
Nutritional Sensor REDD1 in Cancer and Inflammation: Friend or Foe?

 , , , and
, , , and
Abstract
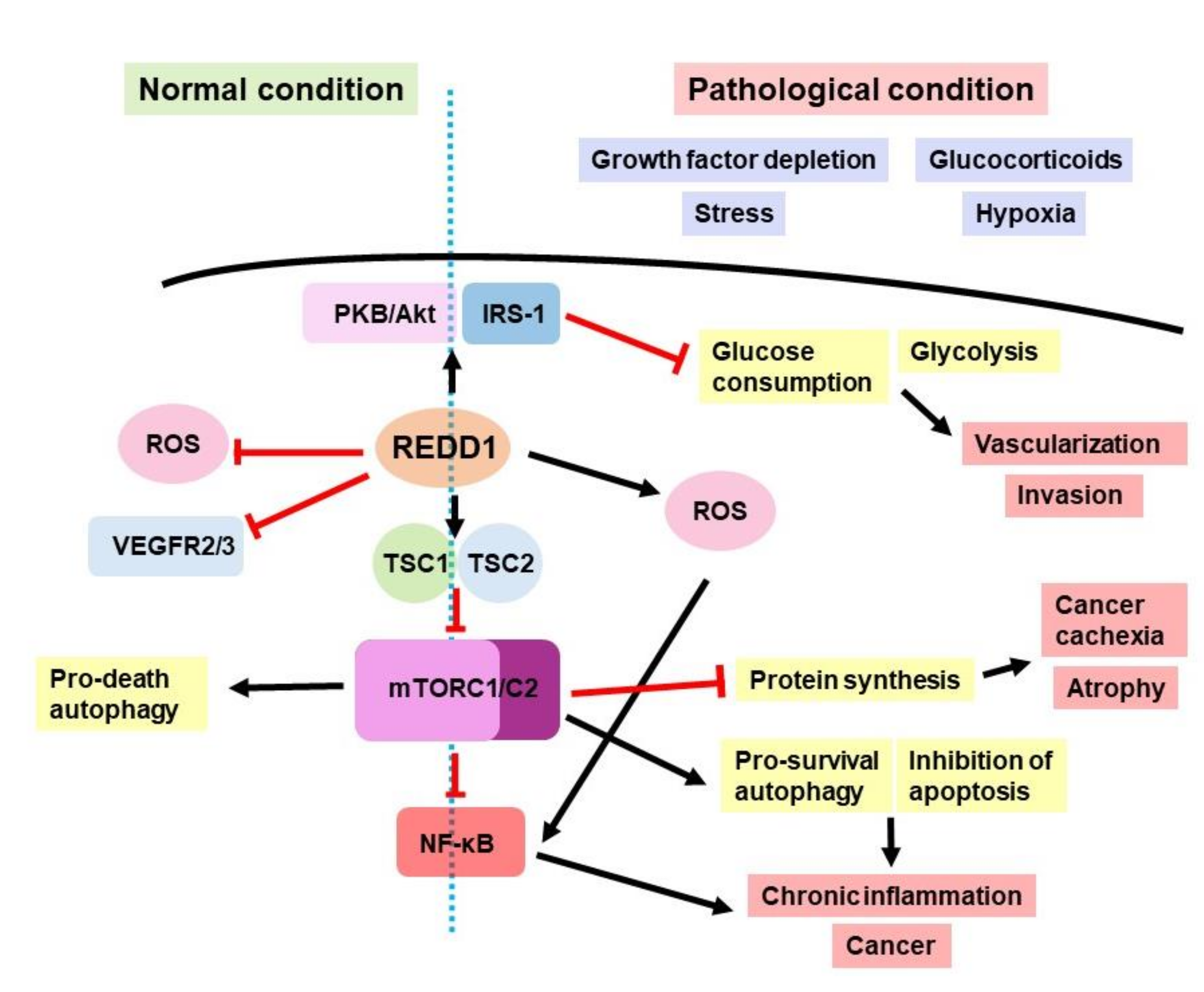
1. REDD1 Involvement in Cell Signaling
2. REDD1 in Cancer
2.1. REDD1 in Proliferation and Apoptosis
2.2. REDD1 in Tumor Microenvironment (TME): Hypoxia, Neoangiogenesis, and Reprogramming of Immune Cells
2.3. REDD1, Immune Cells of TME, and Migration of Cancer Cells
2.4. REDD1 and Cell Senescence
2.5. REDD1 and Epigenetic Changes in Cancer Cells
3. REDD1 in Inflammation
4. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Shoshani, T.; Faerman, A.; Mett, I.; Zelin, E.; Tenne, T.; Gorodin, S.; Moshel, Y.; Elbaz, S.; Budanov, A.; Chajut, A.; et al. Identification of a novel hypoxia-inducible factor 1-responsive gene, RTP801, involved in apoptosis. Mol. Cell. Biol. 2002, 22, 2283–2293. [Google Scholar] [CrossRef] [PubMed]
- Brugarolas, J.; Lei, K.; Hurley, R.L.; Manning, B.D.; Reiling, J.H.; Hafen, E.; Witters, L.A.; Ellisen, L.W.; Kaelin, W.G. Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev. 2004, 18, 2893–2904. [Google Scholar] [CrossRef] [PubMed]
- Reiling, J.H.; Hafen, E. The hypoxia-induced paralogs Scylla and Charybdis inhibit growth by down-regulating S6K activity upstream of TSC in Drosophila. Genes Dev. 2004, 18, 2879–2892. [Google Scholar] [CrossRef] [PubMed]
- Watson, A.; Lipina, C.; McArdle, H.J.; Taylor, P.M.; Hundal, H.S. Iron depletion suppresses mTORC1-directed signalling in intestinal Caco-2 cells via induction of REDD1. Cell Signal. 2016, 28, 412–424. [Google Scholar] [CrossRef] [PubMed]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [PubMed]
- Available online: http://www.signalingpathways.org (accessed on 23 June 2022).
- Ellisen, L.W. Growth control under stress: mTOR regulation through the REDD1-TSC pathway. Cell Cycle 2005, 4, 1500–1502. [Google Scholar] [CrossRef] [PubMed]
- Britto, F.A.; Dumas, K.; Giorgetti-Peraldi, S.; Ollendorff, V.; Favier, F.B. Is REDD1 a metabolic double agent? Lessons from physiology and pathology. Am. J. Physiol. Cell Physiol. 2020, 319, C807–C824. [Google Scholar] [CrossRef]
- Lipina, C.; Hundal, H.S. Is REDD1 a Metabolic Éminence Grise? Trends Endocrinol. Metab. 2016, 27, 868–880. [Google Scholar] [CrossRef]
- Vega-Rubin-de-Celis, S.; Abdallah, Z.; Kinch, L.; Grishin, N.V.; Brugarolas, J.; Zhang, X. Structural analysis and functional implications of the negative mTORC1 regulator REDD1. Biochemistry 2010, 49, 2491–2501. [Google Scholar] [CrossRef]
- Regazzetti, C.; Dumas, K.; Le Marchand-Brustel, Y.; Peraldi, P.; Tanti, J.-F.; Giorgetti-Peraldi, S. Regulated in development and DNA damage responses-1 (REDD1) protein contributes to insulin signaling pathway in adipocytes. PLoS ONE 2012, 7, e52154. [Google Scholar] [CrossRef]
- Schupp, M.; Chen, F.; Briggs, E.R.; Rao, S.; Pelzmann, H.J.; Pessentheiner, A.R.; Bogner-Strauss, J.G.; Lazar, M.A.; Baldwin, D.; Prokesch, A. Metabolite and transcriptome analysis during fasting suggest a role for the p53-Ddit4 axis in major metabolic tissues. BMC Genom. 2013, 14, 758. [Google Scholar] [CrossRef] [PubMed]
- Britto, F.A.; Cortade, F.; Belloum, Y.; Blaquière, M.; Gallot, Y.S.; Docquier, A.; Pagano, A.F.; Jublanc, E.; Bendridi, N.; Koechlin-Ramonatxo, C.; et al. Glucocorticoid-dependent REDD1 expression reduces muscle metabolism to enable adaptation under energetic stress. BMC Biol. 2018, 16, 65. [Google Scholar] [CrossRef] [PubMed]
- Williamson, D.L.; Li, Z.; Tuder, R.M.; Feinstein, E.; Kimball, S.R.; Dungan, C.M. Altered nutrient response of mTORC1 as a result of changes in REDD1 expression: Effect of obesity vs. REDD1 deficiency. J. Appl. Physiol. 2014, 117, 246–256. [Google Scholar] [CrossRef] [PubMed]
- Elam, M.B.; Cowan, G.S.; Rooney, R.J.; Hiler, M.L.; Yellaturu, C.R.; Deng, X.; Howell, G.E.; Park, E.A.; Gerling, I.C.; Patel, D.; et al. Hepatic gene expression in morbidly obese women: Implications for disease susceptibility. Obesity 2009, 17, 1563–1573. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Gonzalez, G.C.; Klopot, A.; Sabin, K.; Baida, G.; Horsley, V.; Budunova, I. Regulated in Development and DNA Damage Responses 1 Prevents Dermal Adipocyte Differentiation and Is Required for Hair Cycle-Dependent Dermal Adipose Expansion. J. Investig. Dermatol. 2020, 140, 1698–1705.e1. [Google Scholar] [CrossRef]
- Sofer, A.; Lei, K.; Johannessen, C.M.; Ellisen, L.W. Regulation of mTOR and cell growth in response to energy stress by REDD1. Mol. Cell. Biol. 2005, 25, 5834–5845. [Google Scholar] [CrossRef]
- Ellisen, L.W.; Ramsayer, K.D.; Johannessen, C.M.; Yang, A.; Beppu, H.; Minda, K.; Oliner, J.D.; McKeon, F.; Haber, D.A. REDD1, a developmentally regulated transcriptional target of p63 and p53, links p63 to regulation of reactive oxygen species. Mol. Cell 2002, 10, 995–1005. [Google Scholar] [CrossRef]
- Malagelada, C.; López-Toledano, M.A.; Willett, R.T.; Jin, Z.H.; Shelanski, M.L.; Greene, L.A. RTP801/REDD1 regulates the timing of cortical neurogenesis and neuron migration. J. Neurosci. 2011, 31, 3186–3196. [Google Scholar] [CrossRef]
- Horak, P.; Crawford, A.R.; Vadysirisack, D.D.; Nash, Z.M.; DeYoung, M.P.; Sgroi, D.; Ellisen, L.W. Negative feedback control of HIF-1 through REDD1-regulated ROS suppresses tumorigenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 4675–4680. [Google Scholar] [CrossRef]
- Qiao, S.; Dennis, M.; Song, X.; Vadysirisack, D.D.; Salunke, D.; Nash, Z.; Yang, Z.; Liesa, M.; Yoshioka, J.; Matsuzawa, S.-I.; et al. A REDD1/TXNIP pro-oxidant complex regulates ATG4B activity to control stress-induced autophagy and sustain exercise capacity. Nat. Commun. 2015, 6, 7014. [Google Scholar] [CrossRef]
- Dungan, C.M.; Wright, D.C.; Williamson, D.L. Lack of REDD1 reduces whole body glucose and insulin tolerance, and impairs skeletal muscle insulin signaling. Biochem. Biophys. Res. Commun. 2014, 453, 778–783. [Google Scholar] [CrossRef] [PubMed]
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef] [PubMed]
- Ben Sahra, I.; Regazzetti, C.; Robert, G.; Laurent, K.; Le Marchand-Brustel, Y.; Auberger, P.; Tanti, J.-F.; Giorgetti-Peraldi, S.; Bost, F. Metformin, independent of AMPK, induces mTOR inhibition and cell-cycle arrest through REDD1. Cancer Res. 2011, 71, 4366–4372. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, N.; Yoshikawa, N.; Ito, N.; Maruyama, T.; Suzuki, Y.; Takeda, S.; Nakae, J.; Tagata, Y.; Nishitani, S.; Takehana, K.; et al. Crosstalk between glucocorticoid receptor and nutritional sensor mTOR in skeletal muscle. Cell Metab. 2011, 13, 170–182. [Google Scholar] [CrossRef] [PubMed]
- Britto, F.A.; Begue, G.; Rossano, B.; Docquier, A.; Vernus, B.; Sar, C.; Ferry, A.; Bonnieu, A.; Ollendorff, V.; Favier, F.B. REDD1 deletion prevents dexamethasone-induced skeletal muscle atrophy. Am. J. Physiol. Endocrinol. Metab. 2014, 307, E983–E993. [Google Scholar] [CrossRef] [PubMed]
- Baida, G.; Bhalla, P.; Kirsanov, K.; Lesovaya, E.; Yakubovskaya, M.; Yuen, K.; Guo, S.; Lavker, R.M.; Readhead, B.; Dudley, J.T.; et al. REDD1 functions at the crossroads between the therapeutic and adverse effects of topical glucocorticoids. EMBO Mol. Med. 2015, 7, 42–58. [Google Scholar] [CrossRef]
- Lesovaya, E.A.; Savinkova, A.V.; Morozova, O.V.; Lylova, E.S.; Zhidkova, E.M.; Kulikov, E.P.; Kirsanov, K.I.; Klopot, A.; Baida, G.; Yakubovskaya, M.G.; et al. A Novel Approach to Safer Glucocorticoid Receptor-Targeted Anti-lymphoma Therapy via REDD1 (Regulated in Development and DNA Damage 1) Inhibition. Mol. Cancer Ther. 2020, 19, 1898–1908. [Google Scholar] [CrossRef]
- Ramamoorthy, S.; Cidlowski, J.A. Corticosteroids: Mechanisms of Action in Health and Disease. Rheum. Dis. Clin. N. Am. 2016, 42, 15–31. [Google Scholar] [CrossRef]
- Pufall, M.A. Glucocorticoids and Cancer. Adv. Exp. Med. Biol. 2015, 872, 315–333. [Google Scholar] [CrossRef]
- Lesovaya, E.; Yemelyanov, A.; Swart, A.C.; Swart, P.; Haegeman, G.; Budunova, I. Discovery of Compound A—A selective activator of the glucocorticoid receptor with anti-inflammatory and anti-cancer activity. Oncotarget 2015, 6, 30730–30744. [Google Scholar] [CrossRef]
- Kleiman, A.; Tuckermann, J.P. Glucocorticoid receptor action in beneficial and side effects of steroid therapy: Lessons from conditional knockout mice. Mol. Cell. Endocrinol. 2007, 275, 98–108. [Google Scholar] [CrossRef] [PubMed]
- Ramamoorthy, S.; Cidlowski, J.A. Ligand-induced repression of the glucocorticoid receptor gene is mediated by an NCoR1 repression complex formed by long-range chromatin interactions with intragenic glucocorticoid response elements. Mol. Cell. Biol. 2013, 33, 1711–1722. [Google Scholar] [CrossRef] [PubMed]
- De Bosscher, K.; Haegeman, G.; Elewaut, D. Targeting inflammation using selective glucocorticoid receptor modulators. Curr. Opin. Pharmacol. 2010, 10, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Schoepe, S.; Schäcke, H.; May, E.; Asadullah, K. Glucocorticoid therapy-induced skin atrophy. Exp. Dermatol. 2006, 15, 406–420. [Google Scholar] [CrossRef] [PubMed]
- De Bosscher, K.; Beck, I.M.; Ratman, D.; Berghe, W.; Vanden Libert, C. Activation of the Glucocorticoid Receptor in Acute Inflammation: The SEDIGRAM Concept. Trends Pharmacol. Sci. 2016, 37, 4–16. [Google Scholar] [CrossRef] [PubMed]
- De Bosscher, K.; Vanden Berghe, W.; Haegeman, G. The interplay between the glucocorticoid receptor and nuclear factor-kappaB or activator protein-1: Molecular mechanisms for gene repression. Endocr. Rev. 2003, 24, 488–522. [Google Scholar] [CrossRef] [PubMed]
- Otulakowski, G.; Duan, W.; Sarangapani, A.; Gandhi, S.; O’Brodovich, H. Glucocorticoid-mediated repression of REDD1 mRNA expression in rat fetal distal lung epithelial cells. Pediatr. Res. 2009, 65, 514–519. [Google Scholar] [CrossRef]
- Agarwal, S.; Mirzoeva, S.; Readhead, B.; Dudley, J.T.; Budunova, I. PI3K inhibitors protect against glucocorticoid-induced skin atrophy. EBioMedicine 2019, 41, 526–537. [Google Scholar] [CrossRef]
- Lesovaya, E.; Agarwal, S.; Readhead, B.; Vinokour, E.; Baida, G.; Bhalla, P.; Kirsanov, K.; Yakubovskaya, M.; Platanias, L.C.; Dudley, J.T.; et al. Rapamycin Modulates Glucocorticoid Receptor Function, Blocks Atrophogene REDD1, and Protects Skin from Steroid Atrophy. J. Investig. Dermatol. 2018, 138, 1935–1944. [Google Scholar] [CrossRef]
- Baida, G.; Agarwal, S.; Readhead, B.; Dudley, J.T.; Budunova, I. Sexual dimorphism in atrophic effects of topical glucocorticoids is driven by differential regulation of atrophogene REDD1 in male and female skin. Oncotarget 2020, 11, 409–418. [Google Scholar] [CrossRef][Green Version]
- Wu, Y.; Zhao, W.; Zhao, J.; Zhang, Y.; Qin, W.; Pan, J.; Bauman, W.A.; Blitzer, R.D.; Cardozo, C. REDD1 is a major target of testosterone action in preventing dexamethasone-induced muscle loss. Endocrinology 2010, 151, 1050–1059. [Google Scholar] [CrossRef]
- White, J.P.; Gao, S.; Puppa, M.J.; Sato, S.; Welle, S.L.; Carson, J.A. Testosterone regulation of Akt/mTORC1/FoxO3a signaling in skeletal muscle. Mol. Cell. Endocrinol. 2013, 365, 174–186. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Malone, M.H.; Thomenius, M.J.; Zhong, F.; Xu, F.; Distelhorst, C.W. Dexamethasone-induced gene 2 (dig2) is a novel pro-survival stress gene induced rapidly by diverse apoptotic signals. J. Biol. Chem. 2003, 278, 27053–27058. [Google Scholar] [CrossRef] [PubMed]
- Sinha, I.; Allen, J.E.; Pinto, J.T.; Sinha, R. Methylseleninic acid elevates REDD1 and inhibits prostate cancer cell growth despite AKT activation and mTOR dysregulation in hypoxia. Cancer Med. 2014, 3, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Koo, J.S.; Kim, H. Hypoxia-related protein expression and its clinicopathologic implication in carcinoma of unknown primary. Tumour Biol. 2011, 32, 893–904. [Google Scholar] [CrossRef] [PubMed]
- Lan, Y.-C.C.; Chang, C.-L.L.; Sung, M.-T.T.; Yin, P.-H.H.; Hsu, C.-C.C.; Wang, K.-C.C.; Lee, H.-C.C.; Tseng, L.-M.M.; Chi, C.-W.W. Zoledronic acid-induced cytotoxicity through endoplasmic reticulum stress triggered REDD1-mTOR pathway in breast cancer cells. Anticancer Res. 2013, 33, 3807–3814. [Google Scholar]
- Yun, S.-M.; Woo, S.H.; Oh, S.T.; Hong, S.-E.; Choe, T.-B.; Ye, S.-K.; Kim, E.-K.; Seong, M.K.; Kim, H.-A.; Noh, W.C.; et al. Melatonin enhances arsenic trioxide-induced cell death via sustained upregulation of Redd1 expression in breast cancer cells. Mol. Cell. Endocrinol. 2016, 422, 64–73. [Google Scholar] [CrossRef]
- Zhidkova, E.M.; Lylova, E.S.; Savinkova, A.V.; Mertsalov, S.A.; Kirsanov, K.I.; Belitsky, G.A.; Yakubovskaya, M.G.; Lesovaya, E.A. A Brief Overview of the Paradoxical Role of Glucocorticoids in Breast Cancer. Breast Cancer 2020, 14, 1178223420974667. [Google Scholar] [CrossRef]
- Tirado-Hurtado, I.; Fajardo, W.; Pinto, J.A. DNA Damage Inducible Transcript 4 Gene: The Switch of the Metabolism as Potential Target in Cancer. Front. Oncol. 2018, 8, 106. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.R.; Xu, X.-X. REDD1, a new Ras oncogenic effector. Cell Cycle 2009, 8, 675–676. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Q.; Liu, J.; Cao, P.; Li, J.J.; Liu, X.; Fan, X.; Liu, L.; Cheng, Y.; Xiong, W.; Li, J.J.; et al. Inhibition of REDD1 Sensitizes Bladder Urothelial Carcinoma to Paclitaxel by Inhibiting Autophagy. Clin. Cancer Res. 2018, 24, 445–459. [Google Scholar] [CrossRef] [PubMed]
- Chang, B.; Meng, J.; Zhu, H.; Du, X.; Sun, L.; Wang, L.; Li, S.; Yang, G. Overexpression of the recently identified oncogene REDD1 correlates with tumor progression and is an independent unfavorable prognostic factor for ovarian carcinoma. Diagn. Pathol. 2018, 13, 87. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Song, K.; Shang, W.; Chen, L.; Wang, C.; Pang, B.; Wang, N. REDD1 overexpression in oral squamous cell carcinoma may predict poor prognosis and correlates with high microvessel density. Oncol. Lett. 2020, 19, 431–441. [Google Scholar] [CrossRef]
- Jia, W.; Chang, B.; Sun, L.; Zhu, H.; Pang, L.; Tao, L.; Zou, H.; Du, J.; Dong, Y.; Qi, Y.; et al. REDD1 and p-AKT over-expression may predict poor prognosis in ovarian cancer. Int. J. Clin. Exp. Pathol. 2014, 7, 5940–5949. [Google Scholar]
- Pinto, J.A.; Rolfo, C.; Raez, L.E.; Prado, A.; Araujo, J.M.; Bravo, L.; Fajardo, W.; Morante, Z.D.; Aguilar, A.; Neciosup, S.P.; et al. In silico evaluation of DNA Damage Inducible Transcript 4 gene (DDIT4) as prognostic biomarker in several malignancies. Sci. Rep. 2017, 7, 1526. [Google Scholar] [CrossRef]
- Pinto, J.A.; Araujo, J.; Cardenas, N.K.; Morante, Z.; Doimi, F.; Vidaurre, T.; Balko, J.M.; Gomez, H.L. A prognostic signature based on three-genes expression in triple-negative breast tumours with residual disease. NPJ Genom. Med. 2016, 1, 15015. [Google Scholar] [CrossRef]
- Du, F.; Sun, L.; Chu, Y.; Li, T.; Lei, C.; Wang, X.; Jiang, M.; Min, Y.; Lu, Y.; Zhao, X.; et al. DDIT4 promotes gastric cancer proliferation and tumorigenesis through the p53 and MAPK pathways. Cancer Commun. 2018, 38, 45. [Google Scholar] [CrossRef]
- Liu, Z.; Li, T.; Zhu, F.; Deng, S.; Li, X.; He, Y. Regulatory roles of miR-22/Redd1-mediated mitochondrial ROS and cellular autophagy in ionizing radiation-induced BMSC injury. Cell Death Dis. 2019, 10, 227. [Google Scholar] [CrossRef]
- Ramachandiran, S.; Adon, A.; Guo, X.; Wang, Y.; Wang, H.; Chen, Z.; Kowalski, J.; Sunay, U.R.; Young, A.N.; Brown, T.; et al. Chromosome instability in diffuse large B cell lymphomas is suppressed by activation of the noncanonical NF-κB pathway. Int. J. Cancer 2015, 136, 2341–2351. [Google Scholar] [CrossRef] [PubMed]
- Sesen, J.; Dahan, P.; Scotland, S.J.; Saland, E.; Dang, V.-T.; Lemarié, A.; Tyler, B.M.; Brem, H.; Toulas, C.; Cohen-Jonathan Moyal, E.; et al. Metformin inhibits growth of human glioblastoma cells and enhances therapeutic response. PLoS ONE 2015, 10, e0123721. [Google Scholar] [CrossRef] [PubMed]
- Molitoris, J.K.; McColl, K.S.; Swerdlow, S.; Matsuyama, M.; Lam, M.; Finkel, T.H.; Matsuyama, S.; Distelhorst, C.W. Glucocorticoid elevation of dexamethasone-induced gene 2 (Dig2/RTP801/REDD1) protein mediates autophagy in lymphocytes. J. Biol. Chem. 2011, 286, 30181–30189. [Google Scholar] [CrossRef] [PubMed]
- Schwarzer, R.; Tondera, D.; Arnold, W.; Giese, K.; Klippel, A.; Kaufmann, J. REDD1 integrates hypoxia-mediated survival signaling downstream of phosphatidylinositol 3-kinase. Oncogene 2005, 24, 1138–1149. [Google Scholar] [CrossRef] [PubMed]
- Chang, B.; Liu, G.; Yang, G.; Mercado-Uribe, I.; Huang, M.; Liu, J. REDD1 is required for RAS-mediated transformation of human ovarian epithelial cells. Cell Cycle 2009, 8, 780–786. [Google Scholar] [CrossRef]
- Feehan, R.P.; Coleman, C.S.; Ebanks, S.; Lang, C.H.; Shantz, L.M. REDD1 interacts with AIF and regulates mitochondrial reactive oxygen species generation in the keratinocyte response to UVB. Biochem. Biophys. Res. Commun. 2022, 616, 56–62. [Google Scholar] [CrossRef]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef]
- Zhu, L.; Zhu, X.; Wu, Y. Effects of Glucose Metabolism, Lipid Metabolism, and Glutamine Metabolism on Tumor Microenvironment and Clinical Implications. Biomolecules 2022, 12, 580. [Google Scholar] [CrossRef]
- Krock, B.L.; Skuli, N.; Simon, M.C. Hypoxia-induced angiogenesis: Good and evil. Genes Cancer 2011, 2, 1117–1133. [Google Scholar] [CrossRef]
- Park, M.; Kim, J.; Kim, T.; Kim, S.; Park, W.; Ha, K.-S.; Cho, S.H.; Won, M.-H.; Lee, J.-H.; Kwon, Y.-G.; et al. REDD1 is a determinant of low-dose metronomic doxorubicin-elicited endothelial cell dysfunction through downregulation of VEGFR-2/3 expression. Exp. Mol. Med. 2021, 53, 1612–1622. [Google Scholar] [CrossRef]
- Park, M.; Kim, J.Y.; Kim, J.; Lee, J.-H.; Kwon, Y.-G.; Kim, Y.-M. Low-dose metronomic doxorubicin inhibits mobilization and differentiation of endothelial progenitor cells through REDD1-mediated VEGFR-2 downregulation. BMB Rep. 2021, 54, 470–475. [Google Scholar] [CrossRef]
- Wenes, M.; Shang, M.; Di Matteo, M.; Goveia, J.; Martín-Pérez, R.; Serneels, J.; Prenen, H.; Ghesquière, B.; Carmeliet, P.; Mazzone, M. Macrophage Metabolism Controls Tumor Blood Vessel Morphogenesis and Metastasis. Cell Metab. 2016, 24, 701–715. [Google Scholar] [CrossRef] [PubMed]
- Jawhari, S.; Ratinaud, M.-H.; Verdier, M. Glioblastoma, hypoxia and autophagy: A survival-prone “ménage-à-trois”. Cell Death Dis. 2016, 7, e2434. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.-L.; DeLay, M.; Jahangiri, A.; Molinaro, A.M.; Rose, S.D.; Carbonell, W.S.; Aghi, M.K. Hypoxia-induced autophagy promotes tumor cell survival and adaptation to antiangiogenic treatment in glioblastoma. Cancer Res. 2012, 72, 1773–1783. [Google Scholar] [CrossRef] [PubMed]
- Di Conza, G.; Trusso Cafarello, S.; Loroch, S.; Mennerich, D.; Deschoemaeker, S.; Di Matteo, M.; Ehling, M.; Gevaert, K.; Prenen, H.; Zahedi, R.P.; et al. The mTOR and PP2A Pathways Regulate PHD2 Phosphorylation to Fine-Tune HIF1α Levels and Colorectal Cancer Cell Survival under Hypoxia. Cell Rep. 2017, 18, 1699–1712. [Google Scholar] [CrossRef]
- Finke, D.; Heckmann, M.B.; Frey, N.; Lehmann, L.H. Cancer—A Major Cardiac Comorbidity With Implications on Cardiovascular Metabolism. Front. Physiol. 2021, 12, 729713. [Google Scholar] [CrossRef]
- Mantovani, A.; Locati, M. Macrophage Metabolism Shapes Angiogenesis in Tumors. Cell Metab. 2016, 24, 653–654. [Google Scholar] [CrossRef]
- Qiao, S.; Koh, S.-B.; Vivekanandan, V.; Salunke, D.; Patra, K.C.; Zaganjor, E.; Ross, K.; Mizukami, Y.; Jeanfavre, S.; Chen, A.; et al. REDD1 loss reprograms lipid metabolism to drive progression of RAS mutant tumors. Genes Dev. 2020, 34, 751–766. [Google Scholar] [CrossRef]
- Zhihua, Y.; Yulin, T.; Yibo, W.; Wei, D.; Yin, C.; Jiahao, X.; Runqiu, J.; Xuezhong, X. Hypoxia decreases macrophage glycolysis and M1 percentage by targeting microRNA-30c and mTOR in human gastric cancer. Cancer Sci. 2019, 110, 2368–2377. [Google Scholar] [CrossRef]
- Casazza, A.; Laoui, D.; Wenes, M.; Rizzolio, S.; Bassani, N.; Mambretti, M.; Deschoemaeker, S.; Van Ginderachter, J.A.; Tamagnone, L.; Mazzone, M. Impeding macrophage entry into hypoxic tumor areas by Sema3A/Nrp1 signaling blockade inhibits angiogenesis and restores antitumor immunity. Cancer Cell 2013, 24, 695–709. [Google Scholar] [CrossRef]
- Kang, C.; Elledge, S.J. How autophagy both activates and inhibits cellular senescence. Autophagy 2016, 12, 898–899. [Google Scholar] [CrossRef] [PubMed]
- Birch, J.; Gil, J. Senescence and the SASP: Many therapeutic avenues. Genes Dev. 2020, 34, 1565–1576. [Google Scholar] [CrossRef] [PubMed]
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular Senescence: Defining a Path Forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef] [PubMed]
- Kowald, A.; Passos, J.F.; Kirkwood, T.B.L. On the evolution of cellular senescence. Aging Cell 2020, 19, e13270. [Google Scholar] [CrossRef]
- Wang, B.; Kohli, J.; Demaria, M. Senescent Cells in Cancer Therapy: Friends or Foes? Trends Cancer 2020, 6, 838–857. [Google Scholar] [CrossRef]
- Hoppe-Seyler, K.; Bossler, F.; Lohrey, C.; Bulkescher, J.; Rösl, F.; Jansen, L.; Mayer, A.; Vaupel, P.; Dürst, M.; Hoppe-Seyler, F. Induction of dormancy in hypoxic human papillomavirus-positive cancer cells. Proc. Natl. Acad. Sci. USA 2017, 114, E990–E998. [Google Scholar] [CrossRef]
- Thienpont, B.; Van Dyck, L.; Lambrechts, D. Tumors smother their epigenome. Mol. Cell. Oncol. 2016, 3, e1240549. [Google Scholar] [CrossRef]
- Hain, B.A.; Xu, H.; VanCleave, A.M.; Gordon, B.S.; Kimball, S.R.; Waning, D.L. REDD1 deletion attenuates cancer cachexia in mice. J. Appl. Physiol. 2021, 131, 1718–1730. [Google Scholar] [CrossRef]
- Niu, M.; Li, L.; Su, Z.; Wei, L.; Pu, W.; Zhao, C.; Ding, Y.; Wazir, J.; Cao, W.; Song, S.; et al. An integrative transcriptome study reveals Ddit4/Redd1 as a key regulator of cancer cachexia in rodent models. Cell Death Dis. 2021, 12, 652. [Google Scholar] [CrossRef]
- Hulmi, J.J.; Nissinen, T.A.; Räsänen, M.; Degerman, J.; Lautaoja, J.H.; Hemanthakumar, K.A.; Backman, J.T.; Ritvos, O.; Silvennoinen, M.; Kivelä, R. Prevention of chemotherapy-induced cachexia by ACVR2B ligand blocking has different effects on heart and skeletal muscle. J. Cachexia Sarcopenia Muscle 2018, 9, 417–432. [Google Scholar] [CrossRef]
- Hachemi, Y.; Rapp, A.E.; Picke, A.-K.; Weidinger, G.; Ignatius, A.; Tuckermann, J. Molecular mechanisms of glucocorticoids on skeleton and bone regeneration after fracture. J. Mol. Endocrinol. 2018, 61, R75–R90. [Google Scholar] [CrossRef]
- Conaway, H.H.; Henning, P.; Lie, A.; Tuckermann, J.; Lerner, U.H. Activation of dimeric glucocorticoid receptors in osteoclast progenitors potentiates RANKL induced mature osteoclast bone resorbing activity. Bone 2016, 93, 43–54. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.Y.; Butler, A.P.; Locniskar, M.F.; Fischer, S.M. Signal transduction pathway(s) involved in phorbol ester and autocrine induction of interleukin-1 alpha mRNA in murine keratinocytes. J. Biol. Chem. 1994, 269, 17971–17980. [Google Scholar] [CrossRef]
- Pastor, F.; Dumas, K.; Barthélémy, M.-A.; Regazzetti, C.; Druelle, N.; Peraldi, P.; Cormont, M.; Tanti, J.-F.; Giorgetti-Peraldi, S. Implication of REDD1 in the activation of inflammatory pathways. Sci. Rep. 2017, 7, 7023. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef]
- Rakoff-Nahoum, S.; Paglino, J.; Eslami-Varzaneh, F.; Edberg, S.; Medzhitov, R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell 2004, 118, 229–241. [Google Scholar] [CrossRef] [PubMed]
- Mariathasan, S.; Weiss, D.S.; Newton, K.; McBride, J.; O’Rourke, K.; Roose-Girma, M.; Lee, W.P.; Weinrauch, Y.; Monack, D.M.; Dixit, V.M. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 2006, 440, 228–232. [Google Scholar] [CrossRef]
- Gould, V.E. Cells, tissues and disease: Principles of general pathology. Hum. Pathol. 1997, 28, 754–755. [Google Scholar] [CrossRef]
- Oakley, R.H.; Cidlowski, J.A. The biology of the glucocorticoid receptor: New signaling mechanisms in health and disease. J. Allergy Clin. Immunol. 2013, 132, 1033–1044. [Google Scholar] [CrossRef]
- Waickman, A.T.; Powell, J.D. mTOR, metabolism, and the regulation of T-cell differentiation and function. Immunol. Rev. 2012, 249, 43–58. [Google Scholar] [CrossRef]
- Reuschel, E.L.; Wang, J.; Shivers, D.K.; Muthumani, K.; Weiner, D.B.; Ma, Z.; Finkel, T.H. REDD1 Is Essential for Optimal T Cell Proliferation and Survival. PLoS ONE 2015, 10, e0136323. [Google Scholar] [CrossRef] [PubMed]
- Angelidou, I.; Chrysanthopoulou, A.; Mitsios, A.; Arelaki, S.; Arampatzioglou, A.; Kambas, K.; Ritis, D.; Tsironidou, V.; Moschos, I.; Dalla, V.; et al. REDD1/Autophagy Pathway Is Associated with Neutrophil-Driven IL-1β Inflammatory Response in Active Ulcerative Colitis. J. Immunol. 2018, 200, 3950–3961. [Google Scholar] [CrossRef] [PubMed]
- Frangou, E.; Chrysanthopoulou, A.; Mitsios, A.; Kambas, K.; Arelaki, S.; Angelidou, I.; Arampatzioglou, A.; Gakiopoulou, H.; Bertsias, G.K.; Verginis, P.; et al. REDD1/autophagy pathway promotes thromboinflammation and fibrosis in human systemic lupus erythematosus (SLE) through NETs decorated with tissue factor (TF) and interleukin-17A (IL-17A). Ann. Rheum. Dis. 2019, 78, 238–248. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Mett, I.; Bhunia, A.K.; Bowman, J.; Perez, M.; Zhang, L.; Gandjeva, A.; Zhen, L.; Chukwueke, U.; Mao, T.; et al. Rtp801, a suppressor of mTOR signaling, is an essential mediator of cigarette smoke-induced pulmonary injury and emphysema. Nat. Med. 2010, 16, 767–773. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Liu, G.; Li, D.; Wei, C.; Hao, J. DDIT4 and Associated lncDDIT4 Modulate Th17 Differentiation through the DDIT4/TSC/mTOR Pathway. J. Immunol. 2018, 200, 1618–1626. [Google Scholar] [CrossRef]
- Hou, X.; Yang, S.; Yin, J. Blocking the REDD1/TXNIP axis ameliorates LPS-induced vascular endothelial cell injury through repressing oxidative stress and apoptosis. Am. J. Physiol. Cell Physiol. 2019, 316, C104–C110. [Google Scholar] [CrossRef]
- Mirzoeva, S.; Yang, Y.; Klopot, A.; Budunova, I.; Brown, M.A. Early Stress-Response Gene REDD1 Controls Oxazolone-Induced Allergic Contact Dermatitis. J. Immunol. 2021, 207, 1747–1754. [Google Scholar] [CrossRef]
- Mata, M.A.; Satterly, N.; Versteeg, G.A.; Frantz, D.; Wei, S.; Williams, N.; Schmolke, M.; Peña-Llopis, S.; Brugarolas, J.; Forst, C.V.; et al. Chemical inhibition of RNA viruses reveals REDD1 as a host defense factor. Nat. Chem. Biol. 2011, 7, 712–719. [Google Scholar] [CrossRef]
- Tiwarekar, V.; Wohlfahrt, J.; Fehrholz, M.; Scholz, C.-J.; Kneitz, S.; Schneider-Schaulies, J. APOBEC3G-Regulated Host Factors Interfere with Measles Virus Replication: Role of REDD1 and Mammalian TORC1 Inhibition. J. Virol. 2018, 92, e00835.18. [Google Scholar] [CrossRef]
- Zeng, H.; Chi, H. mTOR and lymphocyte metabolism. Curr. Opin. Immunol. 2013, 25, 347–355. [Google Scholar] [CrossRef]
- Araki, K.; Turner, A.P.; Shaffer, V.O.; Gangappa, S.; Keller, S.A.; Bachmann, M.F.; Larsen, C.P.; Ahmed, R. mTOR regulates memory CD8 T-cell differentiation. Nature 2009, 460, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Nadon, A.M.; Perez, M.J.; Hernandez-Saavedra, D.; Smith, L.P.; Yang, Y.; Sanders, L.A.; Gandjeva, A.; Chabon, J.; Koyanagi, D.E.; Graham, B.B.; et al. Rtp801 suppression of epithelial mTORC1 augments endotoxin-induced lung inflammation. Am. J. Pathol. 2014, 184, 2382–2389. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Wan, G.; Han, B.; Zhang, Z. Echinacoside alleviated LPS-induced cell apoptosis and inflammation in rat intestine epithelial cells by inhibiting the mTOR/STAT3 pathway. Biomed. Pharmacother. 2018, 104, 622–628. [Google Scholar] [CrossRef]
- Li, X.; Shan, C.; Wu, Z.; Yu, H.; Yang, A.; Tan, B. Emodin alleviated pulmonary inflammation in rats with LPS-induced acute lung injury through inhibiting the mTOR/HIF-1α/VEGF signaling pathway. Inflamm. Res. 2020, 69, 365–373. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.-K.; Kim, J.-H.; Kim, J.; Choi, S.; Park, M.; Park, W.; Kim, S.; Lee, K.-S.; Kim, T.; Jung, J.; et al. REDD-1 aggravates endotoxin-induced inflammation via atypical NF-κB activation. FASEB J. 2018, 32, 4585–4599. [Google Scholar] [CrossRef] [PubMed]
- Reiff, D.D.; Cron, R.Q. Who Would Have Predicted Multisystem Inflammatory Syndrome in Children? Curr. Rheumatol. Rep. 2022, 24, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J. Anti-inflammatory actions of glucocorticoids: Molecular mechanisms. Clin. Sci. 1998, 94, 557–572. [Google Scholar] [CrossRef]
- Adcock, I.M.; Mumby, S. Glucocorticoids. Handb. Exp. Pharmacol. 2017, 237, 171–196. [Google Scholar] [CrossRef]
- Wolff, N.C.; McKay, R.M.; Brugarolas, J. REDD1/DDIT4-independent mTORC1 inhibition and apoptosis by glucocorticoids in thymocytes. Mol. Cancer Res. 2014, 12, 867–877. [Google Scholar] [CrossRef]
- Wang, H.; Kubica, N.; Ellisen, L.W.; Jefferson, L.S.; Kimball, S.R. Dexamethasone represses signaling through the mammalian target of rapamycin in muscle cells by enhancing expression of REDD1. J. Biol. Chem. 2006, 281, 39128–39134. [Google Scholar] [CrossRef]


{kind=link}
| Specific Hallmark of Cancer | REDD1 Role | Effect | References |
|---|---|---|---|
| Evasion from growth inhibition | N/D * | N/D | N/D |
| Sustaining proliferation | REDD1 inhibited proliferation in vitro and in vivo | Anti-cancer | [8,18,24,46,49,50,62,63] |
| Attenuation of apoptosis | REDD1 expression was correlated with abrogation of apoptosis | Pro-oncogenic | [1,64,65,66,67] |
| Stimulation of neo-angiogenesis | REDD1 overexpression was correlated with blood vessel density and slower angiogenesis rate in oral squamous carcinoma | Anti-cancer | [56] |
| REDD1 induced angiogenesis in glioblastoma, colon, and lung cancer cells | Pro-oncogenic | [70,71,72,73,74,75,76] | |
| Escape from immune response | REDD1 promoted tumor escape from immune system | Pro-oncogenic | [73,80] |
| Immortalization | N/D | N/D | N/D |
| Tumor-associated inflammation | REDD1 deficiency blunted response to LPS, attenuated production of pro-inflammatory cytokines, and reduced inflammation | Pro-oncogenic | [94,95] |
| Genetic instability | N/D | N/D | N/D |
| Invasion and metastasis | REDD1 overexpression in TAM was associated with metastasis induction | Pro-oncogenic | [55,78] |
| Metabolic shift | REDD1 depleted intracellular ATP, stimulated ROS-mediated cytotoxicity, and decreased glucose uptake | Anti-cancer | [18,20,21,22] |
| Cell senescence | REDD1 promoted pro-survival autophagy in thymocytes | Pro-oncogenic | [64,87] |
| Cellular plasticity | REDD1 overexpression blocked keratinocyte differentiation | Pro-oncogenic | [1,18] |
| Non-mutational epigenetic reprogramming | REDD1 may cause changes in methylation pattern | Questionable | [88] |
| Microbiome polymorphism | N/D | N/D | N/D |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhidkova, E.M.; Lylova, E.S.; Grigoreva, D.D.; Kirsanov, K.I.; Osipova, A.V.; Kulikov, E.P.; Mertsalov, S.A.; Belitsky, G.A.; Budunova, I.; Yakubovskaya, M.G.; et al. Nutritional Sensor REDD1 in Cancer and Inflammation: Friend or Foe? Int. J. Mol. Sci. 2022, 23, 9686. https://doi.org/10.3390/ijms23179686
Zhidkova EM, Lylova ES, Grigoreva DD, Kirsanov KI, Osipova AV, Kulikov EP, Mertsalov SA, Belitsky GA, Budunova I, Yakubovskaya MG, et al. Nutritional Sensor REDD1 in Cancer and Inflammation: Friend or Foe? International Journal of Molecular Sciences. 2022; 23(17):9686. https://doi.org/10.3390/ijms23179686
Chicago/Turabian StyleZhidkova, Ekaterina M., Evgeniya S. Lylova, Diana D. Grigoreva, Kirill I. Kirsanov, Alena V. Osipova, Evgeny P. Kulikov, Sergey A. Mertsalov, Gennady A. Belitsky, Irina Budunova, Marianna G. Yakubovskaya, and et al. 2022. "Nutritional Sensor REDD1 in Cancer and Inflammation: Friend or Foe?" International Journal of Molecular Sciences 23, no. 17: 9686. https://doi.org/10.3390/ijms23179686
APA StyleZhidkova, E. M., Lylova, E. S., Grigoreva, D. D., Kirsanov, K. I., Osipova, A. V., Kulikov, E. P., Mertsalov, S. A., Belitsky, G. A., Budunova, I., Yakubovskaya, M. G., & Lesovaya, E. A. (2022). Nutritional Sensor REDD1 in Cancer and Inflammation: Friend or Foe? International Journal of Molecular Sciences, 23(17), 9686. https://doi.org/10.3390/ijms23179686