Identification of a Cancer Stem Cell-Related Gene Signature in Hepatocellular Carcinoma Based on Single-Cell RNA-Seq and Bulk RNA-Seq Analysis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
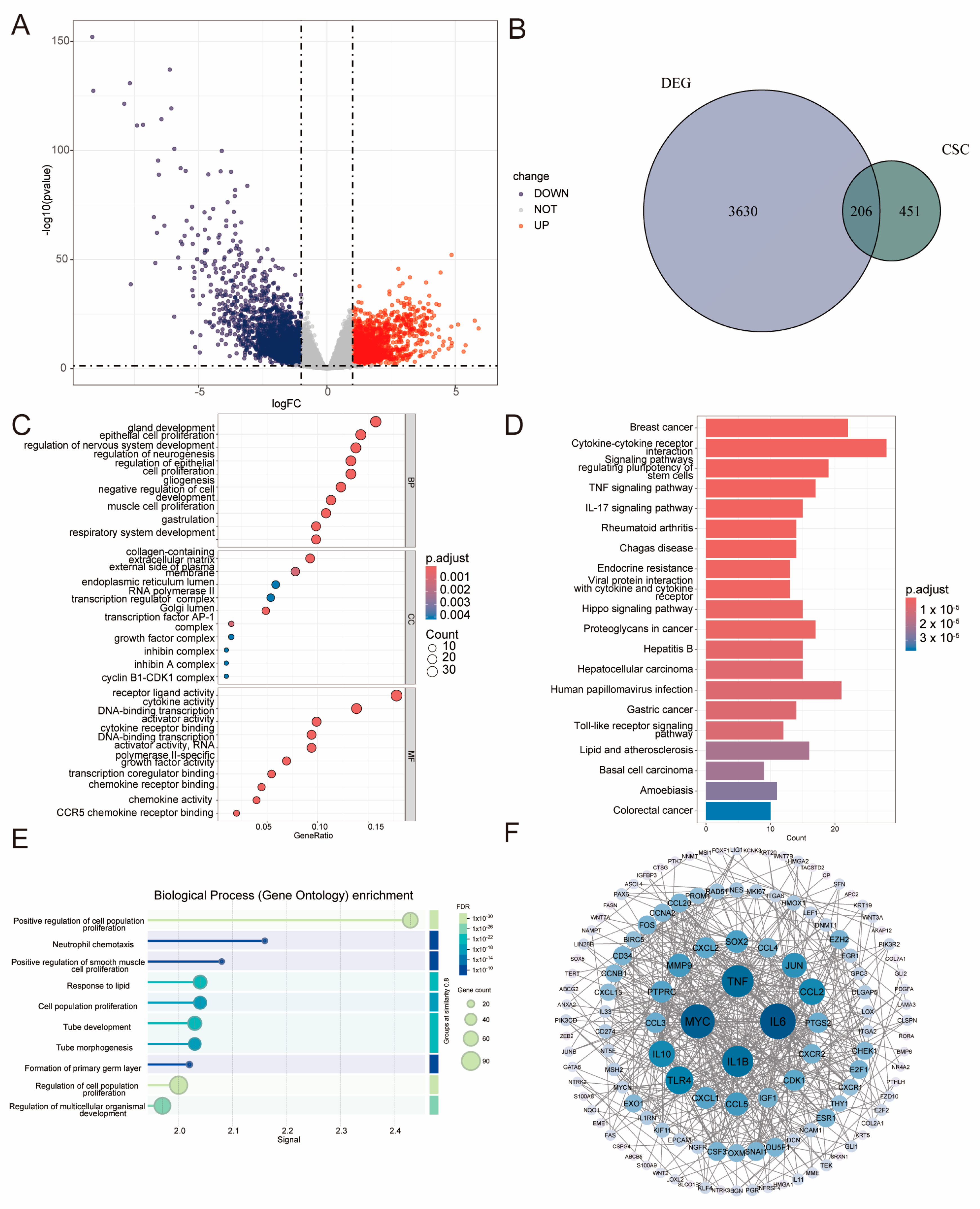
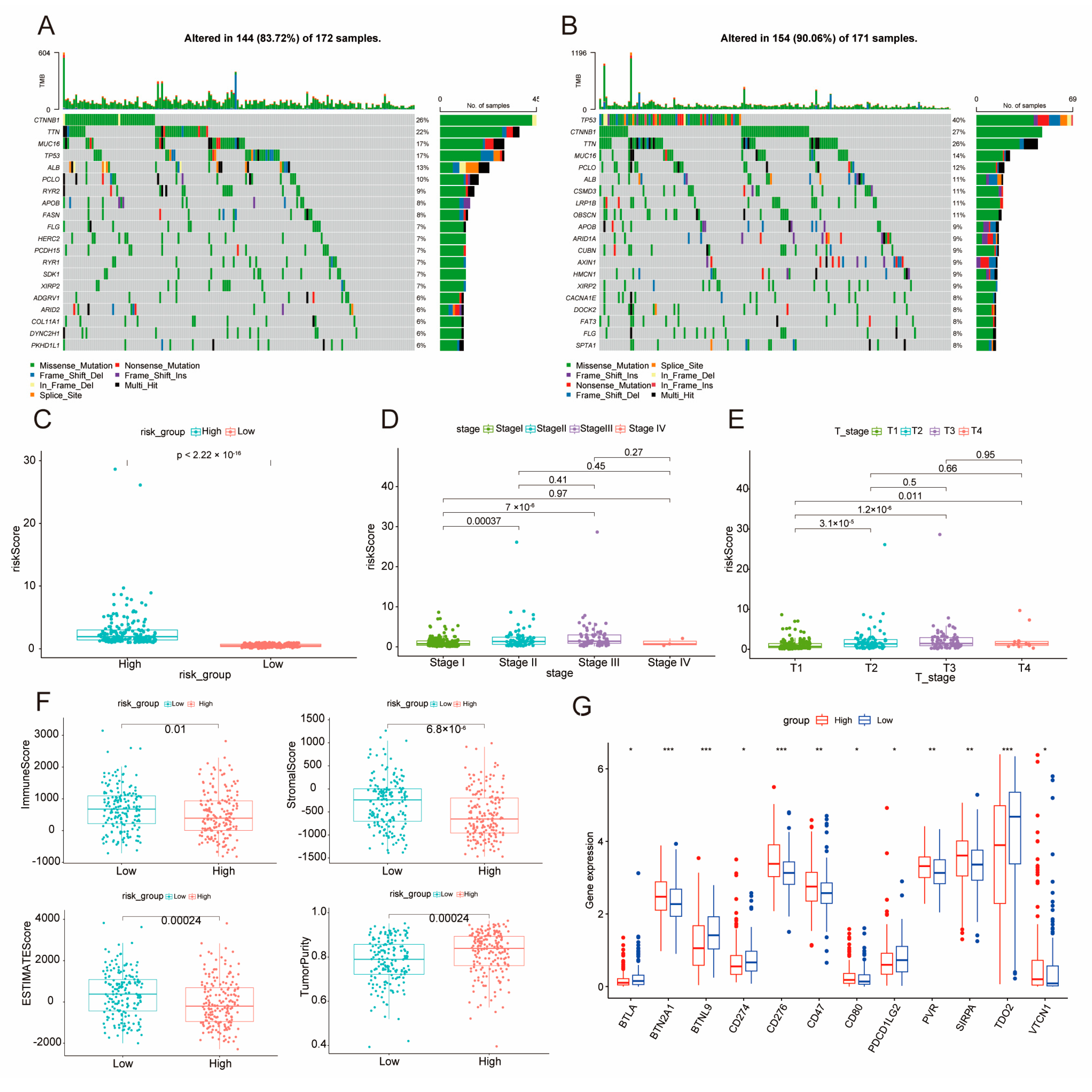
2.1. Identification of LIHC–Specific BCSC–Related Genes and Functional Analysis
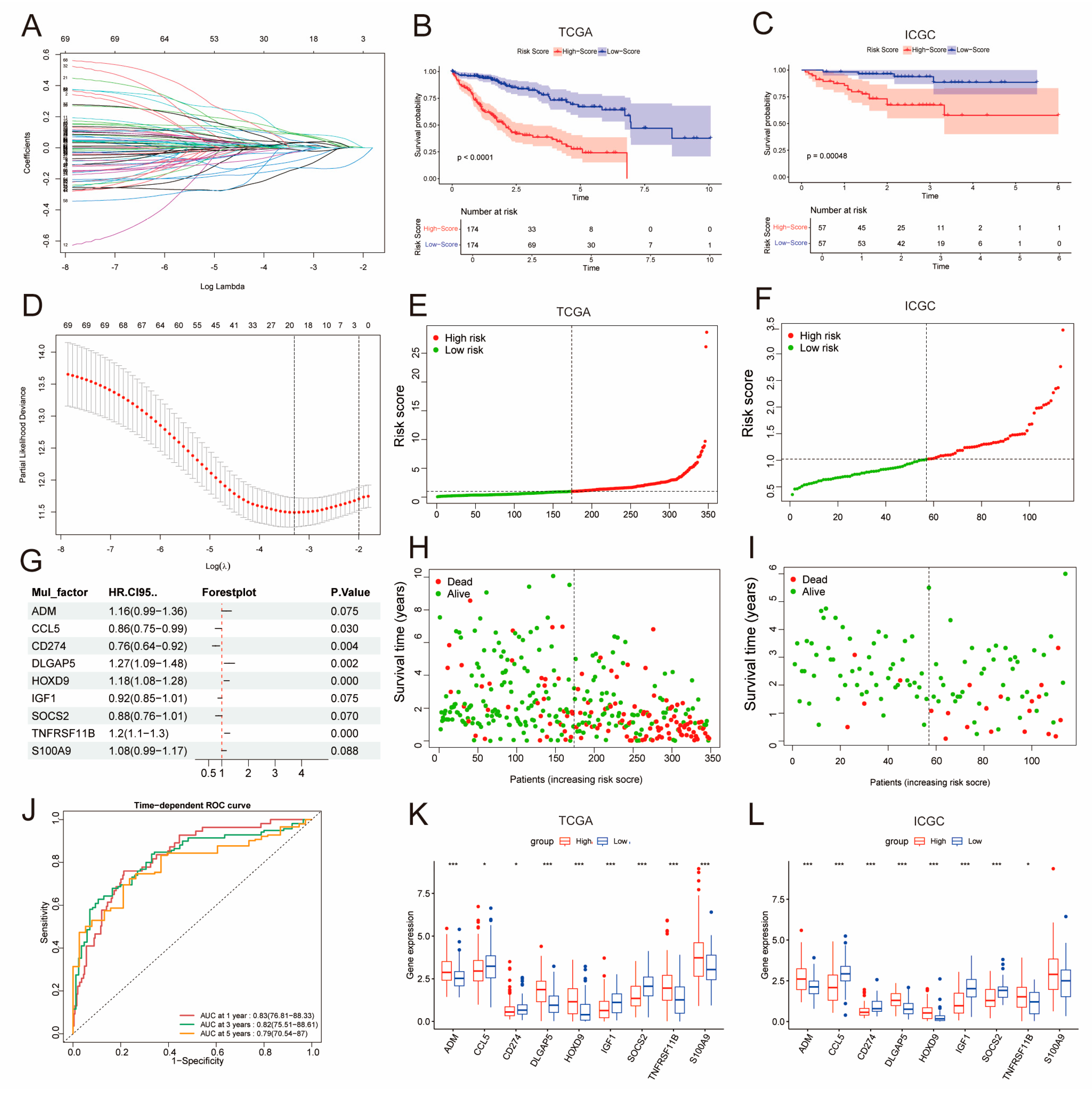
2.2. Construction of a Prognostic Model Using DE-BCSCs
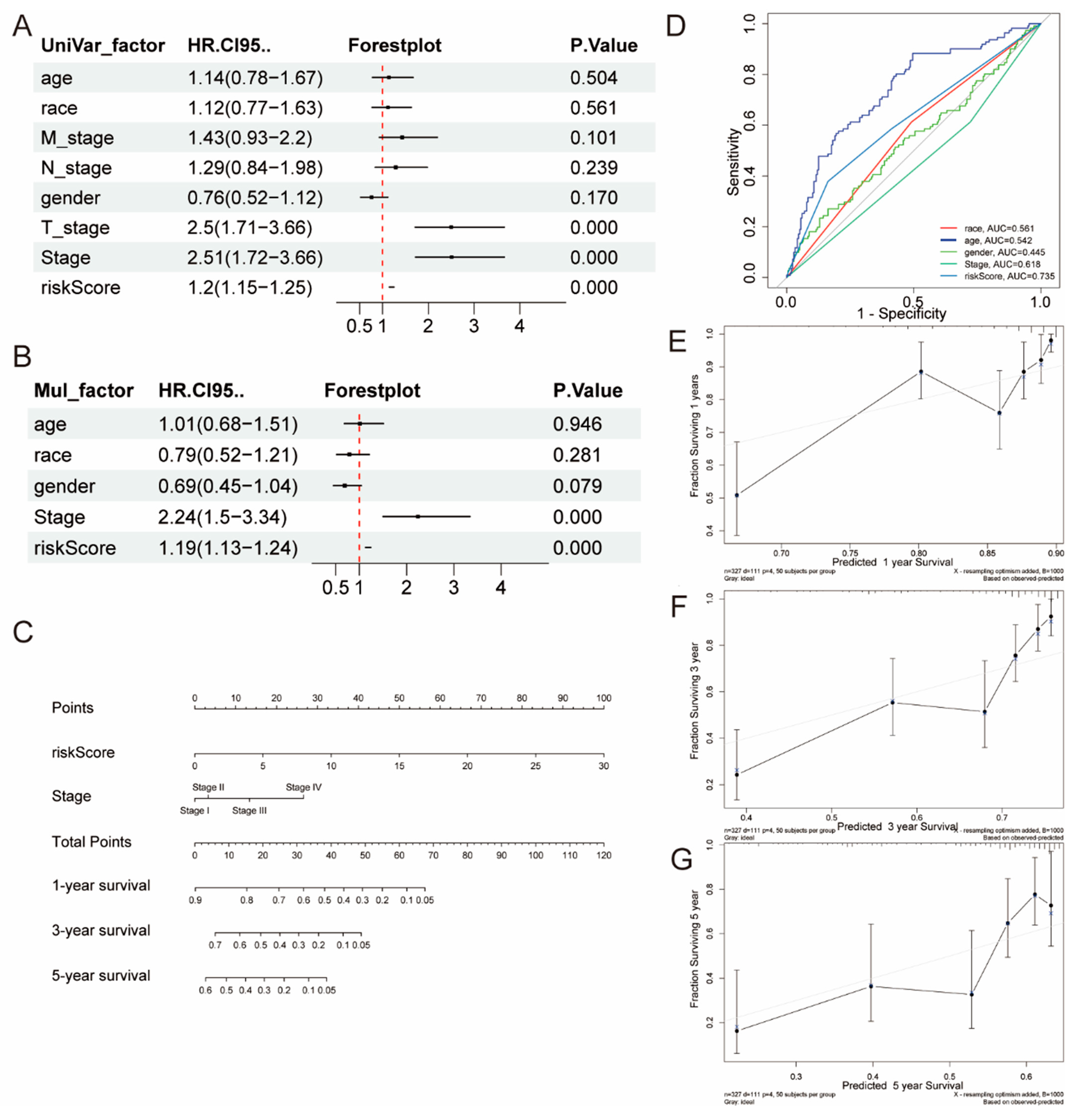
2.3. The Creation and Verification of a Nomogram Model
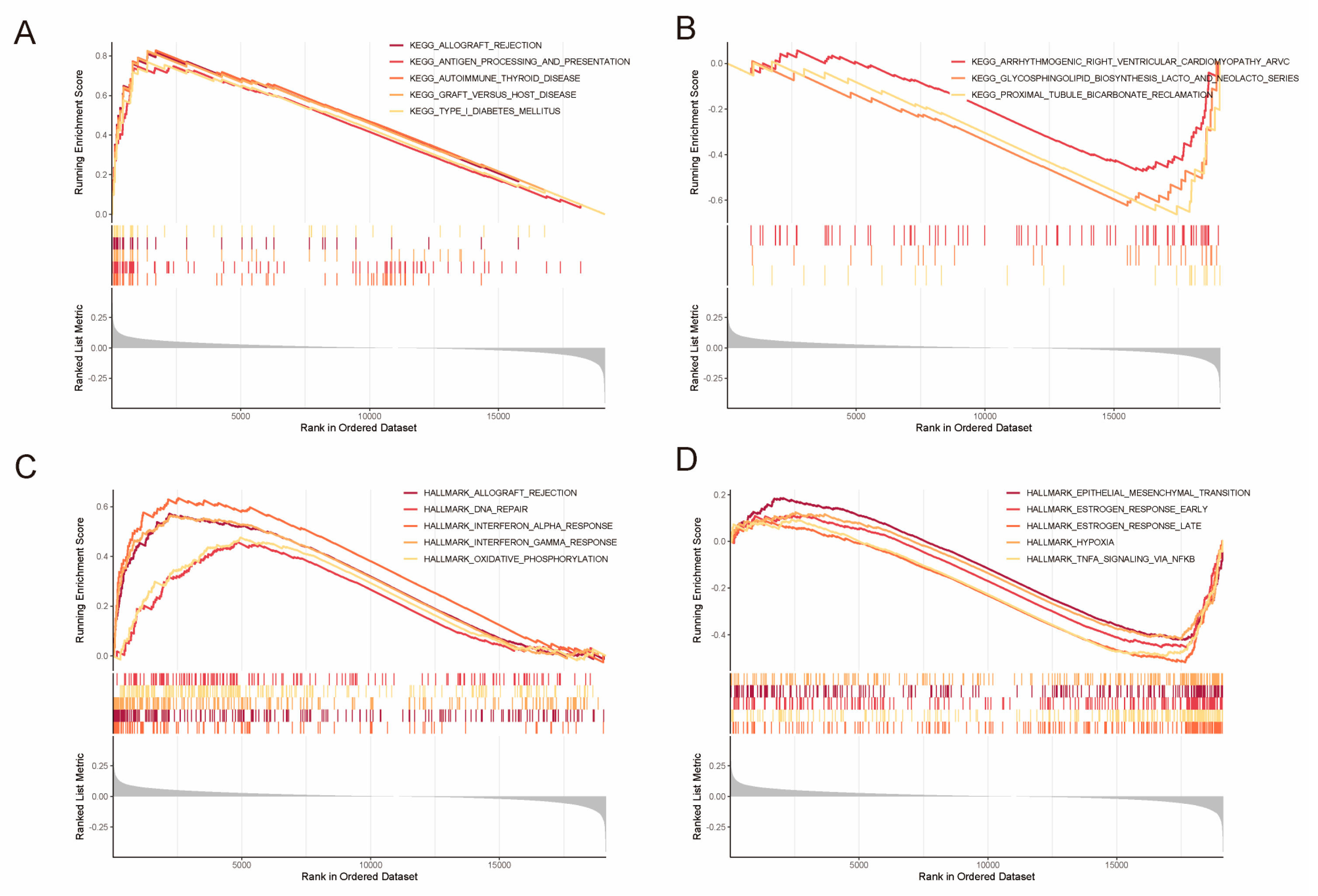
2.4. Identification of DEGs and Gene Set Enrichment Analysis (GSEA)
2.5. Elucidation of DE–BCSCs as Potential Protective Factors
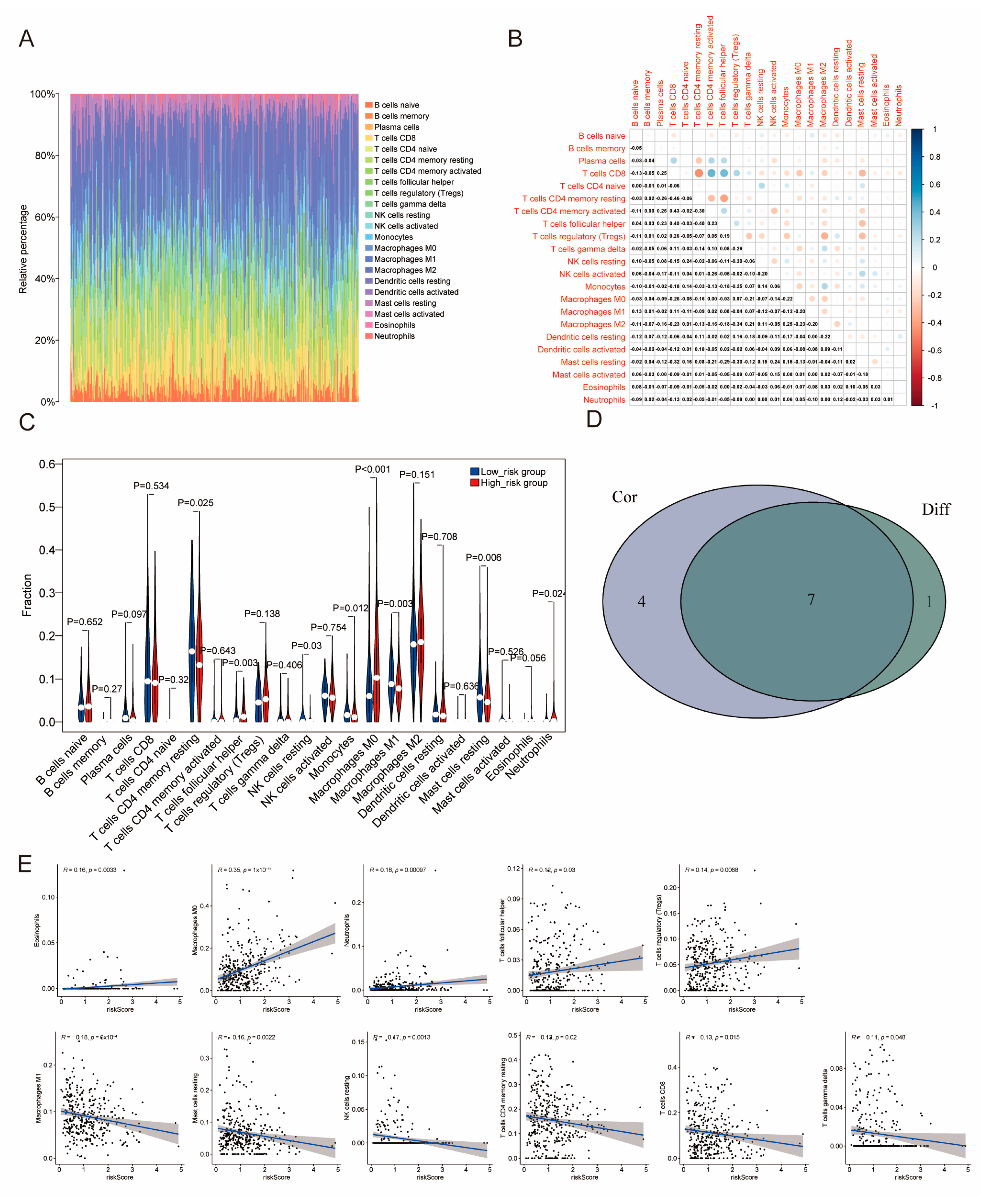
2.6. The Distribution of Tumor-Infiltrating Immune Cells (TICs) Across Two Distinct Subgroups
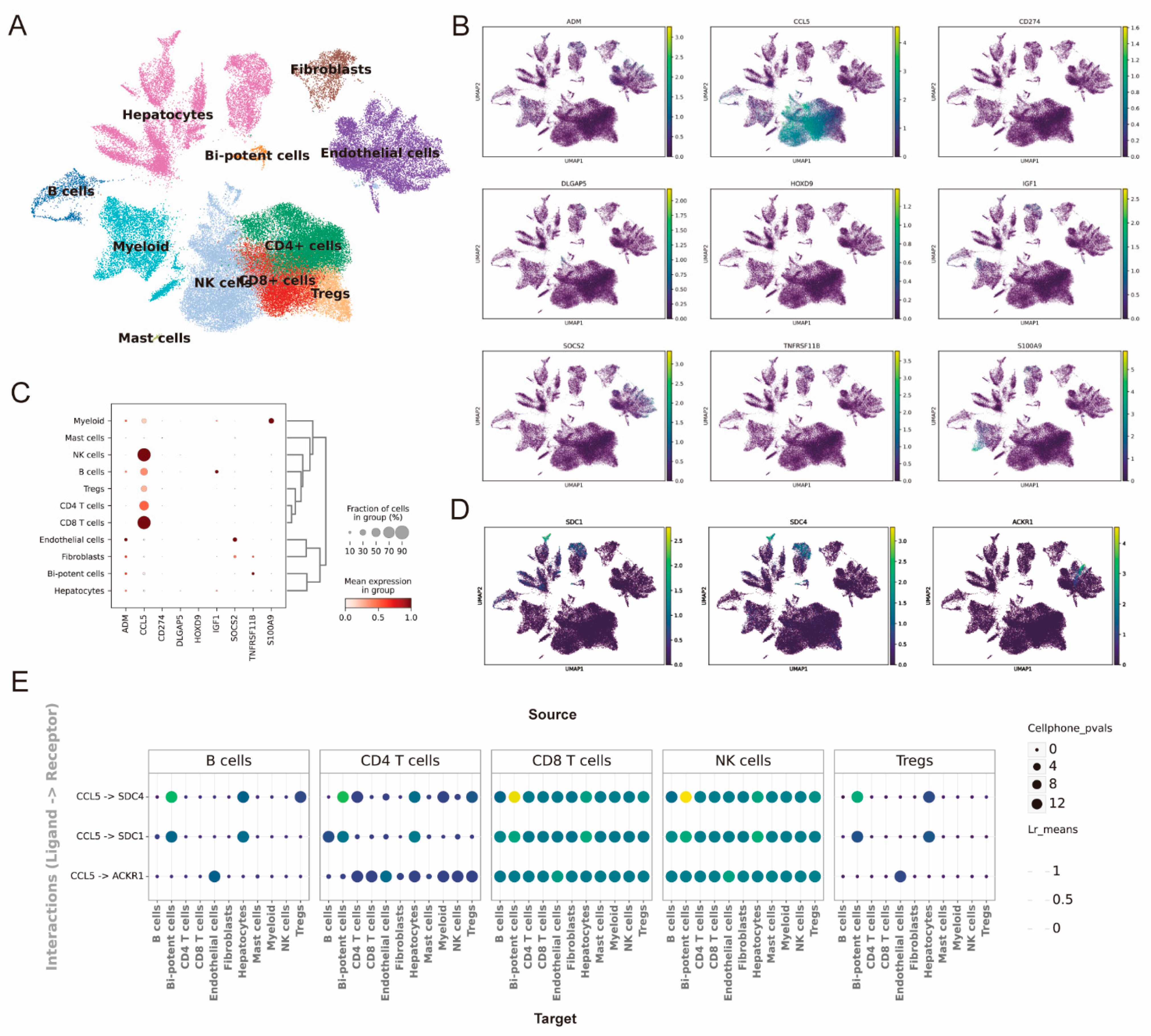
2.7. The Single-Cell Analysis Predicts the Expression Distribution of Model Genes and Intercellular Communication
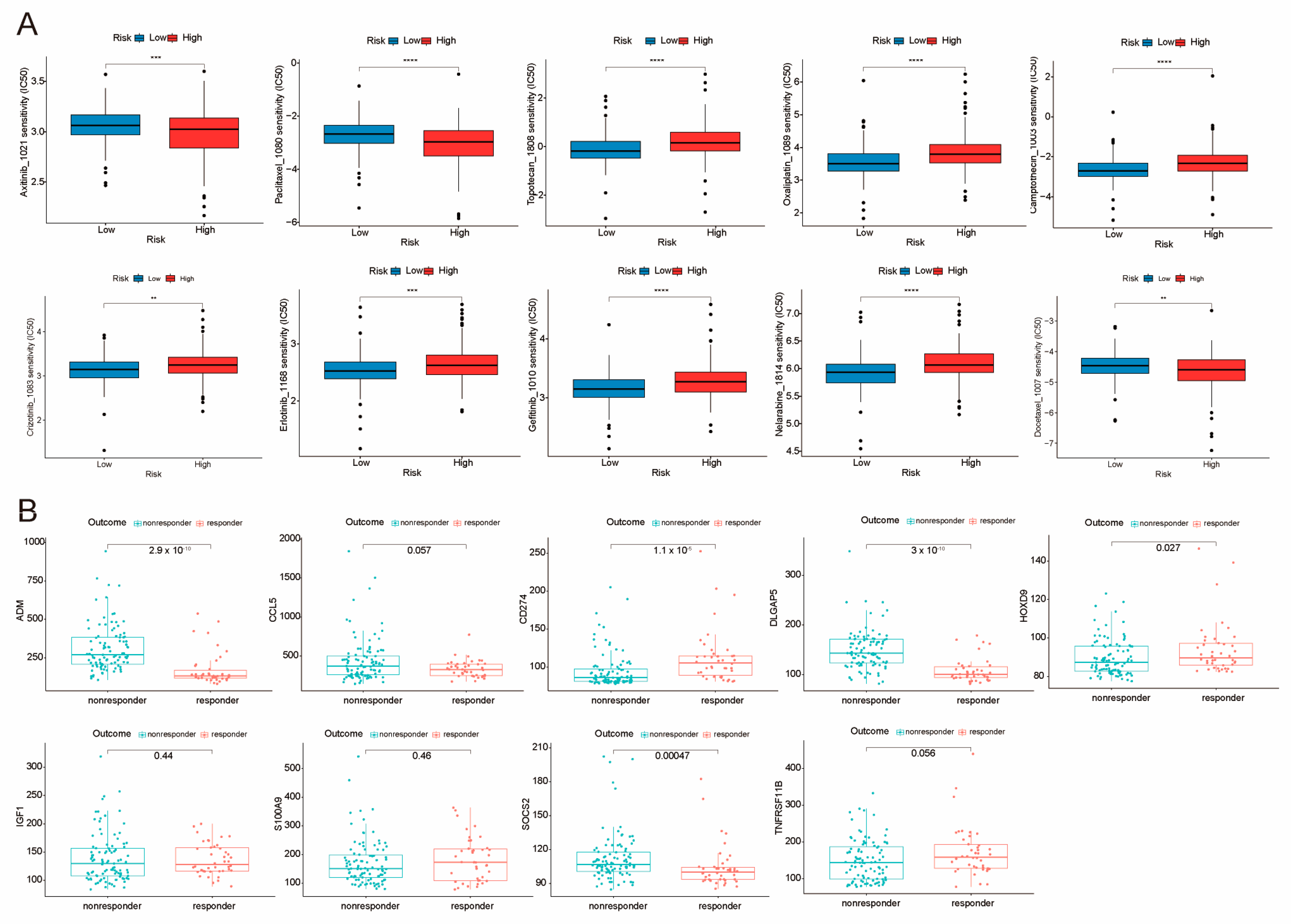
2.8. Sensitivity Analysis of Potential Clinical Drugs Within Groups Identified as High-Risk and Low-Risk
3. Discussion
4. Materials and Methods
4.1. Diagram of Study Flow and Data Collection
4.2. Identification of DE–BCSCs in LIHC
4.3. Designing and Validating Prognostic Models Based on DE–BCSCs
4.4. Examining How the Risk Score Can Help in Clinical Diagnosis
4.5. How Prognostic DE–BCSCs Influence Immune Cell Infiltration in Tumors
4.6. Prediction of Chemotherapy Susceptibility
4.7. ScRNA and Cell Communication Analysis of the GSE156625 Data Set
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CSCs | Cancer stem cells |
| BCSCs | Biomarkers of CSCs |
| DE–BCSCs | Differentially expressed BCSCs |
| TCGA | The Cancer Genome Atlas |
| ICGC | International Cancer Genome Consortium |
| GEO | Gene Expression Omnibus |
| scRNA-seq | single-cell RNA sequencing |
| GO | Gene Ontology |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| LASSO | Least Absolute Shrinkage and Selection Operator |
| OS | Overall Survival |
| TME | Tumor microenvironment |
| LIHC | Liver hepatocellular carcinoma |
| HCC | Hepatocellular carcinoma |
| EMT | Epithelial Mesenchymal Transition |
| UMAP | Uniform Manifold Approximation and Projection |
| TNM | Tumor Node Metastasis |
| TICs | Tumor-Infiltrating Immune Cells |
| GSEA | Gene Set Enrichment Analysis |
| LIANA | LIgand-receptor ANalysis frAmework |
References
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA A Cancer J. Clin. 2024, 74, 229–263. [Google Scholar]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA A Cancer J. Clin. 2021, 71, 209–249. [Google Scholar]
- Zhang, D.; Tang, D.G.; Rycaj, K. Cancer stem cells: Regulation programs, immunological properties and immunotherapy. Semin. Cancer Biol. 2018, 52, 94–106. [Google Scholar] [PubMed]
- Schulte, L.-A.; López-Gil, J.C.; Sainz, B.; Hermann, P.C. The Cancer Stem Cell in Hepatocellular Carcinoma. Cancers 2020, 12, 684. [Google Scholar] [CrossRef]
- Lee, T.K.-W.; Guan, X.-Y.; Ma, S. Cancer stem cells in hepatocellular carcinoma—From origin to clinical implications. Nat. Rev. Gastroenterol Hepatol. 2022, 19, 26–44. [Google Scholar] [PubMed]
- Vermeulen, L.; de Sousa e Melo, F.; Richel, D.J.; Medema, J.P. The developing cancer stem-cell model: Clinical challenges and opportunities. Lancet Oncol. 2012, 13, e83–e89. [Google Scholar]
- Kilmister, E.J.; Koh, S.P.; Weth, F.R.; Gray, C.; Tan, S.T. Cancer Metastasis and Treatment Resistance: Mechanistic Insights and Therapeutic Targeting of Cancer Stem Cells and the Tumor Microenvironment. Biomedicines 2022, 10, 2988. [Google Scholar] [CrossRef]
- Ma, S.; Chan, K.-W.; Hu, L.; Lee, T.K.W.; Wo, J.Y.H.; Ng, I.O.L.; Zheng, B.J.; Guan, X.Y. Identification and Characterization of Tumorigenic Liver Cancer Stem/Progenitor Cells. Gastroenterology 2007, 132, 2542–2556. [Google Scholar]
- Kong, X.; Peng, H.; Liu, P.; Fu, X.; Wang, N.; Zhang, D. Programmed death ligand 1 regulates epithelial-mesenchymal transition and cancer stem cell phenotypes in hepatocellular carcinoma through the serum and glucocorticoid kinase 2/β-catenin signaling pathway. Cancer Sci. 2023, 114, 2265–2276. [Google Scholar]
- Jeng, K.-S.; Chang, C.-F.; Sheen, I.-S.; Jeng, C.-J.; Wang, C.-H. Cellular and Molecular Biology of Cancer Stem Cells of Hepatocellular Carcinoma. Int. J. Mol. Sci. 2023, 24, 1417. [Google Scholar] [CrossRef]
- Wang, H.-C.; Haung, L.-Y.; Wang, C.-J.; Chao, Y.-J.; Hou, Y.-C.; Yen, C.-J.; Shan, Y.-S. Tumor-associated macrophages promote resistance of hepatocellular carcinoma cells against sorafenib by activating CXCR2 signaling. J. Biomed. Sci. 2022, 29, 99. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Liu, J.; Zang, L.; Xiao, T.; Zhang, X.; Li, Z.; Zhu, H.; Gao, W.; Yu, X. Integrated Machine Learning and Bioinformatic Analyses Constructed a Novel Stemness-Related Classifier to Predict Prognosis and Immunotherapy Responses for Hepatocellular Carcinoma Patients. Int. J. Biol. Sci. 2022, 18, 360–373. [Google Scholar] [CrossRef] [PubMed]
- Ruszkowska-Ciastek, B.; Kwiatkowska, K.; Marques-da-Silva, D.; Lagoa, R. Cancer Stem Cells from Definition to Detection and Targeted Drugs. Int. J. Mol. Sci. 2024, 25, 3903. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Du, Y.; Guan, X.-Y.; Yan, Q. The current status of tumor microenvironment and cancer stem cells in sorafenib resistance of hepatocellular carcinoma. Front. Oncol. 2023, 13, 1204513. [Google Scholar] [CrossRef]
- Liang, X.Y.; Zhang, Y.; He, Y.N.; Liu, X.Y.; Ding, Z.H.; Zhang, X.D.; Dong, M.Y.; Du, R.L. A cancer stem cell associated gene signature for predicting overall survival of hepatocellular carcinoma. Front. Genet. 2022, 13, 888601. [Google Scholar] [CrossRef]
- MacLean, M.R.; Walker, O.L.; Arun, R.P.; Fernando, W.; Marcato, P. Informed by Cancer Stem Cells of Solid Tumors: Advances in Treatments Targeting Tumor-Promoting Factors and Pathways. Int. J. Mol. Sci. 2024, 25, 4102. [Google Scholar] [CrossRef]
- Tung-Ping Poon, R.; Fan, S.-T.; Wong, J. Risk Factors, Prevention, and Management of Postoperative Recurrence After Resection of Hepatocellular Carcinoma. Ann. Surg. 2000, 232, 10–24. [Google Scholar] [CrossRef]
- Pons, F.; Varela, M.; Llovet, J.M. Staging systems in hepatocellular carcinoma. HPB 2005, 7, 35–41. [Google Scholar] [CrossRef]
- Mukaida, N.; Nakamoto, Y. Emergence of immunotherapy as a novel way to treat hepatocellular carcinoma. World J. Gastroenterol. 2018, 24, 1839–1858. [Google Scholar] [CrossRef]
- Clara, J.A.; Monge, C.; Yang, Y.; Takebe, N. Targeting signalling pathways and the immune microenvironment of cancer stem cells—A clinical update. Nat. Rev. Clin. Oncol. 2020, 17, 204–232. [Google Scholar] [CrossRef]
- Lv, D.; Chen, L.; Du, L.; Zhou, L.; Tang, H. Emerging Regulatory Mechanisms Involved in Liver Cancer Stem Cell Properties in Hepatocellular Carcinoma. Front. Cell Dev. Biol. 2021, 9, 691410. [Google Scholar]
- Zhou, W.; Ke, S.Q.; Huang, Z.; Flavahan, W.; Fang, X.; Paul, J.; Wu, L.; Sloan, A.E.; McLendon, R.E.; Li, X.; et al. Periostin Secreted by Glioblastoma Stem Cells Recruits M2 Tumor-associated Macrophages and Promotes Malignant Growth. Nat. Cell Biol. 2015, 17, 170–182. [Google Scholar] [PubMed]
- Sun, Y.; Wu, L.; Zhong, Y.; Zhou, K.; Hou, Y.; Wang, Z.; Zhang, Z.; Xie, J.; Wang, C.; Chen, D.; et al. Single-cell landscape of the ecosystem in early-relapse hepatocellular carcinoma. Cell 2021, 184, 404–421.e16. [Google Scholar] [PubMed]
- Li, D.; Zhang, T.; Guo, Y.; Bi, C.; Liu, M.; Wang, G. Biological impact and therapeutic implication of tumor-associated macrophages in hepatocellular carcinoma. Cell Death Dis. 2024, 15, 498. [Google Scholar] [PubMed]
- Oura, K.; Morishita, A.; Tani, J.; Masaki, T. Tumor Immune Microenvironment and Immunosuppressive Therapy in Hepatocellular Carcinoma: A Review. Int. J. Mol. Sci. 2021, 22, 5801. [Google Scholar] [CrossRef]
- Polidoro, M.A.; Mikulak, J.; Cazzetta, V.; Lleo, A.; Mavilio, D.; Torzilli, G.; Donadon, M. Tumor microenvironment in primary liver tumors: A challenging role of natural killer cells. World J. Gastroenterol. 2020, 26, 4900–4918. [Google Scholar]
- Baumjohann, D.; Brossart, P. T follicular helper cells: Linking cancer immunotherapy and immune-related adverse events. J. Immunother. Cancer 2021, 9, e002588. [Google Scholar]
- Wu, L.; Saxena, S.; Singh, R.K. Neutrophils in the Tumor Microenvironment. Adv. Exp. Med. Biol. 2020, 1224, 41–52. [Google Scholar]
- Weiskopf, K. Cancer immunotherapy targeting the CD47/SIRPα axis. Eur. J. Cancer 2017, 76, 100–109. [Google Scholar]
- MacGregor, H.L.; Ohashi, P.S. Molecular Pathways: Evaluating the Potential for B7-H4 as an Immunoregulatory Target. Clin. Cancer Res. 2017, 23, 2934–2941. [Google Scholar]
- Chiang, E.Y.; Mellman, I. TIGIT-CD226-PVR axis: Advancing immune checkpoint blockade for cancer immunotherapy. J. Immunother. Cancer 2022, 10, e004711. [Google Scholar]
- Cabrera-Galván, J.J.; Araujo, E.; de Mirecki-Garrido, M.; Pérez-Rodríguez, D.; Guerra, B.; Aranda-Tavío, H.; Guerra-Rodríguez, M.; Brito-Casillas, Y.; Melián, C.; Martínez-Martín, M.S.; et al. SOCS2 protects against chemical-induced hepatocellular carcinoma progression by modulating inflammation and cell proliferation in the liver. Biomed. Pharmacother. 2023, 157, 114060. [Google Scholar]
- Zhang, G. Regulatory T-cells-related signature for identifying a prognostic subtype of hepatocellular carcinoma with an exhausted tumor microenvironment. Front. Immunol. 2022, 13, 975762. [Google Scholar]
- Roessler, S.; Jia, H.-L.; Budhu, A.; Forgues, M.; Ye, Q.-H.; Lee, J.-S.; Thorgeirsson, S.S.; Sun, Z.; Tang, Z.-Y.; Qin, L.-X.; et al. A Unique Metastasis Gene Signature Enables Prediction of Tumor Relapse in Early Stage Hepatocellular Carcinoma Patients. Cancer Res. 2010, 70, 10202–10212. [Google Scholar] [PubMed]
- Pinyol, R.; Montal, R.; Bassaganyas, L.; Sia, D.; Takayama, T.; Chau, G.-Y.; Mazzaferro, V.; Roayaie, S.; Lee, H.C.; Kokudo, N.; et al. Molecular predictors of prevention of recurrence in HCC with sorafenib as adjuvant treatment and prognostic factors in the phase 3 STORM trial. Gut 2019, 68, 1065–1075. [Google Scholar]
- Sharma, A.; Seow, J.J.W.; Dutertre, C.-A.; Pai, R.; Blériot, C.; Mishra, A.; Wong, R.M.M.; Singh, G.S.N.; Sudhagar, S.; Khalilnezhad, S.; et al. Onco-fetal Reprogramming of Endothelial Cells Drives Immunosuppressive Macrophages in Hepatocellular Carcinoma. Cell 2020, 183, 377–394.e21. [Google Scholar] [PubMed]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevskt, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO. Archive for functional genomics data sets—Update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar]
- Futreal, P.A.; Coin, L.; Marshall, M.; Down, T.; Hubbard, T.; Wooster, R.; Rahman, N.; Stratton, M.R. A census of human cancer genes. Nat. Rev. Cancer 2004, 4, 177–183. [Google Scholar]
- International Cancer Genome Consortium. International network of cancer genome projects. Nature 2010, 464, 993–998. [Google Scholar]
- Firdous, S.; Ghosh, A.; Saha, S. BCSCdb: A database of biomarkers of cancer stem cells. Database 2022, 2022, baac082. [Google Scholar]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [PubMed]
- Kanehisa, M.; Goto, S. KEGG. Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [PubMed]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS J. Integr. Biol. 2012, 16, 284–287. [Google Scholar]
- von Mering, C.; Huynen, M.; Jaeggi, D.; Schmidt, S.; Bork, P.; Snel, B. STRING. A database of predicted functional associations between proteins. Nucleic Acids Res. 2003, 31, 258–261. [Google Scholar] [PubMed]
- Therneau, T.M.; Patricia, M.G. Modeling Survival Data: Extending the Cox Model, 2nd ed.; Springer: New York, NY, USA, 2013. [Google Scholar]
- Kassambara, A.; Kosinski, M.; Biecek, P. Survminer: Drawing Survival Curves Using ‘ggplot2’ (Version 0.4.9) [Computer Software]. 2021. Available online: https://CRAN.R-project.org/package=survminer (accessed on 20 March 2025).
- Huber, W.; Carey, V.J.; Gentleman, R.; Anders, S.; Carlson, M.; Carvalho, B.S.; Bravo, H.C.; Davis, S.; Gatto, L.; Girke, T.; et al. Orchestrating high-throughput genomic analysis with Bioconductor. Nat. Methods 2015, 12, 115–121. [Google Scholar]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar]
- Yoshihara, K.; Shahmoradgoli, M.; Martínez, E.; Vegesna, R.; Kim, H.; Torres-Garcia, W.; Treviño, V.; Shen, H.; Laird, P.W.; Levine, D.A.; et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 2013, 4, 2612. [Google Scholar]
- Newman, A.M.; Liu, C.L.; Green, M.R.; Gentles, A.J.; Feng, W.; Xu, Y.; Hoang, C.D.; Diehn, M. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 2015, 12, 453–457. [Google Scholar]
- Maeser, D.; Gruener, R.F.; Huang, R.S. oncoPredict: An R package for predicting in vivo or cancer patient drug response and biomarkers from cell line screening data. Brief. Bioinform. 2021, 22, bbab260. [Google Scholar]
- Dimitrov, D.; Türei, D.; Garrido-Rodriguez, M.; Burmedi, P.L.; Nagai, J.S.; Boys, C.; Flores, R.O.R.; Kim, H.; Szalai, B.; Costa, I.G.; et al. Comparison of methods and resources for cell-cell communication inference from single-cell RNA-Seq data. Nat. Commun. 2022, 13, 3224. [Google Scholar] [PubMed]
- Efremova, M.; Vento-Tormo, M.; Teichmann, S.A.; Vento-Tormo, R. CellPhoneDB: Inferring cell-cell communication from combined expression of multi-subunit ligand-receptor complexes. Nat. Protoc. 2020, 15, 1484–1506. [Google Scholar] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, J.; Liu, X.; Huang, S.; Liu, W. Identification of a Cancer Stem Cell-Related Gene Signature in Hepatocellular Carcinoma Based on Single-Cell RNA-Seq and Bulk RNA-Seq Analysis. Int. J. Mol. Sci. 2025, 26, 2933. https://doi.org/10.3390/ijms26072933
Wu J, Liu X, Huang S, Liu W. Identification of a Cancer Stem Cell-Related Gene Signature in Hepatocellular Carcinoma Based on Single-Cell RNA-Seq and Bulk RNA-Seq Analysis. International Journal of Molecular Sciences. 2025; 26(7):2933. https://doi.org/10.3390/ijms26072933
Chicago/Turabian StyleWu, Jing, Xu Liu, Sheng Huang, and Wei Liu. 2025. "Identification of a Cancer Stem Cell-Related Gene Signature in Hepatocellular Carcinoma Based on Single-Cell RNA-Seq and Bulk RNA-Seq Analysis" International Journal of Molecular Sciences 26, no. 7: 2933. https://doi.org/10.3390/ijms26072933
APA StyleWu, J., Liu, X., Huang, S., & Liu, W. (2025). Identification of a Cancer Stem Cell-Related Gene Signature in Hepatocellular Carcinoma Based on Single-Cell RNA-Seq and Bulk RNA-Seq Analysis. International Journal of Molecular Sciences, 26(7), 2933. https://doi.org/10.3390/ijms26072933