
Sirtuins Modulation: A Promising Strategy for HIV-Associated Neurocognitive Impairments

 , , , ,
, , , ,
Abstract
:1. Introduction
2. HAND and the Long-Term Exposure to ART
3. Transactivator of Transcription Tat
Secretion and Internalization of Tat
4. Tat and Mitochondria
4.1. Tat and Fusion-Fission Dynamics
4.2. Mitophagy Disruption
4.3. Tat-Induced Apoptosis
5. Sirtuins
5.1. Sirtuins in Neurodegenerative Diseases
5.2. SIRT1
5.2.1. SIRT1 in ER Stress and UPR
5.2.2. SIRT1 and Mitochondrial Dysfunction
5.2.3. SIRT1 and Tat
5.3. SIRT2
5.4. SIRT3
5.4.1. SIRT3 and Antioxidant Response
5.4.2. SIRT3 and Mitochondrial Biogenesis and mtDNA Integrity
5.4.3. SIRT3 and Mitophagy
5.4.4. SIRT3 and Tat
6. SIRT Modulation
6.1. Inhibitors
6.2. Activators
6.2.1. Polyphenols
6.2.2. Other Natural Compounds
6.2.3. Synthetic Drugs
6.2.4. NAD+ as Co-Substrate of SIRTs
7. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- UNAIDS FACT SHEET 2021. Available online: https://www.unaids.org/sites/default/files/media_asset/UNAIDS_FactSheet_en.pdf (accessed on 9 October 2021).
- Brew, B.J.; Barnes, S.L. The impact of HIV central nervous system persistence on pathogenesis. Aids 2019, 33, S113–S121. [Google Scholar] [CrossRef] [PubMed]
- Antinori, A.; Arendt, G.; Becker, J.; Brew, B.; Byrd, D.; Cherner, M.; Clifford, D.; Cinque, P.; Epstein, L.; Goodkin, K. Updated research nosology for HIV-associated neurocognitive disorders. Neurology 2007, 69, 1789–1799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Liu, M.; Lu, Q.; Farrell, M.; Lappin, J.M.; Shi, J.; Lu, L.; Bao, Y. Global prevalence and burden of HIV-associated neurocognitive disorder: A meta-analysis. Neurology 2020, 95, e2610–e2621. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Hou, J.; Su, B.; Jiang, T.; Guo, C.; Wang, W.; Zhang, Y.; Chang, B.; Wu, H.; Zhang, T. The Prevalence of Frascati-Criteria-Based HIV-Associated Neurocognitive Disorder (HAND) in HIV-Infected Adults: A Systematic Review and Meta-Analysis. Front. Neurol. 2020, 11, 581346. [Google Scholar] [CrossRef] [PubMed]
- Carroll, A.; Brew, B. HIV-associated neurocognitive disorders: Recent advances in pathogenesis, biomarkers, and treatment. F1000Research 2017, 6, 1–11. [Google Scholar] [CrossRef]
- Barroso, S.M.; Sousa, K.C.R. Neurocognitive Disorder and Emotional Symptoms in HIV+ Brazilian Elderly: Influence of Gender, Income, Diet, and Sleep. Front. Hum. Neurosci. 2021, 15, 515. [Google Scholar] [CrossRef]
- Diaz, M.M.; Zacarías, M.G.; Sotolongo, P.; Sanes, M.F.; Franklin, D.J.; Marquine, M.J.; Cherner, M.; Cárcamo, C.; Ellis, R.J.; Lanata, S.; et al. Characterization of HIV-Associated Neurocognitive Impairment in Middle-Aged and Older Persons With HIV in Lima, Peru. Front. Neurol. 2021, 12, 629257. [Google Scholar] [CrossRef]
- Irollo, E.; Luchetta, J.; Ho, C.; Nash, B.; Meucci, O. Mechanisms of neuronal dysfunction in HIV-associated neurocognitive disorders. Cell. Mol. Life Sci. 2021, 78, 4283–4303. [Google Scholar] [CrossRef]
- Namagga, J.K.; Rukundo, G.Z.; Niyonzima, V.; Voss, J. Depression and HIV associated neurocognitive disorders among HIV infected adults in rural southwestern Uganda: A cross-sectional quantitative study. BMC Psychiatry 2021, 21, 4–11. [Google Scholar] [CrossRef]
- Forno, G.; Henríquez, F.; Ceballos, M.E.; Gonzalez, M.; Schröder, J.; Toro, P. Neurological soft signs (NSS) and cognitive deficits in HIV associated neurocognitive disorder. Neuropsychologia 2020, 146, 107545. [Google Scholar] [CrossRef]
- Saylor, D.; Dickens, A.M.; Sacktor, N.; Haughey, N.; Slusher, B.; Pletnikov, M.; Mankowski, J.L.; Brown, A.; Volsky, D.J.; McArthur, J.C. HIV-associated neurocognitive disorder—Pathogenesis and prospects for treatment. Nat. Rev. Neurol. 2016, 12, 234–248. [Google Scholar] [CrossRef]
- Filho, S.M.M.F.; de Melo, H.R.L. Frequency and risk factors for HIV-associated neurocognitive disorder and depression in older individuals with HIV in northeastern Brazil. Int. Psychogeriatr. 2012, 24, 1648–1655. [Google Scholar] [CrossRef]
- Robinson-Papp, J. Motor Function and Human Immunodeficiency Virus–Associated Cognitive Impairment in a Highly Active Antiretroviral Therapy–Era Cohort. Arch. Neurol. 2008, 65, 1096. [Google Scholar] [CrossRef] [Green Version]
- Smail, R.C.; Brew, B.J. HIV-associated neurocognitive disorder. In The Neurology of HIV Infection; Elsevier B.V.: Amsterdam, The Netherlands, 2018; Volume 152, pp. 75–97. ISBN 9780444638496. [Google Scholar]
- Chaganti, J.; Marripudi, K.; Staub, L.P.; Rae, C.D.; Gates, T.M.; Moffat, K.J.; Brew, B.J. Imaging correlates of the blood–brain barrier disruption in HIV-associated neurocognitive disorder and therapeutic implications. AIDS 2019, 33, 1843–1852. [Google Scholar] [CrossRef]
- Thangaraj, A.; Periyasamy, P.; Callen, S.; Pendyala, G.; Buch, S. HIV-1 TAT-mediated microglial activation: Role of mitochondrial dysfunction and defective mitophagy. Autophagy 2018, 14, 1596–1619. [Google Scholar] [CrossRef] [Green Version]
- Dong, H.; Ye, X.; Zhong, L.; Xu, J.; Qiu, J.; Wang, J.; Shao, Y.; Xing, H. Role of FOXO3 Activated by HIV-1 Tat in HIV-Associated Neurocognitive Disorder Neuronal Apoptosis. Front. Neurosci. 2019, 13. [Google Scholar] [CrossRef] [Green Version]
- Tang, X.; Lu, H.; Ramratnam, B. Neurotoxicity of HIV-1 Tat is attributed to its penetrating property. Sci. Rep. 2020, 10, 14002. [Google Scholar] [CrossRef]
- Darbinian, N.; Darbinyan, A.; Merabova, N.; Selzer, M.E.; Amini, S. HIV-1 and HIV-1-Tat Induce Mitochondrial DNA Damage in Human Neurons. J. HIV AIDS 2020, 6. [Google Scholar] [CrossRef]
- Hu, G.; Niu, F.; Liao, K.; Periyasamy, P.; Sil, S.; Liu, J.; Dravid, S.M.; Buch, S. HIV-1 Tat-Induced Astrocytic Extracellular Vesicle miR-7 Impairs Synaptic Architecture. J. Neuroimmune Pharmacol. 2020, 15, 538–553. [Google Scholar] [CrossRef]
- Andhavarapu, S.; Katuri, A.; Bryant, J.; Patel, V.; Gupta, U.; Asemu, G.; Makar, T.K. Intersecting roles of ER stress, mitochondrial dysfunction, autophagy, and calcium homeostasis in HIV-associated neurocognitive disorder. J. Neurovirol. 2020, 26, 664–675. [Google Scholar] [CrossRef]
- Campestrini, J.; Silveira, D.B.; Pinto, A.R. HIV-1 Tat-induced bystander apoptosis in Jurkat cells involves unfolded protein responses. Cell Biochem. Funct. 2018, 36, 377–386. [Google Scholar] [CrossRef]
- Rozzi, S.J.; Avdoshina, V.; Fields, J.A.; Mocchetti, I. Human immunodeficiency virus Tat impairs mitochondrial fission in neurons. Cell Death Discov. 2018, 4. [Google Scholar] [CrossRef]
- Norman, J.P.; Perry, S.W.; Kasischke, K.A.; Volsky, D.J.; Gelbard, H.A. HIV-1 Trans Activator of Transcription Protein Elicits Mitochondrial Hyperpolarization and Respiratory Deficit, with Dysregulation of Complex IV and Nicotinamide Adenine Dinucleotide Homeostasis in Cortical Neurons. J. Immunol. 2007, 178, 869–876. [Google Scholar] [CrossRef] [Green Version]
- De Simone, F.I.; Darbinian, N.; Amini, S.; Muniswamy, M.; White, M.K.; Elrod, J.W.; Datta, P.K.; Langford, D.; Khalili, K. HIV-1 Tat and Cocaine Impair Survival of Cultured Primary Neuronal Cells via a Mitochondrial Pathway. J. Neuroimmune Pharmacol. 2016, 11, 358–368. [Google Scholar] [CrossRef]
- Kang, X.; Yang, W.; Wang, R.; Xie, T.; Li, H.; Feng, D.; Jin, X.; Sun, H.; Wu, S. Sirtuin-1 (SIRT1) stimulates growth-plate chondrogenesis by attenuating the PERK–eIF-2α–CHOP pathway in the unfolded protein response. J. Biol. Chem. 2018, 293, 8614–8625. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Y.; Cruzat, V.F.; Newsholme, P.; Cheng, J.; Chen, Y.; Lu, Y. Regulation of SIRT1 in aging: Roles in mitochondrial function and biogenesis. Mech. Ageing Dev. 2016, 155, 10–21. [Google Scholar] [CrossRef] [PubMed]
- Thangaraj, A.; Chivero, E.T.; Tripathi, A.; Singh, S.; Niu, F.; Guo, M.-L.; Pillai, P.; Periyasamy, P.; Buch, S. HIV TAT-mediated microglial senescence: Role of SIRT3-dependent mitochondrial oxidative stress. Redox Biol. 2021, 40, 101843. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.-S.; Brent, M.M.; Getachew, R.; Jayakumar, P.; Chen, L.-F.; Schnolzer, M.; McBurney, M.W.; Marmorstein, R.; Greene, W.C.; Ott, M. Human Immunodeficiency Virus Type 1 Tat Protein Inhibits the SIRT1 Deacetylase and Induces T Cell Hyperactivation. Cell Host Microbe 2008, 3, 158–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pagans, S.; Pedal, A.; North, B.J.; Kaehlcke, K.; Marshall, B.L.; Dorr, A.; Hetzer-Egger, C.; Henklein, P.; Frye, R.; McBurney, M.W.; et al. SIRT1 Regulates HIV Transcription via Tat Deacetylation. PLoS Biol. 2005, 3, e41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, I. Interrogating the impact of combination antiretroviral therapies on HIV-associated neurocognitive disorders. HIV Med. 2021, 22, 783–790. [Google Scholar] [CrossRef] [PubMed]
- Caniglia, E.C.; Cain, L.E.; Justice, A.; Tate, J.; Logan, R.; Sabin, C.; Winston, A.; van Sighem, A.; Miro, J.M.; Podzamczer, D.; et al. Antiretroviral penetration into the CNS and incidence of AIDS-defining neurologic conditions. Neurology 2014, 83, 134–141. [Google Scholar] [CrossRef] [Green Version]
- Ciccarelli, N.; Fabbiani, M.; Di Giambenedetto, S.; Fanti, I.; Baldonero, E.; Bracciale, L.; Tamburrini, E.; Cauda, R.; De Luca, A.; Silveri, M.C. Efavirenz associated with cognitive disorders in otherwise asymptomatic HIV-infected patients. Neurology 2011, 76, 1403–1409. [Google Scholar] [CrossRef]
- Buckley, S.; Byrnes, S.; Cochrane, C.; Roche, M.; Estes, J.D.; Selemidis, S.; Angelovich, T.A.; Churchill, M.J. The role of oxidative stress in HIV-associated neurocognitive disorders. Brain Behav. Immun. Health 2021, 13, 100235. [Google Scholar] [CrossRef]
- Amusan, P.; Power, C.; Gill, M.J.; Gomez, D.; Johnson, E.; Rubin, L.H.; Fujiwara, E. Lifetime antiretroviral exposure and neurocognitive impairment in HIV. J. Neurovirol. 2020, 26, 743–753. [Google Scholar] [CrossRef]
- Lin, S.-P.; Calcagno, A.; Letendre, S.L.; Ma, Q. Clinical Treatment Options and Randomized Clinical Trials for Neurocognitive Complications of HIV Infection: Combination Antiretroviral Therapy, Central Nervous System Penetration Effectiveness, and Adjuvants. Curr. Top. Behav. Neurosci. 2020, 50, 517–545. [Google Scholar]
- Stern, A.L.; Lee, R.N.; Panvelker, N.; Li, J.; Harowitz, J.; Jordan-Sciutto, K.L.; Akay-Espinoza, C. Differential Effects of Antiretroviral Drugs on Neurons In Vitro: Roles for Oxidative Stress and Integrated Stress Response. J. Neuroimmune Pharmacol. 2018, 13, 64–76. [Google Scholar] [CrossRef]
- Roda, R.H.; Hoke, A. Mitochondrial dysfunction in HIV-induced peripheral neuropathy. Int. Rev. Neurobiol. 2019, 145, 67–82. [Google Scholar]
- Ganta, K.K.; Chaubey, B. Endoplasmic reticulum stress leads to mitochondria-mediated apoptosis in cells treated with anti-HIV protease inhibitor ritonavir. Cell Biol. Toxicol. 2019, 35, 189–204. [Google Scholar] [CrossRef]
- Gao, C.; Meng, J.; Xiao, X.; Wang, M.; Williams, A.B.; Wang, H. Antiretroviral therapy improves neurocognitive impairment in people living with HIV? A meta-analysis. Int. J. Nurs. Sci. 2020, 7, 238–247. [Google Scholar] [CrossRef]
- Siangphoe, U.; Archer, K.J.; Nguyen, C.; Lee, K.R. Associations of antiretroviral therapy and comorbidities with neurocognitive outcomes in HIV-1-infected patients. AIDS 2020, 34, 893–902. [Google Scholar] [CrossRef]
- Spector, C.; Mele, A.R.; Wigdahl, B.; Nonnemacher, M.R. Genetic variation and function of the HIV-1 Tat protein. Med. Microbiol. Immunol. 2019, 208, 131–169. [Google Scholar] [CrossRef]
- Rayne, F.; Debaisieux, S.; Bonhoure, A.; Beaumelle, B. HIV-1 Tat is unconventionally secreted through the plasma membrane. Cell Biol. Int. 2010, 34, 409–413. [Google Scholar] [CrossRef]
- Rayne, F.; Debaisieux, S.; Yezid, H.; Lin, Y.-L.; Mettling, C.; Konate, K.; Chazal, N.; Arold, S.T.; Pugnière, M.; Sanchez, F.; et al. Phosphatidylinositol-(4,5)-bisphosphate enables efficient secretion of HIV-1 Tat by infected T-cells. EMBO J. 2010, 29, 1348–1362. [Google Scholar] [CrossRef]
- Mele, A.R.; Marino, J.; Chen, K.; Pirrone, V.; Janetopoulos, C.; Wigdahl, B.; Klase, Z.; Nonnemacher, M.R. Defining the molecular mechanisms of HIV-1 Tat secretion: PtdIns(4,5)P 2 at the epicenter. Traffic 2018, 19, 655–665. [Google Scholar] [CrossRef] [Green Version]
- Rahimian, P.; He, J.J. Exosome-associated release, uptake, and neurotoxicity of HIV-1 Tat protein. J. Neurovirol. 2016, 22, 774–788. [Google Scholar] [CrossRef] [Green Version]
- Vendeville, A.; Rayne, F.; Bonhoure, A.; Bettache, N.; Montcourrier, P.; Beaumelle, B.; Ii, M. HIV-1 Tat Enters T Cells Using Coated Pits before Translocating from Acidified Endosomes and Eliciting. Mol. Biol. Cell 2004, 15, 2347–2360. [Google Scholar] [CrossRef] [Green Version]
- Khan, N.; Halcrow, P.W.; Lakpa, K.L.; Afghah, Z.; Miller, N.M.; Dowdy, S.F.; Geiger, J.D.; Chen, X. Two-pore channels regulate Tat endolysosome escape and Tat-mediated HIV-1 LTR transactivation. FASEB J. 2020, 34, 4147–4162. [Google Scholar] [CrossRef] [Green Version]
- Ruiz, A.P.; Ajasin, D.O.; Ramasamy, S.; DesMarais, V.; Eugenin, E.A.; Prasad, V.R. A Naturally Occurring Polymorphism in the HIV-1 Tat Basic Domain Inhibits Uptake by Bystander Cells and Leads to Reduced Neuroinflammation. Sci. Rep. 2019, 9, 3308. [Google Scholar] [CrossRef] [Green Version]
- Lv, F.; Wang, J.; Chen, H.; Sui, L.; Feng, L.; Liu, Z.; Liu, Y.; Wei, G.; Lu, W. Enhanced mucosal penetration and efficient inhibition efficacy against cervical cancer of PEGylated docetaxel nanocrystals by TAT modification. J. Control. Release 2021, 336, 572–582. [Google Scholar] [CrossRef]
- Wang, Z.; Liu, D.; Varin, A.; Nicolas, V.; Courilleau, D.; Mateo, P.; Caubere, C.; Rouet, P.; Gomez, A.-M.; Vandecasteele, G.; et al. A cardiac mitochondrial cAMP signaling pathway regulates calcium accumulation, permeability transition and cell death. Cell Death Dis. 2016, 7, e2198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Correia-Melo, C.; Marques, F.D.; Anderson, R.; Hewitt, G.; Hewitt, R.; Cole, J.; Carroll, B.M.; Miwa, S.; Birch, J.; Merz, A.; et al. Mitochondria are required for pro-ageing features of the senescent phenotype. EMBO J. 2016, 35, 724–742. [Google Scholar] [CrossRef] [PubMed]
- Bock, F.J.; Tait, S.W.G. Mitochondria as multifaceted regulators of cell death. Nat. Rev. Mol. Cell Biol. 2020, 21, 85–100. [Google Scholar] [CrossRef] [PubMed]
- Fields, J.A.; Ellis, R.J. HIV in the cART era and the mitochondrial: Immune interface in the CNS. Int. Rev. Neurobiol. 2019, 145, 29–65. [Google Scholar]
- Sanna, P.P.; Fu, Y.; Masliah, E.; Lefebvre, C.; Repunte-Canonigo, V. Central nervous system (CNS) transcriptomic correlates of human immunodeficiency virus (HIV) brain RNA load in HIV-infected individuals. Sci. Rep. 2021, 11, 12176. [Google Scholar] [CrossRef]
- Teodorof-Diedrich, C.; Spector, S.A. Human Immunodeficiency Virus Type 1 gp120 and Tat Induce Mitochondrial Fragmentation and Incomplete Mitophagy in Human Neurons. J. Virol. 2018, 92, e00993-18. [Google Scholar] [CrossRef] [Green Version]
- Macho, A.; Calzado, M.A.; Jiménez-Reina, L.; Ceballos, E.; León, J.; Muñoz, E. Susceptibility of HIV-1-TAT transfected cells to undergo apoptosis. Biochemical mechanisms. Oncogene 1999, 18, 7543–7551. [Google Scholar] [CrossRef] [Green Version]
- Roca-Bayerri, C.; Robertson, F.; Pyle, A.; Hudson, G.; Payne, B.A.I. Mitochondrial DNA Damage and Brain Aging in Human Immunodeficiency Virus. Clin. Infect. Dis. 2021, 73, e466–e473. [Google Scholar] [CrossRef]
- Doke, M.; Jeganathan, V.; McLaughlin, J.P.; Samikkannu, T. HIV-1 Tat and cocaine impact mitochondrial epigenetics: Effects on DNA methylation. Epigenetics 2021, 16, 980–999. [Google Scholar] [CrossRef]
- Tahrir, F.G.; Shanmughapriya, S.; Ahooyi, T.M.; Knezevic, T.; Gupta, M.K.; Kontos, C.D.; McClung, J.M.; Madesh, M.; Gordon, J.; Feldman, A.M.; et al. Dysregulation of mitochondrial bioenergetics and quality control by HIV-1 Tat in cardiomyocytes. J. Cell. Physiol. 2018, 233, 748–758. [Google Scholar] [CrossRef]
- El-Amine, R.; Germini, D.; Zakharova, V.V.; Tsfasman, T.; Sheval, E.V.; Louzada, R.A.N.; Dupuy, C.; Bilhou-Nabera, C.; Hamade, A.; Najjar, F.; et al. HIV-1 Tat protein induces DNA damage in human peripheral blood B-lymphocytes via mitochondrial ROS production. Redox Biol. 2018, 15, 97–108. [Google Scholar] [CrossRef]
- Self, R.L.; Mulholland, P.J.; Nath, A.; Harris, B.R.; Prendergast, M.A. The human immunodeficiency virus type-1 transcription factor Tat produces elevations in intracellular Ca2+ that require function of an N-methyl-d-aspartate receptor polyamine-sensitive site. Brain Res. 2004, 995, 39–45. [Google Scholar] [CrossRef]
- Kruman, I.I.; Nath, A.; Mattson, M.P. HIV-1 Protein Tat Induces Apoptosis of Hippocampal Neurons by a Mechanism Involving Caspase Activation, Calcium Overload, and Oxidative Stress. Exp. Neurol. 1998, 154, 276–288. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, Y.; Xu, Q.; Qiu, J.; Zheng, H.; Ye, X.; Xue, Y.; Yin, Y.; Zhang, Z.; Liu, Y.; et al. Menin mediates Tat-induced neuronal apoptosis in brain frontal cortex of SIV-infected macaques and in Tat-treated cells. Oncotarget 2017, 8, 18082–18094. [Google Scholar] [CrossRef] [Green Version]
- Saunders, M.; Eldeen, M.B.; Del Valle, L.; Reiss, K.; Peruzzi, F.; Mameli, G.; Gelman, B.B.; Khalili, K.; Amini, S.; Sawaya, B.E. p73 modulates HIV-1 Tat transcriptional and apoptotic activities in human astrocytes. Apoptosis 2005, 10, 1419–1431. [Google Scholar] [CrossRef]
- Westermann, B. Mitochondrial fusion and fission in cell life and death. Nat. Rev. Mol. Cell Biol. 2010, 11, 872–884. [Google Scholar] [CrossRef]
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat. Cell Biol. 2018, 20, 1013–1022. [Google Scholar] [CrossRef]
- Che, X.; He, F.; Deng, Y.; Xu, S.; Fan, X.; Gu, P.; Wang, Z. HIV-1 Tat-Mediated Apoptosis in Human Blood-Retinal Barrier-Associated Cells. PLoS ONE 2014, 9, e95420. [Google Scholar] [CrossRef]
- Ma, R.; Yang, L.; Niu, F.; Buch, S. HIV Tat-Mediated Induction of Human Brain Microvascular Endothelial Cell Apoptosis Involves Endoplasmic Reticulum Stress and Mitochondrial Dysfunction. Mol. Neurobiol. 2016, 53, 132–142. [Google Scholar] [CrossRef]
- Singh, R.; Letai, A.; Sarosiek, K. Regulation of apoptosis in health and disease: The balancing act of BCL-2 family proteins. Nat. Rev. Mol. Cell Biol. 2019, 20, 175–193. [Google Scholar] [CrossRef]
- Williams, M.E.; Zulu, S.S.; Stein, D.J.; Joska, J.A.; Naudé, P.J.W. Signatures of HIV-1 subtype B and C Tat proteins and their effects in the neuropathogenesis of HIV-associated neurocognitive impairments. Neurobiol. Dis. 2020, 136, 104701. [Google Scholar] [CrossRef]
- Hetz, C.; Zhang, K.; Kaufman, R.J. Mechanisms, regulation and functions of the unfolded protein response. Nat. Rev. Mol. Cell Biol. 2020, 21, 421–438. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.-T. HIV-1 Tat-Mediated Calcium Dysregulation and Neuronal Dysfunction in Vulnerable Brain Regions. Curr. Drug Targets 2015, 17, 4–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martucciello, S.; Masullo, M.; Cerulli, A.; Piacente, S. Natural Products Targeting ER Stress, and the Functional Link to Mitochondria. Int. J. Mol. Sci. 2020, 21, 1905. [Google Scholar] [CrossRef] [Green Version]
- Bartok, A.; Weaver, D.; Golenár, T.; Nichtova, Z.; Katona, M.; Bánsághi, S.; Alzayady, K.J.; Thomas, V.K.; Ando, H.; Mikoshiba, K.; et al. IP3 receptor isoforms differently regulate ER-mitochondrial contacts and local calcium transfer. Nat. Commun. 2019, 10, 3726. [Google Scholar] [CrossRef] [Green Version]
- Haughey, N.J.; Holden, C.P.; Nath, A.; Geiger, J.D. Involvement of Inositol 1,4,5-Trisphosphate-Regulated Stores of Intracellular Calcium in Calcium Dysregulation and Neuron Cell Death Caused by HIV-1 Protein Tat. J. Neurochem. 2002, 73, 1363–1374. [Google Scholar] [CrossRef] [Green Version]
- Carafa, V.; Rotili, D.; Forgione, M.; Cuomo, F.; Serretiello, E.; Hailu, G.S.; Jarho, E.; Lahtela-Kakkonen, M.; Mai, A.; Altucci, L. Sirtuin functions and modulation: From chemistry to the clinic. Clin. Epigenet. 2016, 8, 61. [Google Scholar] [CrossRef]
- Karaman, B.; Jung, M.; Sippl, W. Structure-Based Design and Computational Studies of Sirtuin Inhibitors. In Epi-Informatics; Elsevier: Amsterdam, The Netherlands, 2016; pp. 297–325. [Google Scholar]
- Bheda, P.; Jing, H.; Wolberger, C.; Lin, H. The Substrate Specificity of Sirtuins. Annu. Rev. Biochem. 2016, 85, 405–429. [Google Scholar] [CrossRef]
- Jayasena, T.; Poljak, A.; Braidy, N.; Zhong, L.; Rowlands, B.; Muenchhoff, J.; Grant, R.; Smythe, G.; Teo, C.; Raftery, M.; et al. Application of Targeted Mass Spectrometry for the Quantification of Sirtuins in the Central Nervous System. Sci. Rep. 2016, 6, 35391. [Google Scholar] [CrossRef] [Green Version]
- Donmez, G.; Arun, A.; Chung, C.-Y.; McLean, P.J.; Lindquist, S.; Guarente, L. SIRT1 Protects against -Synuclein Aggregation by Activating Molecular Chaperones. J. Neurosci. 2012, 32, 124–132. [Google Scholar] [CrossRef]
- Outeiro, T.F.; Kontopoulos, E.; Altmann, S.M.; Kufareva, I.; Strathearn, K.E.; Amore, A.M.; Volk, C.B.; Maxwell, M.M.; Rochet, J.-C.; McLean, P.J.; et al. Sirtuin 2 Inhibitors Rescue α-Synuclein-Mediated Toxicity in Models of Parkinson’s Disease. Science 2007, 317, 516–519. [Google Scholar] [CrossRef]
- Fernando, K.K.M.; Wijayasinghe, Y.S. Sirtuins as Potential Therapeutic Targets for Mitigating Neuroinflammation Associated With Alzheimer’s Disease. Front. Cell. Neurosci. 2021, 15. [Google Scholar] [CrossRef] [PubMed]
- Julien, C.; Tremblay, C.; Émond, V.; Lebbadi, M.; Salem, N.; Bennett, D.A.; Calon, F. Sirtuin 1 Reduction Parallels the Accumulation of Tau in Alzheimer Disease. J. Neuropathol. Exp. Neurol. 2009, 68, 48–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Kim, Y.; Liu, T.; Hwang, Y.J.; Hyeon, S.J.; Im, H.; Lee, K.; Alvarez, V.E.; McKee, A.C.; Um, S.-J.; et al. SIRT3 deregulation is linked to mitochondrial dysfunction in Alzheimer’s disease. Aging Cell 2018, 17, e12679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, J.; Han, P.; Song, M.; Nielsen, M.; Beach, T.G.; Serrano, G.E.; Liang, W.S.; Caselli, R.J.; Shi, J. Amyloid-β Increases Tau by Mediating Sirtuin 3 in Alzheimer’s Disease. Mol. Neurobiol. 2018, 55, 8592–8601. [Google Scholar] [CrossRef]
- Hubbard, B.P.; Gomes, A.P.; Dai, H.; Li, J.; Case, A.W.; Considine, T.; Riera, T.V.; Lee, J.E.; E, S.Y.; Lamming, D.W.; et al. Evidence for a Common Mechanism of SIRT1 Regulation by Allosteric Activators. Science 2013, 339, 1216–1219. [Google Scholar] [CrossRef] [Green Version]
- Vaziri, H.; Dessain, S.K.; Eaton, E.N.; Imai, S.; Frye, R.A.; Pandita, T.K.; Guarente, L.; Weinberg, R.A. hSIR2 SIRT1 Functions as an NAD-Dependent p53 Deacetylase. Cell 2001, 107, 149–159. [Google Scholar] [CrossRef] [Green Version]
- Asher, G.; Gatfield, D.; Stratmann, M.; Reinke, H.; Dibner, C.; Kreppel, F.; Mostoslavsky, R.; Alt, F.W.; Schibler, U. SIRT1 Regulates Circadian Clock Gene Expression through PER2 Deacetylation. Cell 2008, 134, 317–328. [Google Scholar] [CrossRef] [Green Version]
- Peng, L.; Yuan, Z.; Ling, H.; Fukasawa, K.; Robertson, K.; Olashaw, N.; Koomen, J.; Chen, J.; Lane, W.S.; Seto, E. SIRT1 Deacetylates the DNA Methyltransferase 1 (DNMT1) Protein and Alters Its Activities. Mol. Cell. Biol. 2011, 31, 4720–4734. [Google Scholar] [CrossRef] [Green Version]
- Vaquero, A.; Scher, M.; Lee, D.; Erdjument-Bromage, H.; Tempst, P.; Reinberg, D. Human SirT1 Interacts with Histone H1 and Promotes Formation of Facultative Heterochromatin. Mol. Cell 2004, 16, 93–105. [Google Scholar] [CrossRef]
- Zhang, W.; Feng, Y.; Guo, Q.; Guo, W.; Xu, H.; Li, X.; Yi, F.; Guan, Y.; Geng, N.; Wang, P.; et al. SIRT1 modulates cell cycle progression by regulating CHK2 acetylation−phosphorylation. Cell Death Differ. 2020, 27, 482–496. [Google Scholar] [CrossRef] [Green Version]
- Liang, D.; Zhuo, Y.; Guo, Z.; He, L.; Wang, X.; He, Y.; Li, L.; Dai, H. SIRT1/PGC-1 pathway activation triggers autophagy/mitophagy and attenuates oxidative damage in intestinal epithelial cells. Biochimie 2020, 170, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Alves-Fernandes, D.K.; Jasiulionis, M.G. The Role of SIRT1 on DNA Damage Response and Epigenetic Alterations in Cancer. Int. J. Mol. Sci. 2019, 20, 3153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mihanfar, A.; Akbarzadeh, M.; Ghazizadeh Darband, S.; Sadighparvar, S.; Majidinia, M. SIRT1: A promising therapeutic target in type 2 diabetes mellitus. Arch. Physiol. Biochem. 2021, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Jiao, F.; Gong, Z. The Beneficial Roles of SIRT1 in Neuroinflammation-Related Diseases. Oxid. Med. Cell. Longev. 2020, 2020, 6782872. [Google Scholar] [CrossRef]
- Xu, J.; Jackson, C.W.; Khoury, N.; Escobar, I.; Perez-pinzon, M.A. Brain SIRT1 Mediates Metabolic Homeostasis and Neuroprotection. Front. Endocrinol. 2018, 9, 702. [Google Scholar] [CrossRef]
- Prola, A.; Pires Da Silva, J.; Guilbert, A.; Lecru, L.; Piquereau, J.; Ribeiro, M.; Mateo, P.; Gressette, M.; Fortin, D.; Boursier, C.; et al. SIRT1 protects the heart from ER stress-induced cell death through eIF2α deacetylation. Cell Death Differ. 2017, 24, 343–356. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, H.S.; Reizis, B.; Robbins, P.D. SIRT1 associates with eIF2-alpha and regulates the cellular stress response. Sci. Rep. 2011, 1, 150. [Google Scholar] [CrossRef]
- Lee, I.C.; Ho, X.Y.; George, S.E.; Goh, C.W.; Sundaram, J.R.; Pang, K.K.L.; Luo, W.; Yusoff, P.; Sze, N.S.K.; Shenolikar, S. Oxidative stress promotes SIRT1 recruitment to the GADD34/PP1α complex to activate its deacetylase function. Cell Death Differ. 2018, 25, 255–267. [Google Scholar] [CrossRef] [Green Version]
- Teertam, S.K.; Prakash Babu, P. Differential role of SIRT1/MAPK pathway during cerebral ischemia in rats and humans. Sci. Rep. 2021, 11, 6339. [Google Scholar] [CrossRef]
- Manjula, R.; Anuja, K.; Alcain, F.J. SIRT1 and SIRT2 Activity Control in Neurodegenerative Diseases. Front. Pharmacol. 2021, 11, 1899. [Google Scholar] [CrossRef]
- Sorrentino, V.; Menzies, K.J.; Auwerx, J. Repairing Mitochondrial Dysfunction in Disease. Annu. Rev. Pharmacol. Toxicol. 2018, 58, 353–389. [Google Scholar] [CrossRef]
- Gupta, P.; Sharma, G.; Lahiri, A.; Barthwal, M.K. FOXO3a acetylation regulates PINK1, mitophagy, inflammasome activation in murine palmitate-conditioned and diabetic macrophages. J. Leukoc. Biol. 2021. [Google Scholar] [CrossRef]
- Das, S.; Mitrovsky, G.; Vasanthi, H.R.; Das, D.K. Antiaging Properties of a Grape-Derived Antioxidant Are Regulated by Mitochondrial Balance of Fusion and Fission Leading to Mitophagy Triggered by a Signaling Network of Sirt1-Sirt3-Foxo3-PINK1-PARKIN. Oxid. Med. Cell. Longev. 2014, 2014, 345105. [Google Scholar] [CrossRef]
- Lovy, A.; Ahumada-Castro, U.; Bustos, G.; Farias, P.; Gonzalez-Billault, C.; Molgó, J.; Cardenas, C. Concerted Action of AMPK and Sirtuin-1 Induces Mitochondrial Fragmentation Upon Inhibition of Ca2+ Transfer to Mitochondria. Front. Cell Dev. Biol. 2020, 8, 378. [Google Scholar] [CrossRef]
- Blazek, D.; Peterlin, B.M. Tat-SIRT1 Tango. Mol. Cell 2008, 29, 539–540. [Google Scholar] [CrossRef]
- Chen, X.; Lu, W.; Wu, D. Sirtuin 2 (SIRT2): Confusing Roles in the Pathophysiology of Neurological Disorders. Front. Neurosci. 2021, 15, 518. [Google Scholar] [CrossRef]
- Li, Z.; Xie, Q.R.; Chen, Z.; Lu, S.; Xia, W. Regulation of SIRT2 levels for human non-small cell lung cancer therapy. Lung Cancer 2013, 82, 9–15. [Google Scholar] [CrossRef]
- Suzuki, K.; Koike, T. Mammalian Sir2-related protein (SIRT) 2–mediated modulation of resistance to axonal degeneration in slow Wallerian degeneration mice: A crucial role of tubulin deacetylation. Neuroscience 2007, 147, 599–612. [Google Scholar] [CrossRef]
- Fourcade, S.; Morató, L.; Parameswaran, J.; Ruiz, M.; Ruiz-Cortés, T.; Jové, M.; Naudí, A.; Martínez-Redondo, P.; Dierssen, M.; Ferrer, I.; et al. Loss of SIRT2 leads to axonal degeneration and locomotor disability associated with redox and energy imbalance. Aging Cell 2017, 16, 1404–1413. [Google Scholar] [CrossRef]
- Liu, G.; Park, S.-H.; Imbesi, M.; Nathan, W.J.; Zou, X.; Zhu, Y.; Jiang, H.; Parisiadou, L.; Gius, D. Loss of NAD-Dependent Protein Deacetylase Sirtuin-2 Alters Mitochondrial Protein Acetylation and Dysregulates Mitophagy. Antioxid. Redox Signal. 2017, 26, 849–863. [Google Scholar] [CrossRef]
- Lemos, V.; de Oliveira, R.M.; Naia, L.; Szegö, É.; Ramos, E.; Pinho, S.; Magro, F.; Cavadas, C.; Rego, A.C.; Costa, V.; et al. The NAD+-dependent deacetylase SIRT2 attenuates oxidative stress and mitochondrial dysfunction and improves insulin sensitivity in hepatocytes. Hum. Mol. Genet. 2017, 26, 4105–4117. [Google Scholar] [CrossRef] [Green Version]
- Singh, P.; Hanson, P.S.; Morris, C.M. Sirtuin-2 Protects Neural Cells from Oxidative Stress and Is Elevated in Neurodegeneration. Parkinsons. Dis. 2017, 2017, 2643587. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.D.; Tan, E.K. Oxidized nicotinamide adenine dinucleotide-dependent mitochondrial deacetylase sirtuin-3 as a potential therapeutic target of Parkinson’s disease. Aging Res. Rev. 2020, 62, 101107. [Google Scholar] [CrossRef]
- Ansari, A.; Rahman, M.S.; Saha, S.K.; Saikot, F.K.; Deep, A.; Kim, K.-H. Function of the SIRT3 mitochondrial deacetylase in cellular physiology, cancer, and neurodegenerative disease. Aging Cell 2017, 16, 4–16. [Google Scholar] [CrossRef]
- Samant, S.A.; Zhang, H.J.; Hong, Z.; Pillai, V.B.; Sundaresan, N.R.; Wolfgeher, D.; Archer, S.L.; Chan, D.C.; Gupta, M.P. SIRT3 Deacetylates and Activates OPA1 To Regulate Mitochondrial Dynamics during Stress. Mol. Cell. Biol. 2014, 34, 807–819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobs, K.M.; Pennington, J.D.; Bisht, K.S.; Aykin-Burns, N.; Kim, H.-S.; Mishra, M.; Sun, L.; Nguyen, P.; Ahn, B.-H.; Leclerc, J.; et al. SIRT3 interacts with the daf-16 homolog FOXO3a in the Mitochondria, as well as increases FOXO3a Dependent Gene expression. Int. J. Biol. Sci. 2008, 291–299. [Google Scholar] [CrossRef] [Green Version]
- Someya, S.; Yu, W.; Hallows, W.C.; Xu, J.; Vann, J.M.; Leeuwenburgh, C.; Tanokura, M.; Denu, J.M.; Prolla, T.A. Sirt3 Mediates Reduction of Oxidative Damage and Prevention of Age-Related Hearing Loss under Caloric Restriction. Cell 2010, 143, 802–812. [Google Scholar] [CrossRef] [Green Version]
- Sundaresan, N.R.; Samant, S.A.; Pillai, V.B.; Rajamohan, S.B.; Gupta, M.P. SIRT3 Is a Stress-Responsive Deacetylase in Cardiomyocytes That Protects Cells from Stress-Mediated Cell Death by Deacetylation of Ku70. Mol. Cell. Biol. 2008, 28, 6384–6401. [Google Scholar] [CrossRef] [Green Version]
- Torrens-Mas, M.; Hernández-López, R.; Pons, D.-G.; Roca, P.; Oliver, J.; Sastre-Serra, J. Sirtuin 3 silencing impairs mitochondrial biogenesis and metabolism in colon cancer cells. Am. J. Physiol. Physiol. 2019, 317, C398–C404. [Google Scholar] [CrossRef]
- Cheng, Y.; Ren, X.; Gowda, A.S.; Shan, Y.; Zhang, L.; Yuan, Y.-S.; Patel, R.; Wu, H.; Huber-Keener, K.; Yang, J.W.; et al. Interaction of Sirt3 with OGG1 contributes to repair of mitochondrial DNA and protects from apoptotic cell death under oxidative stress. Cell Death Dis. 2013, 4, e731. [Google Scholar] [CrossRef] [Green Version]
- Gibellini, L.; Pinti, M.; Beretti, F.; Pierri, C.L.; Onofrio, A.; Riccio, M.; Carnevale, G.; De Biasi, S.; Nasi, M.; Torelli, F.; et al. Sirtuin 3 interacts with Lon protease and regulates its acetylation status. Mitochondrion 2014, 18, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Kunová, N.; Ondrovieòvá, G.; Bauer, J.A.; Bellová, J.; Ambro, L.; Martináková, L.; Kotrasová, V.; Kutejová, E.; Pevala, V. The role of Lon-mediated proteolysis in the dynamics of mitochondrial nucleic acid-protein complexes. Sci. Rep. 2017, 7, 631. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Kan, T.; Hu, X. Sirt3 regulates mitophagy level to promote diabetic corneal epithelial wound healing. Exp. Eye Res. 2019, 181, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Wei, T.; Huang, G.; Gao, J.; Huang, C.; Sun, M.; Wu, J.; Bu, J.; Shen, W. Sirtuin 3 Deficiency Accelerates Hypertensive Cardiac Remodeling by Impairing Angiogenesis. J. Am. Heart Assoc. 2017, 6. [Google Scholar] [CrossRef]
- Li, Y.; Ma, Y.; Song, L.; Yu, L.; Zhang, L.; Zhang, Y.; Xing, Y.; Yin, Y.; Ma, H. SIRT3 deficiency exacerbates p53/Parkin-mediated mitophagy inhibition and promotes mitochondrial dysfunction: Implication for aged hearts. Int. J. Mol. Med. 2018. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Deng, Y.-N.; Zhang, J.-Y.; Liu, J.; Li, Y.-B.; Su, H.; Qu, Q.-M. SIRT3 Protects Rotenone-induced Injury in SH-SY5Y Cells by Promoting Autophagy through the LKB1-AMPK-mTOR Pathway. Aging Dis. 2018, 9, 273. [Google Scholar] [CrossRef] [Green Version]
- Kaplan, L.D.; Wolfe, P.R.; Volberding, P.A.; Feorino, P.; Abrams, D.I.; Levy, J.A.; Wong, R.; Kaufman, L.; Gottlieb, M.S. Lack of response to suramin in patients with AIDS and AIDS-related complex. Am. J. Med. 1987, 82, 615–620. [Google Scholar] [CrossRef]
- Thakur, B.; Chandra, A.; Dittrich, T.; Welte, K.; Chandra, P. Inhibition of SIRT1 by HIV-1 viral protein Tat results in activation of p53 pathway. Biochem. Biophys. Res. Commun. 2012, 424, 245–250. [Google Scholar] [CrossRef]
- Hu, G.; Liao, K.; Yang, L.; Pendyala, G.; Kook, Y.; Fox, H.S.; Buch, S.; Neuroscience, E. Tat-mediated induction of miRs-34a & -138 promotes astrocytic activation via downregulation of SIRT1: Implications for aging in HAND. J. Neuroimmune Pharmacol. 2018, 12, 420–432. [Google Scholar] [CrossRef]
- Zhai, M.; Li, B.; Duan, W.; Jing, L.; Zhang, B.; Zhang, M.; Yu, L.; Liu, Z.; Yu, B.; Ren, K.; et al. Melatonin ameliorates myocardial ischemia reperfusion injury through SIRT3-dependent regulation of oxidative stress and apoptosis. J. Pineal Res. 2017, 63, e12419. [Google Scholar] [CrossRef]
- Ma, S.; Chen, J.; Feng, J.; Zhang, R.; Fan, M.; Han, D.; Li, X.; Li, C.; Ren, J.; Wang, Y.; et al. Melatonin Ameliorates the Progression of Atherosclerosis via Mitophagy Activation and NLRP3 Inflammasome Inhibition. Oxid. Med. Cell. Longev. 2018, 2018, 9286458. [Google Scholar] [CrossRef]
- Pillai, V.B.; Samant, S.; Sundaresan, N.R.; Raghuraman, H.; Kim, G.; Bonner, M.Y.; Arbiser, J.L.; Walker, D.I.; Jones, D.P.; Gius, D.; et al. Honokiol blocks and reverses cardiac hypertrophy in mice by activating mitochondrial Sirt3. Nat. Commun. 2015, 6, 6656. [Google Scholar] [CrossRef]
- Jeong, H.; Cohen, D.E.; Cui, L.; Supinski, A.; Savas, J.N.; Mazzulli, J.R.; Yates, J.R.; Bordone, L.; Guarente, L.; Krainc, D. Sirt1 mediates neuroprotection from mutant huntingtin by activation of the TORC1 and CREB transcriptional pathway. Nat. Med. 2012, 18, 159–165. [Google Scholar] [CrossRef]
- LU, L.; TANG, L.; WEI, W.; HONG, Y.; CHEN, H.; YING, W.; CHEN, S. Nicotinamide mononucleotide improves energy activity and survival rate in an in vitro model of Parkinson’s disease. Exp. Ther. Med. 2014, 8, 943–950. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Han, K.; Wang, C.; Sun, C.; Jia, N. Dioscin Protects against Aβ1–42 Oligomers-Induced Neurotoxicity via the Function of SIRT3 and Autophagy. Chem. Pharm. Bull. 2020, 68, 717–725. [Google Scholar] [CrossRef]
- Maugeri, A.; Barchitta, M.; Mazzone, M.; Giuliano, F.; Basile, G.; Agodi, A. Resveratrol Modulates SIRT1 and DNMT Functions and Restores LINE-1 Methylation Levels in ARPE-19 Cells under Oxidative Stress and Inflammation. Int. J. Mol. Sci. 2018, 19, 2118. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.-S.; Zhou, Y.; Wu, M.-R.; Zhou, H.-S.; Xu, F. Resveratrol inhibited Tat-induced HIV-1 LTR transactivation via NAD+-dependent SIRT1 activity. Life Sci. 2009, 85, 484–489. [Google Scholar] [CrossRef]
- Wang, R.; Wu, Z.; Bai, L.; Liu, R.; Ba, Y.; Zhang, H.; Cheng, X.; Zhou, G.; Huang, H. Resveratrol improved hippocampal neurogenesis following lead exposure in rats through activation of SIRT1 signaling. Environ. Toxicol. 2021, 36, 1664–1673. [Google Scholar] [CrossRef]
- Sun, Q.; Kang, R.; Chen, K.; Liu, K.; Ma, Z.; Liu, C.; Deng, Y.; Liu, W.; Xu, B. Sirtuin 3 is required for the protective effect of Resveratrol on Manganese-induced disruption of mitochondrial biogenesis in primary cultured neurons. J. Neurochem. 2021, 156, 121–135. [Google Scholar] [CrossRef]
- Hosoda, R.; Hamada, H.; Uesugi, D.; Iwahara, N.; Nojima, I.; Horio, Y.; Kuno, A. Different Antioxidative and Antiapoptotic Effects of Piceatannol and Resveratrol. J. Pharmacol. Exp. Ther. 2021, 376, 385–396. [Google Scholar] [CrossRef]
- Mahmoud Moustafa, E.; Rashed, E.R.; Rashed, R.R.; Omar, N.N. Piceatannol promotes hepatic and renal AMPK/SIRT1/PGC-1α mitochondrial pathway in rats exposed to reserpine or gamma-radiation. Int. J. Immunopathol. Pharmacol. 2021, 35, 205873842110161. [Google Scholar] [CrossRef]
- Wang, K.; Zhang, W.; Liu, J.; Cui, Y.; Cui, J. Piceatannol protects against cerebral ischemia/reperfusion-induced apoptosis and oxidative stress via the Sirt1/FoxO1 signaling pathway. Mol. Med. Rep. 2020, 22, 5399–5411. [Google Scholar] [CrossRef]
- Zhang, M.; Zhao, Z.; Shen, M.; Zhang, Y.; Duan, J.; Guo, Y.; Zhang, D.; Hu, J.; Lin, J.; Man, W.; et al. Polydatin protects cardiomyocytes against myocardial infarction injury by activating Sirt3. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1962–1972. [Google Scholar] [CrossRef]
- Zhang, H.; Chen, Y.; Pei, Z.; Gao, H.; Shi, W.; Sun, M.; Xu, Q.; Zhao, J.; Meng, W.; Xiao, K. Protective effects of polydatin against sulfur mustard-induced hepatic injury. Toxicol. Appl. Pharmacol. 2019, 367, 1–11. [Google Scholar] [CrossRef]
- Ye, H.; Meng, Y. Honokiol regulates endoplasmic reticulum stress by promoting the activation of the sirtuin 1-mediated protein kinase B pathway and ameliorates high glucose/high fat-induced dysfunction in human umbilical vein endothelial cells. Endocr. J. 2021, 68, EJ20-0747. [Google Scholar] [CrossRef]
- Zhang, B.; Zhai, M.; Li, B.; Liu, Z.; Li, K.; Jiang, L.; Zhang, M.; Yi, W.; Yang, J.; Yi, D.; et al. Honokiol Ameliorates Myocardial Ischemia/Reperfusion Injury in Type 1 Diabetic Rats by Reducing Oxidative Stress and Apoptosis through Activating the SIRT1-Nrf2 Signaling Pathway. Oxid. Med. Cell. Longev. 2018, 2018, 3159801. [Google Scholar] [CrossRef]
- Song, S.; Chu, L.; Liang, H.; Chen, J.; Liang, J.; Huang, Z.; Zhang, B.; Chen, X. Protective Effects of Dioscin Against Doxorubicin-Induced Hepatotoxicity Via Regulation of Sirt1/FOXO1/NF-κb Signal. Front. Pharmacol. 2019, 10. [Google Scholar] [CrossRef]
- Zhang, X.; Lu, Y.; Li, W.; Tao, T.; Wang, W.; Gao, S.; Zhou, Y.; Guo, Y.; Liu, C.; Zhuang, Z.; et al. Cerebroprotection by dioscin after experimental subarachnoid haemorrhage via inhibiting NLRP3 inflammasome through SIRT1-dependent pathway. Br. J. Pharmacol. 2021, 178, 3648–3666. [Google Scholar] [CrossRef]
- Zhang, J.; Meruvu, S.; Bedi, Y.S.; Chau, J.; Arguelles, A.; Rucker, R.; Choudhury, M. Pyrroloquinoline quinone increases the expression and activity of Sirt1 and -3 genes in HepG2 cells. Nutr. Res. 2015, 35, 844–849. [Google Scholar] [CrossRef] [Green Version]
- Pi, H.; Xu, S.; Reiter, R.J.; Guo, P.; Zhang, L.; Li, Y.; Li, M.; Cao, Z.; Tian, L.; Xie, J.; et al. SIRT3-SOD2-mROS-dependent autophagy in cadmium-induced hepatotoxicity and salvage by melatonin. Autophagy 2015, 11, 1037–1051. [Google Scholar] [CrossRef] [Green Version]
- Arioz, B.I.; Tastan, B.; Tarakcioglu, E.; Tufekci, K.U.; Olcum, M.; Ersoy, N.; Bagriyanik, A.; Genc, K.; Genc, S. Melatonin Attenuates LPS-Induced Acute Depressive-Like Behaviors and Microglial NLRP3 Inflammasome Activation Through the SIRT1/Nrf2 Pathway. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Carloni, S.; Albertini, M.C.; Galluzzi, L.; Buonocore, G.; Proietti, F.; Balduini, W. Melatonin reduces endoplasmic reticulum stress and preserves sirtuin 1 expression in neuronal cells of newborn rats after hypoxia-ischemia. J. Pineal Res. 2014, 57, 192–199. [Google Scholar] [CrossRef] [PubMed]
- Merlo, S.; Luaces, J.P.; Spampinato, S.F.; Toro-Urrego, N.; Caruso, G.I.; D’Amico, F.; Capani, F.; Sortino, M.A. SIRT1 Mediates Melatonin’s Effects on Microglial Activation in Hypoxia: In Vitro and In Vivo Evidence. Biomolecules 2020, 10, 364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niu, Y.; Zhou, W.; Nie, Z.; Shin, K.; Cui, X. Melatonin enhances mitochondrial biogenesis and protects against rotenone-induced mitochondrial deficiency in early porcine embryos. J. Pineal Res. 2020, 68, ee12627. [Google Scholar] [CrossRef]
- Wang, J.; Wang, K.; Huang, C.; Lin, D.; Zhou, Y.; Wu, Y.; Tian, N.; Fan, P.; Pan, X.; Xu, D.; et al. SIRT3 Activation by Dihydromyricetin Suppresses Chondrocytes Degeneration via Maintaining Mitochondrial Homeostasis. Int. J. Biol. Sci. 2018, 14, 1873–1882. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.; Ban, J.-J.; Chung, J.-Y.; Im, W.; Kim, M. Amelioration of Huntington’s disease phenotypes by Beta-Lapachone is associated with increases in Sirt1 expression, CREB phosphorylation and PGC-1α deacetylation. PLoS ONE 2018, 13, e0195968. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Ye, Z.; Lai, W.; Rao, J.; Huang, W.; Zhang, X.; Yao, Z.; Lou, T. Activation of Sirtuin 3 by Silybin Attenuates Mitochondrial Dysfunction in Cisplatin-induced Acute Kidney Injury. Front. Pharmacol. 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- Diaz-Morales, N.; Rovira-Llopis, S.; Bañuls, C.; Lopez-Domenech, S.; Escribano-Lopez, I.; Veses, S.; Jover, A.; Rocha, M.; Hernandez-Mijares, A.; Victor, V.M. Does Metformin Protect Diabetic Patients from Oxidative Stress and Leukocyte-Endothelium Interactions? Antioxid. Redox Signal. 2017, 27, 1439–1445. [Google Scholar] [CrossRef]
- Kitaoka, Y.; Sase, K.; Tsukahara, C.; Fujita, N.; Tokuda, N.; Kogo, J.; Takagi, H. Axonal protection by a small molecule SIRT1 activator, SRT2104, with alteration of autophagy in TNF-induced optic nerve degeneration. Jpn. J. Ophthalmol. 2020, 64, 298–303. [Google Scholar] [CrossRef]
- Kalvala, A.K.; Yerra, V.G.; Kumar, A. LONP1 induction by SRT1720 attenuates mitochondrial dysfunction against high glucose induced neurotoxicity in PC12 cells. Toxicol. In Vitro 2020, 62, 104695. [Google Scholar] [CrossRef]
- Li, X.; Park, S.J.; Jin, F.; Deng, Y.; Yang, J.H.; Chang, J.-H.; Kim, D.-Y.; Kim, J.-A.; Lee, Y.J.; Murakami, M.; et al. Tanshinone IIA suppresses FcεRI-mediated mast cell signaling and anaphylaxis by activation of the Sirt1/LKB1/AMPK pathway. Biochem. Pharmacol. 2018, 152, 362–372. [Google Scholar] [CrossRef]
- Zhong, J.; Ouyang, H.; Sun, M.; Lu, J.; Zhong, Y.; Tan, Y.; Hu, Y. Tanshinone IIA attenuates cardiac microvascular ischemia-reperfusion injury via regulating the SIRT1-PGC1α-mitochondrial apoptosis pathway. Cell Stress Chaperones 2019, 24, 991–1003. [Google Scholar] [CrossRef]
- Zhang, H.-S.; Chen, X.-Y.; Wu, T.-C.; Zhang, F.-J. Tanshinone II A Inhibits Tat-Induced HIV-1 Transactivation Through Redox-Regulated AMPK/Nampt Pathway. J. Cell. Physiol. 2014, 229, 1193–1201. [Google Scholar] [CrossRef]
- Howitz, K.T.; Bitterman, K.J.; Cohen, H.Y.; Lamming, D.W.; Lavu, S.; Wood, J.G.; Zipkin, R.E.; Chung, P.; Kisielewski, A.; Zhang, L.-L.; et al. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature 2003, 425, 191–196. [Google Scholar] [CrossRef]
- Jia, P.; Ji, S.; Zhang, H.; Chen, Y.; Wang, T. Piceatannol Ameliorates Hepatic Oxidative Damage and Mitochondrial Dysfunction of Weaned Piglets Challenged with Diquat. Animals 2020, 10, 1239. [Google Scholar] [CrossRef]
- Wu, J.; Deng, Z.; Sun, M.; Zhang, W.; Yang, Y.; Zeng, Z.; Wu, J.; Zhang, Q.; Liu, Y.; Chen, Z.; et al. Polydatin protects against lipopolysaccharide-induced endothelial barrier disruption via SIRT3 activation. Lab. Investig. 2020, 100, 643–656. [Google Scholar] [CrossRef]
- Woodbury, A.; Yu, S.P.; Wei, L.; García, P. Neuro-Modulating Effects of Honokiol: A Review. Front. Neurol. 2013, 4. [Google Scholar] [CrossRef] [Green Version]
- Giralt, A.; Hondares, E.; Villena, J.A.; Ribas, F.; Díaz-Delfín, J.; Giralt, M.; Iglesias, R.; Villarroya, F. Peroxisome Proliferator-activated Receptor-γ Coactivator-1α Controls Transcription of the Sirt3 Gene, an Essential Component of the Thermogenic Brown Adipocyte Phenotype. J. Biol. Chem. 2011, 286, 16958–16966. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Zhou, J.; Shen, M.; Xu, H.; Yu, S.; Cheng, Q.; Ding, F. Pyrroloquinoline Quinone Inhibits Rotenone-Induced Microglia Inflammation by Enhancing Autophagy. Molecules 2020, 25, 4359. [Google Scholar] [CrossRef]
- Zhang, J.; Xiang, H.; Liu, J.; Chen, Y.; He, R.-R.; Liu, B. Mitochondrial Sirtuin 3: New emerging biological function and therapeutic target. Theranostics 2020, 10, 8315–8342. [Google Scholar] [CrossRef]
- Han, L.; Li, J.; Li, J.; Pan, C.; Xiao, Y.; Lan, X.; Wang, M. Activation of AMPK/Sirt3 pathway by phloretin reduces mitochondrial ROS in vascular endothelium by increasing the activity of MnSOD via deacetylation. Food Funct. 2020, 11, 3073–3083. [Google Scholar] [CrossRef]
- Fu, C.-Y.; Zhong, C.-R.; Yang, Y.-T.; Zhang, M.; Li, W.-A.; Zhou, Q.; Zhang, F. Sirt1 activator SRT2104 protects against oxygen-glucose deprivation/reoxygenation-induced injury via regulating microglia polarization by modulating Sirt1/NF-κB pathway. Brain Res. 2021, 1753, 147236. [Google Scholar] [CrossRef]
- Katsyuba, E.; Romani, M.; Hofer, D.; Auwerx, J. NAD + homeostasis in health and disease. Nat. Metab. 2019, 2, 9–31. [Google Scholar] [CrossRef]
- Yao, Z.; Yang, W.; Gao, Z.; Jia, P. Nicotinamide mononucleotide inhibits JNK activation to reverse Alzheimer disease. Neurosci. Lett. 2017, 647, 133–140. [Google Scholar] [CrossRef]
- Gong, B.; Pan, Y.; Vempati, P.; Zhao, W.; Knable, L.; Ho, L.; Wang, J.; Sastre, M.; Ono, K.; Sauve, A.A.; et al. Nicotinamide riboside restores cognition through an upregulation of proliferator-activated receptor-γ coactivator 1α regulated β-secretase 1 degradation and mitochondrial gene expression in Alzheimer’s mouse models. Neurobiol. Aging 2013, 34, 1581–1588. [Google Scholar] [CrossRef] [Green Version]
- Murray, M.F.; Nghiem, M.; Srinivasan, A. HIV Infection Decreases Intracellular Nicotinamide Adenine Dinucleotide [NAD]. Biochem. Biophys. Res. Commun. 1995, 212, 126–131. [Google Scholar] [CrossRef]
- Chen, X.-Y.; Zhang, H.-S.; Wu, T.-C.; Sang, W.-W.; Ruan, Z. Down-regulation of NAMPT expression by miR-182 is involved in Tat-induced HIV-1 long terminal repeat (LTR) transactivation. Int. J. Biochem. Cell Biol. 2013, 45, 292–298. [Google Scholar] [CrossRef]
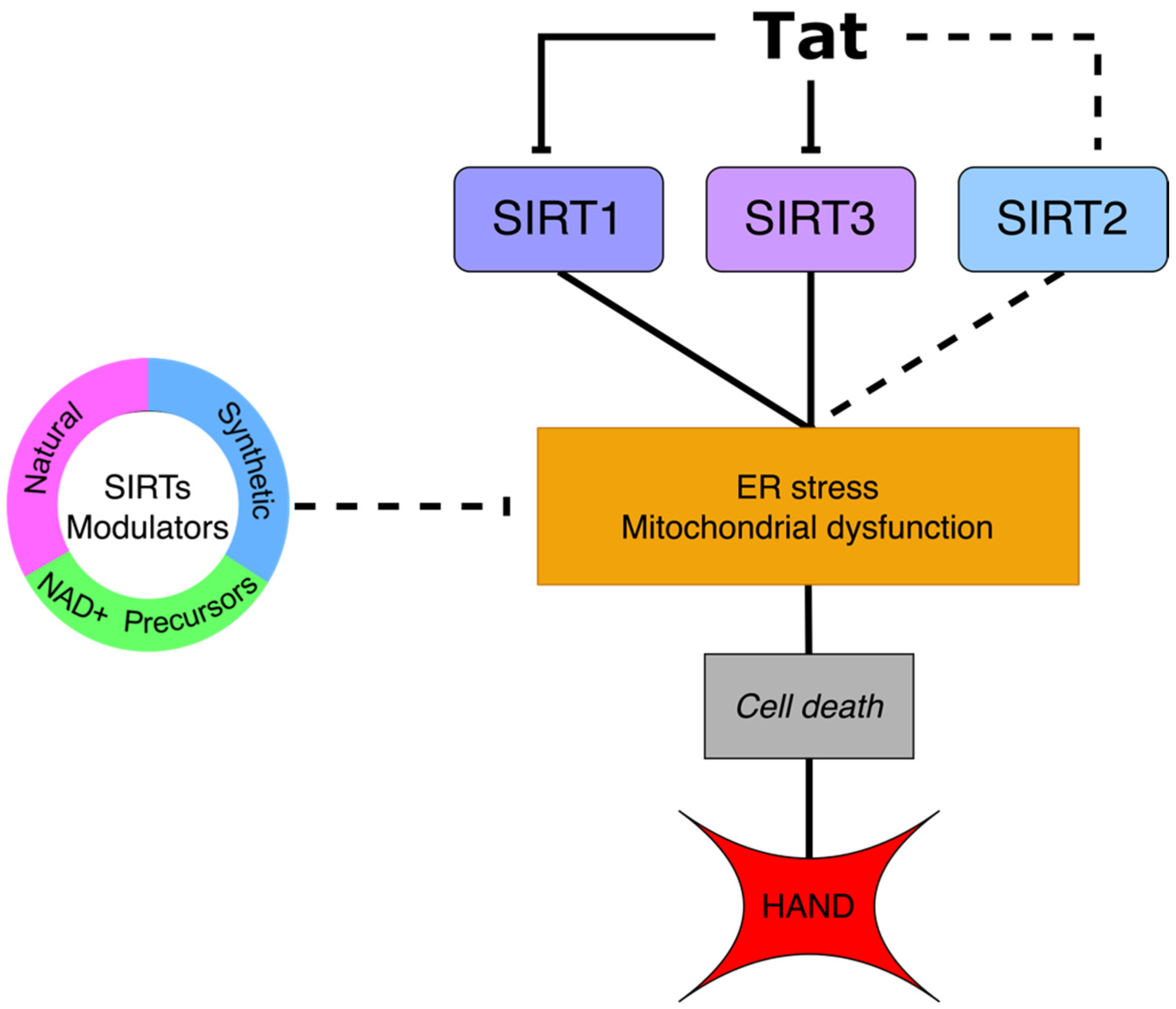

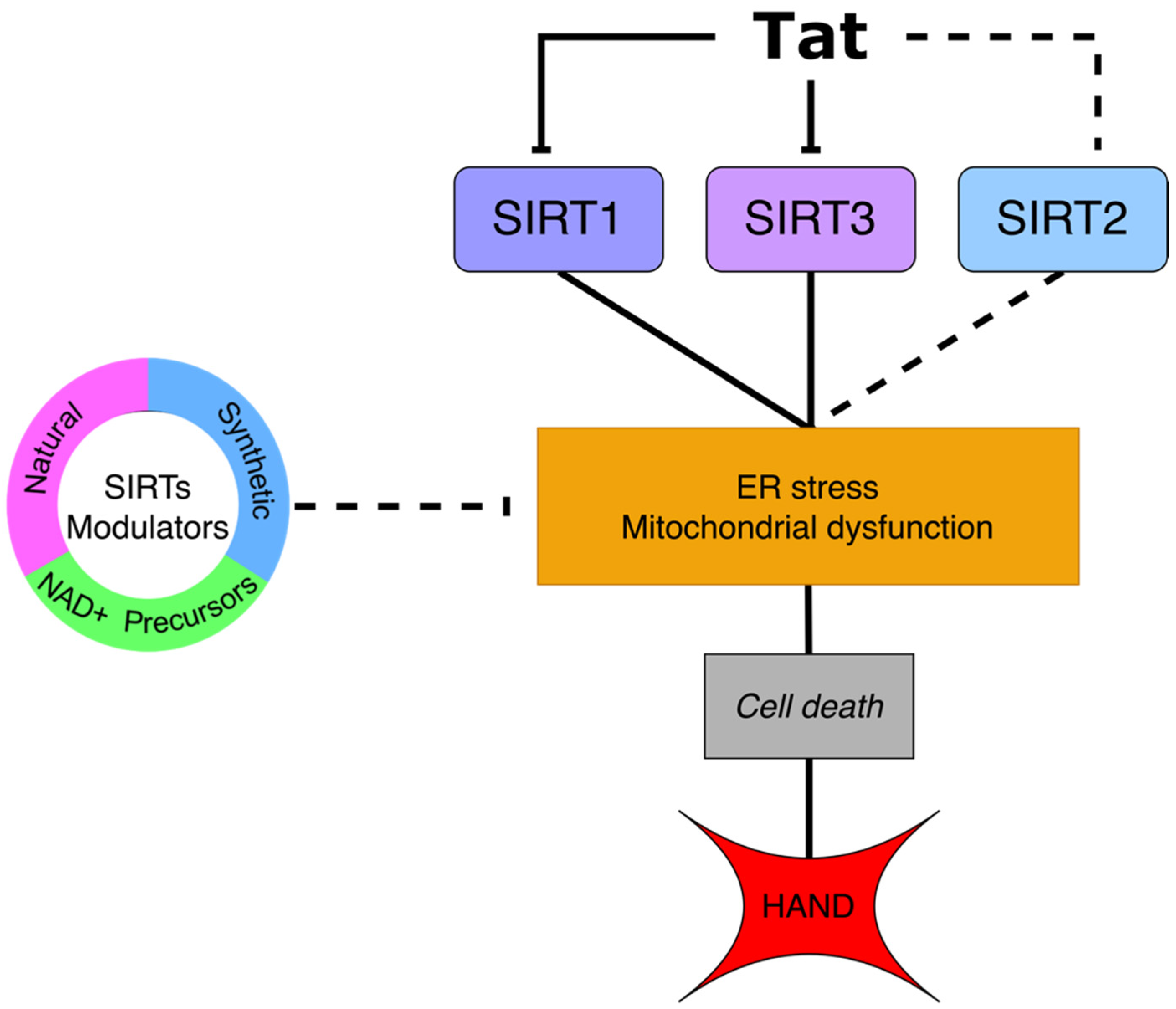{kind=link}
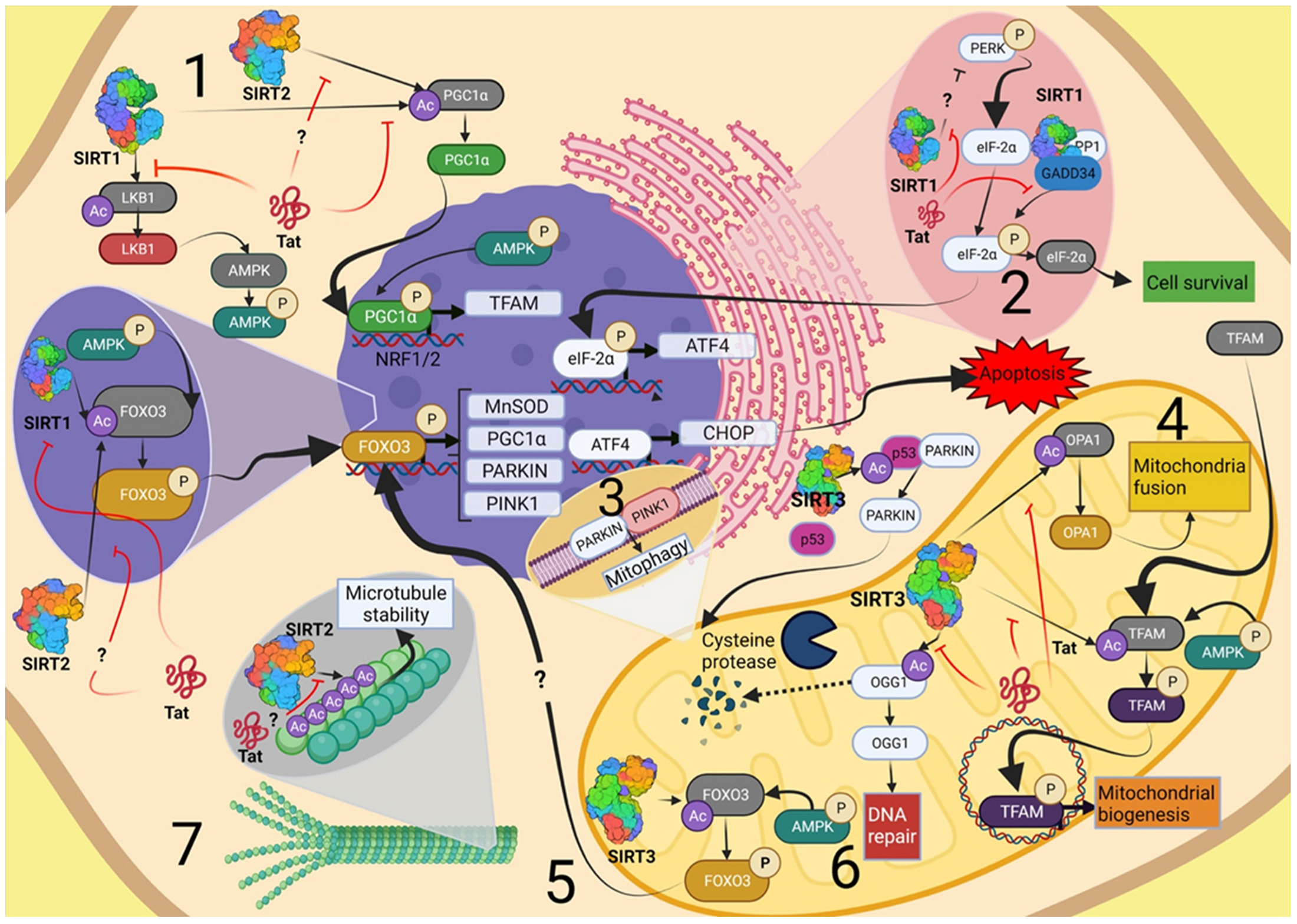
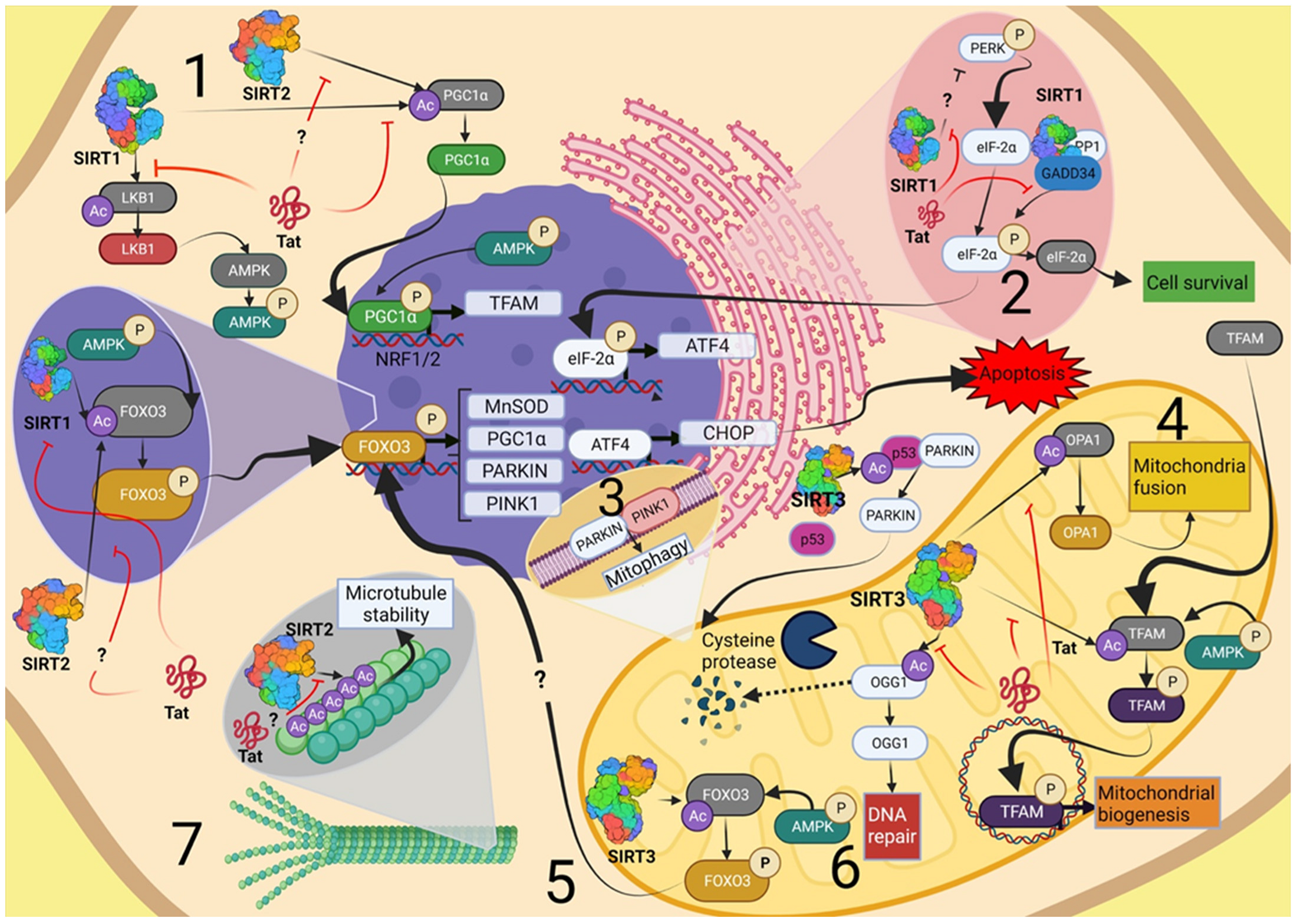{kind=link}
| Compound | Type | Modulation | Sirtuin | Effect | Stressor | Reference |
|---|---|---|---|---|---|---|
| Resveratrol | Natural polyphenol | Expression/ Activation | SIRT1 SIRT3 | Increase: Mitophagy Mitochondria biogenesis Cell proliferation Decrease: Oxidative stress Apoptosis | LPS/GOx | [139] |
| HIV-Tat | [140] | |||||
| Lead (Pb) | [141] | |||||
| Manganese (Mn) | [142] | |||||
| I/R | [106] | |||||
| piceatannol | Natural Polyphenol /Resveratrol analog | Expression/ Activation | SIRT1 | Increase: Mitochondrial biogenesis Decrease: Oxidative stress Apoptosis Inflammation | Antimycin A H2O2 | [143] |
| γ-irradiation | [144] | |||||
| I/R | [145] | |||||
| Polydatin | Natural Polyphenol | Activation | SIRT3 SIRT1 | Increase: Cell viability Antioxidant activity Autophagy Decrease: Oxidative stressApoptosis | Myocardial infarction | [146] |
| Sulfur mustard | [147] | |||||
| Honokiol | Natural polyphenol | Expression/ Activation | SIRT1 SIRT3 | Increase: Mitochondria functions Decrease: Oxidative stress Apoptosis | Cardiac hypertrophy | [135] |
| High glucose/fat diet | [148] | |||||
| I/R | [149] | |||||
| Diosin | Natural saponin | Expression | SIRT1 SIRT3 | Increase: Autophagy Decrease: Oxidative stress Apoptosis | Doxorubicin | [150] |
| Oxyhemoglobin | [151] | |||||
| Aβ | [138] | |||||
| Pyrroloquinoline quinone | Natural quinone | Expression/ Activation | SIRT1 SIRT3 | Increase: Mitochondrial biogenesis NAD+ | Wt | [152] |
| Melatonin | Hormone | Expression/ Activation | SIRT1 SIRT3 | Increase: Antioxidant activity Mitochondria biogenesis Decrease: Oxidative stress Inflammation Apoptosis ER stress | ATH/Ox-LDL | [134] |
| I/R | [133] | |||||
| Cadmium (Cd) | [153] | |||||
| LPS | [154] | |||||
| C/HI | [155] | |||||
| CoCl2 | [156] | |||||
| Rotenone | [157] | |||||
| Dihydromyricetin | Natural | Expression | SIRT3 | Increase: Antioxidant activity Mitochondria dynamics Mitochondria biogenesis | TNF-α | [158] |
| β-lapachonequinone | Natural quinone | Expression | SIRT1 | Increase: Activation of PGC1α Decrease: Oxidative stress | HD | [159] |
| Silybin | Natural flavonoid | Expression | SIRT3 | Increase: Mitochondria function Decrease: Oxidative stress Apoptosis | Cisplatin | [160] |
| Metformin | Synthetic drug | Expression | SIRT3 | Increase: Antioxidant activity Decrease: Oxidative stress | Type 2 diabetes | [161] |
| SRT2104 | Synthetic drug | Expression/ Activation | SIRT1 | Increase: Autophagy | TNF-α | [162] |
| SRT1720 | Synthetic drug | Activation | SIRT1 | Increase: Mitochondria biogenesis Antioxidant activity | High glucose | [163] |
| Tanshinone IIA | natural | Activation | SRT1 | Increase: Energy production Mitochondria biogenesis Antioxidant activity NAD+ Decrease: Oxidative stress Mitochondria dysfunction Apoptosis Inflammation Ca2+ release HIV-1 transactivation | DNP | [164] |
| HR | [165] | |||||
| Rotenone | [164] | |||||
| HIV-Tat | [166] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Figarola-Centurión, I.; Escoto-Delgadillo, M.; González-Enríquez, G.V.; Gutiérrez-Sevilla, J.E.; Vázquez-Valls, E.; Torres-Mendoza, B.M. Sirtuins Modulation: A Promising Strategy for HIV-Associated Neurocognitive Impairments. Int. J. Mol. Sci. 2022, 23, 643. https://doi.org/10.3390/ijms23020643
Figarola-Centurión I, Escoto-Delgadillo M, González-Enríquez GV, Gutiérrez-Sevilla JE, Vázquez-Valls E, Torres-Mendoza BM. Sirtuins Modulation: A Promising Strategy for HIV-Associated Neurocognitive Impairments. International Journal of Molecular Sciences. 2022; 23(2):643. https://doi.org/10.3390/ijms23020643
Chicago/Turabian StyleFigarola-Centurión, Izchel, Martha Escoto-Delgadillo, Gracia Viviana González-Enríquez, Juan Ernesto Gutiérrez-Sevilla, Eduardo Vázquez-Valls, and Blanca Miriam Torres-Mendoza. 2022. "Sirtuins Modulation: A Promising Strategy for HIV-Associated Neurocognitive Impairments" International Journal of Molecular Sciences 23, no. 2: 643. https://doi.org/10.3390/ijms23020643
APA StyleFigarola-Centurión, I., Escoto-Delgadillo, M., González-Enríquez, G. V., Gutiérrez-Sevilla, J. E., Vázquez-Valls, E., & Torres-Mendoza, B. M. (2022). Sirtuins Modulation: A Promising Strategy for HIV-Associated Neurocognitive Impairments. International Journal of Molecular Sciences, 23(2), 643. https://doi.org/10.3390/ijms23020643