Serum Tau Proteins as Potential Biomarkers for the Assessment of Alzheimer’s Disease Progression




Abstract
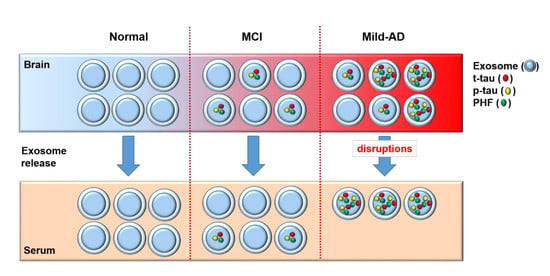
1. Introduction
2. Results
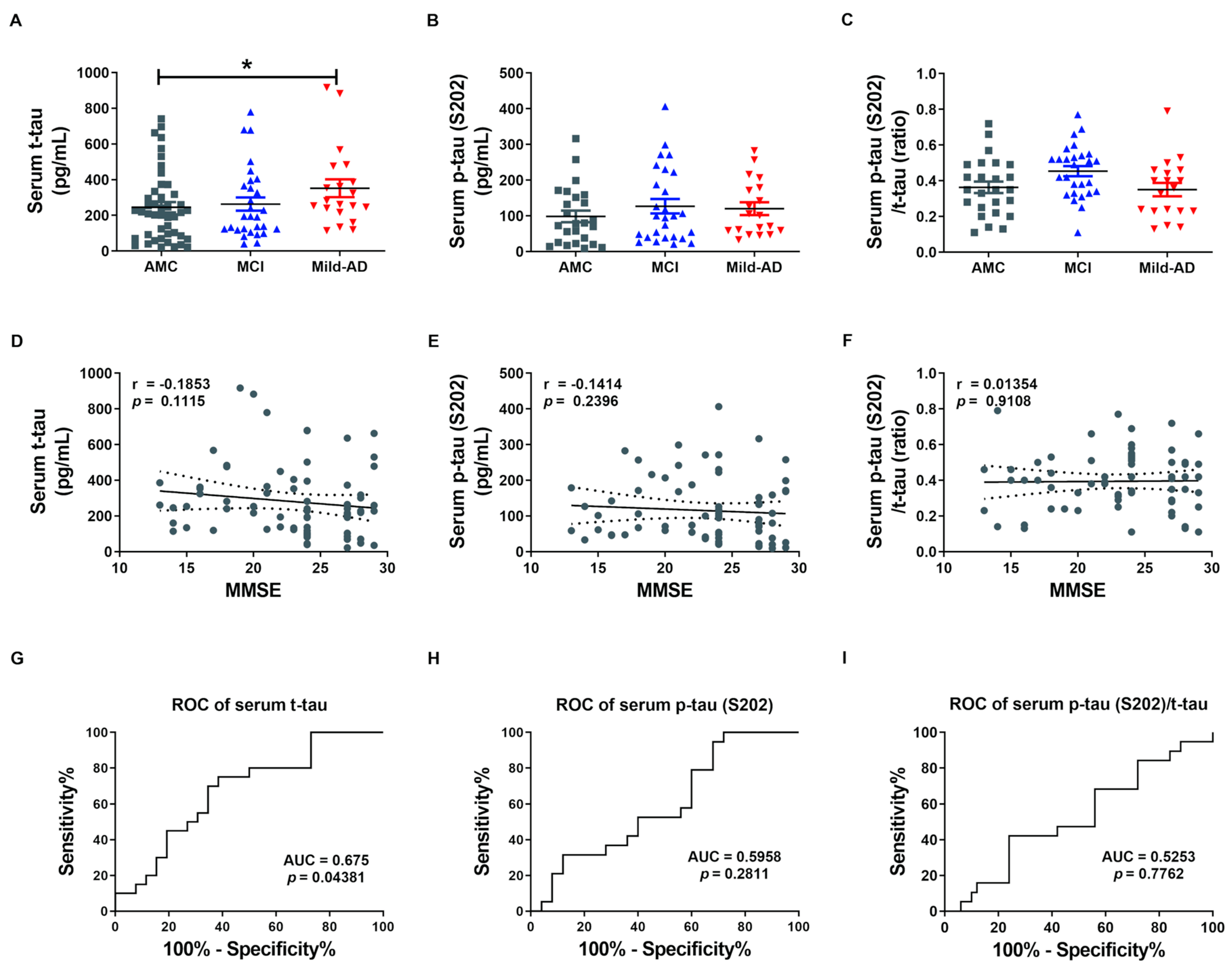
2.1. Serum t-tau and p-tau Protein Levels in Controls, Mild Cognitive Impairments, and Mild-AD Patients
2.2. Characteristics of Neuronal Cell-Derived Exosomes (NEX)
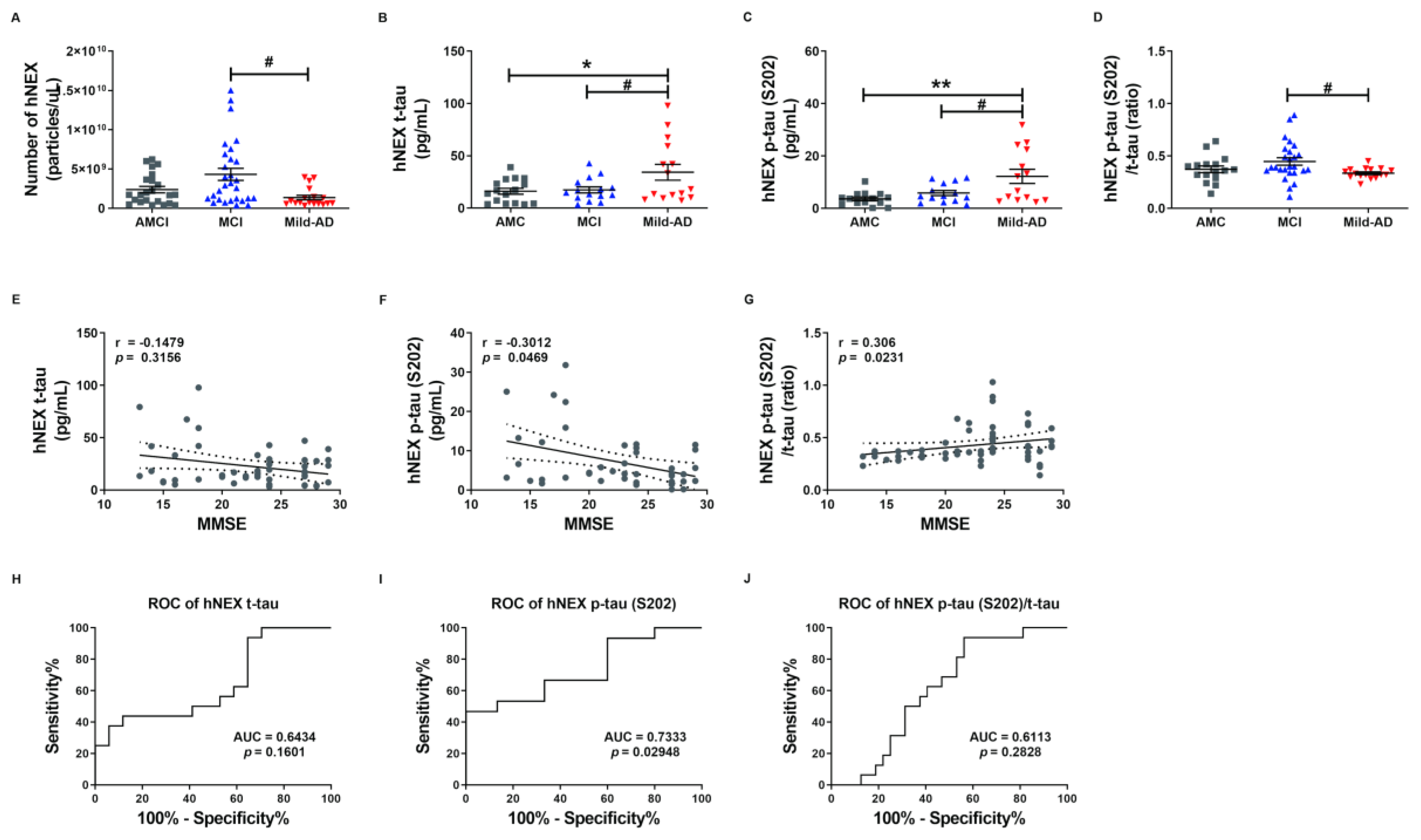
2.3. NEX t-tau and p-tau Protein Levels in Controls, Mild Cognitive Impairments, and Mild-AD Patients
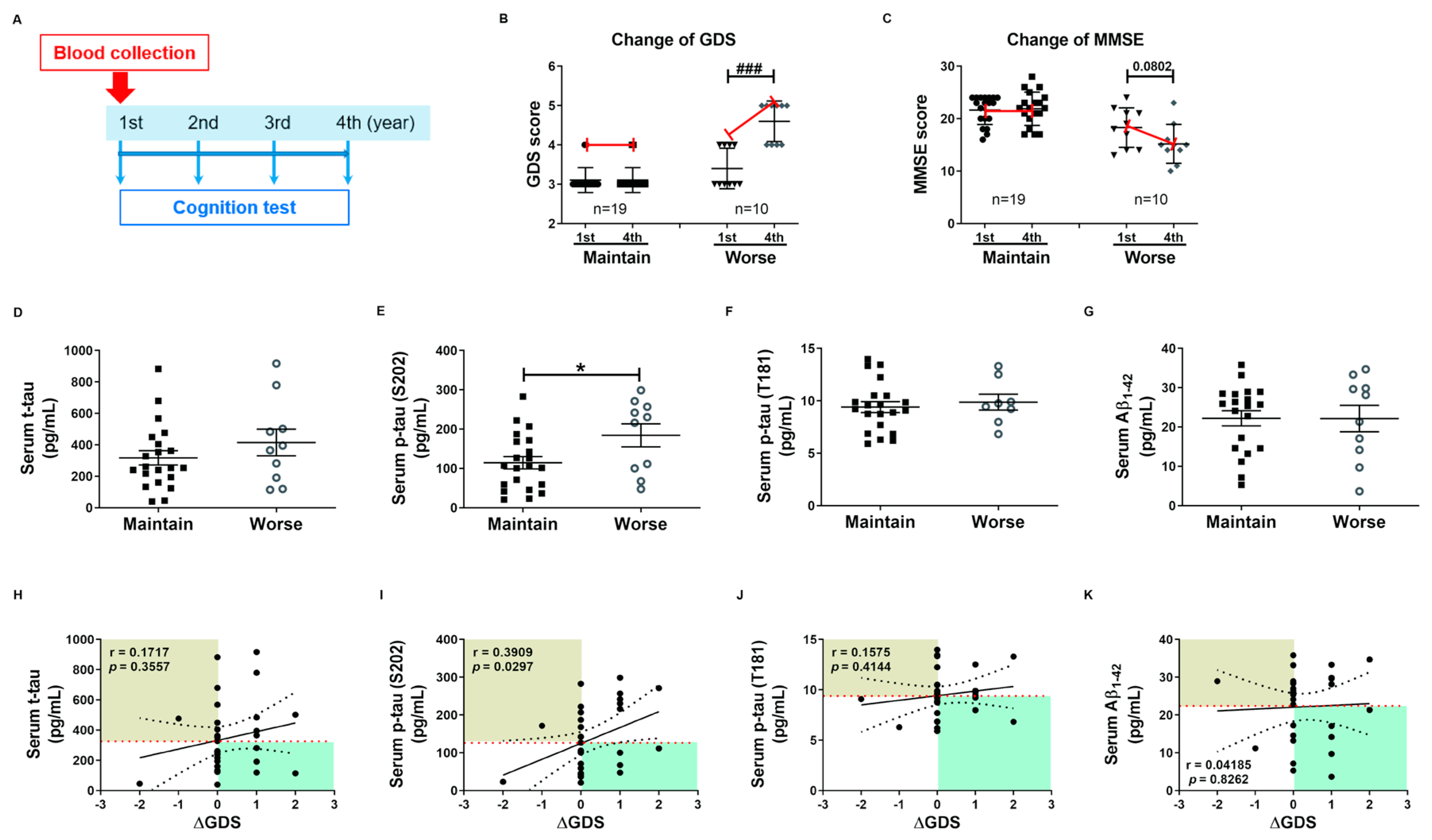
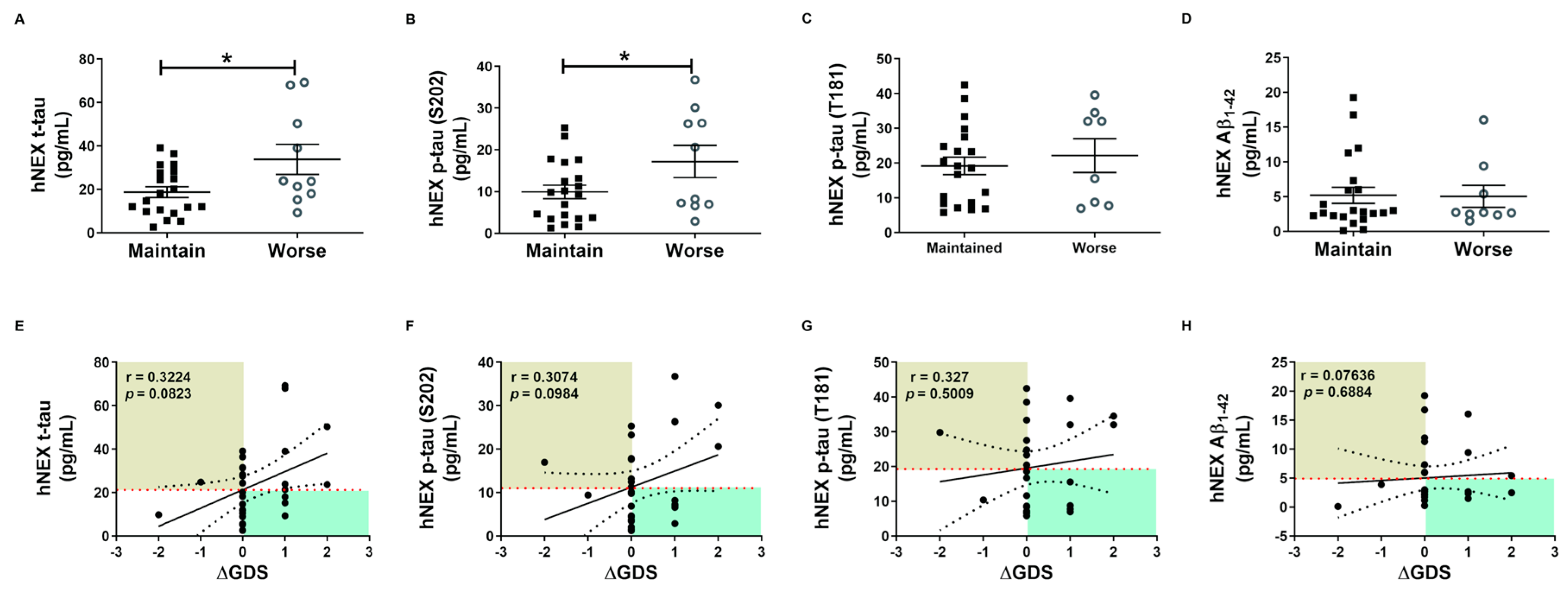
2.4. Phosphorylated tau Protein Levels in Serum and hNEX Predict Cognitive Deterioration
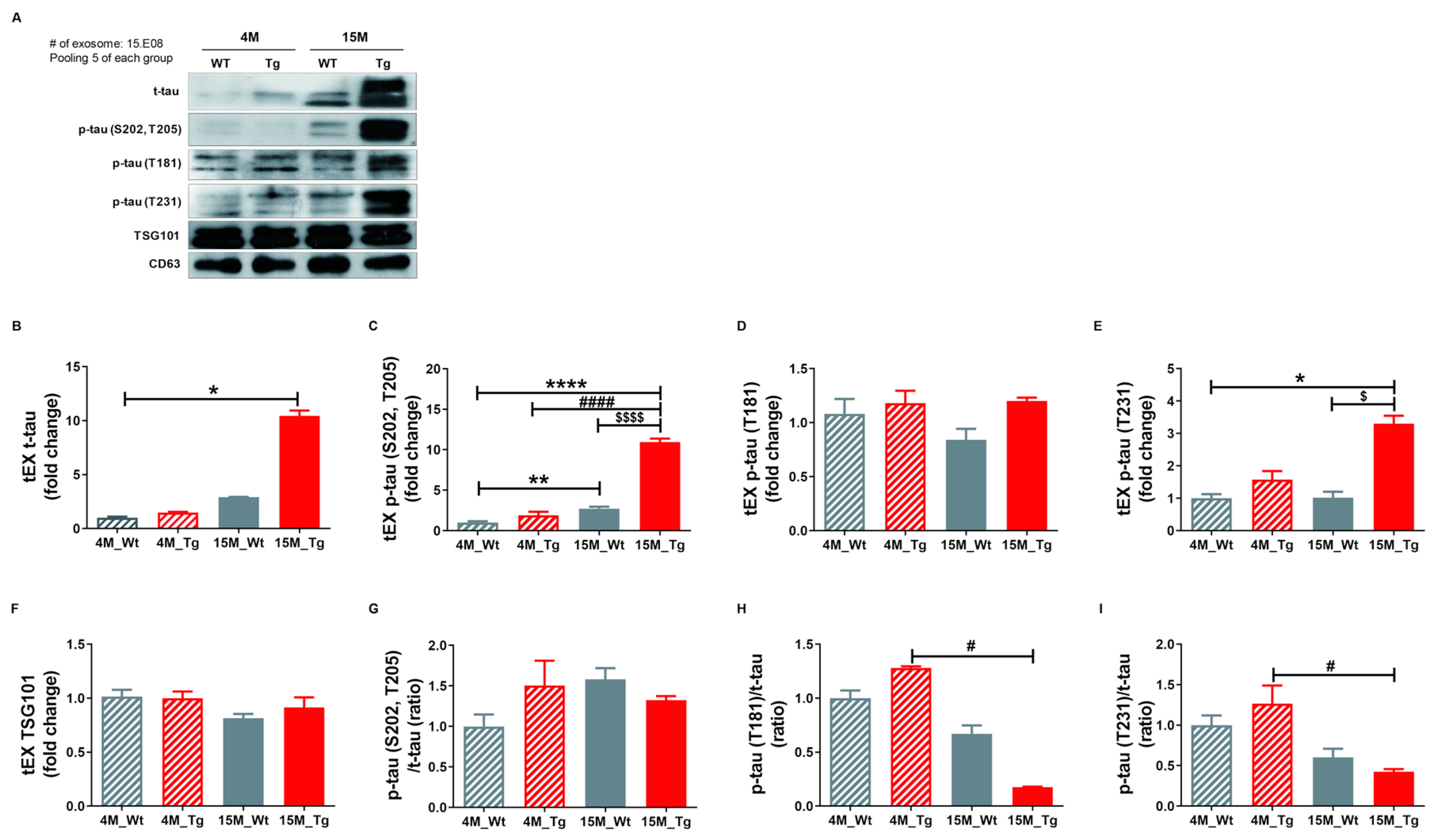
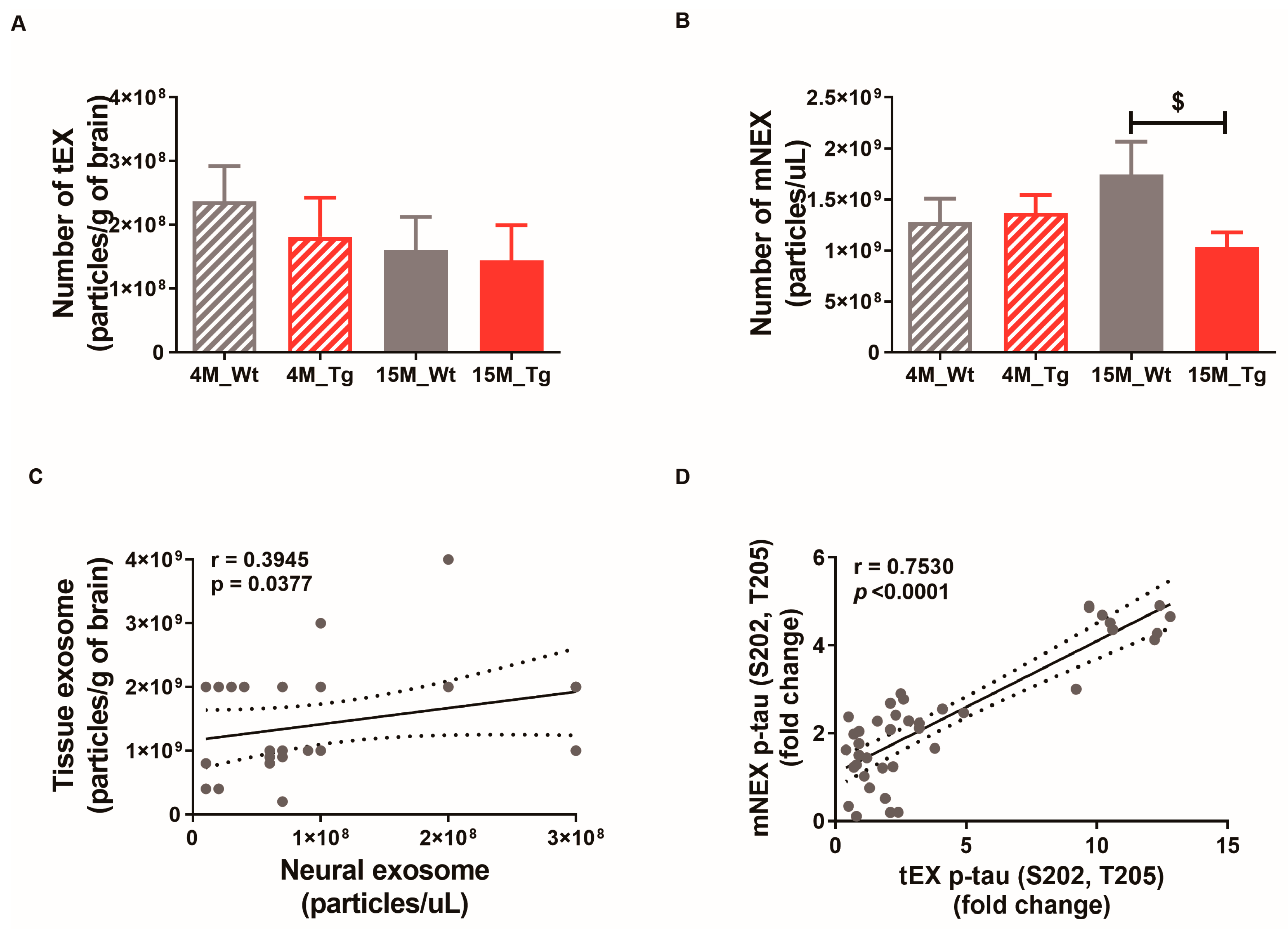
2.5. Phosphorylated Tau Protein Expression in Brain Tissue Exosomes of JNPL3 Mice Increased with The Progression of Pathology
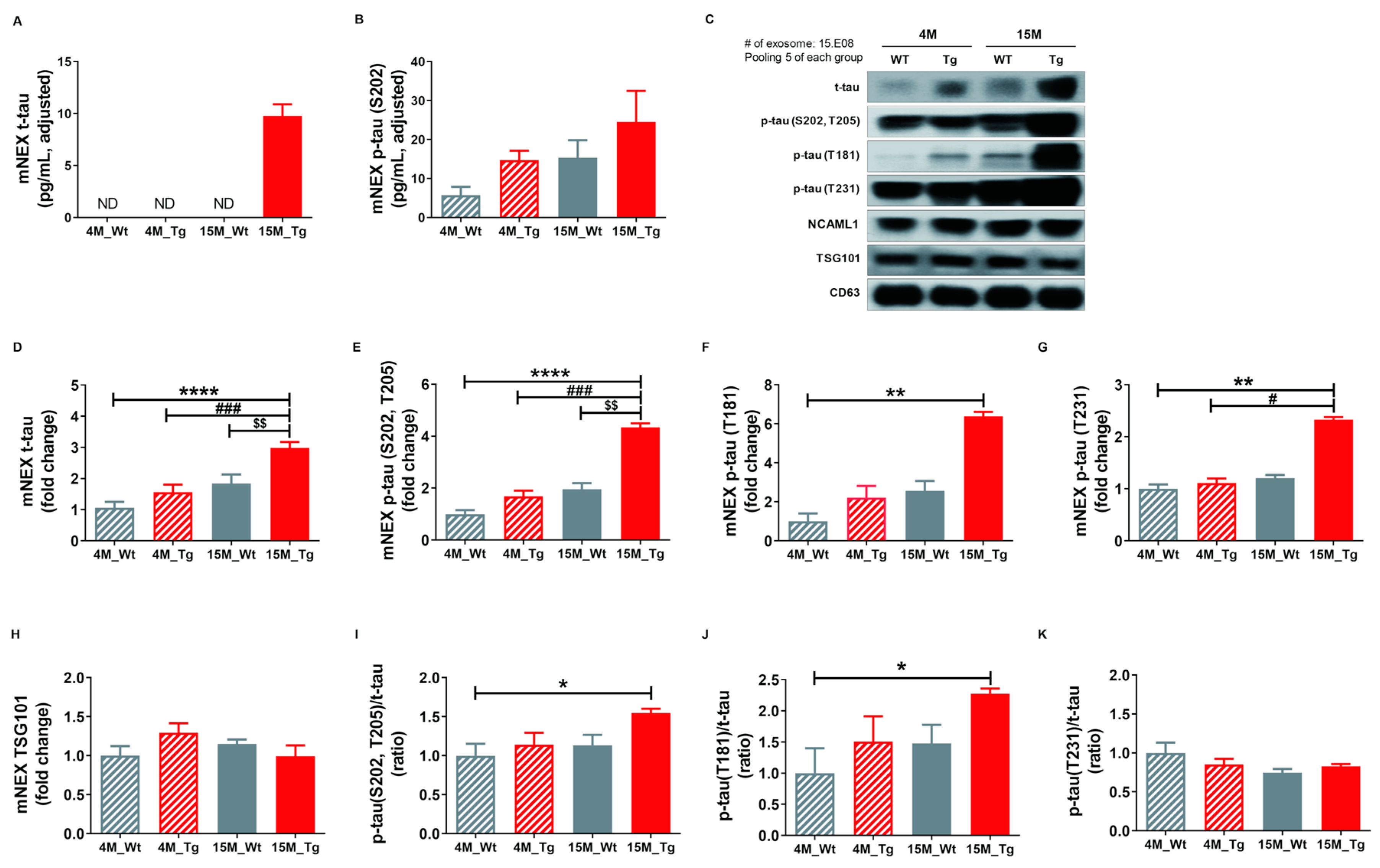
2.6. Total Tau and Phosphorylated Tau Protein Levels in Blood NEX from JNPL3 Mice Increased with The Progression of Pathology
3. Discussion
4. Materials and Methods
4.1. Patients, Controls, and Methods
4.2. Serum Separation
4.3. Animals
4.4. Isolation of NEX from Serum
4.5. Isolation of TEX from Brain Tissue
4.6. Electron Microscopy
4.7. Nanoparticle Tracking Analysis
4.8. Measurements of t-tau and p-tau Protein Levels
4.9. Western Blot Analysis
4.10. Immunofluorescence
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| Aβ | Amyloid-beta |
| AMC | age-matched control |
| APP | Amyloid precursor protein |
| CDR-SOB | Clinical Dementia Rating-Sum of Box |
| CSF | cerebrospinal fluid |
| DAB | 3,3′–diaminobenzidine |
| DAPI | 4′6-diamidino-2-phenylindole |
| ELISA | enzyme-linked immunosorbent assays |
| GDS | Global Deterioration Scale |
| IHC | Immunohistochemistry |
| MCI | Mild cognitive impairment |
| MMSE | Mini-Mental State Examination |
| NEX | neuronal-derived exosomes |
| NFTs | Neurofibrillary tangles |
| NINCDS-ADRDA | National Institute of Neurological and Communicative Disorders and Stroke-Alzheimer’s Disease and Related Disorders Association |
| PBS | Phosphate-buffered saline |
| PFA | Paraformaldehyde |
| p-tau | phosphorylated tau |
| TBS | Tris-buffered saline |
| °C | Celsius |
| TEM | Transmissin electron microscopy |
| t-tau | total tau |
References
- Burns, A.; Iliffe, S. Alzheimer’s disease. BMJ 2009, 338, b158. [Google Scholar] [CrossRef] [PubMed]
- Wortmann, M. Dementia: A global health priority - highlights from an ADI and World Health Organization report. Alzheimers Res. 2012, 4, 40. [Google Scholar] [CrossRef] [PubMed]
- Rapoport, M.; Dawson, H.N.; Binder, L.I.; Vitek, M.P.; Ferreira, A. Tau is essential to beta -amyloid-induced neurotoxicity. Proc. Natl. Acad. Sci. USA 2002, 99, 6364–6369. [Google Scholar] [CrossRef] [PubMed]
- Ballatore, C.; Lee, V.M.; Trojanowski, J.Q. Tau-mediated neurodegeneration in Alzheimer’s disease and related disorders. Nat. Rev. Neurosci. 2007, 8, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Holtzman, D.M.; Carrillo, M.C.; Hendrix, J.A.; Bain, L.J.; Catafau, A.M.; Gault, L.M.; Goedert, M.; Mandelkow, E.; Mandelkow, E.M.; Miller, D.S.; et al. Tau: From research to clinical development. Alzheimers Dement. 2016, 12, 1033–1039. [Google Scholar] [CrossRef]
- Noble, W.; Hanger, D.P.; Miller, C.C.; Lovestone, S. The importance of tau phosphorylation for neurodegenerative diseases. Front. Neurol. 2013, 4, 83. [Google Scholar] [CrossRef] [PubMed]
- Buee, L.; Bussiere, T.; Buee-Scherrer, V.; Delacourte, A.; Hof, P.R. Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res. Brain Res. Rev. 2000, 33, 95–130. [Google Scholar] [CrossRef]
- Mandelkow, E.M.; Mandelkow, E. Biochemistry and cell biology of tau protein in neurofibrillary degeneration. Cold Spring Harb Perspect Med. 2012, 2, a006247. [Google Scholar] [CrossRef] [PubMed]
- Dubois, B.; Feldman, H.H.; Jacova, C.; Dekosky, S.T.; Barberger-Gateau, P.; Cummings, J.; Delacourte, A.; Galasko, D.; Gauthier, S.; Jicha, G.; et al. Research criteria for the diagnosis of Alzheimer’s disease: Revising the NINCDS-ADRDA criteria. Lancet Neurol. 2007, 6, 734–746. [Google Scholar] [CrossRef]
- Naik, M.; Nygaard, H.A. Diagnosing dementia - ICD-10 not so bad after all: A comparison between dementia criteria according to DSM-IV and ICD-10. Int. J. Geriatr. Psychiatry 2008, 23, 279–282. [Google Scholar] [CrossRef]
- Olsson, B.; Lautner, R.; Andreasson, U.; Ohrfelt, A.; Portelius, E.; Bjerke, M.; Holtta, M.; Rosen, C.; Olsson, C.; Strobel, G.; et al. CSF and blood biomarkers for the diagnosis of Alzheimer’s disease: A systematic review and meta-analysis. Lancet Neurol. 2016, 15, 673–684. [Google Scholar] [CrossRef]
- Tapiola, T.; Alafuzoff, I.; Herukka, S.K.; Parkkinen, L.; Hartikainen, P.; Soininen, H.; Pirttila, T. Cerebrospinal fluid β-amyloid 42 and tau proteins as biomarkers of Alzheimer-type pathologic changes in the brain. Arch. Neurol. 2009, 66, 382–389. [Google Scholar] [CrossRef] [PubMed]
- Jack, C.R., Jr.; Dickson, D.W.; Parisi, J.E.; Xu, Y.C.; Cha, R.H.; O’Brien, P.C.; Edland, S.D.; Smith, G.E.; Boeve, B.F.; Tangalos, E.G.; et al. Antemortem MRI findings correlate with hippocampal neuropathology in typical aging and dementia. Neurology 2002, 58, 750–757. [Google Scholar] [CrossRef]
- Hulstaert, F.; Blennow, K.; Ivanoiu, A.; Schoonderwaldt, H.C.; Riemenschneider, M.; De Deyn, P.P.; Bancher, C.; Cras, P.; Wiltfang, J.; Mehta, P.D.; et al. Improved discrimination of AD patients using beta-amyloid(1–42) and tau levels in CSF. Neurology 1999, 52, 1555–1562. [Google Scholar] [CrossRef] [PubMed]
- Zetterberg, H.; Wilson, D.; Andreasson, U.; Minthon, L.; Blennow, K.; Randall, J.; Hansson, O. Plasma tau levels in Alzheimer’s disease. Alzheimers Res. 2013, 5, 9. [Google Scholar] [CrossRef] [PubMed]
- Chiu, M.J.; Chen, Y.F.; Chen, T.F.; Yang, S.Y.; Yang, F.P.; Tseng, T.W.; Chieh, J.J.; Chen, J.C.; Tzen, K.Y.; Hua, M.S.; et al. Plasma tau as a window to the brain-negative associations with brain volume and memory function in mild cognitive impairment and early Alzheimer’s disease. Hum. Brain Mapp. 2014, 35, 3132–3142. [Google Scholar] [CrossRef] [PubMed]
- Yanamandra, K.; Patel, T.K.; Jiang, H.; Schindler, S.; Ulrich, J.D.; Boxer, A.L.; Miller, B.L.; Kerwin, D.R.; Gallardo, G.; Stewart, F.; et al. Anti-tau antibody administration increases plasma tau in transgenic mice and patients with tauopathy. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef]
- Mielke, M.M.; Hagen, C.E.; Xu, J.; Chai, X.; Vemuri, P.; Lowe, V.J.; Airey, D.C.; Knopman, D.S.; Roberts, R.O.; Machulda, M.M.; et al. Plasma phospho-tau181 increases with Alzheimer’s disease clinical severity and is associated with tau- and amyloid-positron emission tomography. Alzheimers Dement. 2018, 14, 989–997. [Google Scholar] [CrossRef]
- Mattsson, N.; Zetterberg, H.; Janelidze, S.; Insel, P.S.; Andreasson, U.; Stomrud, E.; Palmqvist, S.; Baker, D.; Tan Hehir, C.A.; Jeromin, A.; et al. Plasma tau in Alzheimer disease. Neurology 2016, 87, 1827–1835. [Google Scholar] [CrossRef]
- Tatebe, H.; Kasai, T.; Ohmichi, T.; Kishi, Y.; Kakeya, T.; Waragai, M.; Kondo, M.; Allsop, D.; Tokuda, T. Quantification of plasma phosphorylated tau to use as a biomarker for brain Alzheimer pathology: Pilot case-control studies including patients with Alzheimer’s disease and down syndrome. Mol. Neurodegener. 2017, 12, 63. [Google Scholar] [CrossRef]
- Park, J.C.; Han, S.H.; Yi, D.; Byun, M.S.; Lee, J.H.; Jang, S.; Ko, K.; Jeon, S.Y.; Lee, Y.S.; Kim, Y.K.; et al. Plasma tau/amyloid-beta1-42 ratio predicts brain tau deposition and neurodegeneration in Alzheimer’s disease. Brain 2019, 142, 771–786. [Google Scholar] [CrossRef] [PubMed]
- Fruhbeis, C.; Frohlich, D.; Kramer-Albers, E.M. Emerging roles of exosomes in neuron-glia communication. Front. Physiol 2012, 3, 119. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.G.; Grizzle, W.E. Exosomes: A novel pathway of local and distant intercellular communication that facilitates the growth and metastasis of neoplastic lesions. Am. J. Pathol. 2014, 184, 28–41. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, L.; Honsho, M.; Zahn, T.R.; Keller, P.; Geiger, K.D.; Verkade, P.; Simons, K. Alzheimer’s disease beta-amyloid peptides are released in association with exosomes. Proc. Natl. Acad. Sci. USA 2006, 103, 11172–11177. [Google Scholar] [CrossRef] [PubMed]
- Sheinerman, K.S.; Umansky, S.R. Circulating cell-free microRNA as biomarkers for screening, diagnosis and monitoring of neurodegenerative diseases and other neurologic pathologies. Front. Cell Neurosci. 2013, 7, 150. [Google Scholar] [CrossRef] [PubMed]
- Fiandaca, M.S.; Kapogiannis, D.; Mapstone, M.; Boxer, A.; Eitan, E.; Schwartz, J.B.; Abner, E.L.; Petersen, R.C.; Federoff, H.J.; Miller, B.L.; et al. Identification of preclinical Alzheimer’s disease by a profile of pathogenic proteins in neurally derived blood exosomes: A case-control study. Alzheimers Dement. 2015, 11, 600–607.e1. [Google Scholar] [CrossRef] [PubMed]
- Perez-Gonzalez, R.; Gauthier, S.A.; Kumar, A.; Levy, E. The exosome secretory pathway transports amyloid precursor protein carboxyl-terminal fragments from the cell into the brain extracellular space. J. Biol. Chem. 2012, 287, 43108–43115. [Google Scholar] [CrossRef]
- Villemagne, V.L.; Burnham, S.; Bourgeat, P.; Brown, B.; Ellis, K.A.; Salvado, O.; Szoeke, C.; Macaulay, S.L.; Martins, R.; Maruff, P.; et al. Amyloid beta deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer’s disease: A prospective cohort study. Lancet Neurol. 2013, 12, 357–367. [Google Scholar] [CrossRef]
- Hanon, O.; Vidal, J.S.; Lehmann, S.; Bombois, S.; Allinquant, B.; Treluyer, J.M.; Gele, P.; Delmaire, C.; Blanc, F.; Mangin, J.F.; et al. Plasma amyloid levels within the Alzheimer’s process and correlations with central biomarkers. Alzheimers Dement. 2018, 14, 858–868. [Google Scholar] [CrossRef]
- Lambert, J.C.; Schraen-Maschke, S.; Richard, F.; Fievet, N.; Rouaud, O.; Berr, C.; Dartigues, J.F.; Tzourio, C.; Alperovitch, A.; Buee, L.; et al. Association of plasma amyloid beta with risk of dementia: The prospective Three-City Study. Neurology 2009, 73, 847–853. [Google Scholar] [CrossRef]
- Gabelle, A.; Richard, F.; Gutierrez, L.A.; Schraen, S.; Delva, F.; Rouaud, O.; Buee, L.; Dartigues, J.F.; Touchon, J.; Lambert, J.C.; et al. Plasma amyloid-beta levels and prognosis in incident dementia cases of the 3-City Study. J. Alzheimers Dis. 2013, 33, 381–391. [Google Scholar] [CrossRef] [PubMed]
- Lui, J.K.; Laws, S.M.; Li, Q.X.; Villemagne, V.L.; Ames, D.; Brown, B.; Bush, A.I.; De Ruyck, K.; Dromey, J.; Ellis, K.A.; et al. Plasma Amyloid-beta as a Biomarker in Alzheimer’s Disease: The AIBL Study of Aging. J. Alzheimers Dis. 2010, 20, 1233–1242. [Google Scholar] [CrossRef] [PubMed]
- Poljak, A.; Crawford, J.D.; Smythe, G.A.; Brodaty, H.; Slavin, M.J.; Kochan, N.A.; Trollor, J.N.; Wen, W.; Mather, K.A.; Assareh, A.A.; et al. The Relationship Between Plasma Abeta Levels, Cognitive Function and Brain Volumetrics: Sydney Memory and Ageing Study. Curr. Alzheimer Res. 2016, 13, 243–255. [Google Scholar] [CrossRef] [PubMed]
- Lovheim, H.; Elgh, F.; Johansson, A.; Zetterberg, H.; Blennow, K.; Hallmans, G.; Eriksson, S. Plasma concentrations of free amyloid beta cannot predict the development of Alzheimer’s disease. Alzheimers Dement. 2017, 13, 778–782. [Google Scholar] [CrossRef]
- Randall, J.; Mortberg, E.; Provuncher, G.K.; Fournier, D.R.; Duffy, D.C.; Rubertsson, S.; Blennow, K.; Zetterberg, H.; Wilson, D.H. Tau proteins in serum predict neurological outcome after hypoxic brain injury from cardiac arrest: Results of a pilot study. Resuscitation 2013, 84, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Hesse, C.; Rosengren, L.; Andreasen, N.; Davidsson, P.; Vanderstichele, H.; Vanmechelen, E.; Blennow, K. Transient increase in total tau but not phospho-tau in human cerebrospinal fluid after acute stroke. Neurosci. Lett. 2001, 297, 187–190. [Google Scholar] [CrossRef]
- Chang, K.A.; Shin, K.Y.; Nam, E.; Lee, Y.B.; Moon, C.; Suh, Y.H.; Lee, S.H. Plasma soluble neuregulin-1 as a diagnostic biomarker for Alzheimer’s disease. Neurochem. Int. 2016, 97, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.Y.; Lee, J.H.; Ju, Y.S.; Lee, K.U.; Kim, K.W.; Jhoo, J.H.; Yoon, J.C.; Ha, J.; Woo, J.I. The prevalence of dementia in older people in an urban population of Korea: The Seoul study. J. Am. Geriatr. Soc. 2002, 50, 1233–1239. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.H.; Na, D.L.; Lee, B.H.; Kang, S.J.; Ha, C.K.; Han, S.H.; Erzigkeit, H. Validation of the Korean version of the Bayer activities of daily living scale. Hum. Psychopharmacol. 2003, 18, 469–475. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| VARIABLES | AMC | MCI | Mild-AD |
|---|---|---|---|
| Number of subjects | 26 | 30 | 20 |
| Female (F)/Male (M) | 9 (F)/17 (M) | 18 (F)/12 (M) | 17 (F)/3 (M) |
| Age (years) (mean ± SEM) | 73.92 ± 0.88 | 75.13 ± 0.99 | 76.55 ± 1.33 |
| MMSE (mean ± SEM) | 27.69 ± 0.16 | 23.17 ± 0.20 *** | 16.55 ± 0.52 ****,## |
| CDR-SOB (mean ± SEM) | 0.75 ± 0.07 | 2.55 ± 0.03 **** | 4.48 ± 0.28 ****,# |
| GDS (mean ± SEM) | 2.00 ± 0.00 | 3.00 ± 0.00 **** | 3.75 ± 0.14 **** |
| Target | Species | Dilution | Company (Catalog) |
|---|---|---|---|
| For Western blot analysis | |||
| Anti-CD63 IgG | Rabbit | 1:1000 | System Biosciences (EXOAB-CD63A-1) |
| Anti-TSG101 IgG | Rabbit | 1:1000 | Abcam (AB125011) |
| Anti-NCAM-L1 IgG | Goat | 1:1000 | Santa Cruz Biotechnology (SC-1508) |
| Anti-Total tau (C-17) | Goat | 1:500 | Santa Cruz Biotechnology (SC-1995) |
| Anti-Phospho tau (S202, T205) (AT8) | Mouse | 1:500 | Thermo Fisher Scientific (MN1020) |
| Anti-Phospho tau (T181) (AT270) | Mouse | 1:500 | Thermo Fisher Scientific (MN1050) |
| Anti-Phospho tau (T231) (AT180) | Mouse | 1:500 | Thermo Fisher Scientific (MN1040) |
| Anti-mouse IgG HRP | 1:3000 | Biorad (170-6516) | |
| Anti-rabbit IgG HRP | 1:5000 | Biorad (170-6515) | |
| Anti-goat IgG HRP | 1:5000 | Santa cruz (SC-2020) | |
| For ELISA | |||
| Total tau | MyBioSource (MBS022635) | ||
| Phospho tau (S202, T205) | MyBioSource (MBS013458) | ||
| Phospho tau (T181) | MyBioSource (KHB00631) | ||
| Phospho tau (S231) | MyBioSource (KHB8051) | ||
| Amyloid bets1-42 | Thermo Fisher Scientific (KHB3544) | ||
| CD63 | System Biosciences (EXOEL-CD63) | ||
| For immunofluorescence staining | |||
| Anti-Phospho tau (S202, T205) | Mouse | 1:100 | Thermo Fisher Scientific (MN1020) |
| Alexa Fluor 555 Donkey anti mouse IgG | 1:300 | Invitrogen (A-21422) | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nam, E.; Lee, Y.-B.; Moon, C.; Chang, K.-A. Serum Tau Proteins as Potential Biomarkers for the Assessment of Alzheimer’s Disease Progression. Int. J. Mol. Sci. 2020, 21, 5007. https://doi.org/10.3390/ijms21145007
Nam E, Lee Y-B, Moon C, Chang K-A. Serum Tau Proteins as Potential Biomarkers for the Assessment of Alzheimer’s Disease Progression. International Journal of Molecular Sciences. 2020; 21(14):5007. https://doi.org/10.3390/ijms21145007
Chicago/Turabian StyleNam, Eunjoo, Yeong-Bae Lee, Cheil Moon, and Keun-A Chang. 2020. "Serum Tau Proteins as Potential Biomarkers for the Assessment of Alzheimer’s Disease Progression" International Journal of Molecular Sciences 21, no. 14: 5007. https://doi.org/10.3390/ijms21145007
APA StyleNam, E., Lee, Y.-B., Moon, C., & Chang, K.-A. (2020). Serum Tau Proteins as Potential Biomarkers for the Assessment of Alzheimer’s Disease Progression. International Journal of Molecular Sciences, 21(14), 5007. https://doi.org/10.3390/ijms21145007