 CC Genotype (n = 37) | PNPLA3 CG or GG Genotype (n = 80) | p-Value | |
|---|---|---|---|---|
| Age (years) | 71 (65–77) | 59 (51–70) | 0.001 | |
| Vascular age (years) | 78 (69–83) | 63 (54–73) | 0.001 | |
| SBP (mmHg) | 136 (125–150) | 130 (118–140) | 0.039 | |
| DBP (mmHg) | 81 (75–84) | 79 (76–86) | 0.842 | |
| PP (mmHg) | 52 (44–69) | 40 (33–56) | 0.004 | |
| MAP (mmHg) | 101 (95–110) | 99 (93–106) | 0.239 | |
| cSBP (mmHg) | 133 (117–152) | 125 (114–139) | 0.029 | |
| AI (%) | 15.7 (4.5–23.2) | 16.5 (5.9–35.4) | 0.283 | |
| SEVR (%) | 100.1 (82.8–113.4) | 105.4 (93.1–119.3) | 0.250 | |
| LVET (ms) | 301 (290–308) | 306 (282–322) | 0.344 | |
| SysS (mmHg/ms) | 0.92 (0.75–1.07) | 0.64 (0.52–0.73) | 0.001 | |
| PWV (m/sec) | 12.2 (10.9–15.2) | 9.1 (8.0–11.2) | 0.001 | |
| IMT mean (mm) | 1.00 (0.84–1.07) | 0.78 (0.69–0.94) | 0.004 | |
| MAX IMT (mm) | 1.52 (1.31–1.81) | 1.30 (1.04–1.77) | 0.129 | |
| 10-year CVD Risk (%) | Low | 3.5 | 13.2 | 0.001 |
| Moderate | 10.3 | 37.7 | ||
| High | 13.8 | 20.8 | ||
| Very high | 72.4 | 28.3 |
| Predictive Variable | β-Estimate | 95%CI | p-Value |
|---|---|---|---|
| Dependent variable cSBP | |||
| Age | 0.33 | 0.02; 0.63 | 0.03 |
| Sex [M] | −2.03 | −10.35; 6.29 | 0.63 |
| Glucose | −0.03 | −0.20; 0.14 | 0.71 |
| PNPLA3 [CG + GG] | −7.36 | −16.35; 1.63 | 0.11 |
| Dependent variable PP | |||
| Age | 0.43 | 0.19; 0.68 | <0.001 |
| Sex [M] | −0.29 | −7.11; 6.52 | 0.93 |
| Glucose | −0.05 | −0.19; 0.08 | 0.43 |
| PNPLA3 [CG + GG] | −6.66 | −14.03; 0.70 | 0.07 |
| Dependent variable PWV | |||
| Age | 0.15 | 0.07; 0.23 | <0.001 |
| Sex [M] | 1.04 | −1.25; 3.37 | 0.37 |
| Glucose | 0.02 | −0.03; 0.06 | 0.50 |
| PNPLA3 [CG + GG] | −3.04 | −5.52; −0.56 | 0.02 |
| Dependent variable mean IMT | |||
| Age | 0.01 | 0.008; 0.01 | <0.001 |
| Sex [M] | 0.11 | 0.02; 0.21 | 0.02 |
| Glucose | −0.002 | −0.004; 0.001 | 0.08 |
| PNPLA3 [CG + GG] | 0.01 | −0.10; 0.12 | 0.80 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Toffoli, B.; Comar, C.; Grillo, A.; Barbato, V.; Vincis, E.; Baldi, V.; Berti, S.; Volpato, T.; Zorat, F.; Crocè, S.L.; et al. PNPLA3 Polymorphism Is Inversely Correlated with Aortic Stiffness in Patients with Metabolic Dysfunction-Associated Steatotic Liver Disease Without Fibrosis. Int. J. Mol. Sci. 2025, 26, 3256. https://doi.org/10.3390/ijms26073256
Toffoli B, Comar C, Grillo A, Barbato V, Vincis E, Baldi V, Berti S, Volpato T, Zorat F, Crocè SL, et al. PNPLA3 Polymorphism Is Inversely Correlated with Aortic Stiffness in Patients with Metabolic Dysfunction-Associated Steatotic Liver Disease Without Fibrosis. International Journal of Molecular Sciences. 2025; 26(7):3256. https://doi.org/10.3390/ijms26073256
Chicago/Turabian StyleToffoli, Barbara, Consuelo Comar, Andrea Grillo, Vincenzo Barbato, Emanuele Vincis, Veronica Baldi, Silvia Berti, Teresa Volpato, Francesca Zorat, Saveria Lory Crocè, and et al. 2025. "PNPLA3 Polymorphism Is Inversely Correlated with Aortic Stiffness in Patients with Metabolic Dysfunction-Associated Steatotic Liver Disease Without Fibrosis" International Journal of Molecular Sciences 26, no. 7: 3256. https://doi.org/10.3390/ijms26073256
APA StyleToffoli, B., Comar, C., Grillo, A., Barbato, V., Vincis, E., Baldi, V., Berti, S., Volpato, T., Zorat, F., Crocè, S. L., Emmi, G., Fabris, B., Puato, M., & Bernardi, S. (2025). PNPLA3 Polymorphism Is Inversely Correlated with Aortic Stiffness in Patients with Metabolic Dysfunction-Associated Steatotic Liver Disease Without Fibrosis. International Journal of Molecular Sciences, 26(7), 3256. https://doi.org/10.3390/ijms26073256