Pro-Arrhythmic Signaling of Thyroid Hormones and Its Relevance in Subclinical Hyperthyroidism

 ,
,  and
and
Abstract
:1. Introduction
2. Atrial Fibrillation and Pro-Arrhythmic Signaling of TH
2.1. A Short Overview on AF
2.2. Thyroid Status Imbalance Promoting AF
3. Impact of Thyroid Hormones on Ventricular Arrhythmias
3.1. A Brief Overview on VF
3.2. Thyroid Status Imbalance Promotes VF
4. Thyroid Malignancies, Treatment, and Risk for Arrhythmias
4.1. Pathomechanisms, Incidence, and Treatment of Thyroid Carcinoma (TCA)
4.2. Thyroid Carcinoma Suppressive Therapy and Risk for Cardiac Arrhythmias
5. Cellular and Molecular Actions Potentially Involved in Pro-Arrhythmic Signaling of TH
5.1. Targeting Cardiac Ion Channels
5.2. Targeting Cardiac Ca2+ Handling
5.3. Targeting Myocardial Metabolism, Structure, and Intercellular Coupling
5.4. Targeting ANS and RAAS
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Farré, J.; Wellens, H.J. Philippe Coumel: A founding father of modern arrhythmology. Europace 2004, 6, 464–465. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, B.F. Cardiac arrhythmias: What do we need to know about basic mechanisms? J. Cardiovasc. Electrophysiol. 1999, 10, 414–416. [Google Scholar] [PubMed]
- Heijman, J.; Voigt, N.; Nattel, S.; Dobrev, D. Cellular and molecular electrophysiology of atrial fibrillation initiation, maintenance, and progression. Circ. Res. 2014, 114, 1483–1499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voigt, N.; Heijman, J.; Wang, Q.; Chiang, D.; Li, N.; Karck, M.; Wehrens, X.; Nattel, S.; Dobrev, D. Cellular and Molecular Mechanisms of Atrial Arrhythmogenesis in Patients with Paroxysmal Atrial Fibrillation. Circulation 2014, 129, 145–156. [Google Scholar] [CrossRef] [Green Version]
- Tribulova, N.; Egan Benova, T.; Szeiffova Bacova, B.; Viczenczova, C.; Barancik, M. New aspects of pathogenesis of atrial fibrillation: Remodeling of intercalated discs. J. Physiol. Pharmacol. 2015, 66, 625–634. [Google Scholar]
- Tribulova, N.; Szeiffova Bacova, B.; Benova, T.; Viczenczova, C. Can we protect from malignant arrhythmias by modulation of cardiac cell-to-cell coupling? J. Electrocardiol. 2015, 48, 434–440. [Google Scholar] [CrossRef]
- Fabritz, L.; Schotten, U.; Kirchhof, P. ESC CardioMed, 3rd ed.; Camm, A.J., Lüsche, T.F., Maurer, G., Serruys, P.W., Eds.; Oxford University Press: Oxford, UK, 2018; ISBN 9780198784906. [Google Scholar]
- Wijesurendra, R.S.; Casadei, B. Mechanisms of atrial fibrillation. Heart 2019, 105, 1860–1867. [Google Scholar] [CrossRef]
- Roney, C.H.; Wit, A.L.; Peters, N.S. Challenges Associated with Interpreting Mechanisms of AF. Arrhythmia Electrophysiol. Rev. 2019, 8, 273–284. [Google Scholar] [CrossRef]
- Bianco, A.C.; Salvatore, D.; Gereben, B.; Berry, M.J.; Larsen, P.R. Biochemistry, cellular and molecular biology, and physiological roles of the iodothyronine selenodeiodinases. Endocr. Rev. 2002, 23, 38–89. [Google Scholar] [CrossRef]
- Larsen, P. Thyroid-pituitary interaction: Feedback regulation of thyrotropin secretion by thyroid hormones. N. Engl. J. Med. 1982, 306, 23–32. [Google Scholar]
- Franklyn, J.A.; Boelaert, K. Thyrotoxicosis. Lancet 2012, 379, 1155–1166. [Google Scholar] [CrossRef]
- Wang, J.; Zhou, J.J.; Yuan, X.; Li, C.; Sheng, H.; Su, B.; Sheng, C.; Qu, S.; Li, H. Hyperthyroidism caused by acquired immune deficiency syndrome. Eur. Rev. Med. Pharmacol. Sci. Hyperthyroid. 2014, 18, 875–879. [Google Scholar]
- Skelin, M.; Lucijanić, T.; Amidžić Klarić, D.; Rešić, A.; Bakula, M.; Liberati-Čizmek, A.M.; Gharib, H.; Rahelić, D. Factors Affecting Gastrointestinal Absorption of Levothyroxine: A Review. Clin. Ther. 2017, 39, 378–403. [Google Scholar] [CrossRef] [PubMed]
- Tribulova, N.; Knezl, V.; Shainberg, A.; Seki, S.; Soukup, T. Thyroid hormones and cardiac arrhythmias. Vasc. Pharmacol. 2010, 52, 102–112. [Google Scholar] [CrossRef] [PubMed]
- Collet, T.H.; Gussekloo, J.; Bauer, D.C.; Den Elzen, W.P.J.; Cappola, A.R.; Balmer, P.; Iervasi, G.; Åsvold, B.O.; Sgarbi, J.A.; Völzke, H.; et al. Subclinical hyperthyroidism and the risk of coronary heart disease and mortality. Arch. Intern. Med. 2012, 172, 799–809. [Google Scholar] [CrossRef]
- Cappola, A.R.; Desai, A.S.; Medici, M.; Cooper, L.S.; Egan, D.; Sopko, G.; Fishman, G.I.; Goldman, S.; Cooper, D.S.; Mora, S.; et al. Thyroid and Cardiovascular Disease: Research Agenda for Enhancing Knowledge, Prevention, and Treatment. Circulation 2019, 139, 2892–2909. [Google Scholar] [CrossRef]
- Hollowell, J.G.; Staehling, N.W.; Dana Flanders, W.; Harry Hannon, W.; Gunter, E.W.; Spencer, C.A.; Braverman, L.E. Serum TSH, T4, and thyroid antibodies in the United States population (1988 to 1994): National Health and Nutrition Examination Survey (NHANES III). J. Clin. Endocrinol. Metab. 2002, 87, 489–499. [Google Scholar] [CrossRef]
- Portman, M.A. Thyroid hormone regulation of heart metabolism. Thyroid 2008, 18, 217–225. [Google Scholar] [CrossRef]
- Iordanidou, A.; Hadzopoulou-Cladaras, M.; Lazou, A. Non-genomic effects of thyroid hormone in adult cardiac myocytes: Relevance to gene expression and cell growth. Mol. Cell. Biochem. 2010, 340, 291–300. [Google Scholar] [CrossRef]
- Davis, P.J.; Goglia, F.; Leonard, J.L. Nongenomic actions of thyroid hormone. Nat. Rev. Endocrinol. 2016, 12, 111–121. [Google Scholar] [CrossRef]
- Jabbar, A.; Pingitore, A.; Pearce, S.H.S.; Zaman, A.; Iervasi, G.; Razvi, S. Thyroid hormones and cardiovascular disease. Nat. Rev. Cardiol. 2016, 14, 39–55. [Google Scholar] [CrossRef] [PubMed]
- Reddy, V.; Taha, W.; Kundumadam, S.; Khan, M. Atrial fibrillation and hyperthyroidism: A literature review. Indian Heart J. 2017, 69, 545–550. [Google Scholar] [CrossRef] [PubMed]
- Ellervik, C.; Roselli, C.; Christophersen, I.E.; Alonso, A.; Pietzner, M.; Sitlani, C.M.; Trompet, S.; Arking, D.E.; Geelhoed, B.; Guo, X.; et al. Assessment of the relationship between genetic determinants of thyroid function and atrial fibrillation: A mendelian randomization study. JAMA Cardiol. 2019, 4, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Baumgartner, C.; Da Costa, B.R.; Collet, T.H.; Feller, M.; Floriani, C.; Bauer, D.C.; Cappola, A.R.; Heckbert, S.R.; Ceresini, G.; Gussekloo, J.; et al. Thyroid Function Within the Normal Range, Subclinical Hypothyroidism, and the Risk of Atrial Fibrillation. Circulation 2017, 136, 2100–2116. [Google Scholar] [CrossRef]
- Szeiffová Bačova, B.; Egan Beňová, T.; Viczenczová, C.; Soukup, T.; Rauchová, H.; Pavelka, S.; Knezl, V.; Barancík, M.; Tribulová, N. Cardiac connexin-43 and PKC signaling in rats with altered thyroid status without and with omega-3 fatty acids intake. Physiol. Res. 2016, 65, 77–90. [Google Scholar] [CrossRef]
- Bačová, B.S.; Vinczenzová, C.; Žurmanová, J.; Kašparová, D.; Knezl, V.; Beňová, T.E.; Pavelka, S.; Soukup, T.; Tribulová, N. Altered thyroid status affects myocardial expression of connexin-43 and susceptibility of rat heart to malignant arrhythmias that can be partially normalized by red palm oil intake. Histochem. Cell Biol. 2017, 147, 63–73. [Google Scholar] [CrossRef]
- Donzelli, R.; Colligiani, D.; Kusmic, C.; Sabatini, M.; Lorenzini, L.; Accorroni, A.; Nannipieri, M.; Saba, A.; Iervasi, G.; Zucchi, R. Effect of Hypothyroidism and Hyperthyroidism on Tissue Thyroid Hormone Concentrations in Rat. Eur. Thyroid J. 2016, 5, 27–34. [Google Scholar] [CrossRef] [Green Version]
- Gil-Cayuela, C.; Roselló-LLetí, E.; Tarazón, E.; Ortega, A.; Sandoval, J.; Martínez-Dolz, L.; Cinca, J.; Jorge, E.; González-Juanatey, J.R.; Lago, F.; et al. Thyroid hormone biosynthesis machinery is altered in the ischemic myocardium: An epigenomic study. Int. J. Cardiol. 2017, 243, 27–33. [Google Scholar] [CrossRef]
- Heijman, J.; Voigt, N.; Ghezelbash, S.; Schirmer, I.; Dobrev, D. Calcium handling abnormalities as a target for Atrial fibrillation therapeutics: How close to clinical implementation? J. Cardiovasc. Pharmacol. 2015, 66, 515–522. [Google Scholar] [CrossRef]
- Yan, J.; Zhao, W.; Thomson, J.K.; Gao, X.; DeMarco, D.M.; Carrillo, E.; Chen, B.; Wu, X.; Ginsburg, K.S.; Bakhos, M.; et al. Stress signaling JNK2 crosstalk with CaMKII underlies enhanced atrial arrhythmogenesis. Circ. Res. 2018, 122, 821–835. [Google Scholar] [CrossRef]
- Rahmutula, D.; Zhang, H.; Wilson, E.; Olgin, J. Absence of natriuretic peptide clearance receptor attenuates TGF-β1-induced selective atrial fibrosis and atrial fibrillation. Cardiovasc. Res. 2019, 115, 357–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiedmann, F.; Schulte, J.S.; Gomes, B.; Zafeiriou, M.P.; Ratte, A.; Rathjens, F.; Fehrmann, E.; Scholz, B.; Voigt, N.; Müller, F.U.; et al. Atrial fibrillation and heart failure-associated remodeling of two-pore-domain potassium (K2P) channels in murine disease models: Focus on TASK-1. Basic Res. Cardiol. 2018, 113, 27. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Obata, K.; Ohmori, T.; Ishiwata, K.; Abe, M.; Hamaguchi, S.; Namekata, I.; Tanaka, H. Angiotensin II Induces Automatic Activity of the Isolated Guinea Pig Pulmonary Vein Myocardium through Activation of the IP₃ Receptor and the Na+-Ca2+ Exchanger. Int. J. Mol. Sci. 2019, 20, 1768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carballo, S.; Pfenniger, A.; Carballo, D.; Garin, N.; James, R.W.; Mach, F.; Shah, D.; Kwak, B.R. Differential association of Cx37 and Cx40 genetic variants in atrial fibrillation with and without underlying structural heart disease. Int. J. Mol. Sci. 2018, 19, 295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, D. Atrial fibrillation-linked GJA5/connexin40 mutants impaired gap junctions via different mechanisms. FEBS Lett. 2014, 588, 1238–1243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noureldin, M.; Chen, H.; Bai, D. Functional characterization of novel atrial fibrillation-linked GJA5 (CX40) mutants. Int. J. Mol. Sci. 2018, 19, 977. [Google Scholar] [CrossRef] [Green Version]
- Mann, I.; Sandler, B.; Linton, N.; Kanagaratnam, P. Drivers of atrial fibrillation: Theoretical considerations and practical concerns. Arrhythmia Electrophysiol. Rev. 2018, 7, 49–54. [Google Scholar] [CrossRef] [Green Version]
- Hohl, M.; Linz, B.; Böhm, M.; Linz, D. Obstructive sleep apnea and atrial arrhythmogenesis. Curr. Cardiol. Rev. 2014, 10, 362–368. [Google Scholar] [CrossRef] [Green Version]
- Wolke, C.; Bukowska, A.; Goette, A.; Lendeckel, U. Redox control of cardiac remodeling in atrial fibrillation. Biochim. Biophys. Acta Gen. Subj. 2015, 1850, 1555–1565. [Google Scholar] [CrossRef]
- Jalife, J.; Kaur, K. Atrial remodeling, fibrosis, and atrial fibrillation. Trends Cardiovasc. Med. 2015, 25, 475–484. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, C.; Wiedmann, F.; Voigt, N.; Zhou, X.B.; Heijman, J.; Lang, S.; Albert, V.; Kallenberger, S.; Ruhparwar, A.; Szabó, G.; et al. Upregulation of K2P 3.1 K + Current Causes Action Potential Shortening in Patients with Chronic Atrial Fibrillation. Circulation 2015, 132, 82–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seyed Ahmadi, S.; Svensson, A.M.; Pivodic, A.; Rosengren, A.; Lind, M. Risk of atrial fibrillation in persons with type 2 diabetes and the excess risk in relation to glycaemic control and renal function: A Swedish cohort study. Cardiovasc. Diabetol. 2020, 19, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.; Lin, Y. The Pathogenic Role of Very Low Density Lipoprotein on Atrial Remodeling in the Metabolic Syndrome. Int. J. Mol. Sci. 2020, 21, 891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Choi, E.; Cha, M.J.; Hwang, K.C. Looking into a conceptual framework of ROS–miRNA–Atrial fibrillation. Int. J. Mol. Sci. 2014, 15, 21754–21776. [Google Scholar] [CrossRef] [Green Version]
- Liew, R.; Khairunnisa, K.; Gu, Y.; Tee, N.; Yin, N.O.; Naylynn, T.M.; Moe, K.T. Role of tumor necrosis factor-α in the pathogenesis of atrial fibrosis and development of an arrhythmogenic substrate. Circ. J. 2013, 77, 1171–1179. [Google Scholar] [CrossRef] [Green Version]
- Harada, M.; Van Wagoner, D.R.; Nattel, S. Role of inflammation in Atrial fibrillation pathophysiology and management. Circ. J. 2015, 79, 495–502. [Google Scholar] [CrossRef] [Green Version]
- Chiang, D.Y.; Lebesgue, N.; Beavers, D.L.; Alsina, K.M.; Damen, J.M.A.; Voigt, N.; Dobrev, D.; Wehrens, X.H.T.; Scholten, A. Alterations in the interactome of serine/threonine protein phosphatase type-1 in atrial fibrillation patients. J. Am. Coll. Cardiol. 2015, 65, 163–173. [Google Scholar] [CrossRef] [Green Version]
- Ghezelbash, S.; Molina, C.E.; Dobrev, D. Altered atrial metabolism: An underappreciated contributor to the initiation and progression of atrial fibrillation. J. Am. Heart Assoc. 2015, 4, e001808. [Google Scholar] [CrossRef] [Green Version]
- Hu, B.; Sun, Y.; Li, S.; Sun, J.; Liu, T.; Wu, Z.; Feng, L. Association of β1-Adrenergic, M2-Muscarinic Receptor Autoantibody with Occurrence and Development of Nonvalvular Atrial Fibrillation. Pace Pacing Clin. Electrophysiol. 2016, 39, 1379–1387. [Google Scholar] [CrossRef]
- Capecchi, P.L.; Laghi-Pasini, F.; El-Sherif, N.; Qu, Y.; Boutjdir, M.; Lazzerini, P.E. Autoimmune and inflammatory K+ channelopathies in cardiac arrhythmias: Clinical evidence and molecular mechanisms. Heart Rhythm 2019, 16, 1273–1280. [Google Scholar] [CrossRef]
- Lazzerini, P.E.; Capecchi, P.L.; Laghi-Pasini, F.; Boutjdir, M. Autoimmune channelopathies as a novel mechanism in cardiac arrhythmias. Nat. Rev. Cardiol. 2017, 14, 521–535. [Google Scholar] [CrossRef] [PubMed]
- Chera, H.; Nagar, M.; Richler, A.; Pourriahi, M.; Al-Sadawi, M.; Gunsburg, M.; Shoenfeld, Y.; Rosen, Y. Autoantibodies for Cardiac Channels and Sudden Cardiac Death and its Relationship to Autoimmune Disorders. Curr. Cardiol. Rev. 2018, 15, 49–54. [Google Scholar] [CrossRef]
- Qu, Y.S.; Lazzerini, P.E.; Capecchi, P.L.; Laghi-Pasini, F.; El Sherif, N.; Boutjdir, M. Autoimmune Calcium Channelopathies and Cardiac Electrical Abnormalities. Front. Cardiovasc. Med. 2019, 6, 54. [Google Scholar] [CrossRef] [PubMed]
- Packer, M. Effect of catheter ablation on pre-existing abnormalities of left atrial systolic, diastolic, and neurohormonal functions in patients with chronic heart failure and atrial fibrillation. Eur. Heart J. 2019, 40, 1873–1879. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, R.; Perera, T.; Elliott, A.D.; Twomey, D.J.; Kumar, S.; Munwar, D.A.; Khokhar, K.B.; Thiyagarajah, A.; Middeldorp, M.E.; Nalliah, C.J.; et al. Subclinical device-detected atrial fibrillation and stroke risk: A systematic review and meta-analysis. Eur. Heart J. 2018, 39, 1407–1415. [Google Scholar] [CrossRef]
- Brandes, A.; Smit, M.D.; Nguyen, B.O.; Rienstra, M.; Van Gelder, I.C. Risk factor management in atrial fibrillation. Arrhythmia Electrophysiol. Rev. 2018, 7, 118–127. [Google Scholar] [CrossRef] [Green Version]
- Weil, B.; Ozcan, C. Cardiomyocyte Remodeling in Atrial Fibrillation and Hibernating Myocardium: Shared Pathophysiologic Traits Identify Novel Treatment Strategies? BioMed Res. Int. 2015, 2015, 587361. [Google Scholar] [CrossRef]
- Ali, R.L.; Hakim, J.B.; Boyle, P.M.; Zahid, S.; Sivasambu, B.; Marine, J.E.; Calkins, H.; Trayanova, N.A.; Spragg, D.D. Arrhythmogenic propensity of the fibrotic substrate after atrial fibrillation ablation: A longitudinal study using magnetic resonance imaging-based atrial models. Cardiovasc. Res. 2019, 115, 1757–1765. [Google Scholar] [CrossRef]
- Sohinki, D.; Stavrakis, S. New approaches for treating atrial fibrillation: Focus on autonomic modulation. Trends Cardiovasc. Med. 2019. [Google Scholar] [CrossRef]
- Odening, K.E.; Deiß, S.; Dilling-Boer, D.; Didenko, M.; Eriksson, U.; Nedios, S.; Ng, F.S.; Roca Luque, I.; Sanchez Borque, P.; Vernooy, K.; et al. Mechanisms of sex differences in atrial fibrillation: Role of hormones and differences in electrophysiology, structure, function, and remodelling. Europace 2019, 21, 366–376. [Google Scholar] [CrossRef]
- Diederichsen, S.Z.; Haugan, K.J.; Brandes, A.; Lanng, M.B.; Graff, C.; Krieger, D.; Kronborg, C.; Holst, A.G.; Køber, L.; Højberg, S.; et al. Natural History of Subclinical Atrial Fibrillation Detected by Implanted Loop Recorders. J. Am. Coll. Cardiol. 2019, 74, 2771–2781. [Google Scholar] [CrossRef] [PubMed]
- McIntyre, W.F.; Yong, J.H.E.; Sandhu, R.K.; Gladstone, D.J.; Simek, K.; Liu, Y.Y.; Quinn, F.R.; Tytus, R.; Zizzo, D.; Henein, S.; et al. Prevalence of undiagnosed atrial fibrillation in elderly individuals and potential cost-effectiveness of non-invasive ambulatory electrocardiographic screening: The ASSERT-III study. J. Electrocardiol. 2020, 58, 56–60. [Google Scholar] [CrossRef] [PubMed]
- Matusik, P.T. Biomarkers and cardiovascular risk stratification. Eur. Heart J. 2019, 40, 1483–1485. [Google Scholar] [CrossRef] [PubMed]
- Engdahl, J.; Andersson, L.; Mirskaya, M.; Rosenqvist, M. Stepwise screening of atrial fibrillation in a 75-year-old population: Implications for stroke prevention. Circulation 2013, 127, 930–937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chua, W.; Purmah, Y.; Cardoso, V.R.; Gkoutos, G.V.; Tull, S.P.; Neculau, G.; Thomas, M.R.; Kotecha, D.; Lip, G.Y.H.; Kirchhof, P.; et al. Data-driven discovery and validation of circulating blood-based biomarkers associated with prevalent atrial fibrillation. Eur. Heart J. 2019, 40, 1268–1276. [Google Scholar] [CrossRef] [Green Version]
- Patton, K.K.; Heckbert, S.R.; Alonso, A.; Bahrami, H.; Lima, J.A.C.; Burke, G.; Kronmal, R.A. N-terminal pro-B-type natriuretic peptide as a predictor of incident atrial fibrillation in the Multi-Ethnic study of atherosclerosis: The effects of age, sex and ethnicity. Heart 2013, 99, 1832–1836. [Google Scholar] [CrossRef]
- Kemp Gudmundsdottir, K.; Fredriksson, T.; Svennberg, E.; Al-Khalili, F.; Friberg, L.; Frykman, V.; Hijazi, Z.; Rosenqvist, M.; Engdahl, J. Stepwise mass screening for atrial fibrillation using N-terminal B-type natriuretic peptide: The STROKESTOP II study. Europace 2020, 22, 24–32. [Google Scholar] [CrossRef]
- Bielecka-Dabrowa, A.; Mikhailidis, D.P.; Rysz, J.; Banach, M. The mechanisms of atrial fibrillation in hyperthyroidism. Thyroid Res. 2009, 2, 4. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.C.; Chen, S.A.; Chen, Y.J.; Chang, M.S.; Chan, P.; Lin, C.I. Effects of thyroid hormone on the arrhythmogenic activity of pulmonary vein cardiomyocytes. J. Am. Coll. Cardiol. 2002, 39, 366–372. [Google Scholar] [CrossRef] [Green Version]
- Yu, Z.; Huang, C.X.; Wang, S.Y.; Wang, T.; Xu, L. Thyroid hormone predisposes rabbits to atrial arrhythmias by shortening monophasic action period and effective refractory period: Results from an in vivo study. J. Endocrinol. Investig. 2009, 32, 253–257. [Google Scholar] [CrossRef]
- Gen, R.; Akbay, E.; Çamsari, A.; Özcan, T. P-wave dispersion in endogenous and exogenous subclinical hyperthyroidism. J. Endocrinol. Investig. 2010, 33, 88–91. [Google Scholar] [CrossRef] [PubMed]
- Vadiveloo, T.; Donnan, P.T.; Cochrane, L.; Leese, G.P. The Thyroid Epidemiology, Audit, and Research Study (TEARS): Morbidity in patients with endogenous subclinical hyperthyroidism. J. Clin. Endocrinol. Metab. 2011, 96, 1344–1351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaker, L.; Heeringa, J.; Dehghan, A.; Medici, M.; Visser, W.E.; Baumgartner, C.; Hofman, A.; Rodondi, N.; Peeters, R.P.; Franco, O.H. Normal thyroid function and the risk of atrial fibrillation: The Rotterdam study. J. Clin. Endocrinol. Metab. 2015, 100, 3718–3724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turan, E.; Can, I.; Turan, Y.; Uyar, M.; Cakır, M. Comparison of cardiac arrhythmia types between hyperthyroid patients with graves’ disease and toxic nodular goiter. Acta Endocrinol. (Buchar.) 2018, 14, 324–329. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Tan, H.; Huang, J.; Luo, D.; Tang, Y.; Yu, R.; Huang, H. Case report of recurrent atrial fibrillation induced by thyrotropin-secreting pituitary adenoma with Graves’ disease. Med. (Baltim.) 2018, 97, e11047. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, J.M.; Mohamed, H.E.; Noureldine, S.I.; Nazari-Shafti, T.Z.; Thethi, T.K.; Kandil, E. Impact of thyroidectomy on cardiac manifestations of Graves’ disease. Laryngoscope 2016, 126, 1256–1259. [Google Scholar] [CrossRef]
- Weetman, A.P. Graves’ disease following immune reconstitution or immunomodulatory treatment: Should we manage it any differently? Clin. Endocrinol. 2014, 80, 629–632. [Google Scholar] [CrossRef]
- Stavrakis, S.; Yu, X.; Patterson, E.; Huang, S.; Hamlett, S.R.; Chalmers, L.; Pappy, R.; Cunningham, M.W.; Morshed, S.A.; Davies, T.F.; et al. Activating Autoantibodies to the Beta-1 Adrenergic and M2 Muscarinic Receptors Facilitate Atrial Fibrillation in Patients With Graves’ Hyperthyroidism. J. Am. Coll. Cardiol. 2009, 54, 1309–1316. [Google Scholar] [CrossRef] [Green Version]
- Nussinovitch, U.; Shoenfeld, Y. The Diagnostic and clinical significance of anti-muscarinic receptor autoantibodies. Clin. Rev. Allergy Immunol. 2012, 42, 298–308. [Google Scholar] [CrossRef]
- Galloway, A.; Li, H.; Vanderlinde-Wood, M.; Khan, M.; Benbrook, A.; Liles, C.; Zillner, C.; Rao, V.; Cunningham, M.W.; Yu, X.; et al. Activating autoantibodies to the β1/2-adrenergic and M2 muscarinic receptors associate with atrial tachyarrhythmias in patients with hyperthyroidism. Endocrine 2015, 49, 457–463. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Murphy, T.; Zhang, L.; Huang, B.; Veitla, V.; Scherlag, B.J.; Kem, D.C.; Yu, X. β1-Adrenergic and M2 muscarinic autoantibodies and thyroid hormone facilitate induction of atrial fibrillation in male rabbits. Endocrinology 2016, 157, 16–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becker, N.P.; Müller, J.; Göttel, P.; Wallukat, G.; Schimke, I. Cardiomyopathy—An approach to the autoimmune background. Autoimmun. Rev. 2017, 16, 269–286. [Google Scholar] [CrossRef] [PubMed]
- Nussinovitch, U.; Shoenfeld, Y. The clinical significance of Anti-Beta-1 adrenergic receptor autoantibodies in cardiac disease. Clin. Rev. Allergy Immunol. 2013, 44, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Yalcin, M.U.; Gurses, K.M.; Kocyigit, D.; Kesikli, S.A.; Dural, M.; Evranos, B.; Yorgun, H.; Sahiner, L.; Kaya, E.B.; Oto, M.A.; et al. Cardiac autoantibody levels predict recurrence following cryoballoon-based pulmonary vein isolation in paroxysmal atrial fibrillation patients. J. Cardiovasc. Electrophysiol. 2015, 26, 615–621. [Google Scholar] [CrossRef]
- Pfenniger, A.; Arora, R. Beyond beta-blockers: Targeting the sympathetic nervous system for the prevention and treatment of atrial fibrillation. Cardiovasc. Res. 2019, 115, 1940–1942. [Google Scholar] [CrossRef]
- Bacova, B.S.; Radosinska, J.; Wallukat, G.; Barancik, M.; Wallukat, A.; Knezl, V.; Sykora, M.; Paulis, L.; Tribulova, N. Suppression of β1-adrenoceptor autoantibodies is involved in the antiarrhythmic effects of omega-3 fatty acids in male and female hypertensive rats. Int. J. Mol. Sci. 2020, 21, 526. [Google Scholar] [CrossRef] [Green Version]
- Tang, R.B.; Liu, D.L.; Dong, J.Z.; Liu, X.P.; Long, D.Y.; Yu, R.H.; Hu, F.L.; Wu, J.H.; Liu, X.H.; Ma, C.S. High-normal thyroid function and risk of recurrence of atrial fibrillation after catheter ablation. Circ. J. 2010, 74, 1317–1321. [Google Scholar] [CrossRef] [Green Version]
- Sousa, P.A.; Providência, R.; Albenque, J.P.; Khoueiry, Z.; Combes, N.; Combes, S.; Boveda, S. Impact of Free Thyroxine on the Outcomes of Left Atrial Ablation Procedures. Am. J. Cardiol. 2015, 116, 1863–1868. [Google Scholar] [CrossRef]
- Morishima, I.; Okumura, K.; Morita, Y.; Kanzaki, Y.; Takagi, K.; Yoshida, R.; Nagai, H.; Ikai, Y.; Furui, K.; Yoshioka, N.; et al. High-normal thyroid-stimulating hormone shows a potential causal association with arrhythmia recurrence after catheter ablation of atrial fibrillation. J. Am. Heart Assoc. 2018, 7, e009158. [Google Scholar] [CrossRef] [Green Version]
- Wei, S.B.; Wang, W.; Liu, N.; Chen, J.; Guo, X.Y.; Tang, R.B.; Yu, R.H.; Long, D.Y.; Sang, C.H.; Jiang, C.X.; et al. U-shaped association between serum free triiodothyronine and recurrence of atrial fibrillation after catheter ablation. J. Interv. Card. Electrophysiol. 2018, 51, 263–270. [Google Scholar] [CrossRef]
- Kim, K.H.; Mohanty, S.; Mohanty, P.; Trivedi, C.; Morris, E.H.; Santangeli, P.; Bai, R.; Al-Ahmad, A.; Burkhardt, J.D.; Gallinghouse, J.G.; et al. Prevalence of right atrial non-pulmonary vein triggers in atrial fibrillation patients treated with thyroid hormone replacement therapy. J. Interv. Card. Electrophysiol. 2017, 49, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Zipes, D.P. Antiarrhythmic therapy in 2014: Contemporary approaches to treating arrhythmias. Nat. Rev. Cardiol. 2015, 12, 68–69. [Google Scholar] [CrossRef] [PubMed]
- Bacharova, L. Missing link between molecular aspects of ventricular arrhythmias and QRS complex morphology in left ventricular hypertrophy. Int. J. Mol. Sci. 2019, 21, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.H.; Huang, H.X.; Liu, P.; Du, Y.H.; Wang, P.; Wang, W.; Wu, Y.; Wang, L.; Ma, C.S.; Liu, H.R. β1-Adrenoceptor autoantibodies increase the susceptibility to ventricular arrhythmias involving abnormal repolarization in guinea-pigs. Exp. Physiol. 2017, 102, 25–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatterjee, D.; Fatah, M.; Akdis, D.; Spears, D.A.; Koopmann, T.T.; Mittal, K.; Rafiq, M.A.; Cattanach, B.M.; Zhao, Q.; Healey, J.S.; et al. An autoantibody identifies arrhythmogenic right ventricular cardiomyopathy and participates in its pathogenesis. Eur. Heart J. 2018, 39, 3932–3944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isaksen, J.L.; Graff, C.; Ellervik, C.; Kanters, J.K. Electrocardiographic effect of thyroid hormones. J. Electrocardiol. 2019, 57, S108–S109. [Google Scholar] [CrossRef]
- Akar, F.G.; O’Rourke, B. Mitochondria are sources of metabolic sink and arrhythmias. Pharmacol. Ther. 2011, 131, 287–294. [Google Scholar] [CrossRef] [Green Version]
- Akar, F.G. Mitochondrial targets for arrhythmia suppression: Is there a role for pharmacological intervention? J. Interv. Card. Electrophysiol. 2013, 37, 249–258. [Google Scholar] [CrossRef]
- Nadkarni, P.J.; Sharma, M.; Zinsmeister, B.; Wartofsky, L.; Burman, K.D. Thyrotoxicosis-induced ventricular arrhythmias. Thyroid 2008, 18, 1111–1114. [Google Scholar] [CrossRef]
- Eisen, A.; Arnson, Y.; Dovrish, Z.; Hadary, R.; Amital, H. Arrhythmias and Conduction Defects in Rheumatological Diseases-A Comprehensive Review. Semin. Arthritis Rheum. 2009, 39, 145–156. [Google Scholar] [CrossRef]
- Lee, H.C.; Huang, K.T.L.; Wang, X.L.; Shen, W.K. Autoantibodies and cardiac arrhythmias. Heart Rhythm 2011, 8, 1788–1795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brisinda, D.; Sorbo, A.R.; Venuti, A.; Ruggieri, M.P.; Manna, R.; Fenici, P.; Wallukat, G.; Hoebeke, J.; Frustaci, A.; Fenici, R. Anti-β-adrenoceptors autoimmunity causing “idiopathic” arrhythmias and cardiomyopathy. Circ. J. 2012, 76, 1345–1353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryabkova, V.A.; Shubik, Y.V.; Erman, M.V.; Churilov, L.P.; Kanduc, D.; Shoenfeld, Y. Lethal immunoglobulins: Autoantibodies and sudden cardiac death. Autoimmun. Rev. 2019, 18, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Tsai, I.H.; Su, Y.J. Thyrotoxic periodic paralysis with ventricular tachycardia. J. Electrocardiol. 2019, 54, 93–95. [Google Scholar] [CrossRef] [PubMed]
- Xia, H.J.; Dai, D.Z.; Dai, Y. Up-regulated inflammatory factors endothelin, NFκB, TNFα and iNOS involved in exaggerated cardiac arrhythmias in l-thyroxine-induced cardiomyopathy are suppressed by darusentan in rats. Life Sci. 2006, 79, 1812–1819. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Mitasikova, M.; Dlugosova, K.; Okruhlicova, L.; Imanaga, I.; Ogawa, K.; Weismann, P.; Tribulova, N. Thyroid hormones suppress ε-PKC signalling, down-regulate connexin-43 and increase lethal arrhythmia susceptibility in non-diabetic and diabetic rat hearts. J. Physiol. Pharmacol. 2008, 59, 271–285. [Google Scholar]
- Bačová, B.; Viczenczová, C.; Žurmanová, J.; Kašparová, D.; Knezl, V.; Beňová, T.; Pavelka, S.; Soukup, T.; Tribulová, N. Susceptibility of rats with altered thyroid status to malignant arrhythmias is related mainly to myocardial levels of connexin-43 and can be partially ameliorated by supplementation with red palm oil. Exp. Clin. Cardiol. 2013, 18, 41–46. [Google Scholar]
- Knezl, V.; Soukup, T.; Okruhlicová, L.; Slezák, J.; Tribulová, N. Thyroid hormones modulate occurrence and termination of ventricular fibrillation by both long-term and acute actions. Physiol. Res. 2008, 57, 91–96. [Google Scholar]
- Caves, R.E.; Cheng, H.; Choisy, S.C.; Gadeberg, H.C.; Bryant, S.M.; Hancox, J.C.; James, A.F. Atrial-ventricular differences in rabbit cardiac voltage-gated Na+ currents: Basis for atrial-selective block by ranolazine. Heart Rhythm 2017, 14, 1657–1664. [Google Scholar] [CrossRef]
- Lane, J.D.; Montaigne, D.; Tinker, A. Tissue-Level Cardiac Electrophysiology Studied in Murine Myocardium Using a Microelectrode Array: Autonomic and Thermal Modulation. J. Membr. Biol. 2017, 250, 471–481. [Google Scholar] [CrossRef] [Green Version]
- Johnson, E.K.; Matkovich, S.J.; Nerbonne, J.M. Regional Differences in mRNA and lncRNA Expression Profiles in Non-Failing Human Atria and Ventricles. Sci. Rep. 2018, 8, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Meo, S.; De Martino Rosaroll, P.; Piro, M.C.; De Leo, T. Electrophysiological properties of the hyperthyroid rat heart. Arch. Int. Physiol. Biochim. Biophys. 1994, 102, 153–159. [Google Scholar]
- Buscemi, S.; Verga, S.; Cottone, S.; Andronico, G.; D’Orio, L.; Mannino, V.; Panzavecchia, D.; Vitale, F.C.G. Favorable clinical heart and bone effects of anti-thyroid drug therapy in endogenous subclinical hyperthyroidism. J. Endocrinol. Investig. 2007, 30, 230–235. [Google Scholar] [CrossRef] [PubMed]
- Polikar, R.; Burger, A.; Scherrer, U.; Nicod, P. The Thyroid and the Heart. Circulation 1993, 87, 1435–1441. [Google Scholar] [CrossRef] [Green Version]
- Gross, G.; Lues, I. Thyroid-dependent alterations of myocardial adrenoceptors and adrenoceptor-mediated responses in the rat. Naunyn-Schmiedeberg’s Arch. Pharmacol. 1985, 329, 427–439. [Google Scholar] [CrossRef]
- Tielens, E.T.; Forder, J.R.; Chatham, J.C.; Marrelli, S.P.; Ladenson, P.W. Acute L-triiodothyronine administration potentiates inotropic responses to β-adrenergic stimulation in the isolated perfused rat heart. Cardiovasc. Res. 1996, 32, 306–310. [Google Scholar] [CrossRef] [Green Version]
- Gietka-Czernel, M. The thyroid gland in postmenopausal women: Physiology and diseases. Prz. Menopauzalny 2017, 16, 33–37. [Google Scholar] [CrossRef]
- Wang, T.S.; Sosa, J.A. Thyroid surgery for differentiated thyroid cancer—Recent advances and future directions. Nat. Rev. Endocrinol. 2018, 14, 670–683. [Google Scholar] [CrossRef]
- DeSantis, C.E.; Lin, C.C.; Mariotto, A.B.; Siegel, R.L.; Stein, K.D.; Kramer, J.L.; Alteri, R.; Robbins, A.S.; Jemal, A. Cancer treatment and survivorship statistics, 2014. CA Cancer J. Clin. 2014, 64, 252–271. [Google Scholar] [CrossRef]
- Shin, H.J.; Hwang, K.A.; Choi, K.C. Antitumor effect of various phytochemicals on diverse types of thyroid cancers. Nutrients 2019, 11, 125. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Feng, J.F.; Zeng, P.; Yang, Y.H.; Luo, J.; Yang, Y.W. Total oxidant/antioxidant status in sera of patients with thyroid cancers. Endocr. Relat. Cancer 2011, 18, 773–782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosseini-Zijoud, S.M.; Ebadi, S.A.; Goodarzi, M.T.; Hedayati, M.; Abbasalipourkabir, R.; Mahjoob, M.P.; Poorolajal, J.; Zicker, F.; Sheikh, N. Lipid peroxidation and antioxidant status in patients with medullary thyroid carcinoma: A case-control study. J. Clin. Diagn. Res. 2016, 10, BC04–BC07. [Google Scholar] [PubMed]
- Katerji, M.; Filippova, M.; Duerksen-Hughes, P. Approaches and methods to measure oxidative stress in clinical samples: Research applications in the cancer field. Oxidative Med. Cell. Longev. 2019, 2019, 1279250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macejova, D.; Podoba, J.; Toporova, L.; Grigerova, M.; Kajo, K.; Machalekova, K.; Brtko, J. Causal associations of autoimmune thyroiditis and papillary thyroid carcinoma: mRNA expression of selected nuclear receptors and other molecular targets. Oncol. Lett. 2019, 18, 4270–4277. [Google Scholar] [CrossRef] [Green Version]
- Libby, P.; Kobold, S. Inflammation: A common contributor to cancer, aging, and cardiovascular diseases—Expanding the concept of cardio-oncology. Cardiovasc. Res. 2019, 115, 824–829. [Google Scholar] [CrossRef]
- Kebebew, E. Thyroid Cancer: Is It All in the Genes? J. Natl. Cancer Inst. 2018, 110, 327–328. [Google Scholar] [CrossRef]
- Takacsova, E.; Kralik, R.; Waczulikova, I.; Zavodna, K.; Kausitz, J. A different prognostic value of BRAFV600E mutation positivity in various age groups of patients with papillary thyroid cancer. Neoplasma 2017, 64, 156–164. [Google Scholar] [CrossRef] [Green Version]
- Donati, B.; Ciarrocchi, A. Telomerase and telomeres biology in thyroid cancer. Int. J. Mol. Sci. 2019, 20, 2887. [Google Scholar] [CrossRef] [Green Version]
- McIver, B.; Freeman, J.; Shah, J.P.; Shaha, A.R.; Haugen, B.; Cohen, E.; Witterick, I.J.; Randolph, G.W. Summary of the third world congress on thyroid cancer. Thyroid 2018, 28, 1401–1405. [Google Scholar] [CrossRef]
- Callender, G.G.; Carling, T.; Christison-Lagay, E.; Udelsman, R. Surgery for thyroid cancer. Endocrinol. Metab. Clin. N. Am. 2014, 43, 443–458. [Google Scholar] [CrossRef]
- Szujo, S.; Sira, L.; Bajnok, L.; Bodis, B.; Gyory, F.; Nemes, O.; Rucz, K.; Kenyeres, P.; Valkusz, Z.; Sepp, K.; et al. The impact of post-radioiodine therapy SPECT/CT on early risk stratification in differentiated thyroid cancer; a bi-institutional study. Oncotarget 2017, 8, 79825–79834. [Google Scholar] [CrossRef] [Green Version]
- Haugen, B.R.; Alexander, E.K.; Bible, K.C.; Doherty, G.M.; Mandel, S.J.; Nikiforov, Y.E.; Pacini, F.; Randolph, G.W.; Sawka, A.M.; Schlumberger, M.; et al. 2015 American Thyroid Association Management Guidelines for Adult Patients with Thyroid Nodules and Differentiated Thyroid Cancer: The American Thyroid Association Guidelines Task Force on Thyroid Nodules and Differentiated Thyroid Cancer. Thyroid 2016, 26, 1–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carhill, A.A.; Litofsky, D.R.; Ross, D.S.; Jonklaas, J.; Cooper, D.S.; Brierley, J.D.; Ladenson, P.W.; Ain, K.B.; Fein, H.G.; Haugen, B.R.; et al. Long-term outcomes following therapy in differentiated thyroid carcinoma: NTCTCS registry analysis 1987–2012. J. Clin. Endocrinol. Metab. 2015, 100, 3270–3279. [Google Scholar] [CrossRef] [PubMed]
- Ballal, S.; Soundararajan, R.; Garg, A.; Chopra, S.; Bal, C. Intermediate-risk differentiated thyroid carcinoma patients who were surgically ablated do not need adjuvant radioiodine therapy: Long-term outcome study. Clin. Endocrinol. 2016, 84, 408–416. [Google Scholar] [CrossRef] [PubMed]
- Florenzano, P.; Guarda, F.J.; Jaimovich, R.; Droppelmann, N.; González, H.; Domínguez, J.M. Radioactive Iodine Administration Is Associated with Persistent Related Symptoms in Patients with Differentiated Thyroid Cancer. Int. J. Endocrinol. 2016, 2016, 2586512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haymart, M.R.; Banerjee, M.; Stewart, A.K.; Koenig, R.J.; Birkmeyer, J.D.; Griggs, J.J. Use of radioactive iodine for thyroid cancer. JAMA J. Am. Med. Assoc. 2011, 306, 721–728. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.S.; Chang, H.S.; Park, C.S. Changing trends in the management of well-differentiated thyroid carcinoma in Korea. Endocr. J. 2016, 63, 515–521. [Google Scholar] [CrossRef] [Green Version]
- Aschebrook-Kilfoy, B.; James, B.; Nagar, S.; Kaplan, S.; Seng, V.; Ahsan, H.; Angelos, P.; Kaplan, E.L.; Guerrero, M.A.; Kuo, J.H.; et al. Risk Factors for Decreased Quality of Life in Thyroid Cancer Survivors: Initial Findings from the North American Thyroid Cancer Survivorship Study. Thyroid 2015, 25, 1313–1321. [Google Scholar] [CrossRef]
- Hesselink, E.N.K.; Lefrandt, J.D.; Schuurmans, E.P.; Burgerhof, J.G.M.; Groen, B.; Gansevoort, R.T.; Van Der Horst-Schrivers, A.N.A.; Dullaart, R.P.F.; Van Gelder, I.C.; Brouwers, A.H.; et al. Increased risk of atrial fibrillation after treatment for differentiated thyroid carcinoma. J. Clin. Endocrinol. Metab. 2015, 100, 4563–4569. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.C.; Yeh, C.T.; Lin, K.H. Molecular functions of thyroid hormone signaling in regulation of cancer progression and anti-apoptosis. Int. J. Mol. Sci. 2019, 20, 4986. [Google Scholar] [CrossRef] [Green Version]
- Verburg, F.A.; Smit, J.W.A.; Grelle, I.; Visser, T.J.; Peeters, R.P.; Reiners, C. Changes within the thyroid axis after long-term TSH-suppressive levothyroxine therapy. Clin. Endocrinol. 2012, 76, 577–581. [Google Scholar] [CrossRef] [PubMed]
- Selmer, C.; Olesen, J.B.; Hansen, M.L.; Lindhardsen, J.; Olsen, A.M.S.; Madsen, J.C.; Faber, J.; Hansen, P.R.; Pedersen, O.D.; Torp-Pedersen, C.; et al. The spectrum of thyroid disease and risk of new onset atrial fibrillation: A large population cohort study. BMJ (Online) 2012, 345, e7895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, P.J.; Sudha, T.; Lin, H.Y.; Mousa, S.A. Thyroid hormone, hormone analogs, and angiogenesis. Compr. Physiol. 2016, 6, 353–362. [Google Scholar]
- Horne, M.K.; Singh, K.K.; Rosenfeld, K.G.; Wesley, R.; Skarulis, M.C.; Merryman, P.K.; Cullinane, A.; Costello, R.; Patterson, A.; Eggerman, T.; et al. Is thyroid hormone suppression therapy prothrombotic? J. Clin. Endocrinol. Metab. 2004, 89, 4469–4473. [Google Scholar] [CrossRef] [Green Version]
- Miyakawa, M. Effects of thyrotropin-suppressive therapy in patients with well-differentiated thyroid carcinoma. Nihon Rinsho Jpn. J. Clin. Med. 2007, 65, 2073–2077. [Google Scholar]
- Biondi, B.; Cooper, D.S. Benefits of thyrotropin suppression versus the risks of adverse effects in differentiated thyroid cancer. Thyroid 2010, 20, 135–146. [Google Scholar] [CrossRef]
- Klein Hesselink, E.N.; Klein Hesselink, M.S.; De Bock, G.H.; Gansevoort, R.T.; Bakker, S.J.L.; Vredeveld, E.J.; Van Der Horst-Schrivers, A.N.A.; Van Der Horst, I.C.C.; Kamphuisen, P.W.; Plukker, J.T.M.; et al. Long-term cardiovascular mortality in patients with differentiated thyroid carcinoma: An observational study. J. Clin. Oncol. 2013, 31, 4046–4053. [Google Scholar] [CrossRef] [Green Version]
- Abdulrahman, R.M.; Delgado, V.; Hoftijzer, H.C.; Ng, A.C.T.; Ewe, S.H.; Marsan, N.A.; Holman, E.R.; Hovens, G.C.; Corssmit, E.P.; Romijn, J.A.; et al. Both exogenous subclinical hyperthyroidism and short-term overt hypothyroidism affect myocardial strain in patients with differentiated thyroid carcinoma. Thyroid 2011, 21, 471–476. [Google Scholar] [CrossRef]
- Verburg, F.A.; Mäder, U.; Grelle, I.; Visser, T.J.; Peeters, R.P.; Smit, J.W.A.; Reiners, C. The thyroid axis “setpoints” are significantly altered after long-term suppressive LT4 therapy. Horm. Metab. Res. 2014, 46, 794–799. [Google Scholar] [CrossRef] [Green Version]
- Pajamäki, N.; Metso, S.; Hakala, T.; Ebeling, T.; Huhtala, H.; Ryödi, E.; Sand, J.; Jukkola-Vuorinen, A.; Kellokumpu-Lehtinen, P.L.; Jaatinen, P. Long-term cardiovascular morbidity and mortality in patients treated for differentiated thyroid cancer. Clin. Endocrinol. 2018, 88, 303–310. [Google Scholar] [CrossRef] [Green Version]
- Bindi, M.; Moruzzo, D.; Rosada, J.; Castiglioni, M.; Romanelli, A.M. Atrial fibrillation and thyroid nodules. Recenti Progress. Med. 2011, 102, 17–19. [Google Scholar]
- Sawin, C.T.; Geller, A.; Wolf, P.A.; Belanger, A.J.; Baker, E.; Bacharach, P.; Wilson, P.W.; Benjamin, E.J. Low serum thyrotropin concentrations as a risk factor for atrial fibrillation in older persons. N. Engl. J. Med. 1994, 331, 249–252. [Google Scholar] [CrossRef] [PubMed]
- Abonowara, A.; Quraishi, A.; Sapp, J.L.; Alqambar, M.H.; Saric, A.; O’Connell, C.M.; Rajaraman, M.M.; Hart, R.D.; Imran, S.A. Prevalence of atrial fibrillation in patients taking TSH suppression therapy for management of thyroid cancer. Clin. Investig. Med. 2012, 35, 152–156. [Google Scholar] [CrossRef] [Green Version]
- Xia, Q.; Dong, S.; Da Bian, P.; Wang, J.; Li, C.J. Effects of endocrine therapy on the prognosis of elderly patients after surgery for papillary thyroid carcinoma. Eur. Arch. Oto-Rhino-Laryngol. 2016, 273, 1037–1043. [Google Scholar] [CrossRef] [PubMed]
- Toulis, K.A.; Viola, D.; Gkoutos, G.; Keerthy, D.; Boelaert, K.; Nirantharakumar, K. Risk of incident circulatory disease in patients treated for differentiated thyroid carcinoma with no history of cardiovascular disease. Clin. Endocrinol. 2019, 91, 323–330. [Google Scholar] [CrossRef] [PubMed]
- Zoltek, M.; Andersson, T.M.L.; Hedman, C.; Ihre-Lundgren, C.; Nordenvall, C. Cardiovascular Incidence in 6900 Patients with Differentiated Thyroid Cancer: A Swedish Nationwide Study. World J. Surg. 2020, 44, 436–441. [Google Scholar] [CrossRef] [Green Version]
- Parker, W.A.; Edafe, O.; Balasubramanian, S.P. Long-term treatment-related morbidity in differentiated thyroid cancer: A systematic review of the literature. Pragmatic Obs. Res. 2017, 8, 57–67. [Google Scholar] [CrossRef] [Green Version]
- Hodson, D.J.; Legros, C.; Desarménien, M.G.; Guérineau, N.C. Roles of connexins and pannexins in (neuro)endocrine physiology. Cell. Mol. Life Sci. 2015, 72, 2911–2928. [Google Scholar] [CrossRef]
- Darr, E.A.; Patel, A.D.; Yu, G.; Komorowski, Z.; McCormick, S.; Tiwari, R.; Schantz, S.P.; Geliebter, J. Reduced Cx43 gap junction plaque expression differentiates thyroid carcinomas from benign disease. Arch. Otolaryngol. Head Neck Surg. 2011, 137, 1161–1165. [Google Scholar] [CrossRef] [Green Version]
- Dominguez, C.; Karayan-Tapon, L.; Desurmont, T.; Gibelin, H.; Crespin, S.; Fromont, G.; Levillain, P.; Bouche, G.; Cantereau, A.; Mesnil, M.; et al. Altered expression of the gap junction protein Connexin43 Is associated with papillary thyroid carcinomas when compared with other noncancer pathologies of the thyroid. Thyroid 2011, 21, 1057–1066. [Google Scholar] [CrossRef]
- Mitašíková, M.; Lin, H.; Soukup, T.; Imanaga, I.; Tribulová, N. Diabetes and thyroid hormones affect connexin-43 and PKC-ε expression in rat heart atria. Physiol. Res. 2009, 58, 211–217. [Google Scholar] [PubMed]
- Almeida, N.A.S.; Cordeiro, A.; Machado, D.S.; Souza, L.L.; Ortiga-Carvalho, T.M.; Campos-de-Carvalho, A.C.; Wondisford, F.E.; Pazos-Moura, C.C. Connexin40 messenger ribonucleic acid is positively regulated by thyroid hormone (TH) acting in cardiac atria via the TH receptor. Endocrinology 2009, 150, 546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osuna, P.M.; Udovcic, M.; Sharma, M.D. Hyperthyroidism and the heart. Methodist Debakey Cardiovasc. J. 2017, 13, 60–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renaudon, B.; Lenfant, J.; Decressac, S.; Bois, P. Thyroid hormone increases the conductance density of f-channels in rabbit sino-atrial node cells. Recept. Channels 2000, 7, 1–8. [Google Scholar] [PubMed]
- Sun, Z.Q.; Ojamaa, K.; Nakamura, T.Y.; Artman, M.; Klein, I.; Coetzee, W.A. Thyroid hormone increases pacemaker activity in rat neonatal atrial myocytes. J. Mol. Cell. Cardiol. 2001, 33, 811–824. [Google Scholar] [CrossRef] [PubMed]
- Awais, D.; Shao, Y.; Ismail-Beigi, F. Thyroid hormone regulation of myocardial Na/K-ATPase gene expression. J. Mol. Cell. Cardiol. 2000, 32, 1969–1980. [Google Scholar] [CrossRef]
- Shenoy, R.; Klein, I.; Ojamaa, K. Differential regulation of SR calcium transporters by thyroid hormone in rat atria and ventricles. Am. J. Physiol. Heart Circ. Physiol. 2001, 281, 1690–1696. [Google Scholar] [CrossRef]
- Ma, M.L.; Watanabe, K.; Watanabe, H.; Hosaka, Y.; Komura, S.; Aizawa, Y.; Yamamoto, T. Different gene expression of potassium channels by thyroid hormone and an antithyroid drug between the atrium and ventricle of rats. Jpn. Heart J. 2003, 44, 101–110. [Google Scholar] [CrossRef] [Green Version]
- Hoit, B.D.; Khoury, S.F.; Shao, Y.; Gabel, M.; Liggett, S.B.; Walsh, R.A. Effects of thyroid hormone on cardiac β-adrenergic responsiveness in conscious baboons. Circulation 1997, 96, 592–598. [Google Scholar] [CrossRef]
- Morkin, E. Regulation of myosin heavy chain genes in the heart. Circulation 1993, 87, 1451–1460. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, H.; Washizuka, T.; Komura, S.; Yoshida, T.; Hosaka, Y.; Hatada, K.; Aizawa, Y.; Chinushi, M.; Yamamoto, T.; Ma, M.; et al. Genomic and non-genomic regulation of L-type calcium channels in rat ventricle by thyroid hormone. Endocr. Res. 2005, 31, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Sunagawa, M.; Yamakawa, M.; Shimabukuro, M.; Higa, N.; Takasu, N.; Kosugi, T. Electrophysiologic characteristics of atrial myocytes in levo-thyroxine-treated rats. Thyroid 2005, 15, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Hamasaki, Y.; Shinohara, O.; Ishida, H.; Hayashi, Y.; Nakazawa, H. Decreased protein kinase C-epsilon expression in hypertrophied cardiac ventricles induced by triiodothyronine treatment in the rat. Life Sci. 2000, 67, 1859–1868. [Google Scholar] [CrossRef]
- Connelly, T.J.; El-Hayek, R.; Sukhareva, M.; Coronado, R. L-thyroxine activates the intracellular Ca2+ release channel of skeletal muscle sarcoplasmic reticulum. Biochem. Mol. Biol. Int. 1994, 32, 441–448. [Google Scholar] [PubMed]
- Wang, Y.G.; Dedkova, E.N.; Fiening, J.P.; Ojamaa, K.; Blatter, L.A.; Lipsius, S.L. Acute exposure to thyroid hormone increases Na+ current and intracellular Ca2+ in cat atrial myocytes. J. Physiol. 2003, 546, 491–499. [Google Scholar] [CrossRef] [PubMed]
- Dudley, S.C.; Baumgarten, C.M. Bursting of cardiac sodium channels after acute exposure to 3,5,3′-triiodo-L-thyronine. Circ. Res. 1993, 73, 301–313. [Google Scholar] [CrossRef] [Green Version]
- Le Bouter, S.; Demolombe, S.; Chambellan, A.; Bellocq, C.; Aimond, F.; Toumaniantz, G.; Lande, G.; Siavoshian, S.; Baró, I.; Pond, A.L.; et al. Microarray analysis reveals complex remodeling of cardiac ion channel expression with altered thyroid status: Relation to cellular and integrated electrophysiology. Circ. Res. 2003, 92, 234–242. [Google Scholar] [CrossRef]
- Sartiani, L.; Mannaioni, G.; Masi, A.; Romanelli, M.N.; Cerbai, E. The hyperpolarization-activated cyclic nucleotide-gated channels: From biophysics to pharmacology of a unique family of ion channels. Pharmacol. Rev. 2017, 69, 354–395. [Google Scholar] [CrossRef]
- Wustmann, K.; Kucera, J.P.; Zanchi, A.; Burow, A.; Stuber, T.; Chappuis, B.; Diem, P.; Delacrétaz, E. Activation of electrical triggers of atrial fibrillation in hyperthyroidism. J. Clin. Endocrinol. Metab. 2008, 93, 2104–2108. [Google Scholar] [CrossRef] [Green Version]
- Ehrlich, J.R.; Biliczki, P.; Hohnloser, S.H.; Nattel, S. Atrial-Selective Approaches for the Treatment of Atrial Fibrillation. J. Am. Coll. Cardiol. 2008, 51, 787–792. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Jones, S.V.P.; Dillmann, W.H. Effects of hyperthyroidism on delayed rectifier K+ currents in left and right murine atria. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, 1448–1455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komiya, N.; Isomoto, S.; Nakao, K.; Hayano, M.; Yano, K. Electrophysiological abnormalities of the atrial muscle in patients with paroxysmal atrial fibrillation associated with hyperthyroidism. Clin. Endocrinol. 2002, 56, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Katircibasi, M.T.; Deniz, F.; Pamukcu, B.; Binici, S.; Atar, I. Effects of short-term propylthiouracil treatment on P wave duration and P wave dispersion in patients with overt hypertyroidism. Exp. Clin. Endocrinol. Diabetes 2007, 115, 376–379. [Google Scholar] [CrossRef] [PubMed]
- Colkesen, Y.; Acil, T.; Abayli, B.; Yigit, F.; Katircibasi, T.; Kocum, T.; Demircan, S.; Sezgin, A.; Ozin, B.; Muderrisoglu, H. Mean platelet volume is elevated during paroxysmal atrial fibrillation: A marker of increased platelet activation? Blood Coagul. Fibrinolysis 2008, 19, 411. [Google Scholar] [CrossRef]
- Patel, S.; Rauf, A.; Khan, H.; Abu-Izneid, T. Renin-angiotensin-aldosterone (RAAS): The ubiquitous system for homeostasis and pathologies. Biomed. Pharmacother. 2017, 94, 317–325. [Google Scholar] [CrossRef]
- Jones, N.R.; Taylor, C.J.; Hobbs, F.D.R.; Bowman, L.; Casadei, B. Screening for atrial fibrillation: A call for evidence. Eur. Heart J. 2020, 41, 1075–1085. [Google Scholar] [CrossRef]
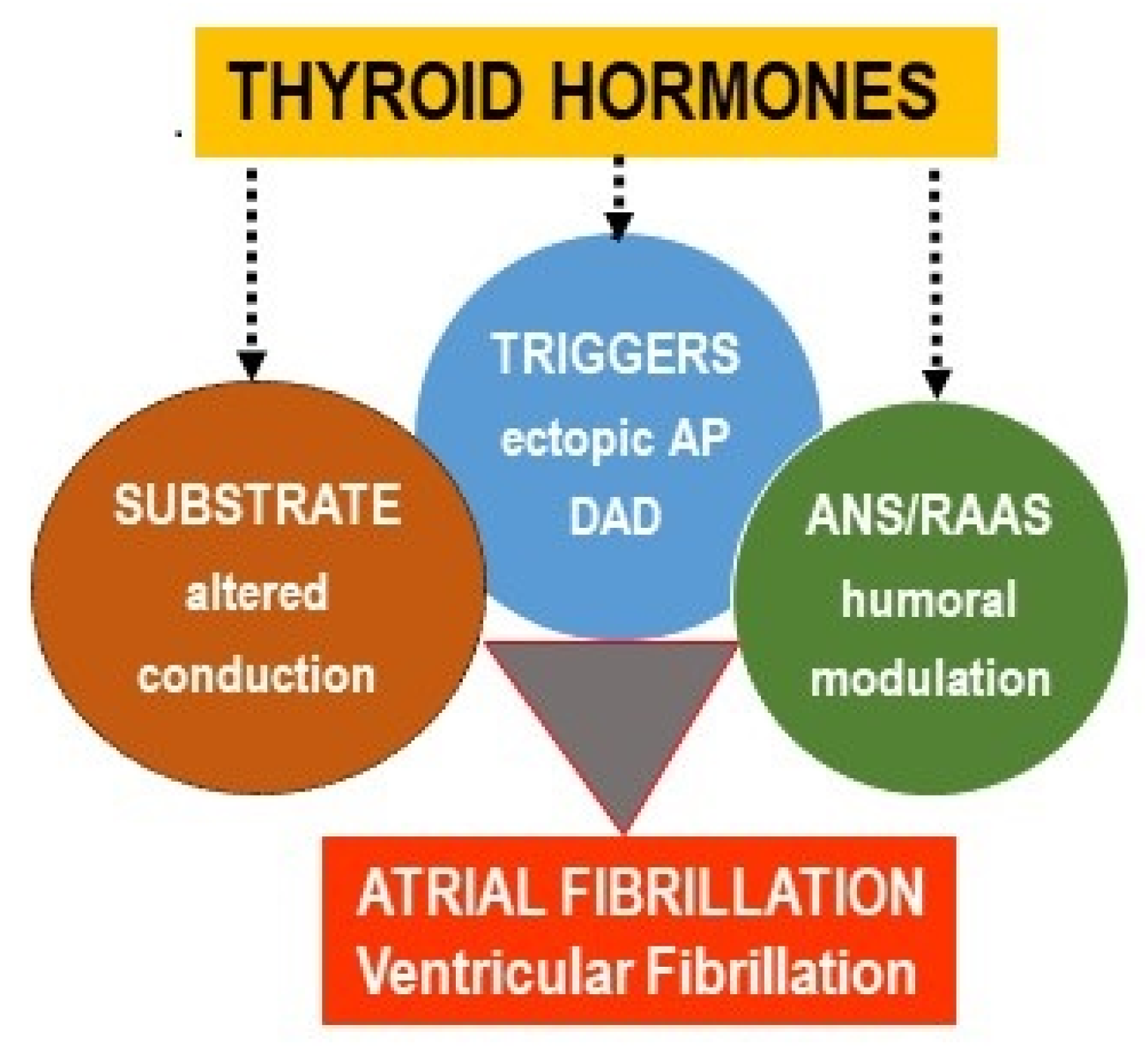

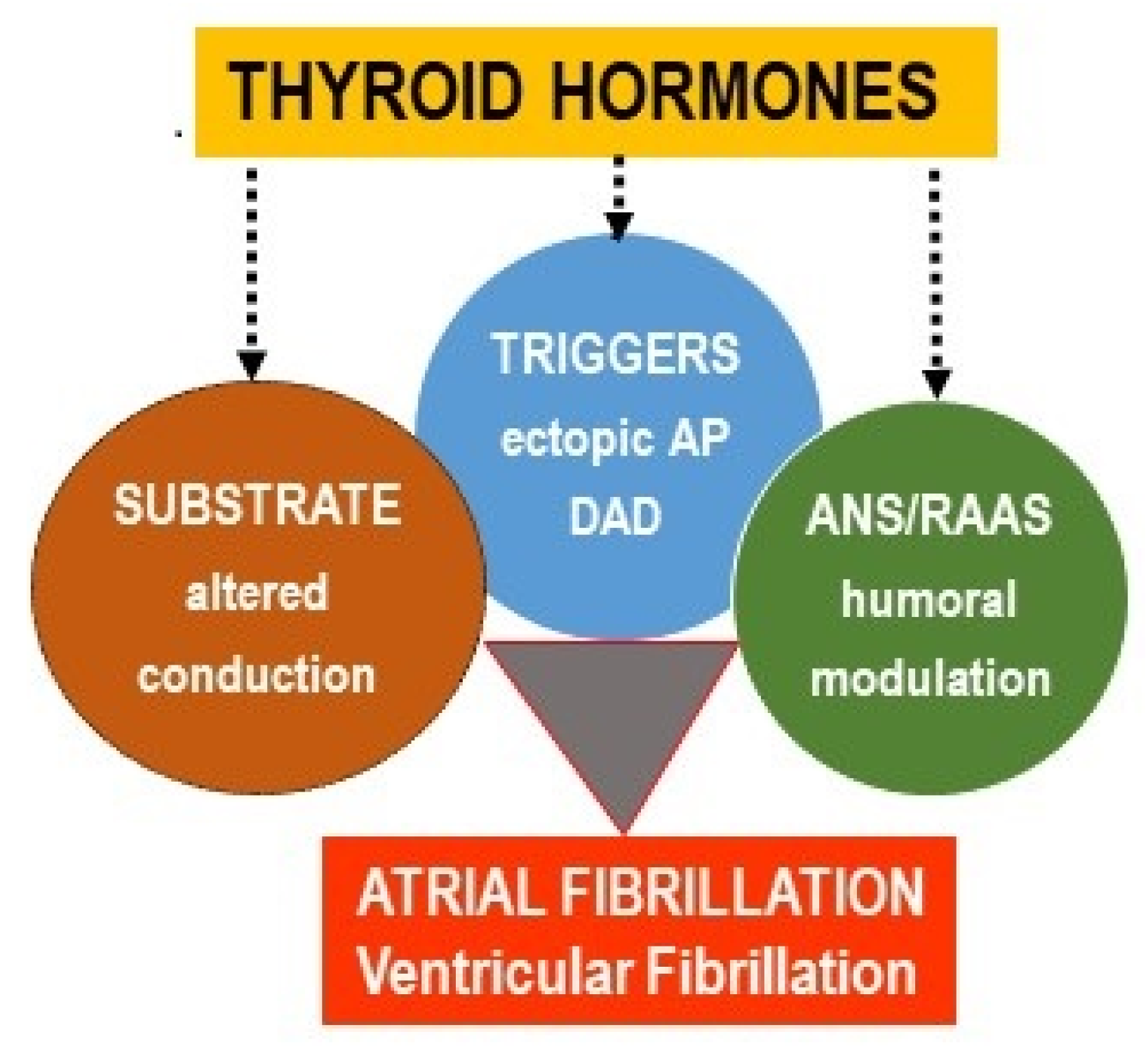{kind=link}
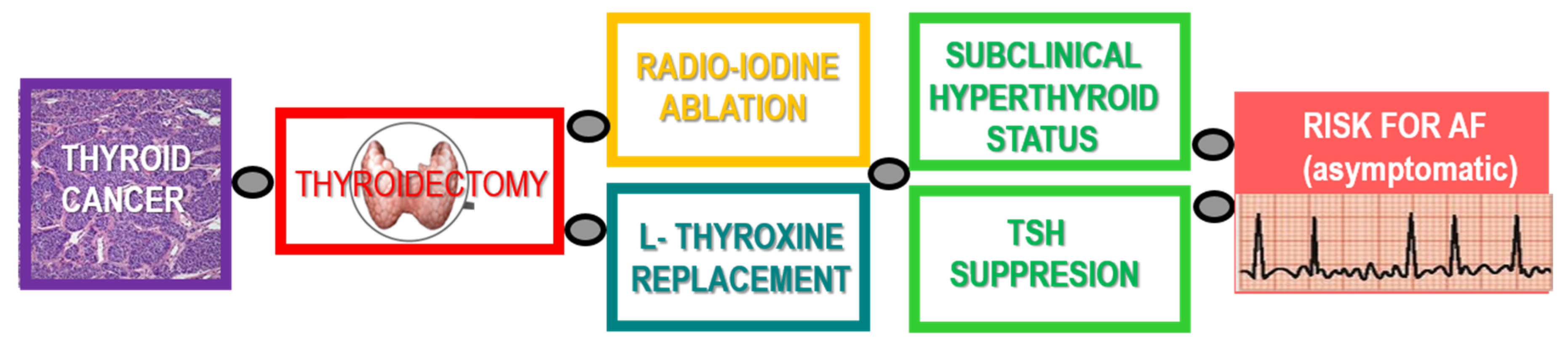
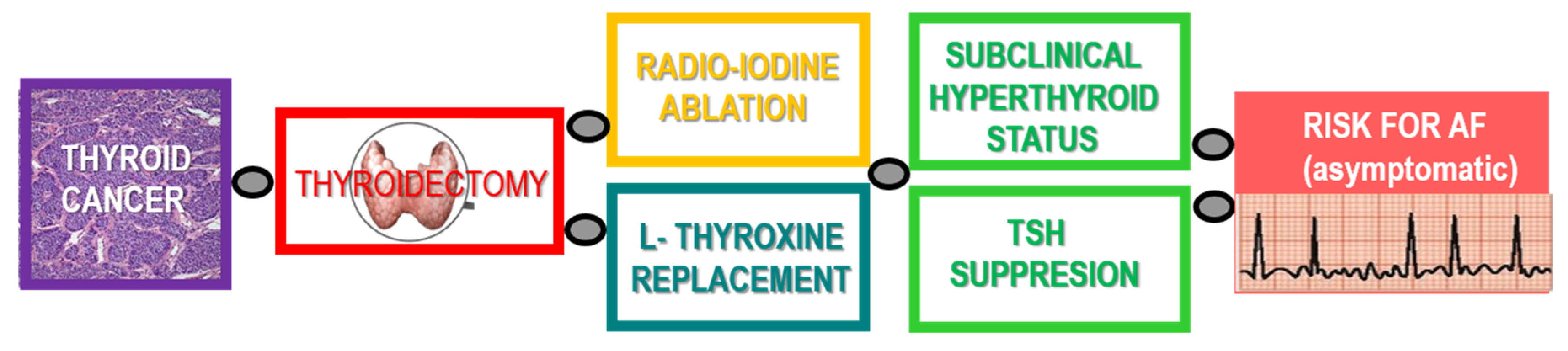{kind=link}
| Target Molecule | Action | Response/Putative Effects |
|---|---|---|
| Nuclear TH receptor-mediated actions | ||
| HCN2 channels | Upregulation [165,166] | Enhanced pacemaker activity |
| Na+ K+ ATPase | Upregulation [167] | Hyperpolarization/Enhanced excitability |
| Ca2+ ATPase | Upregulation [168] | Altered Ca2+ handling/Triggered activity |
| Ryanodine receptor | Upregulation [106] | Spontaneous Ca2+ leak/Triggered activity |
| Kv1.5; 4.2; 4.3 channels | Upregulation [169] | K+ current increase/Shortening APD |
| Connexin-40 | Upregulation [163] | Altered intercellular electrical coupling |
| β1-adrenergic receptor | Upregulation [170] | Sympathetic overdrive |
| α-myosin heavy chain | Upregulation [171] | Structural remodeling/Enhanced contractility |
| Phospholamban | Downregulation [168] | Altered Ca2+ handling/Triggered activity |
| Na/Ca exchanger | Downregulation [168] | Altered Ca2+ handling/Ca2+ overload |
| L-Ca2+ channels | Downregulation [172] | Altered Ca2+ handling/Triggered activity |
| Kv 1.2; Kv 1.4 channels | Downregulation [169,173] | K+ current increase/Shortening APD |
| Connexin-43 | Downregulation [107] | Altered intercellular electrical coupling |
| α1-adrenergic receptor | Downregulation [116] | Sympathetic overdrive |
| Protein kinase C-ε | Downregulation [174] | Reduced protein (Cx) phosphorylation |
| β-myosin heavy chain | Downregulation [171] | Structural remodeling/Enhanced contractility |
| Non-nuclear receptor-mediated actions of TH | ||
| HCN2 (If) current | Activation [165,166] | Enhanced pacemaker activity |
| Ca2+ ATPase | Activation [172] | Altered Ca2+ handling |
| Na+ K+ ATPase | Activation [21,167] | Hyperpolarization/Enhanced excitability |
| L-Ca2+ channels | Suppression [172] | Altered Ca2+ handling |
| Ryanodine channels | Activation [175] | Altered Ca2+ handling |
| Na+ channels | Activation [176,177] | Hyperpolarization |
| Na/Ca exchanger | Activation [21] | Altered Ca2+ handling |
| K+ channels | Activation [21,177] | Shortening APD |
| β-adrenergic receptors | Activation [117] | Sympathetic overdrive |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tribulova, N.; Kurahara, L.H.; Hlivak, P.; Hirano, K.; Szeiffova Bacova, B. Pro-Arrhythmic Signaling of Thyroid Hormones and Its Relevance in Subclinical Hyperthyroidism. Int. J. Mol. Sci. 2020, 21, 2844. https://doi.org/10.3390/ijms21082844
Tribulova N, Kurahara LH, Hlivak P, Hirano K, Szeiffova Bacova B. Pro-Arrhythmic Signaling of Thyroid Hormones and Its Relevance in Subclinical Hyperthyroidism. International Journal of Molecular Sciences. 2020; 21(8):2844. https://doi.org/10.3390/ijms21082844
Chicago/Turabian StyleTribulova, Narcis, Lin Hai Kurahara, Peter Hlivak, Katsuya Hirano, and Barbara Szeiffova Bacova. 2020. "Pro-Arrhythmic Signaling of Thyroid Hormones and Its Relevance in Subclinical Hyperthyroidism" International Journal of Molecular Sciences 21, no. 8: 2844. https://doi.org/10.3390/ijms21082844