
Role of Endocrine-Disrupting Chemicals in the Pathogenesis of Non-Alcoholic Fatty Liver Disease: A Comprehensive Review

 , , , , , and
, , , , , and
Abstract
1. Introduction
2. Endocrine-Disrupting Chemicals: An Overview
3. Endocrine-Disrupting Chemicals Exposure and Liver
4. Mechanisms of Action of Endocrine-Disrupting Chemicals in NAFLD
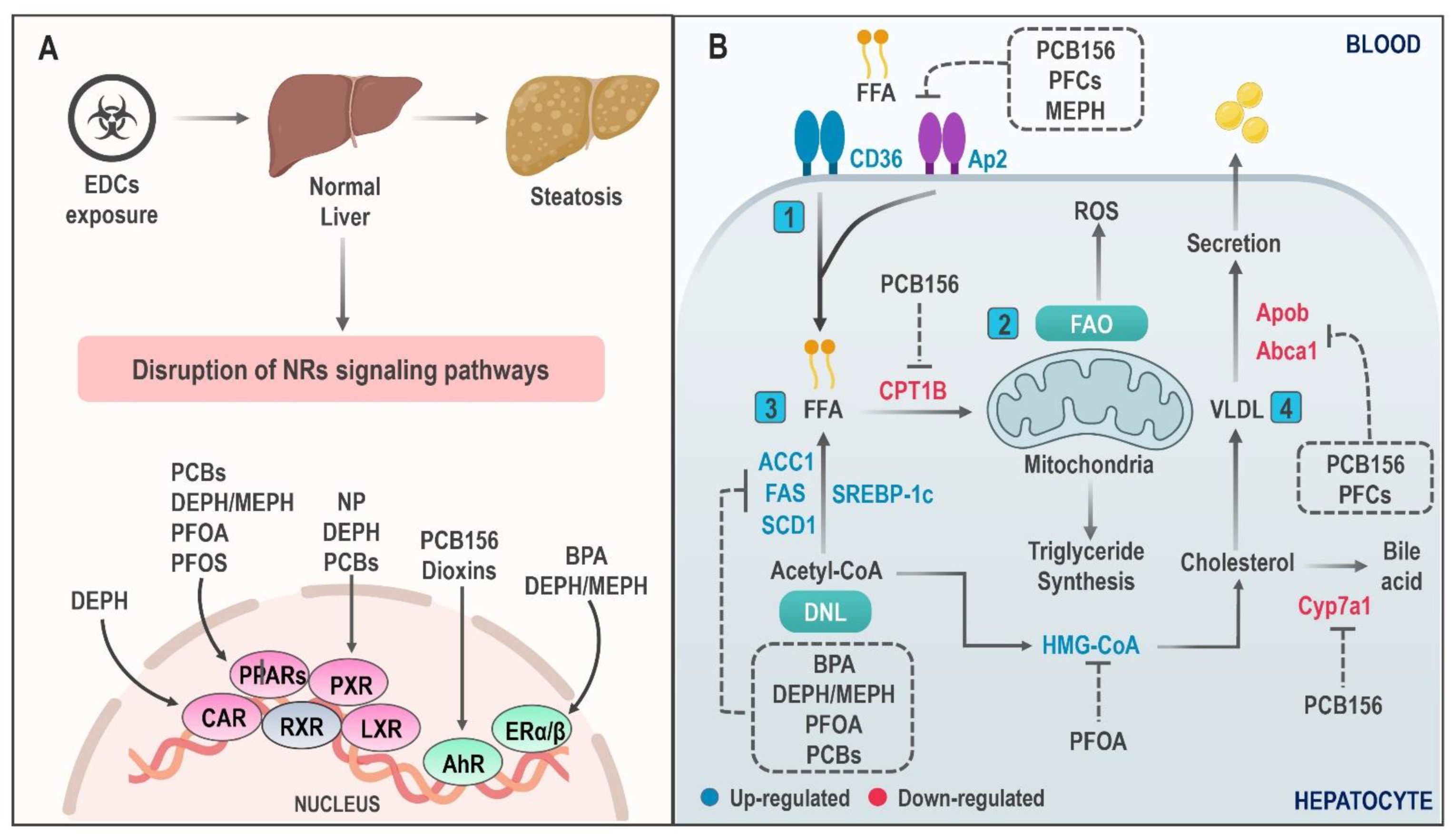
4.1. Hepatic Lipid Accumulation
4.2. Mitochondrial Dysfunction
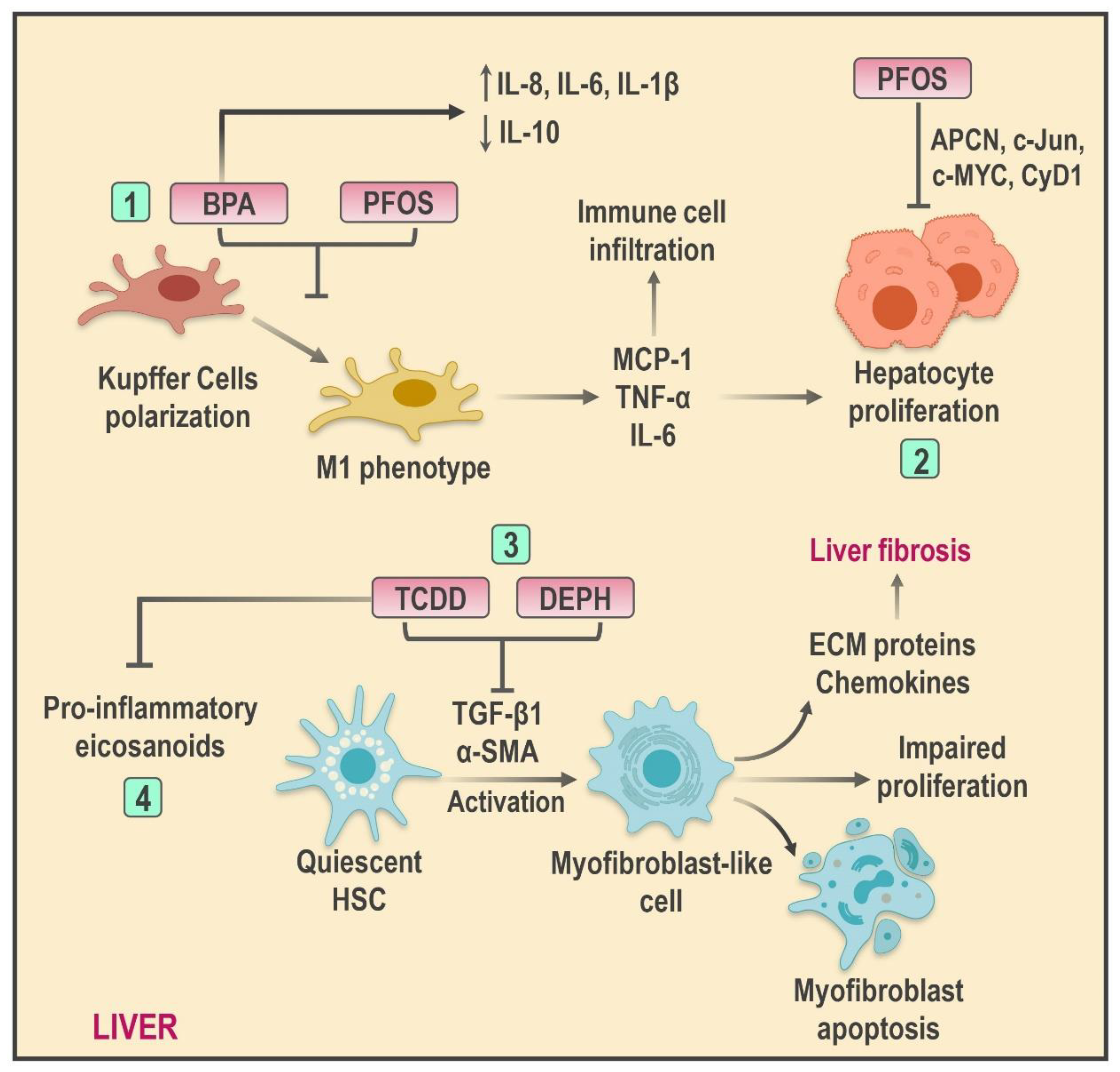
4.3. Mechanisms of Hepatic Inflammation Mediated by EDCs
4.4. Epigenetic Changes and Transgenerational Inheritance
5. Endocrine-Disrupting Chemicals and NAFLD: Clinical Evidence
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The Diagnosis and Management of Nonalcoholic Fatty Liver Disease: Practice Guidance from the American Association for the Study of Liver Diseases. Hepatology 2018, 67, 328–357. [Google Scholar] [CrossRef]
- Amini, M.; Ansari, I.; Yekesadat, S.; Vaseie, M.; Malekhoseyni, M. Response rate to the vaccination with hepatitis b vaccine among cardiovascular health staff in Tehran. Latinoam. Hipertens. 2020, 14, 562–567. [Google Scholar]
- Younossi, Z.; Tacke, F.; Arrese, M.; Chander Sharma, B.; Mostafa, I.; Bugianesi, E.; Wai-Sun Wong, V.; Yilmaz, Y.; George, J.; Fan, J.; et al. Global Perspectives on Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis: Hepatology. Hepatology 2019, 69, 2672–2682. [Google Scholar] [CrossRef] [PubMed]
- Bermudez, V.; Moisés, R.; Lisney, V.; Yettana, L.; Ana, C.; ali, U.; Guerra-Torres, X.; Colmenares, C.A.; Chacín, M.; Rojas Quintero, J.; et al. Pharmacologic Treatment of Obesity: Pitfalls and New Promises. Rev. Latinoam. Hipertens. 2008, 5, 137–147. [Google Scholar]
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global Burden of NAFLD and NASH: Trends, Predictions, Risk Factors and Prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global Epidemiology of Nonalcoholic Fatty Liver Disease-Meta-Analytic Assessment of Prevalence, Incidence, and Outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.-L.; Chen, H.; Wang, C.-L.; Liang, L. Pathogenesis of Non-Alcoholic Fatty Liver Disease in Children and Adolescence: From “Two Hit Theory” to “Multiple Hit Model”. World J. Gastroenterol. 2018, 24, 2974–2983. [Google Scholar] [CrossRef]
- Carr, R.M.; Oranu, A.; Khungar, V. Nonalcoholic Fatty Liver Disease. Gastroenterol. Clin. N. Am. 2016, 45, 639–652. [Google Scholar] [CrossRef]
- Souki-Rincón, A.; Sandoval, M.; Sánchez, G.; Andrade, U.; García-Rondón, D.; Cano-Ponce, C.; Medina, M.; Almarza, J.; Urdaneta, Y.; González, C. Intake of saturated fatty acids and insulin sensitivity in obese young adults from Maracaibo. Rev. Latinoam. Hipertens. 2008, 3, 159–165. [Google Scholar]
- Petta, S.; Gastaldelli, A.; Rebelos, E.; Bugianesi, E.; Messa, P.; Miele, L.; Svegliati-Baroni, G.; Valenti, L.; Bonino, F. Pathophysiology of Non Alcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2016, 17, 2082. [Google Scholar] [CrossRef]
- Masarone, M.; Rosato, V.; Dallio, M.; Gravina, A.G.; Aglitti, A.; Loguercio, C.; Federico, A.; Persico, M. Role of Oxidative Stress in Pathophysiology of Nonalcoholic Fatty Liver Disease. Oxid. Med. Cell. Longev. 2018, 2018, 1–14. [Google Scholar] [CrossRef]
- Arciello, M.; Gori, M.; Maggio, R.; Barbaro, B.; Tarocchi, M.; Galli, A.; Balsano, C. Environmental Pollution: A Tangible Risk for NAFLD Pathogenesis. Int. J. Mol. Sci. 2013, 14, 22052–22066. [Google Scholar] [CrossRef]
- Papalou, O.; Kandaraki, E.A.; Papadakis, G.; Diamanti-Kandarakis, E. Endocrine Disrupting Chemicals: An Occult Mediator of Metabolic Disease. Front. Endocrinol. 2019, 10, 112. [Google Scholar] [CrossRef]
- Bergman, Å.; Heindel, J.J.; Kasten, T.; Kidd, K.A.; Jobling, S.; Neira, M.; Zoeller, R.T.; Becher, G.; Bjerregaard, P.; Bornman, R.; et al. The Impact of Endocrine Disruption: A Consensus Statement on the State of the Science. Environ. Health Perspect. 2013, 121, A104–A106. [Google Scholar] [CrossRef]
- VoPham, T. Environmental Risk Factors for Liver Cancer and Nonalcoholic Fatty Liver Disease. Curr. Epidemiol. Rep. 2019, 6, 50–66. [Google Scholar] [CrossRef] [PubMed]
- Klaunig, J.E.; Li, X.; Wang, Z. Role of Xenobiotics in the Induction and Progression of Fatty Liver Disease. Toxicol. Res. 2018, 7, 664–680. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Sun, X.; Chen, Y.; Li, Y.; Song, L.; Zhou, Z.; Xu, B.; Lin, Y.; Xu, S. Perinatal Exposure to Bisphenol A Exacerbates Nonalcoholic Steatohepatitis-like Phenotype in Male Rat Offspring Fed on a High-Fat Diet. J. Endocrinol. 2014, 222, 313–325. [Google Scholar] [CrossRef] [PubMed]
- Marty, M.S.; Carney, E.W.; Rowlands, J.C. Endocrine Disruption: Historical Perspectives and Its Impact on the Future of Toxicology Testing. Toxicol. Sci. Off. J. Soc. Toxicol. 2011, 120 (Suppl. 1), S93–S108. [Google Scholar] [CrossRef] [PubMed]
- Darbre, P.D. The History of Endocrine-Disrupting Chemicals. Curr. Opin. Endocr. Metab. Res. 2019, 7, 26–33. [Google Scholar] [CrossRef]
- Lee, D.H. Evidence of the Possible Harm of Endocrine-Disrupting Chemicals in Humans: Ongoing Debates and Key Issues. Endocrinol. Metab. Seoul Korea 2018, 33, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Committee, E.S. Scientific Opinion on the Hazard Assessment of Endocrine Disruptors: Scientific Criteria for Identification of Endocrine Disruptors and Appropriateness of Existing Test Methods for Assessing Effects Mediated by These Substances on Human Health and the Environment. EFSA J. 2013, 11, 3132. [Google Scholar] [CrossRef]
- Slama, R.; Bourguignon, J.-P.; Demeneix, B.; Ivell, R.; Panzica, G.; Kortenkamp, A.; Zoeller, R.T. Scientific Issues Relevant to Setting Regulatory Criteria to Identify Endocrine-Disrupting Substances in the European Union. Environ. Health Perspect. 2016, 124, 1497–1503. [Google Scholar] [CrossRef]
- Zoeller, R.T.; Brown, T.R.; Doan, L.L.; Gore, A.C.; Skakkebaek, N.E.; Soto, A.M.; Woodruff, T.J.; Vom Saal, F.S. Endocrine-Disrupting Chemicals and Public Health Protection: A Statement of Principles from The Endocrine Society. Endocrinology 2012, 153, 4097–4110. [Google Scholar] [CrossRef] [PubMed]
- Gore, A.C.; Chappell, V.A.; Fenton, S.E.; Flaws, J.A.; Nadal, A.; Prins, G.S.; Toppari, J.; Zoeller, R.T. Executive Summary to EDC-2: The Endocrine Society’s Second Scientific Statement on Endocrine-Disrupting Chemicals. Endocr. Rev. 2015, 36, 593–602. [Google Scholar] [CrossRef]
- Yang, O.; Kim, H.L.; Weon, J.-I.; Seo, Y.R. Endocrine-Disrupting Chemicals: Review of Toxicological Mechanisms Using Molecular Pathway Analysis. J. Cancer Prev. 2015, 20, 12–24. [Google Scholar] [CrossRef] [PubMed]
- Frye, C.; Bo, E.; Calamandrei, G.; Calzà, L.; Dessì-Fulgheri, F.; Fernández, M.; Fusani, L.; Kah, O.; Kajta, M.; Le Page, Y.; et al. Endocrine Disrupters: A Review of Some Sources, Effects, and Mechanisms of Actions on Behaviour and Neuroendocrine Systems: EDCs-Sources, Effects & Mechanisms. J. Neuroendocrinol. 2012, 24, 144–159. [Google Scholar] [CrossRef]
- Barouki, R. Endocrine Disruptors: Revisiting Concepts and Dogma in Toxicology. Comptes Rendus Biol. 2017, 340, 410–413. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, B.; Terekeci, H.; Sandal, S.; Kelestimur, F. Endocrine Disrupting Chemicals: Exposure, Effects on Human Health, Mechanism of Action, Models for Testing and Strategies for Prevention. Rev. Endocr. Metab. Disord. 2020, 21, 127–147. [Google Scholar] [CrossRef]
- Diamanti-Kandarakis, E.; Palioura, E.; Kandarakis, S.A.; Koutsilieris, M. The Impact of Endocrine Disruptors on Endocrine Targets. Horm. Metab. Res. 2010, 42, 543–552. [Google Scholar] [CrossRef] [PubMed]
- Sargis, R.M.; Heindel, J.J.; Padmanabhan, V. Interventions to Address Environmental Metabolism-Disrupting Chemicals: Changing the Narrative to Empower Action to Restore Metabolic Health. Front. Endocrinol. 2019, 10, 33. [Google Scholar] [CrossRef] [PubMed]
- Kabir, E.R.; Rahman, M.S.; Rahman, I. A Review on Endocrine Disruptors and Their Possible Impacts on Human Health. Environ. Toxicol. Pharmacol. 2015, 40, 241–258. [Google Scholar] [CrossRef]
- Diamanti-Kandarakis, E.; Bourguignon, J.-P.; Giudice, L.C.; Hauser, R.; Prins, G.S.; Soto, A.M.; Zoeller, R.T.; Gore, A.C. Endocrine-Disrupting Chemicals: An Endocrine Society Scientific Statement. Endocr. Rev. 2009, 30, 293–342. [Google Scholar] [CrossRef] [PubMed]
- Lauretta, R.; Sansone, A.; Sansone, M.; Romanelli, F.; Appetecchia, M. Endocrine Disrupting Chemicals: Effects on Endocrine Glands. Front. Endocrinol. 2019, 10, 178. [Google Scholar] [CrossRef] [PubMed]
- Jalal, N.; Surendranath, A.R.; Pathak, J.L.; Yu, S.; Chung, C.Y. Bisphenol A (BPA) the Mighty and the Mutagenic. Toxicol. Rep. 2018, 5, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Rudel, R.A.; Gray, J.M.; Engel, C.L.; Rawsthorne, T.W.; Dodson, R.E.; Ackerman, J.M.; Rizzo, J.; Nudelman, J.L.; Brody, J.G. Food Packaging and Bisphenol A and Bis(2-Ethyhexyl) Phthalate Exposure: Findings from a Dietary Intervention. Environ. Health Perspect. 2011, 119, 914–920. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Li, C.; Song, P.; Yan, B.; Yang, X.; Wu, Y.; Ma, P. Hepatic and Renal Tissue Damage in Balb/c Mice Exposed to Diisodecyl Phthalate: The Role of Oxidative Stress Pathways. Food Chem. Toxicol. 2019, 132, 110600. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Yang, L.; Wang, S.; Zhang, Z.; Yu, Y.; Wang, M.; Cromie, M.; Gao, W.; Wang, S.-L. The Classic EDCs, Phthalate Esters and Organochlorines, in Relation to Abnormal Sperm Quality: A Systematic Review with Meta-Analysis. Sci. Rep. 2016, 6, 19982. [Google Scholar] [CrossRef]
- Qin, W.; Deng, T.; Cui, H.; Zhang, Q.; Liu, X.; Yang, X.; Chen, M. Exposure to Diisodecyl Phthalate Exacerbated Th2 and Th17-Mediated Asthma through Aggravating Oxidative Stress and the Activation of P38 MAPK. Food Chem. Toxicol. 2018, 114, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Amaro, A.A.; Esposito, A.I.; Mirisola, V.; Mehilli, A.; Rosano, C.; Noonan, D.M.; Albini, A.; Pfeffer, U.; Angelini, G. Endocrine Disruptor Agent Nonyl Phenol Exerts An Estrogen-like Transcriptional Activity on Estrogen Receptor Positive Breast Cancer Cells. Curr. Med. Chem. 2014, 21, 630–640. [Google Scholar] [CrossRef]
- Kazemi, S.; Mousavi Kani, S.N.; Ghasemi-Kasman, M.; Aghapour, F.; Khorasani, H.; Moghadamnia, A.A. Nonylphenol Induces Liver Toxicity and Oxidative Stress in Rat. Biochem. Biophys. Res. Commun. 2016, 479, 17–21. [Google Scholar] [CrossRef]
- Monneret, C. What Is an Endocrine Disruptor? Comptes Rendus Biol. 2017, 340, 403–405. [Google Scholar] [CrossRef]
- Bonefeld-Jørgensen, E.C.; Ghisari, M.; Wielsøe, M.; Bjerregaard-Olesen, C.; Kjeldsen, L.S.; Long, M. Biomonitoring and Hormone-Disrupting Effect Biomarkers of Persistent Organic Pollutants In Vitro and Ex Vivo. Basic Clin. Pharmacol. Toxicol. 2014, 115, 118–128. [Google Scholar] [CrossRef]
- Deierlein, A.L.; Rock, S.; Park, S. Persistent Endocrine-Disrupting Chemicals and Fatty Liver Disease. Curr. Environ. Health Rep. 2017, 4, 439–449. [Google Scholar] [CrossRef]
- Hung, H.; Katsoyiannis, A.A.; Guardans, R. Ten Years of Global Monitoring under the Stockholm Convention on Persistent Organic Pollutants (POPs): Trends, Sources and Transport Modelling. Environ. Pollut. 2016, 217, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Magulova, K.; Priceputu, A. Global Monitoring Plan for Persistent Organic Pollutants (POPs) under the Stockholm Convention: Triggering, Streamlining and Catalyzing Global POPs Monitoring. Environ. Pollut. 2016, 217, 82–84. [Google Scholar] [CrossRef] [PubMed]
- Rahman, Z.; Singh, V.P. The Relative Impact of Toxic Heavy Metals (THMs) (Arsenic (As), Cadmium (Cd), Chromium (Cr)(VI), Mercury (Hg), and Lead (Pb)) on the Total Environment: An Overview. Environ. Monit. Assess. 2019, 191, 419. [Google Scholar] [CrossRef] [PubMed]
- Barouki, R. Endocrine disruptor compounds and new mechanisms of toxicity networks. Rev. Prat. 2018, 68, 1069–1074. [Google Scholar]
- Sawada, N. Association between Arsenic Intake and Cancer—From the Viewpoint of Epidemiological Study. Nippon Eiseigaku Zasshi 2018, 73, 265–268. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kim, K.-H.; Jahan, S.A.; Kabir, E.; Brown, R.J.C. A Review of Airborne Polycyclic Aromatic Hydrocarbons (PAHs) and Their Human Health Effects. Environ. Int. 2013, 60, 71–80. [Google Scholar] [CrossRef]
- Alegbeleye, O.O.; Opeolu, B.O.; Jackson, V.A. Polycyclic Aromatic Hydrocarbons: A Critical Review of Environmental Occurrence and Bioremediation. Environ. Manag. 2017, 60, 758–783. [Google Scholar] [CrossRef]
- Lau, C. Perfluorinated Compounds. Exp. Suppl. 2012, 101, 47–86. [Google Scholar] [CrossRef]
- Chen, A.; Zhou, X.; Cheng, Y.; Tang, S.; Liu, M.; Wang, X. Design and Optimization of the Cocktail Assay for Rapid Assessment of the Activity of UGT Enzymes in Human and Rat Liver Microsomes. Toxicol. Lett. 2018, 295, 379–389. [Google Scholar] [CrossRef] [PubMed]
- Rowland, A.; Miners, J.O.; Mackenzie, P.I. The UDP-Glucuronosyltransferases: Their Role in Drug Metabolism and Detoxification. Int. J. Biochem. Cell Biol. 2013, 45, 1121–1132. [Google Scholar] [CrossRef] [PubMed]
- Bissig, K.-D.; Han, W.; Barzi, M.; Kovalchuk, N.; Ding, L.; Fan, X.; Pankowicz, F.P.; Zhang, Q.-Y.; Ding, X. P450-Humanized and Human Liver Chimeric Mouse Models for Studying Xenobiotic Metabolism and Toxicity. Drug Metab. Dispos. 2018, 46, 1734–1744. [Google Scholar] [CrossRef] [PubMed]
- Muncke, J. Exposure to Endocrine Disrupting Compounds via the Food Chain: Is Packaging a Relevant Source? Sci. Total Environ. 2009, 407, 4549–4559. [Google Scholar] [CrossRef]
- Gálvez-Ontiveros, Y.; Páez, S.; Monteagudo, C.; Rivas, A. Endocrine Disruptors in Food: Impact on Gut Microbiota and Metabolic Diseases. Nutrients 2020, 12, 1158. [Google Scholar] [CrossRef] [PubMed]
- Cano-Sancho, G.; Marchand, P.; Le Bizec, B.; Antignac, J.-P. The Challenging Use and Interpretation of Blood Biomarkers of Exposure Related to Lipophilic Endocrine Disrupting Chemicals in Environmental Health Studies. Mol. Cell. Endocrinol. 2020, 499, 110606. [Google Scholar] [CrossRef]
- Nishida, K.; Kobayashi, M.; Miyamoto, H.; Yoshikawa, N.; Fumoto, S.; Sasaki, H.; Nakamura, J. Relationship between Lipophilicity and Absorption from the Liver Surface of Paraben Derivatives and Antipyrine in Rats: Lipophilic Drug Absorption from Liver Surface. J. Pharm. Pharmacol. 2011, 63, 736–740. [Google Scholar] [CrossRef]
- Lonard, D.M.; O’malley, B.W. Nuclear Receptor Coregulators: Judges, Juries, and Executioners of Cellular Regulation. Mol. Cell 2007, 27, 691–700. [Google Scholar] [CrossRef]
- Cave, M.C.; Clair, H.B.; Hardesty, J.E.; Falkner, K.C.; Feng, W.; Clark, B.J.; Sidey, J.; Shi, H.; Aqel, B.A.; McClain, C.J.; et al. Nuclear Receptors and Nonalcoholic Fatty Liver Disease. Biochim. Biophys. Acta 2016, 1859, 1083–1099. [Google Scholar] [CrossRef]
- Cruz-Hurtado, M.; de López-González, M.L.; Mondragón, V.; Sierra-Santoyo, A. In Vitro Phase I Metabolism of Vinclozolin by Human Liver Microsomes. Xenobiotica 2019, 49, 895–904. [Google Scholar] [CrossRef] [PubMed]
- Carrão, D.B.; Habenchus, M.D.; de Albuquerque, N.C.P.; da Silva, R.M.; Lopes, N.P.; de Oliveira, A.R.M. In Vitro Inhibition of Human CYP2D6 by the Chiral Pesticide Fipronil and Its Metabolite Fipronil Sulfone: Prediction of Pesticide-Drug Interactions. Toxicol. Lett. 2019, 313, 196–204. [Google Scholar] [CrossRef] [PubMed]
- Sychev, D.; Ashraf, G.M.; Svistunov, A.; Maksimov, M.; Tarasov, V.; Chubarev, V.N.; Otdelenov, V.A.; Denisenko, N.P.; Barreto, G.E.; Aliev, G. The Cytochrome P450 Isoenzyme and Some New Opportunities for the Prediction of Negative Drug Interaction in Vivo. Drug Des. Devel. Ther. 2018, 12, 1147–1156. [Google Scholar] [CrossRef]
- Docea, A.O.; Vassilopoulou, L.; Fragou, D.; Arsene, A.L.; Fenga, C.; Kovatsi, L.; Petrakis, D.; Rakitskii, V.N.; Nosyrev, A.E.; Izotov, B.N.; et al. CYP Polymorphisms and Pathological Conditions Related to Chronic Exposure to Organochlorine Pesticides. Toxicol. Rep. 2017, 4, 335–341. [Google Scholar] [CrossRef]
- Lee, P.C.; Chakraborty Patra, S.; Stelloh, C.T.; Lee, W.; Struve, M. Interaction of Nonylphenol and Hepatic CYP1A in Rats. Biochem. Pharmacol. 1996, 52, 885–889. [Google Scholar] [CrossRef]
- Ademollo, N.; Patrolecco, L.; Rauseo, J.; Nielsen, J.; Corsolini, S. Bioaccumulation of Nonylphenols and Bisphenol A in the Greenland Shark Somniosus Microcephalus from the Greenland Seawaters. Microchem. J. 2018, 136, 106–112. [Google Scholar] [CrossRef]
- Lv, Y.-Z.; Yao, L.; Wang, L.; Liu, W.-R.; Zhao, J.-L.; He, L.-Y.; Ying, G.-G. Bioaccumulation, Metabolism, and Risk Assessment of Phenolic Endocrine Disrupting Chemicals in Specific Tissues of Wild Fish. Chemosphere 2019, 226, 607–615. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, S.; Masai, E.; Kamimura, N.; Takahashi, K.; Anderson, R.C.; Faisal, P.A. Phthalates Impact Human Health: Epidemiological Evidences and Plausible Mechanism of Action. J. Hazard. Mater. 2017, 340, 360–383. [Google Scholar] [CrossRef] [PubMed]
- Praveena, S.M.; Teh, S.W.; Rajendran, R.K.; Kannan, N.; Lin, C.-C.; Abdullah, R.; Kumar, S. Recent Updates on Phthalate Exposure and Human Health: A Special Focus on Liver Toxicity and Stem Cell Regeneration. Environ. Sci. Pollut. Res. 2018, 25, 11333–11342. [Google Scholar] [CrossRef]
- Larsson, K.; Ljung Björklund, K.; Palm, B.; Wennberg, M.; Kaj, L.; Lindh, C.H.; Jönsson, B.A.G.; Berglund, M. Exposure Determinants of Phthalates, Parabens, Bisphenol A and Triclosan in Swedish Mothers and Their Children. Environ. Int. 2014, 73, 323–333. [Google Scholar] [CrossRef]
- Lee, Y.-M.; Kim, K.-S.; Jacobs, D.R.; Lee, D.-H. Persistent Organic Pollutants in Adipose Tissue Should Be Considered in Obesity Research. Obes. Rev. Off. J. Int. Assoc. Study Obes. 2017, 18, 129–139. [Google Scholar] [CrossRef]
- Shin, M.-Y.; Shin, C.; Choi, J.W.; Lee, J.; Lee, S.; Kim, S. Pharmacokinetic Profile of Propyl Paraben in Humans after Oral Administration. Environ. Int. 2019, 130, 104917. [Google Scholar] [CrossRef]
- Nicolucci, C.; Errico, S.; Federico, A.; Dallio, M.; Loguercio, C.; Diano, N. Human Exposure to Bisphenol A and Liver Health Status: Quantification of Urinary and Circulating Levels by LC–MS/MS. J. Pharm. Biomed. Anal. 2017, 140, 105–112. [Google Scholar] [CrossRef]
- Ulasoglu, C.; Enc, F.Y.; Kaya, E.; Yilmaz, Y. Characterization of Patients with Biopsy-Proven Non-Alcoholic Fatty Liver Disease and Normal Aminotransferase Levels. J. Gastrointestin. Liver Dis. 2019, 28, 427–431. [Google Scholar] [CrossRef]
- Kang, Y.; Park, S.; Kim, S.; Koh, H. Normal Serum Alanine Aminotransferase and Non-Alcoholic Fatty Liver Disease among Korean Adolescents: A Cross-Sectional Study Using Data from KNHANES 2010–2015. BMC Pediatr. 2018, 18, 215. [Google Scholar] [CrossRef] [PubMed]
- Kumar, J.; Lind, L.; Salihovic, S.; van Bavel, B.; Ingelsson, E.; Lind, P.M. Persistent Organic Pollutants and Liver Dysfunction Biomarkers in a Population-Based Human Sample of Men and Women. Environ. Res. 2014, 134, 251–256. [Google Scholar] [CrossRef] [PubMed]
- Baralić, K.; Buha Djordjevic, A.; Živančević, K.; Antonijević, E.; Anđelković, M.; Javorac, D.; Ćurčić, M.; Bulat, Z.; Antonijević, B.; Đukić-Ćosić, D. Toxic Effects of the Mixture of Phthalates and Bisphenol A-Subacute Oral Toxicity Study in Wistar Rats. Int. J. Environ. Res. Public Health 2020, 17, 746. [Google Scholar] [CrossRef]
- Heindel, J.J.; Blumberg, B.; Cave, M.; Machtinger, R.; Mantovani, A.; Mendez, M.A.; Nadal, A.; Palanza, P.; Panzica, G.; Sargis, R.; et al. Metabolism Disrupting Chemicals and Metabolic Disorders. Reprod. Toxicol. Elmsford N 2017, 68, 3–33. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Ahn, S.Y.; Song, I.C.; Chung, M.H.; Jang, H.C.; Park, K.S.; Lee, K.-U.; Pak, Y.K.; Lee, H.K. Chronic Exposure to the Herbicide, Atrazine, Causes Mitochondrial Dysfunction and Insulin Resistance. PLoS ONE 2009, 4, e5186. [Google Scholar] [CrossRef]
- Jin, Y.; Lin, X.; Miao, W.; Wu, T.; Shen, H.; Chen, S.; Li, Y.; Pan, Q.; Fu, Z. Chronic Exposure of Mice to Environmental Endocrine-Disrupting Chemicals Disturbs Their Energy Metabolism. Toxicol. Lett. 2014, 225, 392–400. [Google Scholar] [CrossRef]
- Al-Eryani, L.; Wahlang, B.; Falkner, K.C.; Guardiola, J.J.; Clair, H.B.; Prough, R.A.; Cave, M. Identification of Environmental Chemicals Associated with the Development of Toxicant-Associated Fatty Liver Disease in Rodents. Toxicol. Pathol. 2015, 43, 482–497. [Google Scholar] [CrossRef] [PubMed]
- Maradonna, F.; Carnevali, O. Lipid Metabolism Alteration by Endocrine Disruptors in Animal Models: An Overview. Front. Endocrinol. 2018, 9, 654. [Google Scholar] [CrossRef]
- Armstrong, L.E.; Guo, G.L. Understanding Environmental Contaminants’ Direct Effects on Non-Alcoholic Fatty Liver Disease Progression. Curr. Environ. Health Rep. 2019, 6, 95–104. [Google Scholar] [CrossRef]
- Kawano, Y.; Cohen, D.E. Mechanisms of Hepatic Triglyceride Accumulation in Non-Alcoholic Fatty Liver Disease. J. Gastroenterol. 2013, 48, 434–441. [Google Scholar] [CrossRef]
- Ipsen, D.H.; Lykkesfeldt, J.; Tveden-Nyborg, P. Molecular Mechanisms of Hepatic Lipid Accumulation in Non-Alcoholic Fatty Liver Disease. Cell. Mol. Life Sci. CMLS 2018, 75, 3313–3327. [Google Scholar] [CrossRef]
- Geisler, C.E.; Renquist, B.J. Hepatic Lipid Accumulation: Cause and Consequence of Dysregulated Glucoregulatory Hormones. J. Endocrinol. 2017, 234, R1–R21. [Google Scholar] [CrossRef]
- Polyzos, S.A.; Kountouras, J.; Deretzi, G.; Zavos, C.; Mantzoros, C.S. The Emerging Role of Endocrine Disruptors in Pathogenesis of Insulin Resistance: A Concept Implicating Nonalcoholic Fatty Liver Disease. Curr. Mol. Med. 2012, 12, 68–82. [Google Scholar] [CrossRef] [PubMed]
- Wahlang, B.; Hardesty, J.E.; Jin, J.; Falkner, K.C.; Cave, M.C. Polychlorinated Biphenyls and Nonalcoholic Fatty Liver Disease. Curr. Opin. Toxicol. 2019, 14, 21–28. [Google Scholar] [CrossRef]
- Shan, Q.; Chen, N.; Liu, W.; Qu, F.; Chen, A. Exposure to 2,3,3′,4,4′,5-Hexachlorobiphenyl Promotes Nonalcoholic Fatty Liver Disease Development in C57BL/6 Mice. Environ. Pollut. 2020, 263, 114563. [Google Scholar] [CrossRef] [PubMed]
- Wan, H.T.; Zhao, Y.G.; Wei, X.; Hui, K.Y.; Giesy, J.P.; Wong, C.K.C. PFOS-Induced Hepatic Steatosis, the Mechanistic Actions on β-Oxidation and Lipid Transport. Biochim. Biophys. Acta BBA Gen. Subj. 2012, 1820, 1092–1101. [Google Scholar] [CrossRef] [PubMed]
- Das, K.P.; Wood, C.R.; Lin, M.T.; Starkov, A.A.; Lau, C.; Wallace, K.B.; Corton, J.C.; Abbott, B.D. Perfluoroalkyl Acids-Induced Liver Steatosis: Effects on Genes Controlling Lipid Homeostasis. Toxicology 2017, 378, 37–52. [Google Scholar] [CrossRef]
- Yan, S.; Wang, J.; Dai, J. Activation of Sterol Regulatory Element-Binding Proteins in Mice Exposed to Perfluorooctanoic Acid for 28 Days. Arch. Toxicol. 2015, 89, 1569–1578. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; He, J.; Li, N.; Gao, N.; Du, Q.; Chen, B.; Chen, F.; Shan, X.; Ding, Y.; Zhu, W.; et al. Lipid Accumulation Responses in the Liver of Rana Nigromaculata Induced by Perfluorooctanoic Acid (PFOA). Ecotoxicol. Environ. Saf. 2019, 167, 29–35. [Google Scholar] [CrossRef]
- Chen, H.; Zhang, W.; Rui, B.; Yang, S.; Xu, W.; Wei, W. Di(2-Ethylhexyl) Phthalate Exacerbates Non-Alcoholic Fatty Liver in Rats and Its Potential Mechanisms. Environ. Toxicol. Pharmacol. 2016, 42, 38–44. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, S.; Zhao, T.; Yang, L.; Guo, S.; Shi, Y.; Zhang, X.; Zhou, L.; Ye, L. Mono-2-Ethylhexyl Phthalate (MEHP) Promoted Lipid Accumulation via JAK2/STAT5 and Aggravated Oxidative Stress in BRL-3A Cells. Ecotoxicol. Environ. Saf. 2019, 184, 109611. [Google Scholar] [CrossRef]
- Bai, J.; He, Z.; Li, Y.; Jiang, X.; Yu, H.; Tan, Q. Mono-2-Ethylhexyl Phthalate Induces the Expression of Genes Involved in Fatty Acid Synthesis in HepG2 Cells. Environ. Toxicol. Pharmacol. 2019, 69, 104–111. [Google Scholar] [CrossRef]
- Dallio, M.; Diano, N.; Masarone, M.; Gravina, A.G.; Patanè, V.; Romeo, M.; Di Sarno, R.; Errico, S.; Nicolucci, C.; Abenavoli, L.; et al. Chemical Effect of Bisphenol A on Non-Alcoholic Fatty Liver Disease. Int. J. Environ. Res. Public Health 2019, 16, 3134. [Google Scholar] [CrossRef] [PubMed]
- Marmugi, A.; Ducheix, S.; Lasserre, F.; Polizzi, A.; Paris, A.; Priymenko, N.; Bertrand-Michel, J.; Pineau, T.; Guillou, H.; Martin, P.G.P.; et al. Low Doses of Bisphenol A Induce Gene Expression Related to Lipid Synthesis and Trigger Triglyceride Accumulation in Adult Mouse Liver. Hepatol. Baltim. Md 2012, 55, 395–407. [Google Scholar] [CrossRef] [PubMed]
- Martella, A.; Silvestri, C.; Maradonna, F.; Gioacchini, G.; Allarà, M.; Radaelli, G.; Overby, D.R.; Di Marzo, V.; Carnevali, O. Bisphenol A Induces Fatty Liver by an Endocannabinoid-Mediated Positive Feedback Loop. Endocrinology 2016, 157, 1751–1763. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Rector, R.S.; Thyfault, J.P.; Ibdah, J.A. Nonalcoholic Fatty Liver Disease and Mitochondrial Dysfunction. World J. Gastroenterol. 2008, 14, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Einer, C.; Hohenester, S.; Wimmer, R.; Wottke, L.; Artmann, R.; Schulz, S.; Gosmann, C.; Simmons, A.; Leitzinger, C.; Eberhagen, C.; et al. Mitochondrial Adaptation in Steatotic Mice. Mitochondrion 2018, 40, 1–12. [Google Scholar] [CrossRef]
- Simões, I.C.M.; Fontes, A.; Pinton, P.; Zischka, H.; Wieckowski, M.R. Mitochondria in Non-Alcoholic Fatty Liver Disease. Int. J. Biochem. Cell Biol. 2018, 95, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Mansouri, A.; Gattolliat, C.-H.; Asselah, T. Mitochondrial Dysfunction and Signaling in Chronic Liver Diseases. Gastroenterology 2018, 155, 629–647. [Google Scholar] [CrossRef] [PubMed]
- Marroqui, L.; Tudurí, E.; Alonso-Magdalena, P.; Quesada, I.; Nadal, Á.; Dos Santos, R.S. Mitochondria as Target of Endocrine-Disrupting Chemicals: Implications for Type 2 Diabetes. J. Endocrinol. 2018, 239, R27–R45. [Google Scholar] [CrossRef]
- Lim, S.; Cho, Y.M.; Park, K.S.; Lee, H.K. Persistent Organic Pollutants, Mitochondrial Dysfunction, and Metabolic Syndrome. Ann. N. Y. Acad. Sci. 2010, 1201, 166–176. [Google Scholar] [CrossRef] [PubMed]
- Sagarkar, S.; Gandhi, D.; Devi, S.S.; Sakharkar, A.; Kapley, A. Atrazine Exposure Causes Mitochondrial Toxicity in Liver and Muscle Cell Lines. Indian J. Pharmacol. 2016, 48, 200–207. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Beigh, S.; Chaudhari, B.P.; Sharma, S.; Aliul Hasan Abdi, S.; Ahmad, S.; Ahmad, F.; Parvez, S.; Raisuddin, S. Mitochondrial Dysfunction Induced by Bisphenol A Is a Factor of Its Hepatotoxicity in Rats. Environ. Toxicol. 2016, 31, 1922–1934. [Google Scholar] [CrossRef]
- Jiang, Y.; Xia, W.; Zhu, Y.; Li, X.; Wang, D.; Liu, J.; Chang, H.; Li, G.; Xu, B.; Chen, X.; et al. Mitochondrial Dysfunction in Early Life Resulted from Perinatal Bisphenol A Exposure Contributes to Hepatic Steatosis in Rat Offspring. Toxicol. Lett. 2014, 228, 85–92. [Google Scholar] [CrossRef]
- Yu, J.; Yang, X.; Yang, X.; Yang, M.; Wang, P.; Yang, Y.; Yang, J.; Li, W.; Xu, J. Nonylphenol Aggravates Non-Alcoholic Fatty Liver Disease in High Sucrose-High Fat Diet-Treated Rats. Sci. Rep. 2018, 8, 3232. [Google Scholar] [CrossRef] [PubMed]
- Kourouma, A.; Keita, H.; Duan, P.; Quan, C.; Bilivogui, K.K.; Qi, S.; Christiane, N.A.; Osamuyimen, A.; Yang, K. Effects of 4-Nonylphenol on Oxidant/Antioxidant Balance System Inducing Hepatic Steatosis in Male Rat. Toxicol. Rep. 2015, 2, 1423–1433. [Google Scholar] [CrossRef]
- He, X.; Gao, J.; Hou, H.; Qi, Z.; Chen, H.; Zhang, X.-X. Inhibition of Mitochondrial Fatty Acid Oxidation Contributes to Development of Nonalcoholic Fatty Liver Disease Induced by Environmental Cadmium Exposure. Environ. Sci. Technol. 2019, 53, 13992–14000. [Google Scholar] [CrossRef] [PubMed]
- Ding, R.-B.; Bao, J.; Deng, C.-X. Emerging Roles of SIRT1 in Fatty Liver Diseases. Int. J. Biol. Sci. 2017, 13, 852–867. [Google Scholar] [CrossRef] [PubMed]
- Banks, A.S.; Kon, N.; Knight, C.; Matsumoto, M.; Gutiérrez-Juárez, R.; Rossetti, L.; Gu, W.; Accili, D. SirT1 Gain of Function Increases Energy Efficiency and Prevents Diabetes in Mice. Cell Metab. 2008, 8, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Qiang, L.; Farmer, S.R. Identification of a Domain within Peroxisome Proliferator-Activated Receptor γ Regulating Expression of a Group of Genes Containing Fibroblast Growth Factor 21 That Are Selectively Repressed by SIRT1 in Adipocytes. Mol. Cell. Biol. 2008, 28, 188–200. [Google Scholar] [CrossRef]
- Milne, J.C.; Lambert, P.D.; Schenk, S.; Carney, D.P.; Smith, J.J.; Gagne, D.J.; Jin, L.; Boss, O.; Perni, R.B.; Vu, C.B.; et al. Small Molecule Activators of SIRT1 as Therapeutics for the Treatment of Type 2 Diabetes. Nature 2007, 450, 712–716. [Google Scholar] [CrossRef]
- Li, X.; Zhang, S.; Blander, G.; Tse, J.G.; Krieger, M.; Guarente, L. SIRT1 Deacetylates and Positively Regulates the Nuclear Receptor LXR. Mol. Cell 2007, 28, 91–106. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Sun, M.; Cao, Y.; Ma, L.; Shen, Y.; Velikanova, A.A.; Li, X.; Sun, C.; Zhao, Y. MiR-34a Regulates Lipid Metabolism by Targeting SIRT1 in Non-Alcoholic Fatty Liver Disease with Iron Overload. Arch. Biochem. Biophys. 2020, 695, 108642. [Google Scholar] [CrossRef]
- Yoshizaki, T.; Milne, J.C.; Imamura, T.; Schenk, S.; Sonoda, N.; Babendure, J.L.; Lu, J.-C.; Smith, J.J.; Jirousek, M.R.; Olefsky, J.M. SIRT1 Exerts Anti-Inflammatory Effects and Improves Insulin Sensitivity in Adipocytes. Mol. Cell. Biol. 2009, 29, 1363–1374. [Google Scholar] [CrossRef]
- Yamazaki, Y.; Usui, I.; Kanatani, Y.; Matsuya, Y.; Tsuneyama, K.; Fujisaka, S.; Bukhari, A.; Suzuki, H.; Senda, S.; Imanishi, S.; et al. Treatment with SRT1720, a SIRT1 Activator, Ameliorates Fatty Liver with Reduced Expression of Lipogenic Enzymes in MSG Mice. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E1179–E1186. [Google Scholar] [CrossRef]
- Colak, Y.; Ozturk, O.; Senates, E.; Tuncer, I.; Yorulmaz, E.; Adali, G.; Doganay, L.; Enc, F.Y. SIRT1 as a Potential Therapeutic Target for Treatment of Nonalcoholic Fatty Liver Disease. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2011, 17, HY5–HY9. [Google Scholar] [CrossRef]
- Bai, P.; Cantó, C.; Oudart, H.; Brunyánszki, A.; Cen, Y.; Thomas, C.; Yamamoto, H.; Huber, A.; Kiss, B.; Houtkooper, R.H.; et al. PARP-1 Inhibition Increases Mitochondrial Metabolism through SIRT1 Activation. Cell Metab. 2011, 13, 461–468. [Google Scholar] [CrossRef]
- Peverill, W.; Powell, L.W.; Skoien, R. Evolving Concepts in the Pathogenesis of NASH: Beyond Steatosis and Inflammation. Int. J. Mol. Sci. 2014, 15, 8591–8638. [Google Scholar] [CrossRef]
- Luedde, T.; Schwabe, R.F. NF-ΚB in the Liver--Linking Injury, Fibrosis and Hepatocellular Carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Huc, L.; Lemarié, A.; Guéraud, F.; Héliès-Toussaint, C. Low Concentrations of Bisphenol A Induce Lipid Accumulation Mediated by the Production of Reactive Oxygen Species in the Mitochondria of HepG2 Cells. Toxicol. Vitro Int. J. Publ. Assoc. BIBRA 2012, 26, 709–717. [Google Scholar] [CrossRef] [PubMed]
- Han, R.; Hu, M.; Zhong, Q.; Wan, C.; Liu, L.; Li, F.; Zhang, F.; Ding, W. Perfluorooctane Sulphonate Induces Oxidative Hepatic Damage via Mitochondria-Dependent and NF-ΚB/TNF-α-Mediated Pathway. Chemosphere 2018, 191, 1056–1064. [Google Scholar] [CrossRef] [PubMed]
- Deng, P.; Barney, J.; Petriello, M.C.; Morris, A.J.; Wahlang, B.; Hennig, B. Hepatic Metabolomics Reveals That Liver Injury Increases PCB 126-Induced Oxidative Stress and Metabolic Dysfunction. Chemosphere 2019, 217, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Park, C.G.; Sung, B.; Ryu, C.S.; Kim, Y.J. Mono-(2-Ethylhexyl) Phthalate Induces Oxidative Stress and Lipid Accumulation in Zebrafish Liver Cells. Comp. Biochem. Physiol. Part C Toxicol. Pharmacol. 2020, 230, 108704. [Google Scholar] [CrossRef] [PubMed]
- Arrese, M.; Cabrera, D.; Kalergis, A.M.; Feldstein, A.E. Innate Immunity and Inflammation in NAFLD/NASH. Dig. Dis. Sci. 2016, 61, 1294–1303. [Google Scholar] [CrossRef]
- Schuster, S.; Cabrera, D.; Arrese, M.; Feldstein, A.E. Triggering and Resolution of Inflammation in NASH. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 349–364. [Google Scholar] [CrossRef]
- Alisi, A.; Carpino, G.; Oliveira, F.L.; Panera, N.; Nobili, V.; Gaudio, E. The Role of Tissue Macrophage-Mediated Inflammation on NAFLD Pathogenesis and Its Clinical Implications. Mediat. Inflamm. 2017, 2017, 8162421. [Google Scholar] [CrossRef] [PubMed]
- Katsarou, A.; Moustakas, I.I.; Pyrina, I.; Lembessis, P.; Koutsilieris, M.; Chatzigeorgiou, A. Metabolic Inflammation as an Instigator of Fibrosis during Non-Alcoholic Fatty Liver Disease. World J. Gastroenterol. 2020, 26, 1993–2011. [Google Scholar] [CrossRef]
- Meyer, S.K.; Probert, P.M.E.; Lakey, A.F.; Axon, A.R.; Leitch, A.C.; Williams, F.M.; Jowsey, P.A.; Blain, P.G.; Kass, G.E.N.; Wright, M.C. Hepatic Effects of Tartrazine (E 102) after Systemic Exposure Are Independent of Oestrogen Receptor Interactions in the Mouse. Toxicol. Lett. 2017, 273, 55–68. [Google Scholar] [CrossRef]
- Acaroz, U.; Ince, S.; Arslan-Acaroz, D.; Gurler, Z.; Demirel, H.H.; Kucukkurt, I.; Eryavuz, A.; Kara, R.; Varol, N.; Zhu, K. Bisphenol-A Induced Oxidative Stress, Inflammatory Gene Expression, and Metabolic and Histopathological Changes in Male Wistar Albino Rats: Protective Role of Boron. Toxicol. Res. 2019, 8, 262–269. [Google Scholar] [CrossRef]
- Lv, Q.; Gao, R.; Peng, C.; Yi, J.; Liu, L.; Yang, S.; Li, D.; Hu, J.; Luo, T.; Mei, M.; et al. Bisphenol A Promotes Hepatic Lipid Deposition Involving Kupffer Cells M1 Polarization in Male Mice. J. Endocrinol. 2017, 234, 143–154. [Google Scholar] [CrossRef]
- Han, R.; Zhang, F.; Wan, C.; Liu, L.; Zhong, Q.; Ding, W. Effect of Perfluorooctane Sulphonate-Induced Kupffer Cell Activation on Hepatocyte Proliferation through the NF-ΚB/TNF-α/IL-6-Dependent Pathway. Chemosphere 2018, 200, 283–294. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.-B.; Ji, K.; Shen, X.-Y.; Zhang, W.-W.; Wang, R.; Xu, W.-P.; Wei, W. Di(2-Ethylhexyl) Phthalate Promotes Hepatic Fibrosis by Regulation of Oxidative Stress and Inflammation Responses in Rats. Environ. Toxicol. Pharmacol. 2019, 68, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Gaitantzi, H.; Hakenberg, P.; Theobald, J.; Heinlein, H.; Cai, C.; Loff, S.; Wölfl, S.; Ebert, M.P.; Breitkopf-Heinlein, K.; Subotic, U. Di (2-Ethylhexyl) Phthalate and Its Role in Developing Cholestasis: An In Vitro Study on Different Liver Cell Types. J. Pediatr. Gastroenterol. Nutr. 2018, 66, e28–e35. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-Y.; Suk, F.-M.; Twu, Y.-C.; Liao, Y.-J. Long-Term Exposure to Low-Dose Di-(2-Ethylhexyl) Phthalate Impairs Cholesterol Metabolism in Hepatic Stellate Cells and Exacerbates Liver Librosis. Int. J. Environ. Res. Public Health 2020, 17, 3802. [Google Scholar] [CrossRef] [PubMed]
- Harvey, W.A.; Jurgensen, K.; Pu, X.; Lamb, C.L.; Cornell, K.A.; Clark, R.J.; Klocke, C.; Mitchell, K.A. Exposure to 2,3,7,8-Tetrachlorodibenzo-p-Dioxin (TCDD) Increases Human Hepatic Stellate Cell Activation. Toxicology 2016, 344–346, 26–33. [Google Scholar] [CrossRef]
- Doskey, C.M.; Fader, K.A.; Nault, R.; Lydic, T.; Matthews, J.; Potter, D.; Sharratt, B.; Williams, K.; Zacharewski, T. 2,3,7,8-Tetrachlorodibenzo-p-Dioxin (TCDD) Alters Hepatic Polyunsaturated Fatty Acid Metabolism and Eicosanoid Biosynthesis in Female Sprague-Dawley Rats. Toxicol. Appl. Pharmacol. 2020, 398, 115034. [Google Scholar] [CrossRef] [PubMed]
- Foulds, C.E.; Treviño, L.S.; York, B.; Walker, C.L. Endocrine-Disrupting Chemicals and Fatty Liver Disease. Nat. Rev. Endocrinol. 2017, 13, 445–457. [Google Scholar] [CrossRef] [PubMed]
- Walker, C.L. Minireview: Epigenomic Plasticity and Vulnerability to EDC Exposures. Mol. Endocrinol. Baltim. Md. 2016, 30, 848–855. [Google Scholar] [CrossRef] [PubMed]
- Treviño, L.S.; Dong, J.; Kaushal, A.; Katz, T.A.; Jangid, R.K.; Robertson, M.J.; Grimm, S.L.; Ambati, C.S.R.; Putluri, V.; Cox, A.R.; et al. Epigenome Environment Interactions Accelerate Epigenomic Aging and Unlock Metabolically Restricted Epigenetic Reprogramming in Adulthood. Nat. Commun. 2020, 11, 2316. [Google Scholar] [CrossRef]
- Skinner, M.K.; Manikkam, M.; Guerrero-Bosagna, C. Epigenetic Transgenerational Actions of Environmental Factors in Disease Etiology. Trends Endocrinol. Metab. TEM 2010, 21, 214–222. [Google Scholar] [CrossRef]
- Rissman, E.F.; Adli, M. Minireview: Transgenerational Epigenetic Inheritance: Focus on Endocrine Disrupting Compounds. Endocrinology 2014, 155, 2770–2780. [Google Scholar] [CrossRef] [PubMed]
- Skinner, M.K. What Is an Epigenetic Transgenerational Phenotype? F3 or F2. Reprod. Toxicol. Elmsford N 2008, 25, 2–6. [Google Scholar] [CrossRef]
- Patterson, T.A.; Twaddle, N.C.; Roegge, C.S.; Callicott, R.J.; Fisher, J.W.; Doerge, D.R. Concurrent Determination of Bisphenol A Pharmacokinetics in Maternal and Fetal Rhesus Monkeys. Toxicol. Appl. Pharmacol. 2013, 267, 41–48. [Google Scholar] [CrossRef]
- Dolinoy, D.C.; Huang, D.; Jirtle, R.L. Maternal Nutrient Supplementation Counteracts Bisphenol A-Induced DNA Hypomethylation in Early Development. Proc. Natl. Acad. Sci. USA 2007, 104, 13056–13061. [Google Scholar] [CrossRef] [PubMed]
- Doshi, T.; Mehta, S.S.; Dighe, V.; Balasinor, N.; Vanage, G. Hypermethylation of Estrogen Receptor Promoter Region in Adult Testis of Rats Exposed Neonatally to Bisphenol A. Toxicology 2011, 289, 74–82. [Google Scholar] [CrossRef]
- Zhu, L.; Liu, Y.; Xue, X.; Yuan, C.; Wang, Z. BPA’s Transgenerational Disturbance to Transcription of Ovarian Steroidogenic Genes in Rare Minnow Gobiocypris Rarus via DNA and Histone Methylation. Sci. Total Environ. 2021, 762, 143055. [Google Scholar] [CrossRef]
- Qin, T.; Zhang, X.; Guo, T.; Yang, T.; Gao, Y.; Hao, W.; Xiao, X. Epigenetic Alteration Shaped by the Environmental Chemical Bisphenol A. Front. Genet. 2020, 11, 618966. [Google Scholar] [CrossRef] [PubMed]
- Longo, M.; Zatterale, F.; Naderi, J.; Nigro, C.; Oriente, F.; Formisano, P.; Miele, C.; Beguinot, F. Low-Dose Bisphenol-A Promotes Epigenetic Changes at Pparγ Promoter in Adipose Precursor Cells. Nutrients 2020, 12, 3498. [Google Scholar] [CrossRef]
- McCabe, C.F.; Padmanabhan, V.; Dolinoy, D.C.; Domino, S.E.; Jones, T.R.; Bakulski, K.M.; Goodrich, J.M. Maternal Environmental Exposure to Bisphenols and Epigenome-Wide DNA Methylation in Infant Cord Blood. Environ. Epigenetics 2020, 6, dvaa021. [Google Scholar] [CrossRef]
- Cimmino, I.; Fiory, F.; Perruolo, G.; Miele, C.; Beguinot, F.; Formisano, P.; Oriente, F. Potential Mechanisms of Bisphenol A (BPA) Contributing to Human Disease. Int. J. Mol. Sci. 2020, 21, 5761. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Xia, W.; Wang, D.Q.; Wan, Y.J.; Xu, B.; Chen, X.; Li, Y.Y.; Xu, S.Q. Hepatic DNA Methylation Modifications in Early Development of Rats Resulting from Perinatal BPA Exposure Contribute to Insulin Resistance in Adulthood. Diabetologia 2013, 56, 2059–2067. [Google Scholar] [CrossRef]
- Ke, Z.-H.; Pan, J.-X.; Jin, L.-Y.; Xu, H.-Y.; Yu, T.-T.; Ullah, K.; Rahman, T.U.; Ren, J.; Cheng, Y.; Dong, X.-Y.; et al. Bisphenol A Exposure May Induce Hepatic Lipid Accumulation via Reprogramming the DNA Methylation Patterns of Genes Involved in Lipid Metabolism. Sci. Rep. 2016, 6, 31331. [Google Scholar] [CrossRef] [PubMed]
- Chen, H. Chronic Inorganic Arsenic Exposure Induces Hepatic Global and Individual Gene Hypomethylation: Implications for Arsenic Hepatocarcinogenesis. Carcinogenesis 2004, 25, 1779–1786. [Google Scholar] [CrossRef]
- Ditzel, E.J.; Nguyen, T.; Parker, P.; Camenisch, T.D. Effects of Arsenite Exposure during Fetal Development on Energy Metabolism and Susceptibility to Diet-Induced Fatty Liver Disease in Male Mice. Environ. Health Perspect. 2016, 124, 201–209. [Google Scholar] [CrossRef]
- Gu, H.; Liu, Y.; Wang, W.; Ding, L.; Teng, W.; Liu, L. In Utero Exposure to Di-(2-Ethylhexyl) Phthalate Induces Metabolic Disorder and Increases Fat Accumulation in Visceral Depots of C57BL/6J Mice Offspring. Exp. Ther. Med. 2016, 12, 3806–3812. [Google Scholar] [CrossRef]
- Manikkam, M.; Tracey, R.; Guerrero-Bosagna, C.; Skinner, M.K. Plastics Derived Endocrine Disruptors (BPA, DEHP and DBP) Induce Epigenetic Transgenerational Inheritance of Obesity, Reproductive Disease and Sperm Epimutations. PLoS ONE 2013, 8, e55387. [Google Scholar] [CrossRef]
- King, S.E.; McBirney, M.; Beck, D.; Sadler-Riggleman, I.; Nilsson, E.; Skinner, M.K. Sperm Epimutation Biomarkers of Obesity and Pathologies Following DDT Induced Epigenetic Transgenerational Inheritance of Disease. Environ. Epigenetics 2019, 5, dvz008. [Google Scholar] [CrossRef]
- Strakovsky, R.S.; Wang, H.; Engeseth, N.J.; Flaws, J.A.; Helferich, W.G.; Pan, Y.-X.; Lezmi, S. Developmental Bisphenol A (BPA) Exposure Leads to Sex-Specific Modification of Hepatic Gene Expression and Epigenome at Birth That May Exacerbate High-Fat Diet-Induced Hepatic Steatosis. Toxicol. Appl. Pharmacol. 2015, 284, 101–112. [Google Scholar] [CrossRef]
- Maranghi, F.; Lorenzetti, S.; Tassinari, R.; Moracci, G.; Tassinari, V.; Marcoccia, D.; Di Virgilio, A.; Eusepi, A.; Romeo, A.; Magrelli, A.; et al. In Utero Exposure to Di-(2-Ethylhexyl) Phthalate Affects Liver Morphology and Metabolism in Post-Natal CD-1 Mice. Reprod. Toxicol. Elmsford N 2010, 29, 427–432. [Google Scholar] [CrossRef]
- Chamorro-García, R.; Sahu, M.; Abbey, R.J.; Laude, J.; Pham, N.; Blumberg, B. Transgenerational Inheritance of Increased Fat Depot Size, Stem Cell Reprogramming, and Hepatic Steatosis Elicited by Prenatal Exposure to the Obesogen Tributyltin in Mice. Environ. Health Perspect. 2013, 121, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Z.; Chen, S.; Wu, T.; Zhang, J.; Su, Y.; Chen, Y.; Wang, C. Tributyltin Causes Obesity and Hepatic Steatosis in Male Mice. Environ. Toxicol. 2011, 26, 79–85. [Google Scholar] [CrossRef]
- Boverhof, D.R.; Burgoon, L.D.; Tashiro, C.; Sharratt, B.; Chittim, B.; Harkema, J.R.; Mendrick, D.L.; Zacharewski, T.R. Comparative Toxicogenomic Analysis of the Hepatotoxic Effects of TCDD in Sprague Dawley Rats and C57BL/6 Mice. Toxicol. Sci. Off. J. Soc. Toxicol. 2006, 94, 398–416. [Google Scholar] [CrossRef]
- Tomaszewski, K.E.; Montgomery, C.A.; Melnick, R.L. Modulation of 2,3,7,8-Tetrachlorodibenzo-p-Dioxin Toxicity in F344 Rats by Di(2-Ethylhexyl)Phthalate. Chem. Biol. Interact. 1988, 65, 205–222. [Google Scholar] [CrossRef]
- Jones, G.; Greig, J.B. Pathological Changes in the Liver of Mice given 2,3,7,8-Tetrachlorodibenzo-p-Dioxin. Experientia 1975, 31, 1315–1317. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Alcalá, L.M.; Sá, C.; Pimentel, L.L.; Pestana, D.; Teixeira, D.; Faria, A.; Calhau, C.; Gomes, A. Endocrine Disruptor DDE Associated with a High-Fat Diet Enhances the Impairment of Liver Fatty Acid Composition in Rats. J. Agric. Food Chem. 2015, 63, 9341–9348. [Google Scholar] [CrossRef]
- Tan, X.; Xie, G.; Sun, X.; Li, Q.; Zhong, W.; Qiao, P.; Sun, X.; Jia, W.; Zhou, Z. High Fat Diet Feeding Exaggerates Perfluorooctanoic Acid-Induced Liver Injury in Mice via Modulating Multiple Metabolic Pathways. PLoS ONE 2013, 8, e61409. [Google Scholar] [CrossRef] [PubMed]
- Lv, Z.; Li, G.; Li, Y.; Ying, C.; Chen, J.; Chen, T.; Wei, J.; Lin, Y.; Jiang, Y.; Wang, Y.; et al. Glucose and Lipid Homeostasis in Adult Rat Is Impaired by Early-Life Exposure to Perfluorooctane Sulfonate. Environ. Toxicol. 2013, 28, 532–542. [Google Scholar] [CrossRef]
- Cave, M.; Appana, S.; Patel, M.; Falkner, K.C.; McClain, C.J.; Brock, G. Polychlorinated Biphenyls, Lead, and Mercury Are Associated with Liver Disease in American Adults: NHANES 2003-2004. Environ. Health Perspect. 2010, 118, 1735–1742. [Google Scholar] [CrossRef]
- Kim, M.-J.; Marchand, P.; Henegar, C.; Antignac, J.-P.; Alili, R.; Poitou, C.; Bouillot, J.-L.; Basdevant, A.; Le Bizec, B.; Barouki, R.; et al. Fate and Complex Pathogenic Effects of Dioxins and Polychlorinated Biphenyls in Obese Subjects before and after Drastic Weight Loss. Environ. Health Perspect. 2011, 119, 377–383. [Google Scholar] [CrossRef]
- Pazderova-Vejlupková, J.; Lukás, E.; Nĕmcova, M.; Pícková, J.; Jirásek, L. The Development and Prognosis of Chronic Intoxication by Tetrachlordibenzo-p-Dioxin in Men. Arch. Environ. Health 1981, 36, 5–11. [Google Scholar] [CrossRef]
- Lee, C.-C.; Yao, Y.-J.; Chen, H.-L.; Guo, Y.-L.; Su, H.-J. Fatty Liver and Hepatic Function for Residents with Markedly High Serum PCDD/Fs Levels in Taiwan. J. Toxicol. Environ. Health A 2006, 69, 367–380. [Google Scholar] [CrossRef] [PubMed]
- Mocarelli, P.; Marocchi, A.; Brambilla, P.; Gerthoux, P.; Young, D.S.; Mantel, N. Clinical Laboratory Manifestations of Exposure to Dioxin in Children. A Six-Year Study of the Effects of an Environmental Disaster near Seveso, Italy. JAMA 1986, 256, 2687–2695. [Google Scholar] [CrossRef]
- Lin, C.-Y.; Lin, L.-Y.; Chiang, C.-K.; Wang, W.-J.; Su, Y.-N.; Hung, K.-Y.; Chen, P.-C. Investigation of the Associations between Low-Dose Serum Perfluorinated Chemicals and Liver Enzymes in US Adults. Am. J. Gastroenterol. 2010, 105, 1354–1363. [Google Scholar] [CrossRef] [PubMed]
- Gleason, J.A.; Post, G.B.; Fagliano, J.A. Associations of Perfluorinated Chemical Serum Concentrations and Biomarkers of Liver Function and Uric Acid in the US Population (NHANES), 2007-2010. Environ. Res. 2015, 136, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Jin, R.; McConnell, R.; Catherine, C.; Xu, S.; Walker, D.I.; Stratakis, N.; Jones, D.P.; Miller, G.W.; Peng, C.; Conti, D.V.; et al. Perfluoroalkyl Substances and Severity of Nonalcoholic Fatty Liver in Children: An Untargeted Metabolomics Approach. Environ. Int. 2020, 134, 105220. [Google Scholar] [CrossRef] [PubMed]
- Lang, I.A.; Galloway, T.S.; Scarlett, A.; Henley, W.E.; Depledge, M.; Wallace, R.B.; Melzer, D. Association of Urinary Bisphenol A Concentration with Medical Disorders and Laboratory Abnormalities in Adults. JAMA 2008, 300, 1303–1310. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.-R.; Park, H.; Bae, S.; Lim, Y.-H.; Kim, J.H.; Cho, S.-H.; Hong, Y.-C. Urinary Bisphenol A Concentrations Are Associated with Abnormal Liver Function in the Elderly: A Repeated Panel Study. J. Epidemiol. Community Health 2014, 68, 312–317. [Google Scholar] [CrossRef] [PubMed]
- Tarantino, G.; Valentino, R.; Di Somma, C.; D’Esposito, V.; Passaretti, F.; Pizza, G.; Brancato, V.; Orio, F.; Formisano, P.; Colao, A.; et al. Bisphenol A in Polycystic Ovary Syndrome and Its Association with Liver-Spleen Axis. Clin. Endocrinol. 2013, 78, 447–453. [Google Scholar] [CrossRef] [PubMed]
- Khalil, N.; Ebert, J.R.; Wang, L.; Belcher, S.; Lee, M.; Czerwinski, S.A.; Kannan, K. Bisphenol A and Cardiometabolic Risk Factors in Obese Children. Sci. Total Environ. 2014, 470–471, 726–732. [Google Scholar] [CrossRef] [PubMed]
- Medic-Stojanoska, M.; Milosevic, N.; Milanovic, M.; Stojanoski, S.; Vukovic, B.; Icin, T.; Bajkin, I.; Stepanovic, K.; Sudji, J.; Milic, N. Can Phthalates Impair Liver Function? Endocr. Abstr. 2019. [Google Scholar] [CrossRef]
- Milošević, N.; Milić, N.; Živanović Bosić, D.; Bajkin, I.; Perčić, I.; Abenavoli, L.; Medić Stojanoska, M. Potential Influence of the Phthalates on Normal Liver Function and Cardiometabolic Risk in Males. Environ. Monit. Assess. 2017, 190, 17. [Google Scholar] [CrossRef]
- Motamed, N.; Sohrabi, M.; Ajdarkosh, H.; Hemmasi, G.; Maadi, M.; Sayeedian, F.S.; Pirzad, R.; Abedi, K.; Aghapour, S.; Fallahnezhad, M.; et al. Fatty Liver Index vs. Waist Circumference for Predicting Non-Alcoholic Fatty Liver Disease. World J. Gastroenterol. 2016, 22, 3023–3030. [Google Scholar] [CrossRef]
- Lee, S.B.; Kim, M.K.; Kang, S.; Park, K.; Kim, J.H.; Baik, S.J.; Nam, J.S.; Ahn, C.W.; Park, J.S. Triglyceride Glucose Index Is Superior to the Homeostasis Model Assessment of Insulin Resistance for Predicting Nonalcoholic Fatty Liver Disease in Korean Adults. Endocrinol. Metab. Seoul Korea 2019, 34, 179–186. [Google Scholar] [CrossRef]
- Xu, C.; Ma, Z.; Wang, Y.; Liu, X.; Tao, L.; Zheng, D.; Guo, X.; Yang, X. Visceral Adiposity Index as a Predictor of NAFLD: A Prospective Study with 4-Year Follow-Up. Liver Int. Off. J. Int. Assoc. Study Liver 2018, 38, 2294–2300. [Google Scholar] [CrossRef]
- Özcabı, B.; Demirhan, S.; Akyol, M.; Öztürkmen Akay, H.; Güven, A. Lipid Accumulation Product Is a Predictor of Nonalcoholic Fatty Liver Disease in Childhood Obesity. Korean J. Pediatr. 2019, 62, 450–455. [Google Scholar] [CrossRef]
- Mansour-Ghanaei, R.; Mansour-Ghanaei, F.; Naghipour, M.; Joukar, F.; Atrkar-Roushan, Z.; Tabatabaii, M.; Ghorani, N. The Role of Anthropometric Indices in the Prediction of Non-Alcoholic Fatty Liver Disease in the PERSIAN Guilan Cohort Study (PGCS). J. Med. Life 2018, 11, 194–202. [Google Scholar] [CrossRef]
- Hatch, E.E.; Nelson, J.W.; Qureshi, M.M.; Weinberg, J.; Moore, L.L.; Singer, M.; Webster, T.F. Association of Urinary Phthalate Metabolite Concentrations with Body Mass Index and Waist Circumference: A Cross-Sectional Study of NHANES Data, 1999–2002. Environ. Health Glob. Access Sci. Source 2008, 7, 27. [Google Scholar] [CrossRef]
- Stahlhut, R.W.; van Wijngaarden, E.; Dye, T.D.; Cook, S.; Swan, S.H. Concentrations of Urinary Phthalate Metabolites Are Associated with Increased Waist Circumference and Insulin Resistance in Adult U.S. Males. Environ. Health Perspect. 2007, 115, 876–882. [Google Scholar] [CrossRef]
- Lee, D.-H.; Lee, I.-K.; Jin, S.-H.; Steffes, M.; Jacobs, D.R. Association between Serum Concentrations of Persistent Organic Pollutants and Insulin Resistance among Nondiabetic Adults: Results from the National Health and Nutrition Examination Survey 1999–2002. Diabetes Care 2007, 30, 622–628. [Google Scholar] [CrossRef] [PubMed]
- La Merrill, M.A.; Johnson, C.L.; Smith, M.T.; Kandula, N.R.; Macherone, A.; Pennell, K.D.; Kanaya, A.M. Exposure to Persistent Organic Pollutants (POPs) and Their Relationship to Hepatic Fat and Insulin Insensitivity among Asian Indian Immigrants in the United States. Environ. Sci. Technol. 2019, 53, 13906–13918. [Google Scholar] [CrossRef]
- Geiger, S.D.; Yao, P.; Vaughn, M.G.; Qian, Z. PFAS Exposure and Overweight/Obesity among Children in a Nationally Representative Sample. Chemosphere 2021, 268, 128852. [Google Scholar] [CrossRef]
- Carter, D.; Dieterich, D.T.; Chang, C. Nonalcoholic Fatty Liver Disease/Nonalcoholic Steatohepatitis in Liver Transplantation. Clin. Liver Dis. 2018, 22, 213–227. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Type | Chemical Name | Abbreviation | Introduction Date | Restricted/ Banned | Source |
|---|---|---|---|---|---|
| Residential | |||||
| Phenols | Bisphenol A Bisphenol S | BPA BPS | 1960 | Restricted | Polycarbonate plastics, epoxy resins, plastic toys and bottles, lining of food cans |
| Phthalates | Mono-(2-ethylhexyl)-phthalate Di-(2-ethylhexyl)-phthalate Dibutyl-phthalate Dicyclohexyl phthalate | MEHP DEHP DBP DCHP | 1920 | Restricted | PVC: lubricants, perfumes, cosmetics, medical tubing, wood finishes, adhesives, paints, toys, emulsifiers in food, flooring, personal care products |
| Perfluorinated chemicals | Perfluorooctanoic acid Perfluoroctanesulfonates Perfluorononanoic acid Perfluorohexanesulfonic Acid | PFOA PFOS PFNA PFHxS | 1940 | Restricted | Contaminated food and water, dust, floor waxes, firefighting foam, electrical wiring, lining of food wrappers, stain resistant carpeting |
| Industrialist | |||||
| Dioxins | Polychlorinated Dibenzo P | PCDD | 1872 | Restricted | By-product of chlorinated herbicide production, smelting, chlorine bleaching of paper |
| Polychlorinated biphenyls | Polychlorinated biphenyls Polybrominated biphenyls Polychlorinated terphenyls Polychlorinated naphthalenes | PCBs PBBs PCTs PCNs | 1927 | Banned | Contaminated air and food, skin contact with old electrical equipment |
| Polycyclic aromatic hydrocarbons | Benzo[a]pyrene, anthracene, acenaphtylene, fluorene | PAH | – | Restricted | Products of fuel burning |
| Alkylphenols | Nonylphenol Octylphenol | NP OP | – | Restricted and banned in certain areas of use in the USA | Surfactants, detergents, emulsifiers; fish, drinking water, personal care products |
| Heavy metals | Arsenic | As | – | Restricted | Pesticides, smelting, industrial waste, drinking water, soil, seafood, rice, mushrooms, poultry |
| Mercury | Hg | – | Restricted | Mining, waste incineration, manufacturing; fish, shellfish, medical/dental procedures | |
| Cadmium | Cd | – | Restricted | Soil, water, air; leafy vegetables, peanuts, soybeans, sunflower seeds; inhalation products of mining, combustion, waste incineration | |
| Agricultural | |||||
| Dicarboximide | Vinclozolin | Vnz | 1981 | Banned | Diet and occupational |
| Organotins | Tributyltin oxide Triphenyltin | TBT TPT | – | Banned by many countries | Used as a biocide (fungicide and molluscicide), especially as a wood preservative |
| Organochloride | Dichlorodiphenyltrichloroethane Dichlorodiphenyldichloroethylene | DDT DDE | 1940 | Banned | Contaminated water, soil crops, fish, pesticides |
| Chlorotriazine | Atrazine | ATR | 1959 | Banned | Pesticide/herbicide, contaminated water and soil |
| Pharmaceutics | |||||
| Parabens | Butylparaben, methylparaben, ethylparaben, propylparaben, benzylparaben | Parabens | 1924 | Restricted | Antimicrobial agents for the preservation of food, paper products, and pharmaceutical products |
| Non-steroidal synthetic estrogen | Diethylstilbestro | DES | 1941–1947 | Restricted | Pharmaceutical |
| Author [Ref] | EDC | Methodology | Results |
|---|---|---|---|
| Cave et al. [153] | PCBs | Cross-sectional cohort study evaluating the influence of environmental pollutants in serum ALT in 436 adults. | 20 PCBs were positively associated with subjects that had elevated ALT levels (p ≤ 0.05). |
| Lee et al. [156] | Dioxins | Cross-sectional study which evaluated the associations between serum PCDD/Fs levels and adverse hepatic-related health outcomes in adults. | In comparison to the control group, the risk of fatty liver increased significantly in adults with higher BMI and higher serum PCDD/Fs (OR = 27.00, 95% CI = 4.47–229.58). |
| Jin et al. [160] | PFAS | Cross-sectional study assessing the relationship of PFAS to histologic severity of NAFLD in 74 children. | The odds of having NASH significantly increased with the increase in plasma concentrations of PFOS (OR: 3.32, 95% CI: 1.40–7.87), PFHxS (OR: 4.18, 95% CI: 1.64–10.7), and PFAS composite variable (OR: 4.89, 95% CI: 1.86–12.8). |
| Lin et al. [158] | PFOA | Cross-sectional cohort study examining the relationship between serum levels of PFOA and the levels of liver enzymes in 2216 adults. | When PFOA concentration increased by one unit, the serum levels of ALT and GGT increased by 1.86 (95% CI, 1.24–2.48; p = 0.005) and 0.08 units (95% CI, 0.05–0.11; p = 0.019), respectively. |
| Tarantino et al. [163] | BPA | Cross-sectional study that evaluated the effects of increased serum BPA levels on low-grade chronic inflammation and hepatic steatosis in women with polycystic ovary syndrome. | Higher serum levels of BPA were associated with higher grades of hepatic steatosis and AST, ALT, and GGT (p ≤ 0.05). |
| Milošević et al. [166] | Phthalates | Cross-sectional study with 102 male participants assessing the influence of MEP and MEHP on the liver function and cardiometabolic risk factors. | MEP+ normal weight group had statistically significant elevated transaminase serum levels. Moreover, there were correlations found between MEP concentration in urine samples and TAG serum levels (r2 = 0.33; p < 0.01), VAI (r2 = 0.41; p < 0.01), LAP (r2 = 0.32; p < 0.01), and TAG-to-HDL ratio (r2 = 0.40, p < 0.01) among obese subjects. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cano, R.; Pérez, J.L.; Dávila, L.A.; Ortega, Á.; Gómez, Y.; Valero-Cedeño, N.J.; Parra, H.; Manzano, A.; Véliz Castro, T.I.; Albornoz, M.P.D.; et al. Role of Endocrine-Disrupting Chemicals in the Pathogenesis of Non-Alcoholic Fatty Liver Disease: A Comprehensive Review. Int. J. Mol. Sci. 2021, 22, 4807. https://doi.org/10.3390/ijms22094807
Cano R, Pérez JL, Dávila LA, Ortega Á, Gómez Y, Valero-Cedeño NJ, Parra H, Manzano A, Véliz Castro TI, Albornoz MPD, et al. Role of Endocrine-Disrupting Chemicals in the Pathogenesis of Non-Alcoholic Fatty Liver Disease: A Comprehensive Review. International Journal of Molecular Sciences. 2021; 22(9):4807. https://doi.org/10.3390/ijms22094807
Chicago/Turabian StyleCano, Raquel, José L. Pérez, Lissé Angarita Dávila, Ángel Ortega, Yosselin Gómez, Nereida Josefina Valero-Cedeño, Heliana Parra, Alexander Manzano, Teresa Isabel Véliz Castro, María P. Díaz Albornoz, and et al. 2021. "Role of Endocrine-Disrupting Chemicals in the Pathogenesis of Non-Alcoholic Fatty Liver Disease: A Comprehensive Review" International Journal of Molecular Sciences 22, no. 9: 4807. https://doi.org/10.3390/ijms22094807
APA StyleCano, R., Pérez, J. L., Dávila, L. A., Ortega, Á., Gómez, Y., Valero-Cedeño, N. J., Parra, H., Manzano, A., Véliz Castro, T. I., Albornoz, M. P. D., Cano, G., Rojas-Quintero, J., Chacín, M., & Bermúdez, V. (2021). Role of Endocrine-Disrupting Chemicals in the Pathogenesis of Non-Alcoholic Fatty Liver Disease: A Comprehensive Review. International Journal of Molecular Sciences, 22(9), 4807. https://doi.org/10.3390/ijms22094807