
Selective Modulation of PAR-2-Driven Inflammatory Pathways by Oleocanthal: Attenuation of TNF-α and Calcium Dysregulation in Colorectal Cancer Models


Abstract
:1. Introduction
2. Results
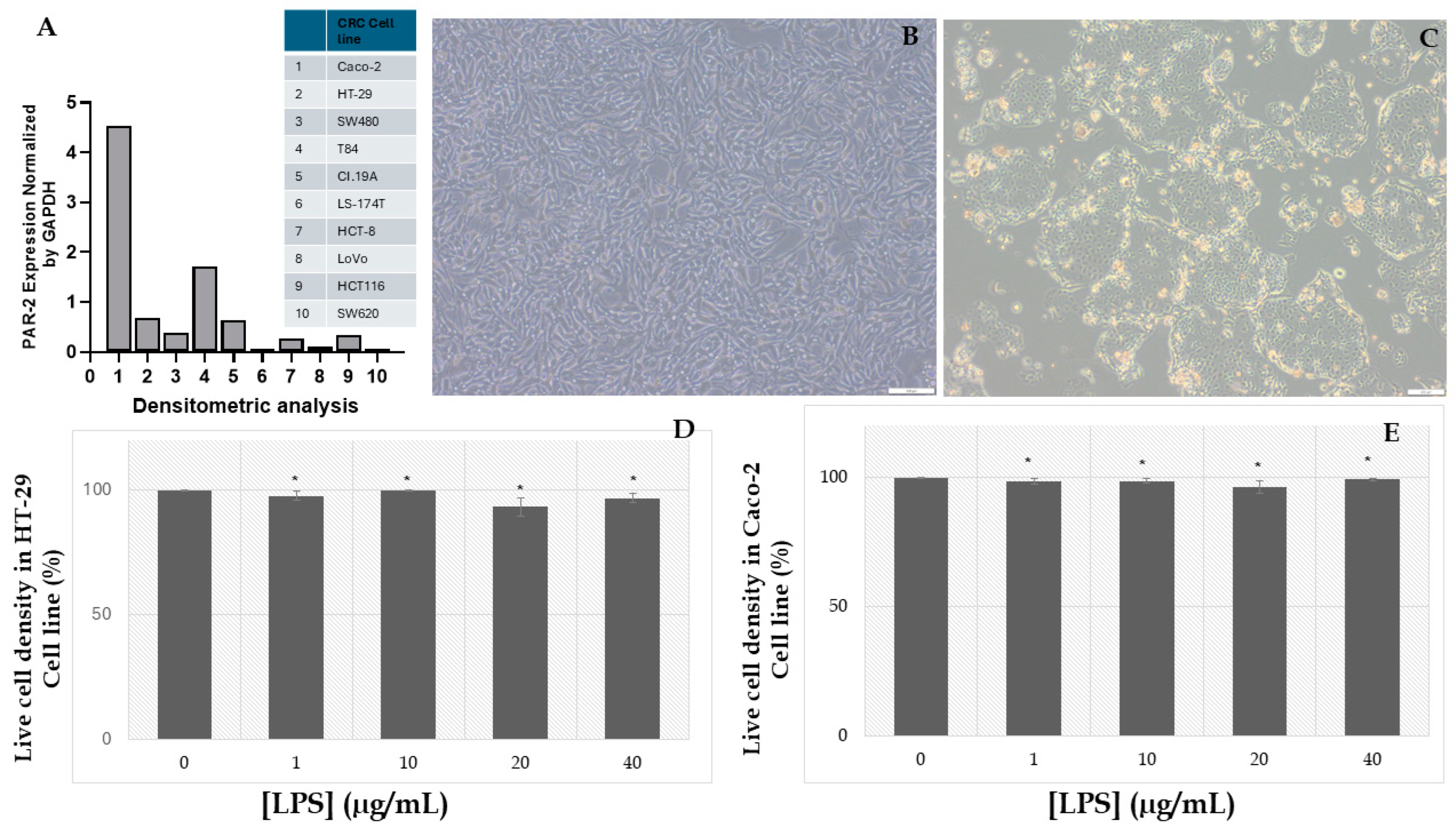
2.1. Assessment of LPS-Induced Cytotoxicity and the Establishment of an Inflammatory Cancer Model in HT-29 and Caco-2 Cell Lines
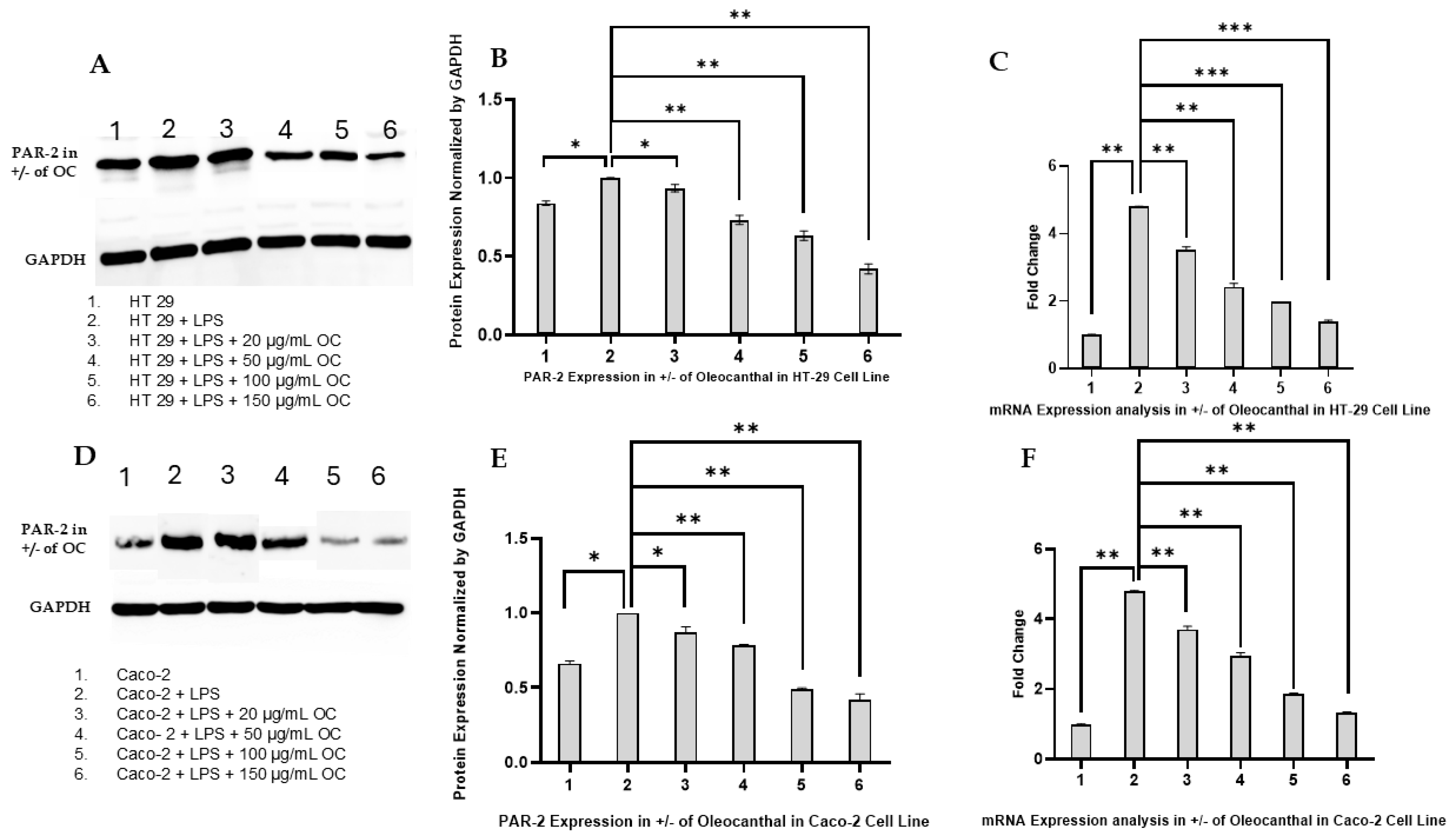
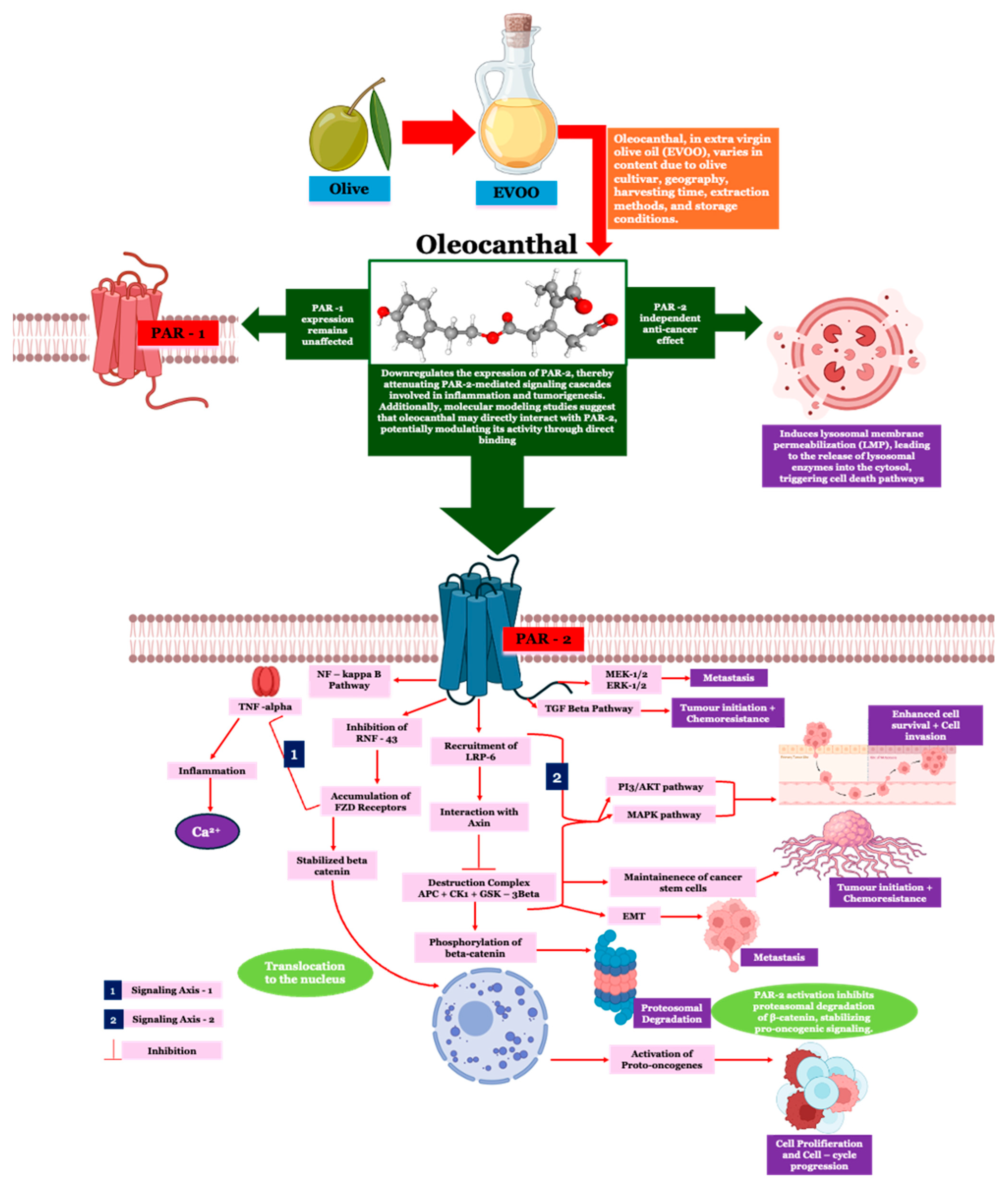
2.2. Effect of OC on PAR-2 Expression in HT-29 and Caco-2 Cells
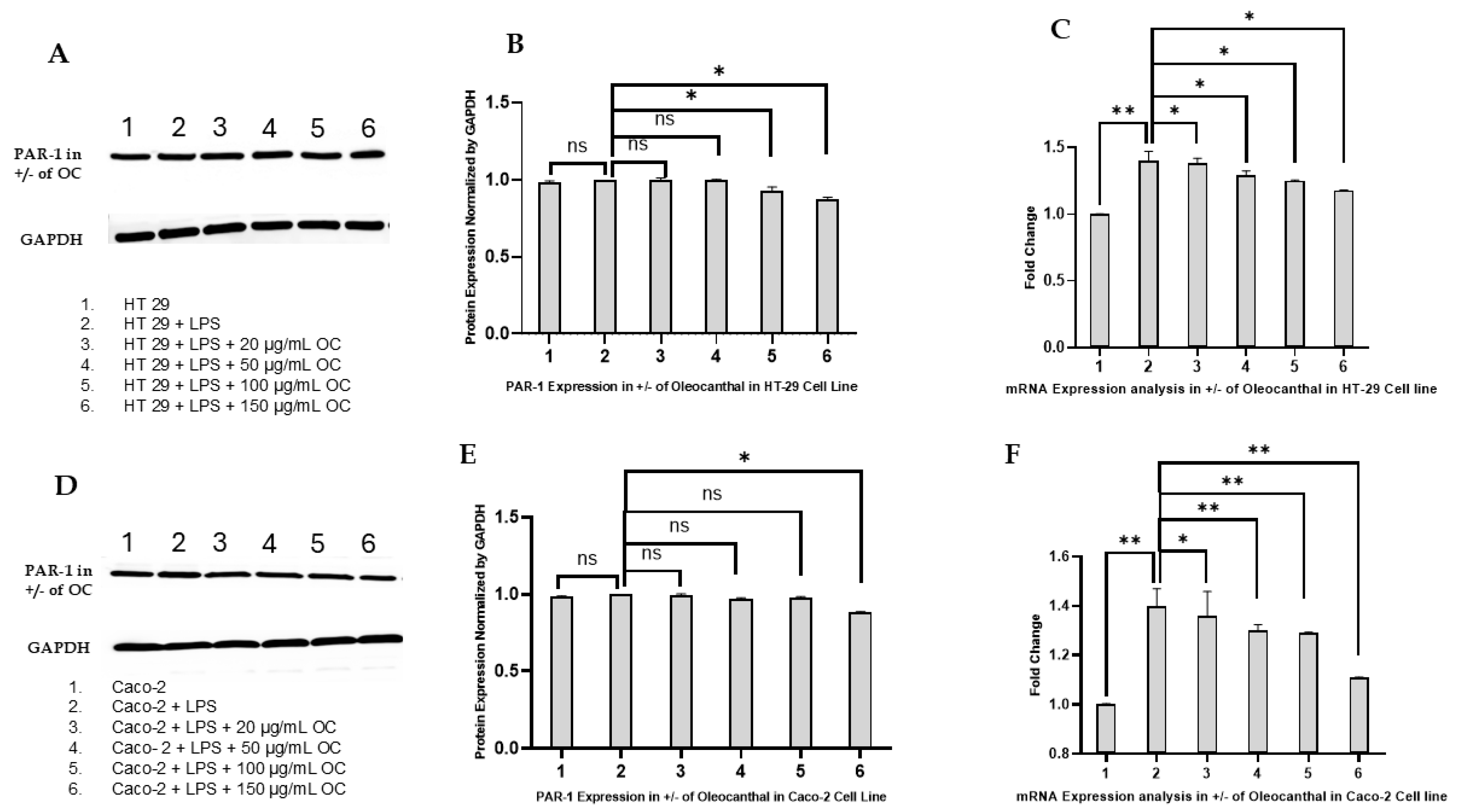
2.3. Effect of OC on PAR-1 Expression in HT-29 and Caco-2 Cells
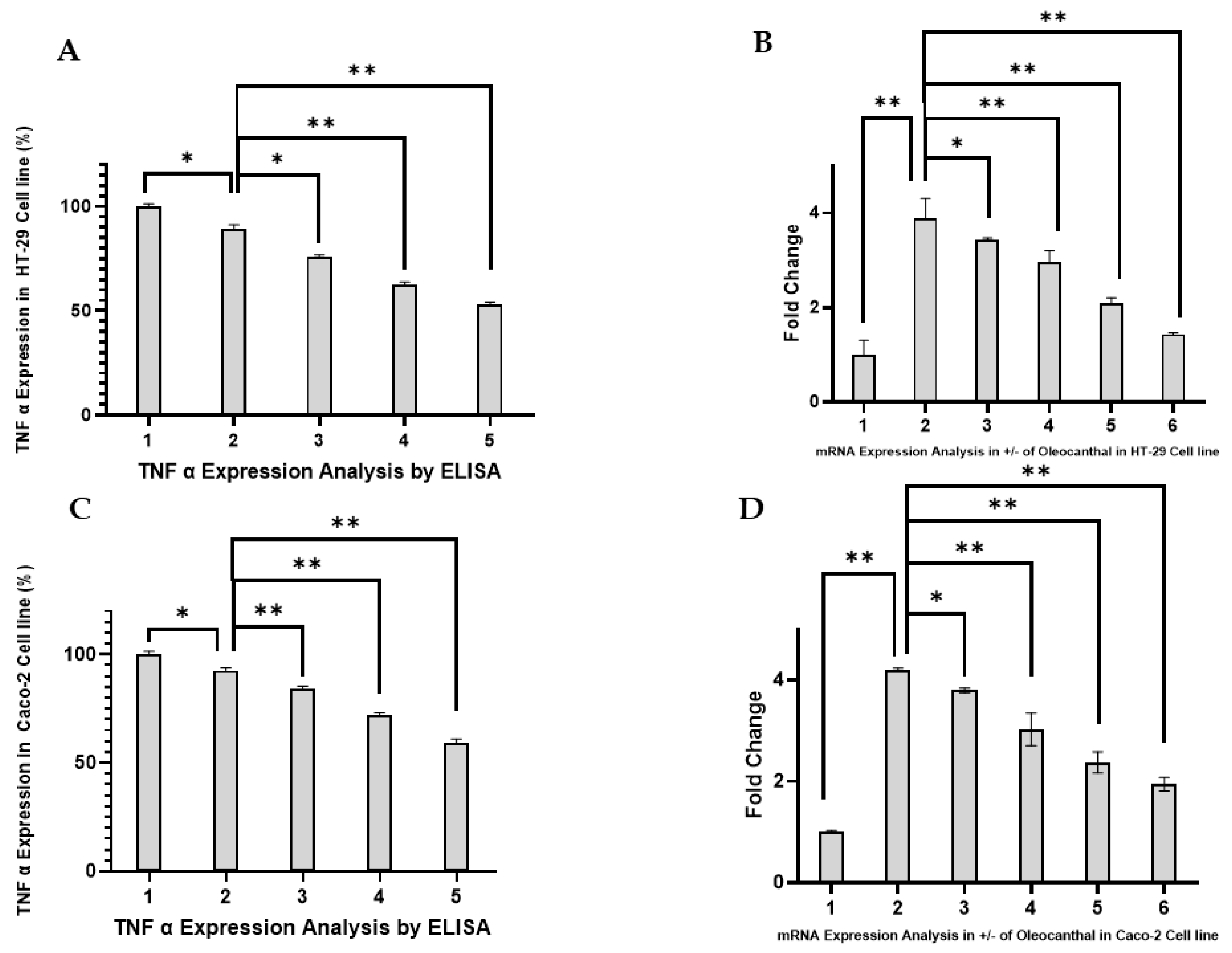
2.4. Effect of OC on TNF-α Expression in HT-29 and Caco-2 Cells
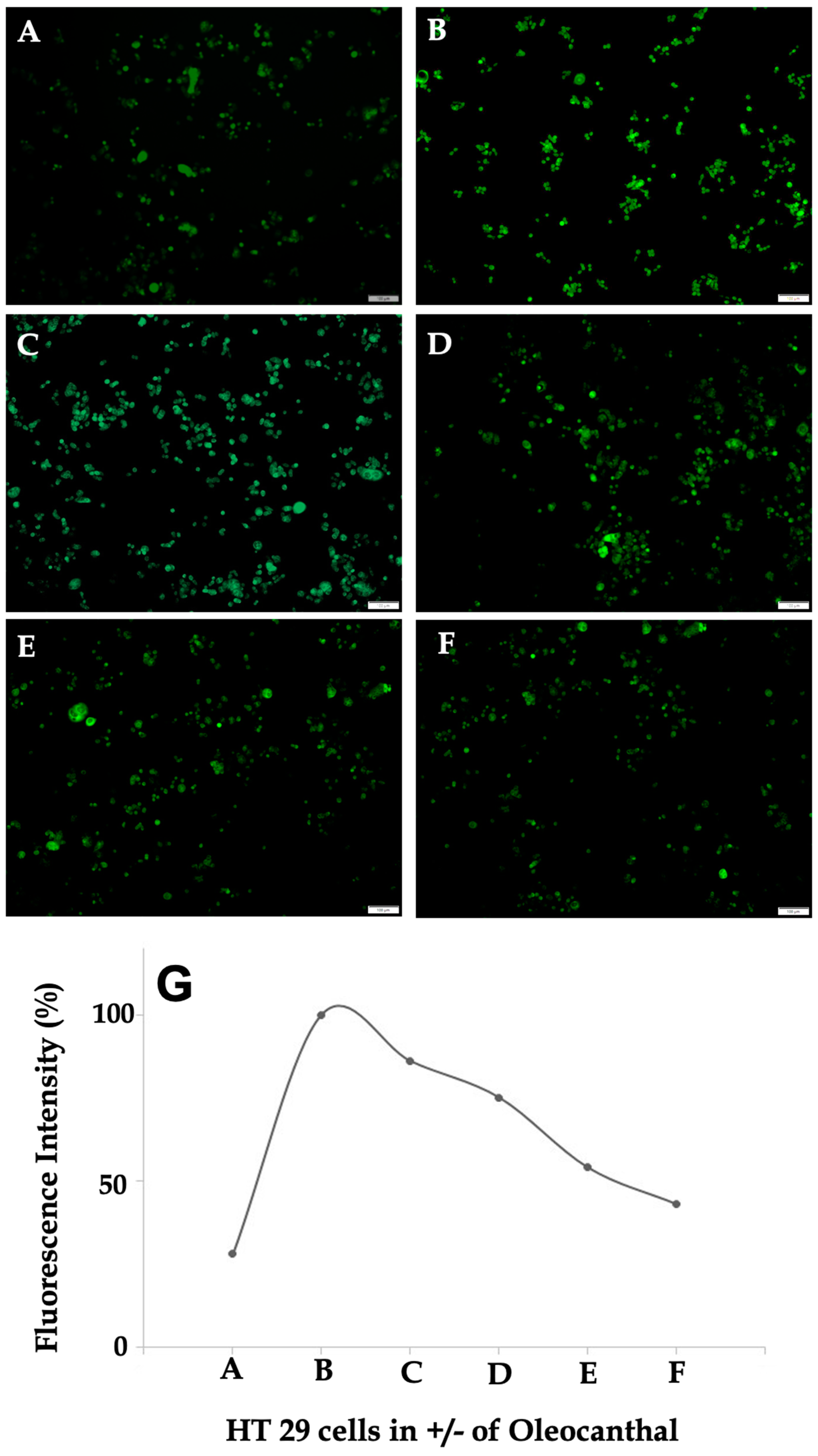
2.5. Effect of OC on Intracellular Calcium Levels in HT-29 Cells
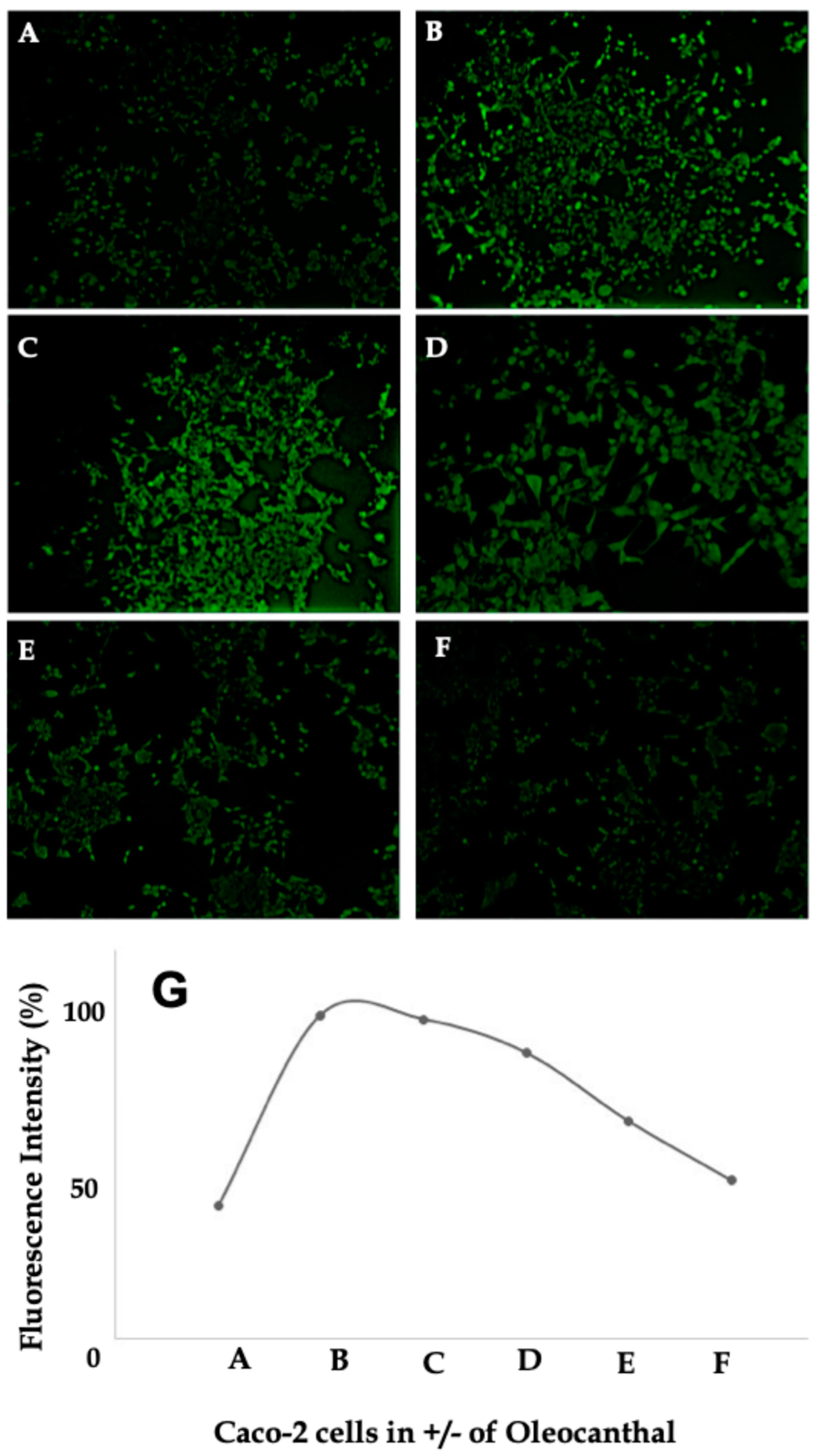
2.6. Effect of OC on Intracellular Calcium Levels in Caco-2 Cells
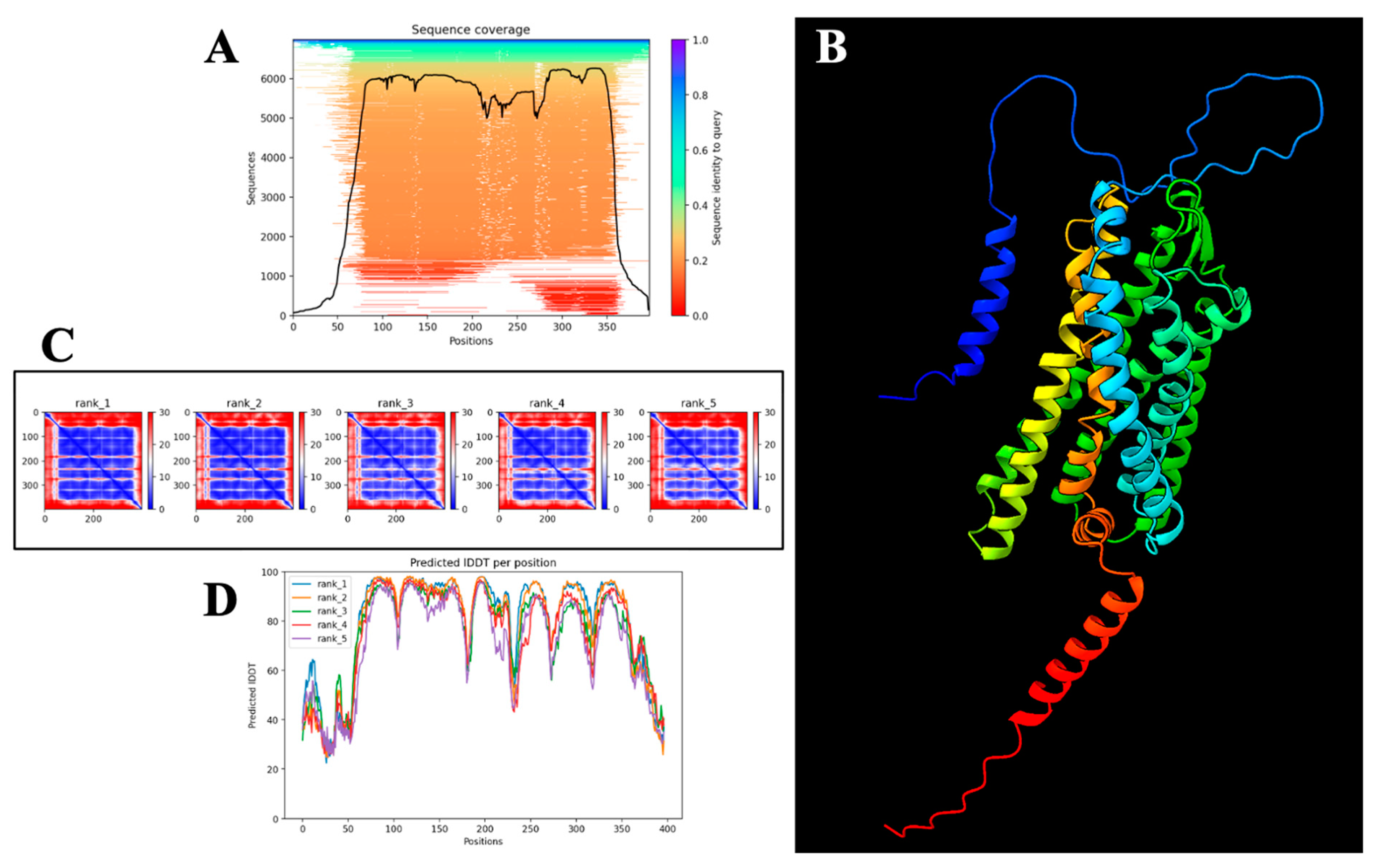
2.7. Computational Structural Validation and Confidence Assessment of the AlphaFold-Predicted PAR-2 Model
Structural Reliability Assessment
2.8. Predicted Binding Mode and Structural Insights into OC Interaction with PAR-2
3. Discussion
4. Materials and Methods
4.1. Study Design
4.2. Ethics Considerations
4.3. Cell Line Selection
4.4. Cell Culture and Treatment
4.5. Cell Culture Methodology
4.6. Subculturing Protocol
4.7. Cryopreservation of HT-29 and Caco-2 Cells
4.8. Assessment of Cytotoxicity Using the MTT Assay
4.9. Inducing Inflammation in HT-29 and Caco-2 Cells Using LPS and OC Treatment
- (a)
- Olive cultivar: certain cultivars, such as Coratina, exhibit substantially higher OC concentrations (78.2 µg/mL), whereas others, such as Taggiasca, contain significantly lower levels (8.3 µg/mL);
- (b)
- Geographical location: Italian EVOOs have been reported to contain up to 191.8 µg/mL of OC, whereas EVOOs from the United States typically exhibit lower levels (~22.6 µg/mL);
- (c)
- Agricultural techniques: increased irrigation reduces OC content, suggesting that water stress conditions enhance OC biosynthesis;
- (d)
- Olive maturity and harvest time: OC concentration is dependent on the degree of ripeness at harvest, with early harvests yielding EVOO with higher phenolic content;
- (e)
- Processing methods: EVOO extraction techniques influence OC retention, with two-phase centrifugation preserving higher OC levels compared to three-phase methods;
- (f)
- Storage conditions and thermal stability: the chemical stability of OC is affected by exposure to oxygen, light, and temperature fluctuations, though OC remains relatively stable when initially present in high concentrations in EVOO.
4.10. Assessment of TNF-α Secretion Using ELISA
4.11. Western Blot Analysis for the Expression of PAR-1 and PAR-2
4.11.1. Protein Extraction and Quantification
4.11.2. Experimental Design
4.11.3. Blocking and Antibody Incubation
4.11.4. Secondary Antibody and Detection
4.11.5. Quantification
4.12. RNA Extraction and cDNA Synthesis
4.13. Real-Time PCR for Quantification (qPCR)
4.14. Calcium Signaling Assay
4.15. Computational Modeling of PAR-2
4.16. Molecular Docking of OC with AlphaFold-Predicted PAR-2 Structure
5. Conclusions
5.1. Limitations
5.2. Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2024, 74, 229–263. [Google Scholar] [PubMed]
- Al Abdouli, L.; Dalmook, H.; Akram Abdo, M.; Carrick, F.R.; Abdul Rahman, M. Colorectal Cancer Risk Awareness and Screening Uptake among Adults in the United Arab Emirates. Asian Pac. J. Cancer Prev. 2018, 19, 2343–2349. [Google Scholar] [PubMed]
- Alsaad, L.N.; Sreedharan, J. Practice of colorectal cancer screening in the United Arab Emirates and factors associated—A cross-sectional study. BMC Public Health 2023, 23, 2015. [Google Scholar]
- Makhlouf, N.A.; Abdel-Gawad, M.; Mahros, A.M.; Lashen, S.A.; Zaghloul, M.; Eliwa, A.; Elshemy, E.E.; Ali-Eldin, Z.; Abdeltawab, D.; El-Raey, F.; et al. Colorectal cancer in Arab world: A systematic review. World J. Gastrointest. Oncol. 2021, 13, 1791–1798. [Google Scholar]
- Ramadan, M.; Ghamdi, H.; Aboalola, D.; Alorainan, N.; Alsalmi, R.; Afash, A.; Hariri, A.; Alboloshi, A.; Samkari, A.; Alsiary, R. Disease burden and projection of total and early-onset colorectal cancer in Gulf cooperation council countries from 1990 to 2019. Neoplasia 2024, 51, 100988. [Google Scholar]
- Mamdouh, H.; Hussain, H.Y.; Ibrahim, G.M.; Alawadi, F.; Hassanein, M.; Zarooni, A.A.; Suwaidi, H.A.; Hassan, A.; Alsheikh-Ali, A.; Alnakhi, W.K. Prevalence and associated risk factors of overweight and obesity among adult population in Dubai: A population-based cross-sectional survey in Dubai, the United Arab Emirates. BMJ Open 2023, 13, e062053. [Google Scholar]
- Mahmoud, I.; Sulaiman, N. Dyslipidaemia prevalence and associated risk factors in the United Arab Emirates: A population-based study. BMJ Open 2019, 9, e031969. [Google Scholar]
- Mezhal, F.; Oulhaj, A.; Abdulle, A.; AlJunaibi, A.; Alnaeemi, A.; Ahmad, A.; Leinberger-Jabari, A.; Al Dhaheri, A.S.; AlZaabi, E.; Al-Maskari, F.; et al. High prevalence of cardiometabolic risk factors amongst young adults in the United Arab Emirates: The UAE Healthy Future Study. BMC Cardiovasc. Disord. 2023, 23, 137. [Google Scholar]
- Sharma, C.; Suliman, A.; Al Hamad, S.M.; Yasin, J.; Abuzakouk, M.; AlKaabi, J.; Aburawi, E.H. Association of Biomarkers for Dyslipidemia, Inflammation, and Oxidative Stress with Endothelial Dysfunction in Obese Youths: A Case-Control Study. Diabetes Metab. Syndr. Obes. 2024, 17, 2533–2545. [Google Scholar] [CrossRef]
- Molla, M.D.; Symonds, E.L.; Winter, J.M.; Debie, A.; Wassie, M.M. Metabolic risk factors of colorectal cancer: Umbrella review. Crit. Rev. Oncol. Hematol. 2024, 204, 104502. [Google Scholar]
- Kirichenko, T.V.; Markina, Y.V.; Bogatyreva, A.I.; Tolstik, T.V.; Varaeva, Y.R.; Starodubova, A.V. The Role of Adipokines in Inflammatory Mechanisms of Obesity. Int. J. Mol. Sci. 2022, 23, 14982. [Google Scholar] [CrossRef] [PubMed]
- Masenga, S.K.; Kabwe, L.S.; Chakulya, M.; Kirabo, A. Mechanisms of Oxidative Stress in Metabolic Syndrome. Int. J. Mol. Sci. 2023, 24, 7898. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.Y.; Tsai, M.C.; Tu, W.; Yeh, H.C.; Wang, S.C.; Huang, S.P.; Li, C.Y. Role of the NLRP3 Inflammasome: Insights Into Cancer Hallmarks. Front. Immunol. 2020, 11, 610492. [Google Scholar]
- Bahrami, A.; Khalaji, A.; Bahri Najafi, M.; Sadati, S.; Raisi, A.; Abolhassani, A.; Eshraghi, R.; Khaksary Mahabady, M.; Rahimian, N.; Mirzaei, H. NF-kappaB pathway and angiogenesis: Insights into colorectal cancer development and therapeutic targets. Eur. J. Med. Res. 2024, 29, 610. [Google Scholar] [CrossRef]
- Quaglio, A.E.V.; Grillo, T.G.; De Oliveira, E.C.S.; Di Stasi, L.C.; Sassaki, L.Y. Gut microbiota, inflammatory bowel disease and colorectal cancer. World J. Gastroenterol. 2022, 28, 4053–4060. [Google Scholar]
- Yang, M.; Zhang, Q.; Ge, Y.Z.; Tang, M.; Zhang, X.; Song, M.M.; Ruan, G.T.; Zhang, X.W.; Zhang, K.P.; Shi, H.P. Association between insulin resistance related indicators with the prognosis of patients with colorectal cancer. Cancer Epidemiol. 2023, 87, 102478. [Google Scholar]
- Colloca, A.; Donisi, I.; Anastasio, C.; Balestrieri, M.L.; D’Onofrio, N. Metabolic Alteration Bridging the Prediabetic State and Colorectal Cancer. Cells 2024, 13, 663. [Google Scholar] [CrossRef]
- Kasprzak, A. Insulin-Like Growth Factor 1 (IGF-1) Signaling in Glucose Metabolism in Colorectal Cancer. Int. J. Mol. Sci. 2021, 22, 6434. [Google Scholar] [CrossRef]
- Xu, J.; Ye, Y.; Wu, H.; Duerksen-Hughes, P.; Zhang, H.; Li, P.; Huang, J.; Yang, J.; Wu, Y.; Xia, D. Association between markers of glucose metabolism and risk of colorectal cancer. BMJ Open 2016, 6, e011430. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, Q.; Huang, C.; Wu, J.; Wen, Y. Association Between Metabolic Syndrome and the Risk of Lung Cancer: A Meta-Analysis. Horm. Metab. Res. 2023, 55, 846–854. [Google Scholar]
- Jannati, S.; Patnaik, R.; Banerjee, Y. Beyond Anticoagulation: A Comprehensive Review of Non-Vitamin K Oral Anticoagulants (NOACs) in Inflammation and Protease-Activated Receptor Signaling. Int. J. Mol. Sci. 2024, 25, 8727. [Google Scholar] [CrossRef]
- Reches, G.; Piran, R. Par2-mediated responses in inflammation and regeneration: Choosing between repair and damage. Inflamm. Regen. 2024, 44, 26. [Google Scholar] [CrossRef] [PubMed]
- Malfettone, A.; Silvestris, N.; Saponaro, C.; Ranieri, G.; Russo, A.; Caruso, S.; Popescu, O.; Simone, G.; Paradiso, A.; Mangia, A. High density of tryptase-positive mast cells in human colorectal cancer: A poor prognostic factor related to protease-activated receptor 2 expression. J. Cell Mol. Med. 2013, 17, 1025–1037. [Google Scholar] [CrossRef]
- Johnson, J.J.; Miller, D.L.; Jiang, R.; Liu, Y.; Shi, Z.; Tarwater, L.; Williams, R.; Balsara, R.; Sauter, E.R.; Stack, M.S. Protease-activated Receptor-2 (PAR-2)-mediated Nf-kappaB Activation Suppresses Inflammation-associated Tumor Suppressor MicroRNAs in Oral Squamous Cell Carcinoma. J. Biol. Chem. 2016, 291, 6936–6945. [Google Scholar] [CrossRef]
- Vetvicka, D.; Suhaj, P.; Olejar, T.; Sivak, L.; Benes, J.; Pouckova, P. Proteinase-activated Receptor 2: Springboard of Tumors. Anticancer Res. 2024, 44, 1–12. [Google Scholar] [CrossRef]
- Nag, J.K.; Kancharla, A.; Maoz, M.; Turm, H.; Agranovich, D.; Gupta, C.L.; Uziely, B.; Bar-Shavit, R. Low-density lipoprotein receptor-related protein 6 is a novel coreceptor of protease-activated receptor-2 in the dynamics of cancer-associated beta-catenin stabilization. Oncotarget 2017, 8, 38650–38667. [Google Scholar] [CrossRef]
- Patti, A.M.; Carruba, G.; Cicero, A.F.G.; Banach, M.; Nikolic, D.; Giglio, R.V.; Terranova, A.; Soresi, M.; Giannitrapani, L.; Montalto, G.; et al. Daily Use of Extra Virgin Olive Oil with High Oleocanthal Concentration Reduced Body Weight, Waist Circumference, Alanine Transaminase, Inflammatory Cytokines and Hepatic Steatosis in Subjects with the Metabolic Syndrome: A 2-Month Intervention Study. Metabolites 2020, 10, 392. [Google Scholar] [CrossRef]
- Elnagar, A.Y.; Sylvester, P.W.; El Sayed, K.A. (−)-Oleocanthal as a c-Met inhibitor for the control of metastatic breast and prostate cancers. Planta Med. 2011, 77, 1013–1019. [Google Scholar] [CrossRef]
- Goren, L.; Zhang, G.; Kaushik, S.; Breslin, P.A.S.; Du, Y.N.; Foster, D.A. (−)-Oleocanthal and (−)-oleocanthal-rich olive oils induce lysosomal membrane permeabilization in cancer cells. PLoS ONE 2019, 14, e0216024. [Google Scholar] [CrossRef]
- Pei, T.; Meng, Q.; Han, J.; Sun, H.; Li, L.; Song, R.; Sun, B.; Pan, S.; Liang, D.; Liu, L. (−)-Oleocanthal inhibits growth and metastasis by blocking activation of STAT3 in human hepatocellular carcinoma. Oncotarget 2016, 7, 43475–43491. [Google Scholar] [CrossRef]
- Cusimano, A.; Balasus, D.; Azzolina, A.; Augello, G.; Emma, M.R.; Di Sano, C.; Gramignoli, R.; Strom, S.C.; McCubrey, J.A.; Montalto, G.; et al. Oleocanthal exerts antitumor effects on human liver and colon cancer cells through ROS generation. Int. J. Oncol. 2017, 51, 533–544. [Google Scholar] [PubMed]
- Siddique, A.B.; Ebrahim, H.Y.; Tajmim, A.; King, J.A.; Abdelwahed, K.S.; Abd Elmageed, Z.Y.; El Sayed, K.A. Oleocanthal Attenuates Metastatic Castration-Resistant Prostate Cancer Progression and Recurrence by Targeting SMYD2. Cancers 2022, 14, 3542. [Google Scholar] [CrossRef] [PubMed]
- Patnaik, R.; Varghese, R.; Jannati, S.; Naidoo, N.; Banerjee, Y. Targeting PAR2-mediated inflammation in osteoarthritis: A comprehensive in vitro evaluation of oleocanthal’s potential as a functional food intervention for chondrocyte protection and anti-inflammatory effects. BMC Musculoskelet. Disord. 2024, 25, 769. [Google Scholar] [CrossRef]
- Darmoul, D.; Marie, J.C.; Devaud, H.; Gratio, V.; Laburthe, M. Initiation of human colon cancer cell proliferation by trypsin acting at protease-activated receptor-2. Br. J. Cancer 2001, 85, 772–779. [Google Scholar]
- Perico, L.L.; Vegso, A.J.; Baggio, C.H.; MacNaughton, W.K. Protease-activated receptor 2 drives migration in a colon cancer cell line but not in noncancerous human epithelial cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2024, 326, G525–G542. [Google Scholar]
- Wang, Z.; Chang, Y.; Sun, H.; Li, Y.; Tang, T. Advances in molecular mechanisms of inflammatory bowel disease-associated colorectal cancer (Review). Oncol. Lett. 2024, 27, 257. [Google Scholar]
- Rayees, S.; Rochford, I.; Joshi, J.C.; Joshi, B.; Banerjee, S.; Mehta, D. Macrophage TLR4 and PAR2 Signaling: Role in Regulating Vascular Inflammatory Injury and Repair. Front. Immunol. 2020, 11, 2091. [Google Scholar]
- Guo, H.B.; Perminov, A.; Bekele, S.; Kedziora, G.; Farajollahi, S.; Varaljay, V.; Hinkle, K.; Molinero, V.; Meister, K.; Hung, C.; et al. AlphaFold2 models indicate that protein sequence determines both structure and dynamics. Sci. Rep. 2022, 12, 10696. [Google Scholar]
- Sadler, F.; Ma, N.; Ritt, M.; Sharma, Y.; Vaidehi, N.; Sivaramakrishnan, S. Autoregulation of GPCR signalling through the third intracellular loop. Nature 2023, 615, 734–741. [Google Scholar]
- Aranda-Garcia, D.; Stepniewski, T.M.; Torrens-Fontanals, M.; Garcia-Recio, A.; Lopez-Balastegui, M.; Medel-Lacruz, B.; Morales-Pastor, A.; Peralta-Garcia, A.; Dieguez-Eceolaza, M.; Sotillo-Nunez, D.; et al. Large scale investigation of GPCR molecular dynamics data uncovers allosteric sites and lateral gateways. Nat. Commun. 2025, 16, 2020. [Google Scholar] [CrossRef]
- Covic, L.; Gresser, A.L.; Talavera, J.; Swift, S.; Kuliopulos, A. Activation and inhibition of G protein-coupled receptors by cell-penetrating membrane-tethered peptides. Proc. Natl. Acad. Sci. USA 2002, 99, 643–648. [Google Scholar] [CrossRef] [PubMed]
- Gnanasambandam, R.; Ghatak, C.; Yasmann, A.; Nishizawa, K.; Sachs, F.; Ladokhin, A.S.; Sukharev, S.I.; Suchyna, T.M. GsMTx4: Mechanism of Inhibiting Mechanosensitive Ion Channels. Biophys. J. 2017, 112, 31–45. [Google Scholar] [CrossRef] [PubMed]
- Szlenk, C.T.; Gc, J.B.; Natesan, S. Does the Lipid Bilayer Orchestrate Access and Binding of Ligands to Transmembrane Orthosteric/Allosteric Sites of G Protein-Coupled Receptors? Mol. Pharmacol. 2019, 96, 527–541. [Google Scholar] [CrossRef] [PubMed]
- Ayoub, N.M.; Siddique, A.B.; Ebrahim, H.Y.; Mohyeldin, M.M.; El Sayed, K.A. The olive oil phenolic (−)-oleocanthal modulates estrogen receptor expression in luminal breast cancer in vitro and in vivo and synergizes with tamoxifen treatment. Eur. J. Pharmacol. 2017, 810, 100–111. [Google Scholar] [CrossRef]
- Quan, Q.; Zhong, F.; Wang, X.; Chen, K.; Guo, L. PAR2 Inhibition Enhanced the Sensitivity of Colorectal Cancer Cells to 5-FU and Reduced EMT Signaling. Oncol. Res. 2019, 27, 779–788. [Google Scholar] [CrossRef]
- Lidfeldt, J.; Bendahl, P.O.; Forsare, C.; Malmstrom, P.; Ferno, M.; Belting, M. Protease Activated Receptors 1 and 2 Correlate Differently with Breast Cancer Aggressiveness Depending on Tumor ER Status. PLoS ONE 2015, 10, e0134932. [Google Scholar] [CrossRef]
- Huo, H.; Feng, Y.; Tang, Q. Inhibition of proteinase-activated receptor 2 (PAR2) decreased the malignant progression of lung cancer cells and increased the sensitivity to chemotherapy. Cancer Chemother. Pharmacol. 2024, 93, 397–410. [Google Scholar] [CrossRef]
- Nag, J.K.; Appasamy, P.; Malka, H.; Sedley, S.; Bar-Shavit, R. New Target(s) for RNF43 Regulation: Implications for Therapeutic Strategies. Int. J. Mol. Sci. 2024, 25, 8083. [Google Scholar] [CrossRef]
- Ma, Y.; He, L.; Zhao, X.; Li, W.; Lv, X.; Zhang, X.; Peng, J.; Yang, L.; Xu, Q.; Wang, H. Protease activated receptor 2 signaling promotes self-renewal and metastasis in colorectal cancer through beta-catenin and periostin. Cancer Lett. 2021, 521, 130–141. [Google Scholar] [CrossRef]
- Tsai, C.C.; Chou, Y.T.; Fu, H.W. Protease-activated receptor 2 induces migration and promotes Slug-mediated epithelial-mesenchymal transition in lung adenocarcinoma cells. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 486–503. [Google Scholar] [CrossRef]
- Han, X.; Nieman, M.T.; Kerlin, B.A. Protease-activated receptors: An illustrated review. Res. Pract. Thromb. Haemost. 2021, 5, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Thrasivoulou, C.; Millar, M.; Ahmed, A. Activation of intracellular calcium by multiple Wnt ligands and translocation of beta-catenin into the nucleus: A convergent model of Wnt/Ca2+ and Wnt/beta-catenin pathways. J. Biol. Chem. 2013, 288, 35651–35659. [Google Scholar] [CrossRef] [PubMed]
- Oguma, K.; Oshima, H.; Aoki, M.; Uchio, R.; Naka, K.; Nakamura, S.; Hirao, A.; Saya, H.; Taketo, M.M.; Oshima, M. Activated macrophages promote Wnt signalling through tumour necrosis factor-alpha in gastric tumour cells. EMBO J. 2008, 27, 1671–1681. [Google Scholar] [CrossRef]
- Diarra, D.; Stolina, M.; Polzer, K.; Zwerina, J.; Ominsky, M.S.; Dwyer, D.; Korb, A.; Smolen, J.; Hoffmann, M.; Scheinecker, C.; et al. Dickkopf-1 is a master regulator of joint remodeling. Nat. Med. 2007, 13, 156–163. [Google Scholar] [CrossRef]
- Zhang, X.; Fang, Q.; Ma, Y.; Zou, S.; Liu, Q.; Wang, H. Protease activated receptor 2 mediates tryptase-induced cell migration through MYO10 in colorectal cancer. Am. J. Cancer Res. 2019, 9, 1995–2006. [Google Scholar]
- Guo, Q.; Jin, Y.; Chen, X.; Ye, X.; Shen, X.; Lin, M.; Zeng, C.; Zhou, T.; Zhang, J. NF-kappaB in biology and targeted therapy: New insights and translational implications. Signal Transduct. Target. Ther. 2024, 9, 53. [Google Scholar] [CrossRef]
- Borzi, A.M.; Biondi, A.; Basile, F.; Luca, S.; Vicari, E.S.D.; Vacante, M. Olive Oil Effects on Colorectal Cancer. Nutrients 2018, 11, 32. [Google Scholar] [CrossRef]
- Cariello, M.; Contursi, A.; Gadaleta, R.M.; Piccinin, E.; De Santis, S.; Piglionica, M.; Spaziante, A.F.; Sabba, C.; Villani, G.; Moschetta, A. Extra-Virgin Olive Oil from Apulian Cultivars and Intestinal Inflammation. Nutrients 2020, 12, 1084. [Google Scholar] [CrossRef]
- Vrdoljak, J.; Kumric, M.; Vilovic, M.; Martinovic, D.; Tomic, I.J.; Krnic, M.; Ticinovic Kurir, T.; Bozic, J. Effects of Olive Oil and Its Components on Intestinal Inflammation and Inflammatory Bowel Disease. Nutrients 2022, 14, 757. [Google Scholar] [CrossRef]
- Qusa, M.H.; Abdelwahed, K.S.; Hill, R.A.; El Sayed, K.A. S-(−)-Oleocanthal Ex Vivo Modulatory Effects on Gut Microbiota. Nutrients 2023, 15, 618. [Google Scholar] [CrossRef]
- Sain, A.; Sahu, S.; Naskar, D. Potential of olive oil and its phenolic compounds as therapeutic intervention against colorectal cancer: A comprehensive review. Br. J. Nutr. 2022, 128, 1257–1273. [Google Scholar] [PubMed]
- Pang, K.L.; Chin, K.Y. The Biological Activities of Oleocanthal from a Molecular Perspective. Nutrients 2018, 10, 570. [Google Scholar] [CrossRef] [PubMed]
- Kotsiou, K.; Tasioula-Margari, M. Monitoring the phenolic compounds of Greek extra-virgin olive oils during storage. Food Chem. 2016, 200, 255–262. [Google Scholar]
- Hu, L.; Xia, L.; Zhou, H.; Wu, B.; Mu, Y.; Wu, Y.; Yan, J. TF/FVIIa/PAR2 promotes cell proliferation and migration via PKCalpha and ERK-dependent c-Jun/AP-1 pathway in colon cancer cell line SW620. Tumour Biol. 2013, 34, 2573–2581. [Google Scholar]
- Agrawal, K.; Arora, N. Serine protease allergen favours Th2 responses via PAR-2 and STAT3 activation in murine model. Allergy 2018, 73, 569–575. [Google Scholar]
- Fan, Y.; Mao, R.; Yang, J. NF-kappaB and STAT3 signaling pathways collaboratively link inflammation to cancer. Protein Cell 2013, 4, 176–185. [Google Scholar]
- Bollrath, J.; Greten, F.R. IKK/NF-kappaB and STAT3 pathways: Central signalling hubs in inflammation-mediated tumour promotion and metastasis. EMBO Rep. 2009, 10, 1314–1319. [Google Scholar]
- Carpi, S.; Scoditti, E.; Massaro, M.; Polini, B.; Manera, C.; Digiacomo, M.; Esposito Salsano, J.; Poli, G.; Tuccinardi, T.; Doccini, S.; et al. The Extra-Virgin Olive Oil Polyphenols Oleocanthal and Oleacein Counteract Inflammation-Related Gene and miRNA Expression in Adipocytes by Attenuating NF-kappaB Activation. Nutrients 2019, 11, 2855. [Google Scholar]
- Del Saz-Lara, A.; Lopez de Las Hazas, M.C.; Visioli, F.; Davalos, A. Nutri-Epigenetic Effects of Phenolic Compounds from Extra Virgin Olive Oil: A Systematic Review. Adv. Nutr. 2022, 13, 2039–2060. [Google Scholar]
- Hasdemir, B.; Murphy, J.E.; Cottrell, G.S.; Bunnett, N.W. Endosomal deubiquitinating enzymes control ubiquitination and down-regulation of protease-activated receptor 2. J. Biol. Chem. 2009, 284, 28453–28466. [Google Scholar]
- Shenoy, S.K.; Lefkowitz, R.J. beta-Arrestin-mediated receptor trafficking and signal transduction. Trends Pharmacol. Sci. 2011, 32, 521–533. [Google Scholar] [PubMed]
- Nag, S.; Goswami, B.; Das Mandal, S.; Ray, P.S. Cooperation and competition by RNA-binding proteins in cancer. Semin. Cancer Biol. 2022, 86 Pt 3, 286–297. [Google Scholar]
- Behm-Ansmant, I.; Rehwinkel, J.; Doerks, T.; Stark, A.; Bork, P.; Izaurralde, E. mRNA degradation by miRNAs and GW182 requires both CCR4:NOT deadenylase and DCP1:DCP2 decapping complexes. Genes Dev. 2006, 20, 1885–1898. [Google Scholar] [PubMed]
- Herman, A.B.; Autieri, M.V. Inflammation-regulated mRNA stability and the progression of vascular inflammatory diseases. Clin. Sci. 2017, 131, 2687–2699. [Google Scholar]
- Fogh, J.; Fogh, J.M.; Orfeo, T. One hundred and twenty-seven cultured human tumor cell lines producing tumors in nude mice. J. Natl. Cancer Inst. 1977, 59, 221–226. [Google Scholar]
- Cohen, E.; Ophir, I.; Shaul, Y.B. Induced differentiation in HT29, a human colon adenocarcinoma cell line. J. Cell Sci. 1999, 112 Pt 16, 2657–2666. [Google Scholar]
- Nautiyal, J.; Kanwar, S.S.; Yu, Y.; Majumdar, A.P. Combination of dasatinib and curcumin eliminates chemo-resistant colon cancer cells. J. Mol. Signal 2011, 6, 7. [Google Scholar]
- Lea, T. Caco-2 Cell Line. In The Impact of Food Bioactives on Health: In Vitro and Ex Vivo Models; Verhoeckx, K., Cotter, P., Lopez-Exposito, I., Kleiveland, C., Lea, T., Mackie, A., Requena, T., Swiatecka, D., Wichers, H., Eds.; Springer: Cham, Switzerland, 2015; pp. 103–111. [Google Scholar]
- Fernando, E.H.; Gordon, M.H.; Beck, P.L.; MacNaughton, W.K. Inhibition of Intestinal Epithelial Wound Healing through Protease-Activated Receptor-2 Activation in Caco2 Cells. J. Pharmacol. Exp. Ther. 2018, 367, 382–392. [Google Scholar]
- Han, L.L.; Lu, Q.Q.; Zheng, W.W.; Li, Y.L.; Song, Y.Y.; Zhang, X.Z.; Long, S.R.; Liu, R.D.; Wang, Z.Q.; Cui, J. A novel trypsin of Trichinella spiralis mediates larval invasion of gut epithelium via binding to PAR2 and activating ERK1/2 pathway. PLoS Negl. Trop. Dis. 2024, 18, e0011874. [Google Scholar]
- Dharmsathaphorn, K.; McRoberts, J.A.; Mandel, K.G.; Tisdale, L.D.; Masui, H. A human colonic tumor cell line that maintains vectorial electrolyte transport. Am. J. Physiol. 1984, 246 Pt 1, G204–G208. [Google Scholar]
- Barrett, K.E.; Keely, S.J. Chloride secretion by the intestinal epithelium: Molecular basis and regulatory aspects. Annu. Rev. Physiol. 2000, 62, 535–572. [Google Scholar] [CrossRef] [PubMed]
- Leibovitz, A.; Stinson, J.C.; McCombs, W.B., III; McCoy, C.E.; Mazur, K.C.; Mabry, N.D. Classification of human colorectal adenocarcinoma cell lines. Cancer Res. 1976, 36, 4562–4569. [Google Scholar] [PubMed]
- Ge, L.; Shenoy, S.K.; Lefkowitz, R.J.; DeFea, K. Constitutive protease-activated receptor-2-mediated migration of MDA MB-231 breast cancer cells requires both beta-arrestin-1 and -2. J. Biol. Chem. 2004, 279, 55419–55424. [Google Scholar] [CrossRef]
- Hsu, R.Y.; Chan, C.H.; Spicer, J.D.; Rousseau, M.C.; Giannias, B.; Rousseau, S.; Ferri, L.E. LPS-induced TLR4 signaling in human colorectal cancer cells increases beta1 integrin-mediated cell adhesion and liver metastasis. Cancer Res. 2011, 71, 1989–1998. [Google Scholar] [CrossRef]
- Rousset, M. The human colon carcinoma cell lines HT-29 and Caco-2: Two in vitro models for the study of intestinal differentiation. Biochimie 1986, 68, 1035–1040. [Google Scholar] [CrossRef]
- Chantret, I.; Barbat, A.; Dussaulx, E.; Brattain, M.G.; Zweibaum, A. Epithelial polarity, villin expression, and enterocytic differentiation of cultured human colon carcinoma cells: A survey of twenty cell lines. Cancer Res. 1988, 48, 1936–1942. [Google Scholar]
- Augeron, C.; Laboisse, C.L. Emergence of permanently differentiated cell clones in a human colonic cancer cell line in culture after treatment with sodium butyrate. Cancer Res. 1984, 44, 3961–3969. [Google Scholar]
- Kolios, G.; Wright, K.L.; Jordan, N.J.; Leithead, J.B.; Robertson, D.A.; Westwick, J. C-X-C and C-C chemokine expression and secretion by the human colonic epithelial cell line, HT-29: Differential effect of T lymphocyte-derived cytokines. Eur. J. Immunol. 1999, 29, 530–536. [Google Scholar] [CrossRef]
- Househam, J.; Heide, T.; Cresswell, G.D.; Spiteri, I.; Kimberley, C.; Zapata, L.; Lynn, C.; James, C.; Mossner, M.; Fernandez-Mateos, J.; et al. Phenotypic plasticity and genetic control in colorectal cancer evolution. Nature 2022, 611, 744–753. [Google Scholar] [CrossRef]
- Verhoeckx, K.; Cotter, P.; Lopez-Exposito, I.; Kleiveland, C.; Lea, T.; Mackie, A.; Requena, T.; Swiatecka, D.; Wichers, H. (Eds.) The Impact of Food Bioactives on Health: In Vitro and Ex Vivo Models; Springer: Cham, Switzerland, 2015. [Google Scholar]
- Behrendt, I.; Roder, I.; Will, F.; Mostafa, H.; Gonzalez-Dominguez, R.; Merono, T.; Andres-Lacueva, C.; Fasshauer, M.; Rudloff, S.; Kuntz, S. Influence of Plasma-Isolated Anthocyanins and Their Metabolites on Cancer Cell Migration (HT-29 and Caco-2) In Vitro: Results of the ATTACH Study. Antioxidants 2022, 11, 1341. [Google Scholar] [CrossRef]
- Gheytanchi, E.; Naseri, M.; Karimi-Busheri, F.; Atyabi, F.; Mirsharif, E.S.; Bozorgmehr, M.; Ghods, R.; Madjd, Z. Morphological and molecular characteristics of spheroid formation in HT-29 and Caco-2 colorectal cancer cell lines. Cancer Cell Int. 2021, 21, 204. [Google Scholar] [PubMed]
- Patnaik, R.; Jannati, S.; Sivani, B.M.; Rizzo, M.; Naidoo, N.; Banerjee, Y. Efficient Generation of Chondrocytes From Bone Marrow-Derived Mesenchymal Stem Cells in a 3D Culture System: Protocol for a Practical Model for Assessing Anti-Inflammatory Therapies. JMIR Res. Protoc. 2023, 12, e42964. [Google Scholar] [PubMed]
- Patnaik, R.; Riaz, S.; Sivani, B.M.; Faisal, S.; Naidoo, N.; Rizzo, M.; Banerjee, Y. Evaluating the potential of Vitamin D and curcumin to alleviate inflammation and mitigate the progression of osteoarthritis through their effects on human chondrocytes: A proof-of-concept investigation. PLoS ONE 2023, 18, e0290739. [Google Scholar] [CrossRef]
- Rallabhandi, P.; Nhu, Q.M.; Toshchakov, V.Y.; Piao, W.; Medvedev, A.E.; Hollenberg, M.D.; Fasano, A.; Vogel, S.N. Analysis of proteinase-activated receptor 2 and TLR4 signal transduction: A novel paradigm for receptor cooperativity. J. Biol. Chem. 2008, 283, 24314–24325. [Google Scholar] [CrossRef]
- Nhu, Q.M.; Shirey, K.; Teijaro, J.R.; Farber, D.L.; Netzel-Arnett, S.; Antalis, T.M.; Fasano, A.; Vogel, S.N. Novel signaling interactions between proteinase-activated receptor 2 and Toll-like receptors in vitro and in vivo. Mucosal Immunol. 2010, 3, 29–39. [Google Scholar] [CrossRef]
- Huang, C.; Wong, G.W.; Ghildyal, N.; Gurish, M.F.; Sali, A.; Matsumoto, R.; Qiu, W.T.; Stevens, R.L. The tryptase, mouse mast cell protease 7, exhibits anticoagulant activity in vivo and in vitro due to its ability to degrade fibrinogen in the presence of the diverse array of protease inhibitors in plasma. J. Biol. Chem. 1997, 272, 31885–31893. [Google Scholar]
- Kuwahara, I.; Lillehoj, E.P.; Lu, W.; Singh, I.S.; Isohama, Y.; Miyata, T.; Kim, K.C. Neutrophil elastase induces IL-8 gene transcription and protein release through p38/NF-kappaB activation via EGFR transactivation in a lung epithelial cell line. Am. J. Physiol. Lung Cell Mol. Physiol. 2006, 291, L407–L416. [Google Scholar]
- Yoo, H.G.; Shin, B.A.; Park, J.S.; Lee, K.H.; Chay, K.O.; Yang, S.Y.; Ahn, B.W.; Jung, Y.D. IL-1beta induces MMP-9 via reactive oxygen species and NF-kappaB in murine macrophage RAW 264.7 cells. Biochem. Biophys. Res. Commun. 2002, 298, 251–256. [Google Scholar]
- Greten, F.R.; Eckmann, L.; Greten, T.F.; Park, J.M.; Li, Z.W.; Egan, L.J.; Kagnoff, M.F.; Karin, M. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell 2004, 118, 285–296. [Google Scholar]
- Nystedt, S.; Emilsson, K.; Wahlestedt, C.; Sundelin, J. Molecular cloning of a potential proteinase activated receptor. Proc. Natl. Acad. Sci. USA 1994, 91, 9208–9212. [Google Scholar]
- Ishihara, H.; Connolly, A.J.; Zeng, D.; Kahn, M.L.; Zheng, Y.W.; Timmons, C.; Tram, T.; Coughlin, S.R. Protease-activated receptor 3 is a second thrombin receptor in humans. Nature 1997, 386, 502–506. [Google Scholar] [PubMed]
- Xu, W.F.; Andersen, H.; Whitmore, T.E.; Presnell, S.R.; Yee, D.P.; Ching, A.; Gilbert, T.; Davie, E.W.; Foster, D.C. Cloning and characterization of human protease-activated receptor 4. Proc. Natl. Acad. Sci. USA 1998, 95, 6642–6646. [Google Scholar]
- Bohm, S.K.; Kong, W.; Bromme, D.; Smeekens, S.P.; Anderson, D.C.; Connolly, A.; Kahn, M.; Nelken, N.A.; Coughlin, S.R.; Payan, D.G.; et al. Molecular cloning, expression and potential functions of the human proteinase-activated receptor-2. Biochem. J. 1996, 314 Pt 3, 1009–1016. [Google Scholar]
- Dery, O.; Corvera, C.U.; Steinhoff, M.; Bunnett, N.W. Proteinase-activated receptors: Novel mechanisms of signaling by serine proteases. Am. J. Physiol. 1998, 274, C1429–C1452. [Google Scholar]
- DeFea, K.A.; Zalevsky, J.; Thoma, M.S.; Dery, O.; Mullins, R.D.; Bunnett, N.W. beta-arrestin-dependent endocytosis of proteinase-activated receptor 2 is required for intracellular targeting of activated ERK1/2. J. Cell Biol. 2000, 148, 1267–1281. [Google Scholar] [PubMed]
- Roosterman, D.; Schmidlin, F.; Bunnett, N.W. Rab5a and rab11a mediate agonist-induced trafficking of protease-activated receptor 2. Am. J. Physiol. Cell Physiol. 2003, 284, C1319–C1329. [Google Scholar] [PubMed]
- Travis, J.; Salvesen, G.S. Human plasma proteinase inhibitors. Annu. Rev. Biochem. 1983, 52, 655–709. [Google Scholar]
- Liu, Q.; Weng, H.J.; Patel, K.N.; Tang, Z.; Bai, H.; Steinhoff, M.; Dong, X. The distinct roles of two GPCRs, MrgprC11 and PAR2, in itch and hyperalgesia. Sci. Signal 2011, 4, ra45. [Google Scholar]
- Kawabata, A.; Kuroda, R.; Nishikawa, H.; Kawai, K. Modulation by protease-activated receptors of the rat duodenal motility in vitro: Possible mechanisms underlying the evoked contraction and relaxation. Br. J. Pharmacol. 1999, 128, 865–872. [Google Scholar]
- Ramachandran, R.; Mihara, K.; Chung, H.; Renaux, B.; Lau, C.S.; Muruve, D.A.; DeFea, K.A.; Bouvier, M.; Hollenberg, M.D. Neutrophil elastase acts as a biased agonist for proteinase-activated receptor-2 (PAR2). J. Biol. Chem. 2011, 286, 24638–24648. [Google Scholar]
- Lohman, R.J.; Cotterell, A.J.; Barry, G.D.; Liu, L.; Suen, J.Y.; Vesey, D.A.; Fairlie, D.P. An antagonist of human protease activated receptor-2 attenuates PAR2 signaling, macrophage activation, mast cell degranulation, and collagen-induced arthritis in rats. FASEB J. 2012, 26, 2877–2887. [Google Scholar] [PubMed]
- LeGendre, O.; Breslin, P.A.; Foster, D.A. (−)-Oleocanthal rapidly and selectively induces cancer cell death via lysosomal membrane permeabilization. Mol. Cell Oncol. 2015, 2, e1006077. [Google Scholar]
- Rojas Gil, A.P.; Kodonis, I.; Ioannidis, A.; Nomikos, T.; Dimopoulos, I.; Kosmidis, G.; Katsa, M.E.; Melliou, E.; Magiatis, P. The Effect of Dietary Intervention With High-Oleocanthal and Oleacein Olive Oil in Patients With Early-Stage Chronic Lymphocytic Leukemia: A Pilot Randomized Trial. Front. Oncol. 2021, 11, 810249. [Google Scholar]
- Gonzalez-Rodriguez, M.; Ait Edjoudi, D.; Cordero-Barreal, A.; Farrag, M.; Varela-Garcia, M.; Torrijos-Pulpon, C.; Ruiz-Fernandez, C.; Capuozzo, M.; Ottaiano, A.; Lago, F.; et al. Oleocanthal, an Antioxidant Phenolic Compound in Extra Virgin Olive Oil (EVOO): A Comprehensive Systematic Review of Its Potential in Inflammation and Cancer. Antioxidants 2023, 12, 2112. [Google Scholar] [CrossRef]
- Akl, M.R.; Ayoub, N.M.; Mohyeldin, M.M.; Busnena, B.A.; Foudah, A.I.; Liu, Y.Y.; Sayed, K.A. Olive phenolics as c-Met inhibitors: (−)-Oleocanthal attenuates cell proliferation, invasiveness, and tumor growth in breast cancer models. PLoS ONE 2014, 9, e97622. [Google Scholar]
- Olmo-Cunillera, A.; Perez, M.; Lopez-Yerena, A.; Abuhabib, M.M.; Ninot, A.; Romero-Aroca, A.; Vallverdu-Queralt, A.; Lamuela-Raventos, R.M. Oleacein and Oleocanthal: Key Metabolites in the Stability of Extra Virgin Olive Oil. Antioxidants 2023, 12, 1776. [Google Scholar] [CrossRef]
- Lanza, B.; Ninfali, P. Antioxidants in Extra Virgin Olive Oil and Table Olives: Connections between Agriculture and Processing for Health Choices. Antioxidants 2020, 9, 41. [Google Scholar] [CrossRef]
- Olmo-Cunillera, A.; Lozano-Castellon, J.; Perez, M.; Miliarakis, E.; Tresserra-Rimbau, A.; Ninot, A.; Romero-Aroca, A.; Lamuela-Raventos, R.M.; Vallverdu-Queralt, A. Optimizing the Malaxation Conditions to Produce an Arbequina EVOO with High Content of Bioactive Compounds. Antioxidants 2021, 10, 1819. [Google Scholar] [CrossRef]
- Kou, J.; Gao, L.; Ni, L.; Shao, T.; Ding, J. Mechanism of Hirudin-Mediated Inhibition of Proliferation in Ovarian Cancer Cells. Mol. Biotechnol. 2024, 66, 1062–1070. [Google Scholar]
- Lin, H.; Liu, A.P.; Smith, T.H.; Trejo, J. Cofactoring and dimerization of proteinase-activated receptors. Pharmacol. Rev. 2013, 65, 1198–1213. [Google Scholar]
- Liu, X.; Yu, J.; Song, S.; Yue, X.; Li, Q. Protease-activated receptor-1 (PAR-1): A promising molecular target for cancer. Oncotarget 2017, 8, 107334–107345. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Jiang, P.; Zhang, C.; Lee, S.; Wang, W.; Zou, H. PAR4 overexpression promotes colorectal cancer cell proliferation and migration. Oncol. Lett. 2018, 16, 5745–5752. [Google Scholar] [CrossRef] [PubMed]
- Vermani, L.; Kumar, R.; Senthil Kumar, N. GAPDH and PUM1: Optimal Housekeeping Genes for Quantitative Polymerase Chain Reaction-Based Analysis of Cancer Stem Cells and Epithelial-Mesenchymal Transition Gene Expression in Rectal Tumors. Cureus 2020, 12, e12020. [Google Scholar] [CrossRef] [PubMed]
- Barber, R.D.; Harmer, D.W.; Coleman, R.A.; Clark, B.J. GAPDH as a housekeeping gene: Analysis of GAPDH mRNA expression in a panel of 72 human tissues. Physiol. Genom. 2005, 21, 389–395. [Google Scholar] [CrossRef]
- Liu, K.; Tang, Z.; Huang, A.; Chen, P.; Liu, P.; Yang, J.; Lu, W.; Liao, J.; Sun, Y.; Wen, S.; et al. Glyceraldehyde-3-phosphate dehydrogenase promotes cancer growth and metastasis through upregulation of SNAIL expression. Int. J. Oncol. 2017, 50, 252–262. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Kim, H.Y. Statistical notes for clinical researchers: Post-hoc multiple comparisons. Restor. Dent. Endod. 2015, 40, 172–176. [Google Scholar] [CrossRef]
- Ramey-Ward, A.N.; Walthall, H.P.; Smith, S.; Barrows, T.H. Human keratin matrices promote wound healing by modulating skin cell expression of cytokines and growth factors. Wound Repair. Regen. 2024, 32, 257–267. [Google Scholar] [CrossRef]
- Moon, D.O. Calcium’s Role in Orchestrating Cancer Apoptosis: Mitochondrial-Centric Perspective. Int. J. Mol. Sci. 2023, 24, 8982. [Google Scholar] [CrossRef]
- Macfarlane, S.R.; Sloss, C.M.; Cameron, P.; Kanke, T.; McKenzie, R.C.; Plevin, R. The role of intracellular Ca2+ in the regulation of proteinase-activated receptor-2 mediated nuclear factor kappa B signalling in keratinocytes. Br. J. Pharmacol. 2005, 145, 535–544. [Google Scholar] [CrossRef]
- UniProt, C. UniProt: The Universal Protein Knowledgebase in 2025. Nucleic Acids Res. 2025, 53, D609–D617. [Google Scholar]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [PubMed]
- Mirdita, M.; Schutze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold: Making protein folding accessible to all. Nat. Methods 2022, 19, 679–682. [Google Scholar] [PubMed]
- Han, S.; Kwak, I.Y. Mastering data visualization with Python: Practical tips for researchers. J. Minim. Invasive Surg. 2023, 26, 167–175. [Google Scholar]
- Wroblewski, K.; Kmiecik, S. Integrating AlphaFold pLDDT Scores into CABS-flex for enhanced protein flexibility simulations. Comput. Struct. Biotechnol. J. 2024, 23, 4350–4356. [Google Scholar]
- Meng, E.C.; Goddard, T.D.; Pettersen, E.F.; Couch, G.S.; Pearson, Z.J.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Tools for structure building and analysis. Protein Sci. 2023, 32, e4792. [Google Scholar]
- Zhang, C.; Wang, Q.; Li, Y.; Teng, A.; Hu, G.; Wuyun, Q.; Zheng, W. The Historical Evolution and Significance of Multiple Sequence Alignment in Molecular Structure and Function Prediction. Biomolecules 2024, 14, 1531. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, X.; Gan, J.; Chen, S.; Xiao, Z.X.; Cao, Y. CB-Dock2: Improved protein-ligand blind docking by integrating cavity detection, docking and homologous template fitting. Nucleic Acids Res. 2022, 50, W159–W164. [Google Scholar]
- Liu, Y.; Cao, Y. Protein-Ligand Blind Docking Using CB-Dock2. Methods Mol. Biol. 2024, 2714, 113–125. [Google Scholar]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar]
- He, X.; Jiang, Y.; Zhang, L.; Li, Y.; Hu, X.; Hua, G.; Cai, S.; Mo, S.; Peng, J. Patient-derived organoids as a platform for drug screening in metastatic colorectal cancer. Front. Bioeng. Biotechnol. 2023, 11, 1190637. [Google Scholar]
- Guenot, D.; Guerin, E.; Aguillon-Romain, S.; Pencreach, E.; Schneider, A.; Neuville, A.; Chenard, M.P.; Duluc, I.; Du Manoir, S.; Brigand, C.; et al. Primary tumour genetic alterations and intra-tumoral heterogeneity are maintained in xenografts of human colon cancers showing chromosome instability. J. Pathol. 2006, 208, 643–652. [Google Scholar] [PubMed]
- Ahmad Zawawi, S.S.; Salleh, E.A.; Musa, M. Spheroids and organoids derived from colorectal cancer as tools for in vitro drug screening. Explor. Target. Antitumor Ther. 2024, 5, 409–431. [Google Scholar] [PubMed]
- Guinney, J.; Dienstmann, R.; Wang, X.; de Reynies, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar]
- Valdeolivas, A.; Amberg, B.; Giroud, N.; Richardson, M.; Galvez, E.J.C.; Badillo, S.; Julien-Laferriere, A.; Turos, D.; Voith von Voithenberg, L.; Wells, I.; et al. Profiling the heterogeneity of colorectal cancer consensus molecular subtypes using spatial transcriptomics. npj Precis. Oncol. 2024, 8, 10. [Google Scholar]
- Joanito, I.; Wirapati, P.; Zhao, N.; Nawaz, Z.; Yeo, G.; Lee, F.; Eng, C.L.P.; Macalinao, D.C.; Kahraman, M.; Srinivasan, H.; et al. Single-cell and bulk transcriptome sequencing identifies two epithelial tumor cell states and refines the consensus molecular classification of colorectal cancer. Nat. Genet. 2022, 54, 963–975. [Google Scholar]
- Kundu, S.; Ali, M.A.; Handin, N.; Conway, L.P.; Rendo, V.; Artursson, P.; He, L.; Globisch, D.; Sjoblom, T. Common and mutation specific phenotypes of KRAS and BRAF mutations in colorectal cancer cells revealed by integrative -omics analysis. J. Exp. Clin. Cancer Res. 2021, 40, 225. [Google Scholar] [CrossRef]
- Alfarouk, K.O.; Stock, C.M.; Taylor, S.; Walsh, M.; Muddathir, A.K.; Verduzco, D.; Bashir, A.H.; Mohammed, O.Y.; Elhassan, G.O.; Harguindey, S.; et al. Resistance to cancer chemotherapy: Failure in drug response from ADME to P-gp. Cancer Cell Int. 2015, 15, 71. [Google Scholar]
- Komatsu, H.; Enjouji, S.; Ito, A.; Ohama, T.; Sato, K. Prostaglandin E2 inhibits proteinase-activated receptor 2-signal transduction through regulation of receptor internalization. J. Vet. Med. Sci. 2013, 75, 255–261. [Google Scholar]
- Bakele, M.; Lotz-Havla, A.S.; Jakowetz, A.; Carevic, M.; Marcos, V.; Muntau, A.C.; Gersting, S.W.; Hartl, D. An interactive network of elastase, secretases, and PAR-2 protein regulates CXCR1 receptor surface expression on neutrophils. J. Biol. Chem. 2014, 289, 20516–20525. [Google Scholar]
- Yung, Y.C.; Stoddard, N.C.; Chun, J. LPA receptor signaling: Pharmacology, physiology, and pathophysiology. J. Lipid Res. 2014, 55, 1192–1214. [Google Scholar] [PubMed]
- Lopez-Yerena, A.; Vallverdu-Queralt, A.; Mols, R.; Augustijns, P.; Lamuela-Raventos, R.M.; Escribano-Ferrer, E. Absorption and Intestinal Metabolic Profile of Oleocanthal in Rats. Pharmaceutics 2020, 12, 134. [Google Scholar] [PubMed]
- Schwobel, J.A.; Madden, J.C.; Cronin, M.T. Examination of Michael addition reactivity towards glutathione by transition-state calculations. SAR QSAR Environ. Res. 2010, 21, 693–710. [Google Scholar]
- Gerritsen, J.S.; White, F.M. Phosphoproteomics: A valuable tool for uncovering molecular signaling in cancer cells. Expert. Rev. Proteomics 2021, 18, 661–674. [Google Scholar]
- Cook, A.L.; Wyhs, N.; Sur, S.; Ptak, B.; Popoli, M.; Dobbyn, L.; Papadopoulos, T.; Bettegowda, C.; Papadopoulos, N.; Vogelstein, B.; et al. An isogenic cell line panel for sequence-based screening of targeted anticancer drugs. iScience 2022, 25, 104437. [Google Scholar] [CrossRef]
- Shah, H.; Hill, T.A.; Lim, J.; Fairlie, D.P. Protease-activated receptor 2 attenuates doxorubicin-induced apoptosis in colon cancer cells. J. Cell Commun. Signal 2023, 17, 1293–1307. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer Type | Sequence | Accession | E-Value | Bit Score |
|---|---|---|---|---|---|
| GAPDH | Forward (5′-3′) Reverse (5′-3′) | GTCTCCTCTGACTTCAACAGCG ACCACCCTGTTGCTGTAGCCAA | NM_002046 | 0.0 | 2374 |
| PAR-2 | Forward (5′-3′) Reverse (5′-3’) | CTCCTCTCTGTCATCTGGTTCC TGCACACTGAGGCAGGTCATGA | NM-005242 | 0.0 | 2861 |
| PAR-1 | Forward (5’-3’) Reverse (5’-3’) | GCTGTCCTACTGCTTGGAAGAC CTGCATCAGCACATACTCCTCC | NM_022002 | 0.0 | 2745 |
| TNF-α | Forward (5’-3’) Reverse (5’-3’) | CTCTTCTGCCTGCTGCACTTTG ATGGGCTACAGGCTTGTCACTC | NM_000594 | 0.0 | 2449 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patnaik, R.; Varghese, R.L.; Banerjee, Y. Selective Modulation of PAR-2-Driven Inflammatory Pathways by Oleocanthal: Attenuation of TNF-α and Calcium Dysregulation in Colorectal Cancer Models. Int. J. Mol. Sci. 2025, 26, 2934. https://doi.org/10.3390/ijms26072934
Patnaik R, Varghese RL, Banerjee Y. Selective Modulation of PAR-2-Driven Inflammatory Pathways by Oleocanthal: Attenuation of TNF-α and Calcium Dysregulation in Colorectal Cancer Models. International Journal of Molecular Sciences. 2025; 26(7):2934. https://doi.org/10.3390/ijms26072934
Chicago/Turabian StylePatnaik, Rajashree, Riah Lee Varghese, and Yajnavalka Banerjee. 2025. "Selective Modulation of PAR-2-Driven Inflammatory Pathways by Oleocanthal: Attenuation of TNF-α and Calcium Dysregulation in Colorectal Cancer Models" International Journal of Molecular Sciences 26, no. 7: 2934. https://doi.org/10.3390/ijms26072934
APA StylePatnaik, R., Varghese, R. L., & Banerjee, Y. (2025). Selective Modulation of PAR-2-Driven Inflammatory Pathways by Oleocanthal: Attenuation of TNF-α and Calcium Dysregulation in Colorectal Cancer Models. International Journal of Molecular Sciences, 26(7), 2934. https://doi.org/10.3390/ijms26072934