
Mitochondria-Associated Membranes as Key Regulators in Cellular Homeostasis and the Potential Impact of Exercise on Insulin Resistance


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
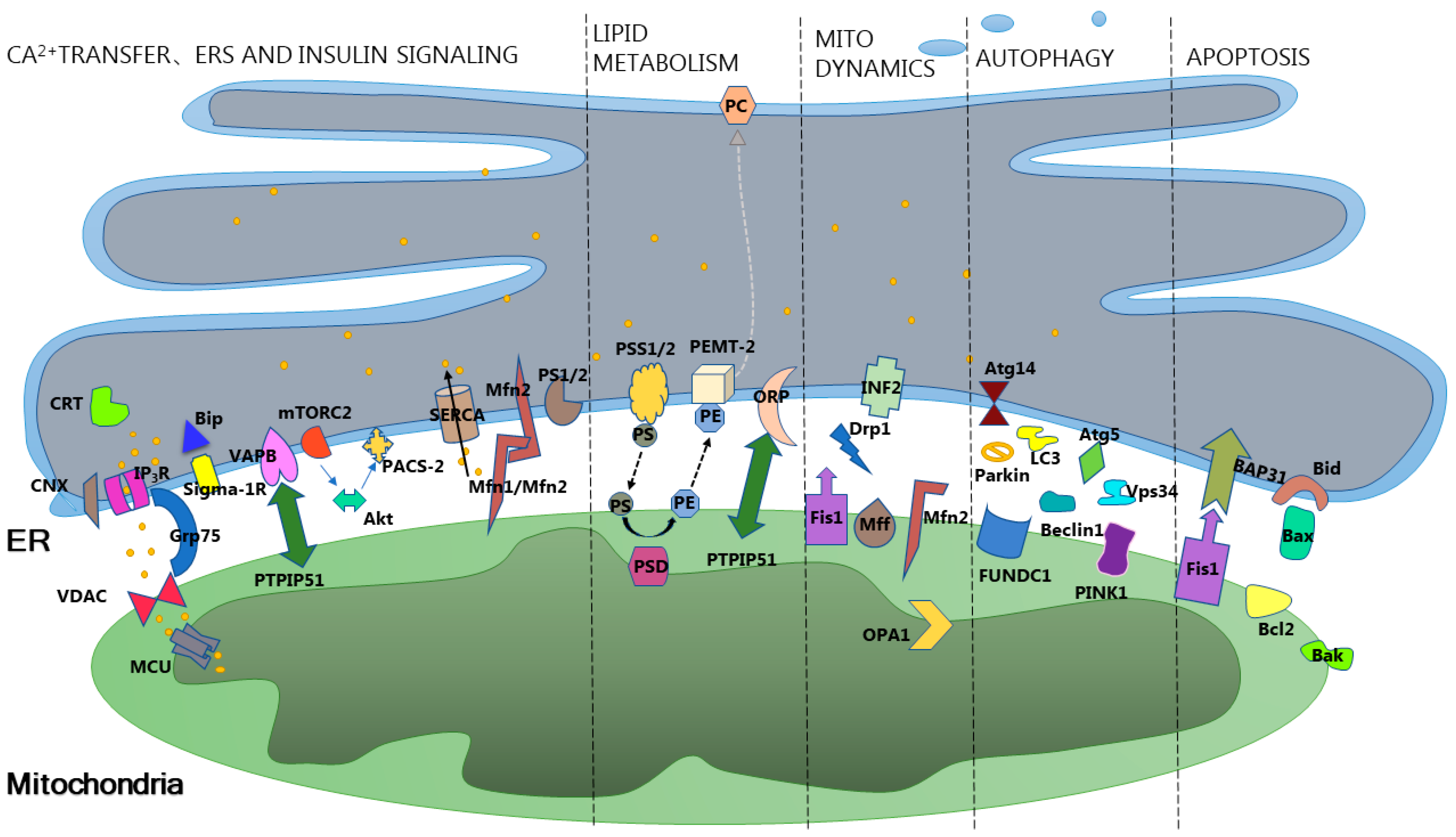
2. Overview of Mitochondria-Associated Membranes
3. Mitochondria-Associated Membranes Intervene in Insulin Resistance through Cellular Homeostasis
3.1. Mitochondria-Associated Membranes Intervene in Insulin Resistance through Calcium Homeostasis
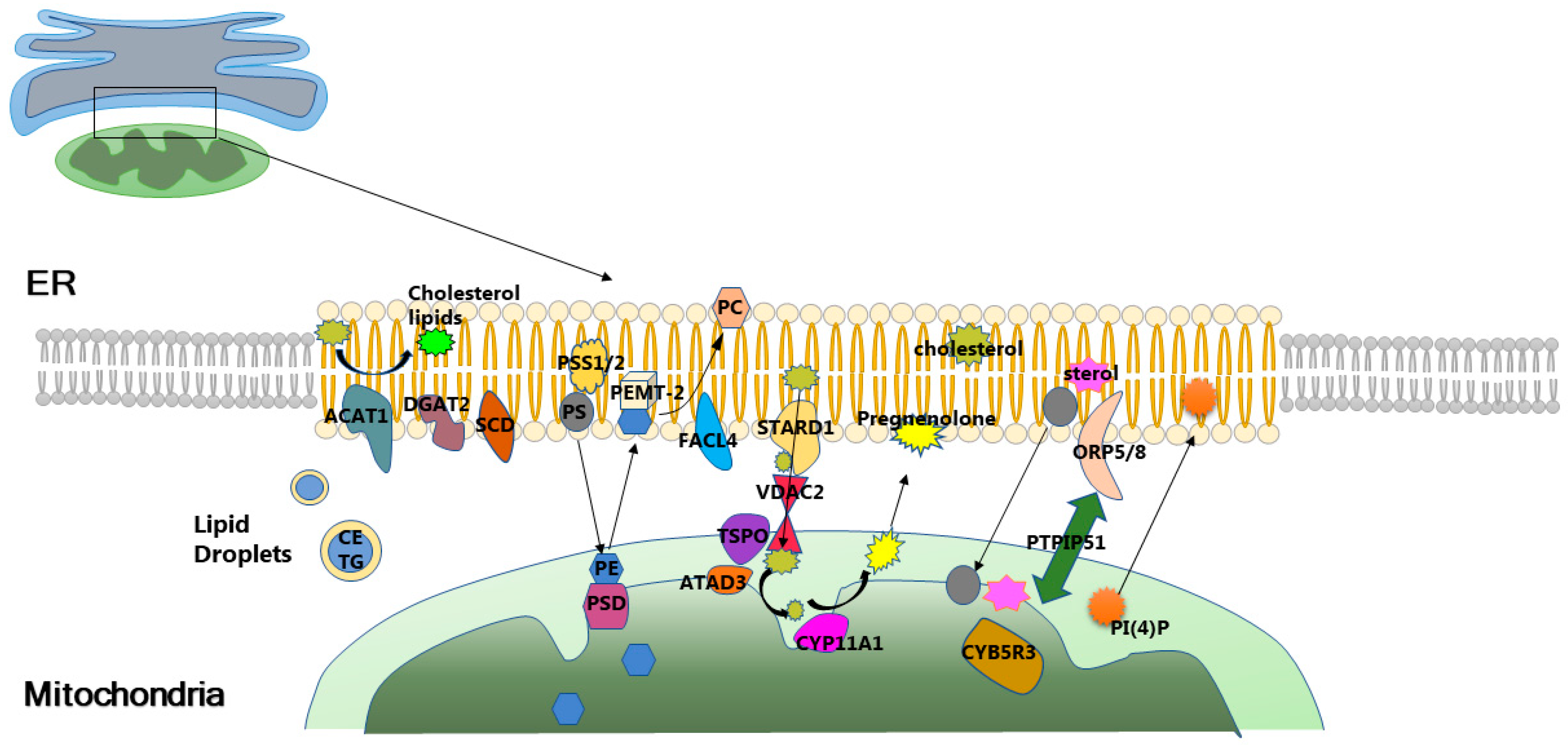
3.2. Mitochondria-Associated Membranes Modulate Insulin Resistance through Lipid Homeostasis
3.3. The Role and Mechanism of Mitochondrial Quality Control in Mitochondria-Associated Membranes Intervention in Insulin Resistance
3.3.1. Mitochondrial Biogenesis, Mitochondria-Associated Membranes, and Insulin Resistance
3.3.2. Mitochondrial Dynamics, Mitochondria-Associated Membranes, and Insulin Resistance
3.3.3. Mitophagy, Mitochondria-Associated Membranes, and Insulin Resistance
3.3.4. Endoplasmic Reticulum Stress, Mitochondria-Associated Membranes, and Insulin Resistance
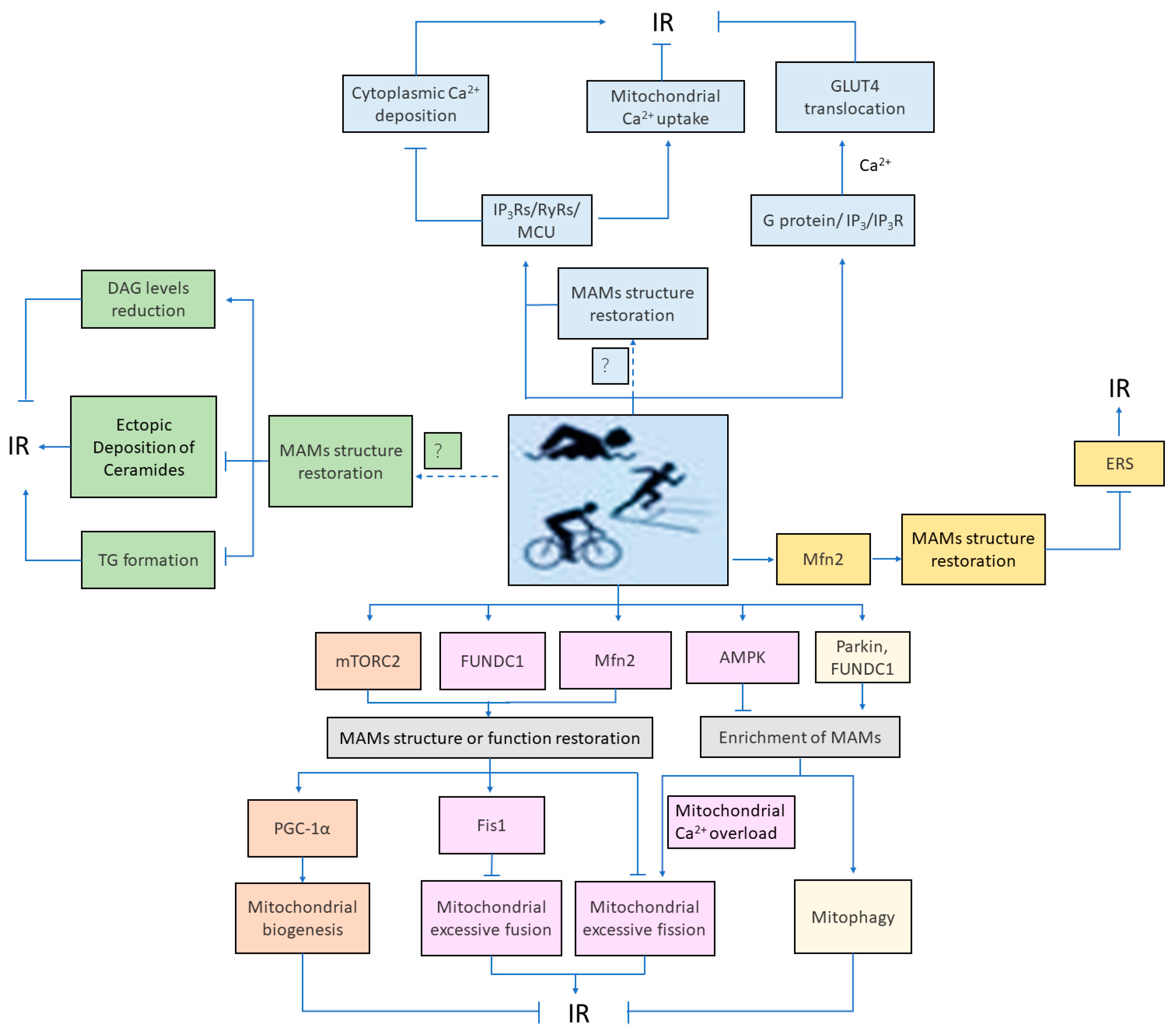
4. Role and Mechanism of Mitochondria-Associated Membranes-Mediated Cellular Homeostasis in Exercise Intervention in Insulin Resistance
4.1. Mitochondria-Associated Membranes-Associated Calcium Ion Signaling Mediates Exercise Intervention in Insulin Resistance
4.2. Mitochondria-Associated Membranes-Associated Lipid Metabolism Mediates Exercise Intervention in Insulin Resistance
4.3. Mitochondria-Associated Membranes-Associated Mitochondrial Quality Control Mediates Exercise Intervention Insulin Resistance
4.4. Mitochondria-Associated Membranes-Associated Endoplasmic Reticulum Stress Mediates Exercise Intervention in Insulin Resistance
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cheng, H.; Gang, X.; Liu, Y.; Wang, G.; Zhao, X.; Wang, G. Mitochondrial dysfunction plays a key role in the development of neurodegenerative diseases in diabetes. Am. J. Physiol.-Endocrinol. Metab. 2020, 318, E750–E764. [Google Scholar] [CrossRef]
- Cheng, H.; Gang, X.; He, G.; Liu, Y.; Wang, Y.; Zhao, X.; Wang, G. The Molecular Mechanisms Underlying Mitochondria-Associated Endoplasmic Reticulum Membrane-Induced Insulin Resistance. Front. Endocrinol. 2020, 11, 592129. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Ding, S. ER-Mitochondria Contacts and Insulin Resistance Modulation through Exercise Intervention. Int. J. Mol. Sci. 2020, 21, 9587. [Google Scholar] [CrossRef] [PubMed]
- Betz, C.; Stracka, D.; Prescianotto-Baschong, C.; Frieden, M.; Demaurex, N.; Hall, M.N. Feature Article: mTOR complex 2-Akt signaling at mitochondria-associated endoplasmic reticulum membranes (MAM) regulates mitochondrial physiology. Proc. Natl. Acad. Sci. USA 2013, 110, 12526–12534. [Google Scholar] [CrossRef] [PubMed]
- Bononi, A.; Bonora, M.; Marchi, S.; Missiroli, S.; Poletti, F.; Giorgi, C.; Pandolfi, P.P.; Pinton, P. Identification of PTEN at the ER and MAMs and its regulation of Ca2+ signaling and apoptosis in a protein phosphatase-dependent manner. Cell Death Differ. 2013, 20, 1631–1643. [Google Scholar] [CrossRef] [PubMed]
- Vance, J.E. Phospholipid synthesis in a membrane fraction associated with mitochondria. J. Biol. Chem. 1990, 265, 7248–7256. [Google Scholar] [CrossRef] [PubMed]
- Csordas, G.; Renken, C.; Varnai, P.; Walter, L.; Weaver, D.; Buttle, K.F.; Balla, T.; Mannella, C.A.; Hajnoczky, G. Structural and functional features and significance of the physical linkage between ER and mitochondria. J. Cell Biol. 2006, 174, 915–921. [Google Scholar] [CrossRef] [PubMed]
- Kornmann, B.; Currie, E.; Collins, S.R.; Schuldiner, M.; Nunnari, J.; Weissman, J.S.; Walter, P. An ER-mitochondria tethering complex revealed by a synthetic biology screen. Science 2009, 325, 477–481. [Google Scholar] [CrossRef]
- Raturi, A.; Simmen, T. Where the endoplasmic reticulum and the mitochondrion tie the knot: The mitochondria-associated membrane (MAM). Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2013, 1833, 213–224. [Google Scholar] [CrossRef]
- Marchi, S.; Patergnani, S.; Pinton, P. The endoplasmic reticulum-mitochondria connection: One touch, multiple functions. Biochim. Biophys. Acta (BBA) Bioenerg. 2014, 1837, 461–469. [Google Scholar] [CrossRef]
- Zhu, Z.; Zhang, J.; Sun, X. Progress in the Study of the Relationship between Mitochondria Ca2+ Intake Mediated by Mitochondria-Associated Endoplasmic Reticulum Membranes and Tumorigenesis. Chin. J. Cell Biol. 2018, 40, 2072–2076. [Google Scholar]
- Doghman-Bouguerra, M.; Lalli, E. ER-mitochondria interactions: Both strength and weakness within cancer cells. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2019, 1866, 650–662. [Google Scholar] [CrossRef]
- Zhao, L.; Lu, T.; Gao, L.; Fu, X.; Zhu, S.; Hou, Y. Enriched endoplasmic reticulum-mitochondria interactions result in mitochondrial dysfunction and apoptosis in oocytes from obese mice. J. Anim. Sci. Biotechnol. 2017, 8, 62. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Lu, Q.; Ding, Y.; Wu, Y.; Qiu, Y.; Wang, P.; Mao, X.; Huang, K.; Xie, Z.; Zou, M.H. Hyperglycemia-Driven Inhibition of AMP-Activated Protein Kinase α2 Induces Diabetic Cardiomyopathy by Promoting Mitochondria-Associated Endoplasmic Reticulum Membranes in vivo. Circulation 2019, 139, 1913–1936. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Tian, D.; Song, G.; Ming, Q.; Liu, J.; Shen, J.; Liu, Q.H.; Yang, X. Neferine Promotes GLUT4 Expression and Fusion With the Plasma Membrane to Induce Glucose Uptake in L6 Cells. Front. Pharmacol. 2019, 10, 999. [Google Scholar] [CrossRef] [PubMed]
- Campana, M.; Bellini, L.; Rouch, C.; Rachdi, L.; Coant, N.; Butin, N.; Bandet, C.L.; Philippe, E.; Meneyrol, K.; Kassis, N.; et al. Inhibition of central de novo ceramide synthesis restores insulin signaling in hypothalamus and enhances beta-cell function of obese Zucker rats. Mol. Metab. 2018, 8, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Sajan, M.P.; Ivey, R.A.; Lee, M.C.; Farese, R.V. Hepatic insulin resistance in ob/ob mice involves increases in ceramide, aPKC activity, and selective impairment of Akt-dependent FoxO1 phosphorylation. J. Lipid Res. 2015, 56, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Vance, J.E. MAM (mitochondria-associated membranes) in mammalian cells: Lipids and beyond. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2014, 1841, 595–609. [Google Scholar] [CrossRef] [PubMed]
- Lowell, B.B.; Shulman, G.I. Mitochondrial dysfunction and type 2 diabetes. Science 2005, 307, 384–387. [Google Scholar] [CrossRef]
- Wu, B.X.; Rajagopalan, V.; Roddy, P.L.; Clarke, C.J.; Hannun, Y.A. Identification and characterization of murine mitochondria-associated neutral sphingomyelinase (MA-nSMase), the mammalian sphingomyelin phosphodiesterase 5. J. Biol. Chem. 2010, 285, 17993–18002. [Google Scholar] [CrossRef]
- Reidy, P.T.; Mahmassani, Z.S.; McKenzie, A.I.; Petrocelli, J.J.; Summers, S.A.; Drummond, M.J. Influence of Exercise Training on Skeletal Muscle Insulin Resistance in Aging: Spotlight on Muscle Ceramides. Int. J. Mol. Sci. 2020, 21, 1514. [Google Scholar] [CrossRef]
- Zhang, H.; Liang, J.; Qian, S. Research progress of resveratrol in mitochondrial quality control. J. Biol. 2020, 37, 99–103. [Google Scholar]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstrale, M.; Laurila, E.; et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar] [CrossRef]
- Wu, Z.; Huang, X.; Feng, Y.; Handschin, C.; Feng, Y.; Gullicksen, P.S.; Bare, O.; Labow, M.; Spiegelman, B.; Stevenson, S.C. Transducer of regulated CREB-binding proteins (TORCs) induce PGC-1alpha transcription and mitochondrial biogenesis in muscle cells. Proc. Natl. Acad. Sci. USA 2006, 103, 14379–14384. [Google Scholar] [CrossRef]
- Tang, L.L.; Wang, J.D.; Xu, T.T.; Zhao, Z.; Zheng, J.J.; Ge, R.S.; Zhu, D.Y. Mitochondrial toxicity of perfluorooctane sulfonate in mouse embryonic stem cell-derived cardiomyocytes. Toxicology 2017, 382, 108–116. [Google Scholar] [CrossRef]
- Ortiz-Sandoval, C.G.; Hughes, S.C.; Dacks, J.B.; Simmen, T. Interaction with the effector dynamin-related protein 1 (Drp1) is an ancient function of Rab32 subfamily proteins. Cell Logist. 2014, 4, e986399. [Google Scholar] [CrossRef]
- Schreiner, B.; Ankarcrona, M. Isolation of Mitochondria-Associated Membranes (MAM) from Mouse Brain Tissue. Methods Mol. Biol. 2017, 1567, 53–68. [Google Scholar]
- Friedman, J.R.; Lackner, L.L.; West, M.; DiBenedetto, J.R.; Nunnari, J.; Voeltz, G.K. ER tubules mark sites of mitochondrial division. Science 2011, 334, 358–362. [Google Scholar] [CrossRef] [PubMed]
- Abrisch, R.G.; Gumbin, S.C.; Wisniewski, B.T.; Lackner, L.L.; Voeltz, G.K. Fission and fusion machineries converge at ER contact sites to regulate mitochondrial morphology. J. Cell Biol. 2020, 219, e201911122. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Lu, Q.; Wang, Q.; Ding, Y.; Ma, Z.; Mao, X.; Huang, K.; Xie, Z.; Zou, M.H. Binding of FUN14 Domain Containing 1 With Inositol 1,4,5-Trisphosphate Receptor in Mitochondria-Associated Endoplasmic Reticulum Membranes Maintains Mitochondrial Dynamics and Function in Hearts in vivo. Circulation 2017, 136, 2248–2266. [Google Scholar] [CrossRef] [PubMed]
- de Brito, O.M.; Scorrano, L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 2008, 456, 605–610. [Google Scholar] [CrossRef] [PubMed]
- Sebastian, D.; Hernandez-Alvarez, M.I.; Segales, J.; Sorianello, E.; Munoz, J.P.; Sala, D.; Waget, A.; Liesa, M.; Paz, J.C.; Gopalacharyulu, P.; et al. Mitofusin 2 (Mfn2) links mitochondrial and endoplasmic reticulum function with insulin signaling and is essential for normal glucose homeostasis. Proc. Natl. Acad. Sci. USA 2012, 109, 5523–5528. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.K.; Howie, J.; Petrie, J.R.; Lang, C.C. AMP-activated protein kinase pathway: A potential therapeutic target in cardiometabolic disease. Clin. Sci. 2009, 116, 607–620. [Google Scholar] [CrossRef]
- Twig, G.; Elorza, A.; Molina, A.J.; Mohamed, H.; Wikstrom, J.D.; Walzer, G.; Stiles, L.; Haigh, S.E.; Katz, S.; Las, G.; et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. Embo J. 2008, 27, 433–446. [Google Scholar] [CrossRef] [PubMed]
- Cang, X.; Wang, X.; Liu, P.; Wu, X.; Yan, J.; Chen, J.; Wu, G.; Jin, Y.; Xu, F.; Su, J.; et al. PINK1 alleviates palmitate induced insulin resistance in HepG2 cells by suppressing ROS mediated MAPK pathways. Biochem. Biophys. Res. Commun. 2016, 478, 431–438. [Google Scholar] [CrossRef]
- Missiroli, S.; Patergnani, S.; Caroccia, N.; Pedriali, G.; Perrone, M.; Previati, M.; Wieckowski, M.R.; Giorgi, C. Mitochondria-associated membranes (MAMs) and inflammation. Cell Death Dis. 2018, 9, 329. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Li, Y.; Qian, Z.; Zhao, T.; Feng, Z.; Weng, X.; Yu, L. Role of the inflammasome in insulin resistance and type 2 diabetes mellitus. Front. Immunol. 2023, 14, 1052756. [Google Scholar] [CrossRef]
- Bockler, S.; Westermann, B. Mitochondrial ER contacts are crucial for mitophagy in yeast. Dev. Cell. 2014, 28, 450–458. [Google Scholar] [CrossRef]
- Murley, A.; Lackner, L.L.; Osman, C.; West, M.; Voeltz, G.K.; Walter, P.; Nunnari, J. ER-associated mitochondrial division links the distribution of mitochondria and mitochondrial DNA in yeast. eLife 2013, 2, e422. [Google Scholar] [CrossRef]
- Wu, W.; Lin, C.; Wu, K.; Jiang, L.; Wang, X.; Li, W.; Zhuang, H.; Zhang, X.; Chen, H.; Li, S.; et al. FUNDC1 regulates mitochondrial dynamics at the ER-mitochondrial contact site under hypoxic conditions. EMBO J. 2016, 35, 1368–1384. [Google Scholar] [CrossRef]
- Wu, H.; Wang, Y.; Li, W.; Chen, H.; Du, L.; Liu, D.; Wang, X.; Xu, T.; Liu, L.; Chen, Q. Deficiency of mitophagy receptor FUNDC1 impairs mitochondrial quality and aggravates dietary-induced obesity and metabolic syndrome. Autophagy 2019, 15, 1882–1898. [Google Scholar] [CrossRef] [PubMed]
- Laybutt, D.R.; Preston, A.M.; Akerfeldt, M.C.; Kench, J.G.; Busch, A.K.; Biankin, A.V.; Biden, T.J. Endoplasmic reticu-lum stress contributes to beta cell apoptosis in type 2 diabetes. Diabetologia 2007, 50, 752–763. [Google Scholar] [CrossRef] [PubMed]
- Lebeaupin, C.; Vallee, D.; Hazari, Y.; Hetz, C.; Chevet, E.; Bailly-Maitre, B. Endoplasmic reticulum stress signalling and the pathogenesis of non-alcoholic fatty liver disease. J. Hepatol. 2018, 69, 927–947. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Rieusset, J.; Fauconnier, J.; Paillard, M.; Belaidi, E.; Tubbs, E.; Chauvin, M.A.; Durand, A.; Bravard, A.; Teixeira, G.; Bartosch, B.; et al. Disruption of calcium transfer from ER to mitochondria links alterations of mitochondria-associated ER membrane integrity to hepatic insulin resistance. Diabetologia 2016, 59, 614–623. [Google Scholar] [CrossRef] [PubMed]
- Tubbs, E.; Chanon, S.; Robert, M.; Bendridi, N.; Bidaux, G.; Chauvin, M.A.; Ji-Cao, J.; Durand, C.; Gauvrit-Ramette, D.; Vidal, H.; et al. Disruption of Mitochondria-Associated Endoplasmic Reticulum Membrane (MAM) Integrity Contributes to Muscle Insulin Resistance in Mice and Humans. Diabetes 2018, 67, 636–650. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.H.; Lee, H.J.; Ho, J.M.; Song, J. Coupling mitochondrial dysfunction to endoplasmic reticulum stress response: A molecular mechanism leading to hepatic insulin resistance. Cell. Signal. 2009, 21, 169–177. [Google Scholar] [CrossRef]
- Bravo, R.; Vicencio, J.M.; Parra, V.; Troncoso, R.; Munoz, J.P.; Bui, M.; Quiroga, C.; Rodriguez, A.E.; Verdejo, H.E.; Ferreira, J.; et al. Increased ER-mitochondrial coupling promotes mitochondrial respiration and bioenergetics during early phases of ER stress. J. Cell Sci. 2011, 124, 2143–2152. [Google Scholar] [CrossRef]
- Simmen, T.; Aslan, J.E.; Blagoveshchenskaya, A.D.; Thomas, L.; Wan, L.; Xiang, Y.; Feliciangeli, S.F.; Hung, C.H.; Crump, C.M.; Thomas, G. PACS-2 controls endoplasmic reticulum-mitochondria communication and Bid-mediated apoptosis. EMBO J. 2005, 24, 717–729. [Google Scholar] [CrossRef]
- Li, C.; Wu, Y.; Wang, Y. Advance in Exercise of Different Styles for Insulin Resistance. Chin. J. Rehabil. Theory Pract. 2017, 23, 1077–1080. [Google Scholar]
- Diaz-Vegas, A.R.; Cordova, A.; Valladares, D.; Llanos, P.; Hidalgo, C.; Gherardi, G.; De Stefani, D.; Mammucari, C.; Rizzuto, R.; Contreras-Ferrat, A.; et al. Mitochondrial Calcium Increase Induced by RyR1 and IP3R Channel Activation After Membrane Depolarization Regulates Skeletal Muscle Metabolism. Front. Physiol. 2018, 9, 791. [Google Scholar] [CrossRef]
- De Stefani, D.; Rizzuto, R.; Pozzan, T. Enjoy the Trip: Calcium in Mitochondria Back and Forth. Annu. Rev. Biochem. 2016, 85, 161–192. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Liu, J.; Nguyen, T.; Liu, C.; Sun, J.; Teng, Y.; Fergusson, M.M.; Rovira, I.I.; Allen, M.; Springer, D.A.; et al. The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat. Cell Biol. 2013, 15, 1464–1472. [Google Scholar] [CrossRef] [PubMed]
- Richter, E.A.; Hargreaves, M. Exercise, GLUT4, and skeletal muscle glucose uptake. Physiol. Rev. 2013, 93, 993–1017. [Google Scholar] [CrossRef] [PubMed]
- Bergman, B.C.; Goodpaster, B.H. Exercise and Muscle Lipid Content, Composition, and Localization: Influence on Muscle Insulin Sensitivity. Diabetes 2020, 69, 848–858. [Google Scholar] [CrossRef] [PubMed]
- Dube, J.J.; Amati, F.; Stefanovic-Racic, M.; Toledo, F.G.; Sauers, S.E.; Goodpaster, B.H. Exercise-induced alterations in intramyocellular lipids and insulin resistance: The athlete’s paradox revisited. Am. J. Physiol.-Endocrinol. Metab. 2008, 294, E882–E888. [Google Scholar] [CrossRef] [PubMed]
- Beaulant, A.; Dia, M.; Pillot, B.; Chauvin, M.A.; Ji-Cao, J.; Durand, C.; Bendridi, N.; Chanon, S.; Vieille-Marchiset, A.; Da, S.C.; et al. Endoplasmic reticulum-mitochondria miscommunication is an early and causal trigger of hepatic insulin resistance and steatosis. J. Hepatol. 2022, 77, 710–722. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Cui, D.; Zhang, T.; Sun, Y.; Ding, S. Swimming Differentially Affects T2DM-Induced Skeletal Muscle ER Stress and Mitochondrial Dysfunction Related to MAM. Diabetes Metab. Syndr. Obes. 2020, 13, 1417–1428. [Google Scholar] [CrossRef]
- Kasumov, T.; Solomon, T.P.; Hwang, C.; Huang, H.; Haus, J.M.; Zhang, R.; Kirwan, J.P. Improved insulin sensitivity after exercise training is linked to reduced plasma C14:0 ceramide in obesity and type 2 diabetes. Obesity 2015, 23, 1414–1421. [Google Scholar] [CrossRef]
- Shepherd, S.O.; Cocks, M.; Meikle, P.J.; Mellett, N.A.; Ranasinghe, A.M.; Barker, T.A.; Wagenmakers, A.; Shaw, C.S. Lipid droplet remodelling and reduced muscle ceramides following sprint interval and moderate-intensity continuous exercise training in obese males. Int. J. Obes. 2017, 41, 1745–1754. [Google Scholar] [CrossRef]
- Ruegsegger, G.N.; Creo, A.L.; Cortes, T.M.; Dasari, S.; Nair, K.S. Altered mitochondrial function in insulin-deficient and insulin-resistant states. J. Clin. Investig. 2018, 128, 3671–3681. [Google Scholar] [CrossRef]
- Kleinert, M.; Parker, B.L.; Fritzen, A.M.; Knudsen, J.R.; Jensen, T.E.; Kjobsted, R.; Sylow, L.; Ruegg, M.; James, D.E.; Richter, E.A. Mammalian target of rapamycin complex 2 regulates muscle glucose uptake during exercise in mice. J. Physiol. 2017, 595, 4845–4855. [Google Scholar] [CrossRef]
- Moore, T.M.; Zhou, Z.; Cohn, W.; Norheim, F.; Lin, A.J.; Kalajian, N.; Strumwasser, A.R.; Cory, K.; Whitney, K.; Ho, T.; et al. The impact of exercise on mitochondrial dynamics and the role of Drp1 in exercise performance and training adaptations in skeletal muscle. Mol. Metab. 2019, 21, 51–67. [Google Scholar] [CrossRef]
- Zhang, Z.; Ding, S. Mitochondrial Fission 1 Protein Regulates Mitochondrial Quality of Skeletal Muscle in Resting State and Exhaustive Exercise. J. Beijing Sport. Univ. 2020, 43, 104–113. [Google Scholar]
- Ding, H.; Jiang, N.; Liu, H.; Liu, X.; Liu, D.; Zhao, F.; Wen, L.; Liu, S.; Ji, L.L.; Zhang, Y. Response of mitochondrial fusion and fission protein gene expression to exercise in rat skeletal muscle. Biochim. Biophys. Acta (BBA) Gen. Subj. 2010, 1800, 250–256. [Google Scholar] [CrossRef]
- Huertas, J.R.; Ruiz-Ojeda, F.J.; Plaza-Diaz, J.; Nordsborg, N.B.; Martin-Albo, J.; Rueda-Robles, A.; Casuso, R.A. Human muscular mitochondrial fusion in athletes during exercise. FASEB J. 2019, 33, 12087–12098. [Google Scholar] [CrossRef]
- Zhang, T.; Sun, Y.; Ding, S. Role of Mitophagy in Exercise-Induced Adaption of Insulin Resistance. J. Shanghai Univ. Sport 2017, 41, 50–54. [Google Scholar]
- Gautier, C.A.; Erpapazoglou, Z.; Mouton-Liger, F.; Muriel, M.P.; Cormier, F.; Bigou, S.; Duffaure, S.; Girard, M.; Foret, B.; Iannielli, A.; et al. The endoplasmic reticulum-mitochondria interface is perturbed in PARK2 knockout mice and patients with PARK2 mutations. Hum. Mol. Genet. 2016, 25, 2972–2984. [Google Scholar] [CrossRef] [PubMed]
- Cui, D.; Qiu, S.; Wang, H. The Effect of Endurance Exercise on Autophagy and Mitophagy in the Skeletal Muscle of Mice with Alimentary Obesity. China Sport Sci. 2014, 34, 63–71. [Google Scholar]
- Yu, L.; Shi, X.Y.; Liu, Z.M.; Wang, Z.; Li, L.; Gao, J.X.; Liu, X.R.; Wang, R.Y. Effects of exercises with different durations and intensities on mitochondrial autophagy and FUNDC1 expression in rat skeletal muscles. Sheng Li Xue Bao 2020, 72, 631–642. [Google Scholar] [PubMed]
- Cuellar, J.; Martin-Benito, J.; Scheres, S.H.; Sousa, R.; Moro, F.; Lopez-Vinas, E.; Gomez-Puertas, P.; Muga, A.; Carrascosa, J.L.; Valpuesta, J.M. The structure of CCT-Hsc70 NBD suggests a mechanism for Hsp70 delivery of substrates to the chaperonin. Nat. Struct. Mol. Biol. 2008, 15, 858–864. [Google Scholar] [CrossRef] [PubMed]
- Llorca, O.; McCormack, E.A.; Hynes, G.; Grantham, J.; Cordell, J.; Carrascosa, J.L.; Willison, K.R.; Fernandez, J.J.; Valpuesta, J.M. Eukaryotic type II chaperonin CCT interacts with actin through specific subunits. Nature 1999, 402, 693–696. [Google Scholar] [CrossRef] [PubMed]
- Da, L.G.; Frederico, M.J.; Da, S.S.; Vitto, M.F.; Cesconetto, P.A.; de Pinho, R.A.; Pauli, J.R.; Silva, A.S.; Cintra, D.E.; Ropelle, E.R.; et al. Endurance exercise training ameliorates insulin resistance and reticulum stress in adipose and hepatic tissue in obese rats. Eur. J. Appl. Physiol. 2011, 111, 2015–2023. [Google Scholar]
- Williamson, D.; Gallagher, P.; Harber, M.; Hollon, C.; Trappe, S. Mitogen-activated protein kinase (MAPK) pathway activation: Effects of age and acute exercise on human skeletal muscle. J. Physiol. 2003, 547, 977–987. [Google Scholar] [CrossRef] [PubMed]
- Little, J.P.; Gillen, J.B.; Percival, M.E.; Safdar, A.; Tarnopolsky, M.A.; Punthakee, Z.; Jung, M.E.; Gibala, M.J. Low-volume high-intensity interval training reduces hyperglycemia and increases muscle mitochondrial capacity in patients with type 2 diabetes. J. Appl. Physiol. 2011, 111, 1554–1560. [Google Scholar] [CrossRef]
- Cartoni, R.; Leger, B.; Hock, M.B.; Praz, M.; Crettenand, A.; Pich, S.; Ziltener, J.L.; Luthi, F.; Deriaz, O.; Zorzano, A.; et al. Mitofusins 1/2 and ERRalpha expression are increased in human skeletal muscle after physical exercise. J. Physiol. 2005, 567, 349–358. [Google Scholar] [CrossRef]
- Sabaratnam, R.; Pedersen, A.; Kristensen, J.M.; Handberg, A.; Wojtaszewski, J.; Hojlund, K. Intact regulation of muscle expression and circulating levels of myokines in response to exercise in patients with type 2 diabetes. Physiol. Rep. 2018, 6, e13723. [Google Scholar] [CrossRef]
- Pinto, A.P.; Da, R.A.; Kohama, E.B.; Gaspar, R.C.; Simabuco, F.M.; Frantz, F.G.; de Moura, L.P.; Pauli, J.R.; Cintra, D.E.; Ropelle, E.R.; et al. Exhaustive acute exercise-induced ER stress is attenuated in IL-6-knockout mice. J. Endocrinol. 2019, 240, 181–193. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, X.; Yang, Y.; Shi, X.; Zhang, Z.; Ding, S. Mitochondria-Associated Membranes as Key Regulators in Cellular Homeostasis and the Potential Impact of Exercise on Insulin Resistance. Int. J. Mol. Sci. 2024, 25, 3196. https://doi.org/10.3390/ijms25063196
Li X, Yang Y, Shi X, Zhang Z, Ding S. Mitochondria-Associated Membranes as Key Regulators in Cellular Homeostasis and the Potential Impact of Exercise on Insulin Resistance. International Journal of Molecular Sciences. 2024; 25(6):3196. https://doi.org/10.3390/ijms25063196
Chicago/Turabian StyleLi, Xi, Yangjun Yang, Xiaoyu Shi, Zhe Zhang, and Shuzhe Ding. 2024. "Mitochondria-Associated Membranes as Key Regulators in Cellular Homeostasis and the Potential Impact of Exercise on Insulin Resistance" International Journal of Molecular Sciences 25, no. 6: 3196. https://doi.org/10.3390/ijms25063196