The Interplay between Diabetes and Alzheimer’s Disease—In the Hunt for Biomarkers


{kind=link}
{kind=link}
Abstract
1. Alzheimer’s Disease vs. Type 2 Diabetes Mellitus
1.1. Alzheimer Disease Characteristic
1.2. Carbohydrate Metabolism
1.2.1. Glucose Metabolism in the Brain
1.2.2. Insulin Role in the Brain
1.3. Brain Energy Metabolism and AD Onset
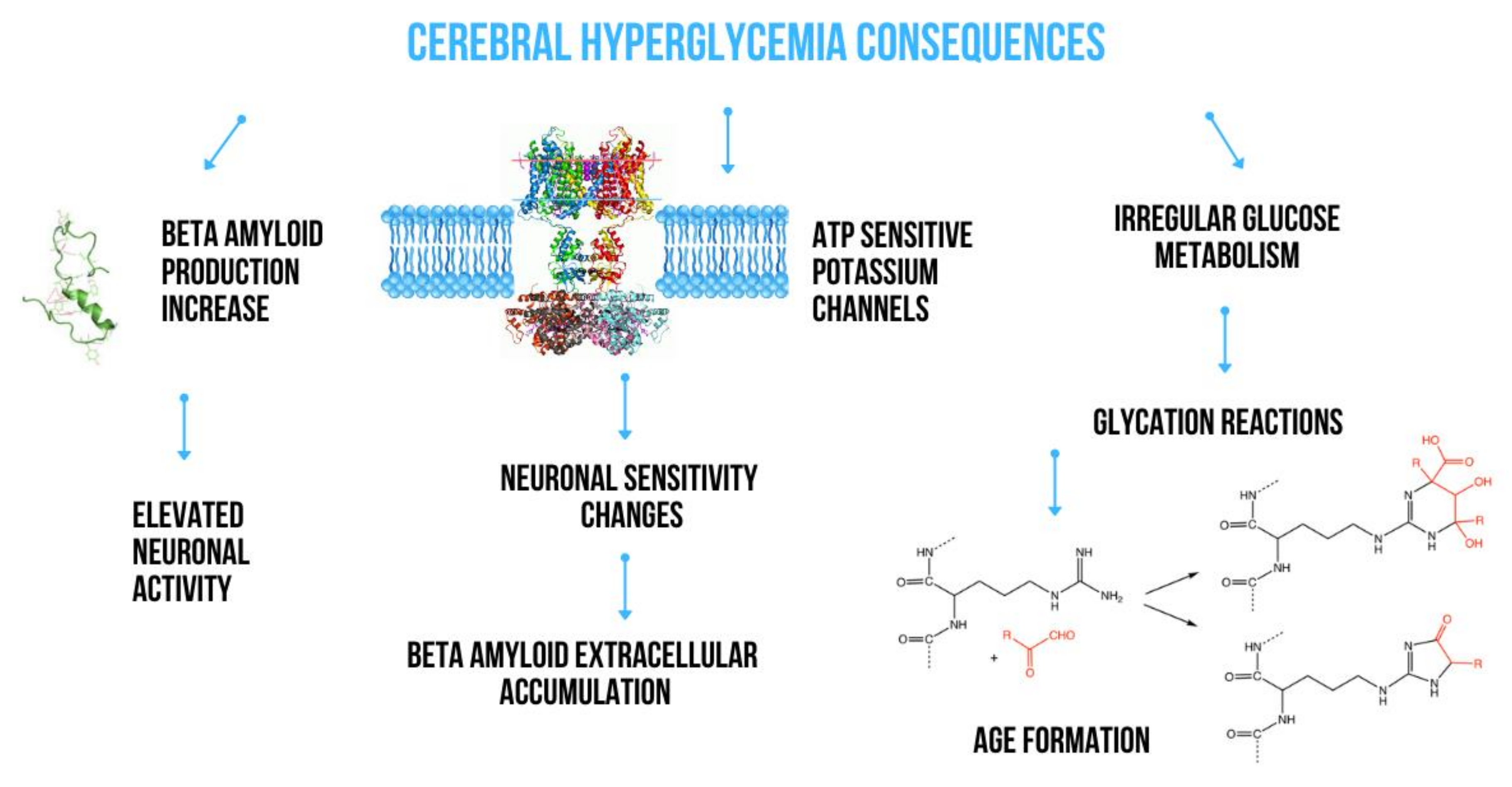
2. The Effect of Metabolic Disturbances on Amyloid Production
2.1. Impact of Glucose Metabolism on Amyloidogenesis
2.2. The Role of Insulin Signaling in the Amyloid β Cascade
3. The Effect of Metabolic Disturbances on Tau Protein Metabolism
3.1. Impact of Glucose Metabolism on Tau Protein Aggregation
3.2. Improper Insulin Metabolism and Tau Protein Synthesis
4. Biomarkers for Alzheimer’s Disease Diagnosis
4.1. T2DM-Related Biomarkers in AD
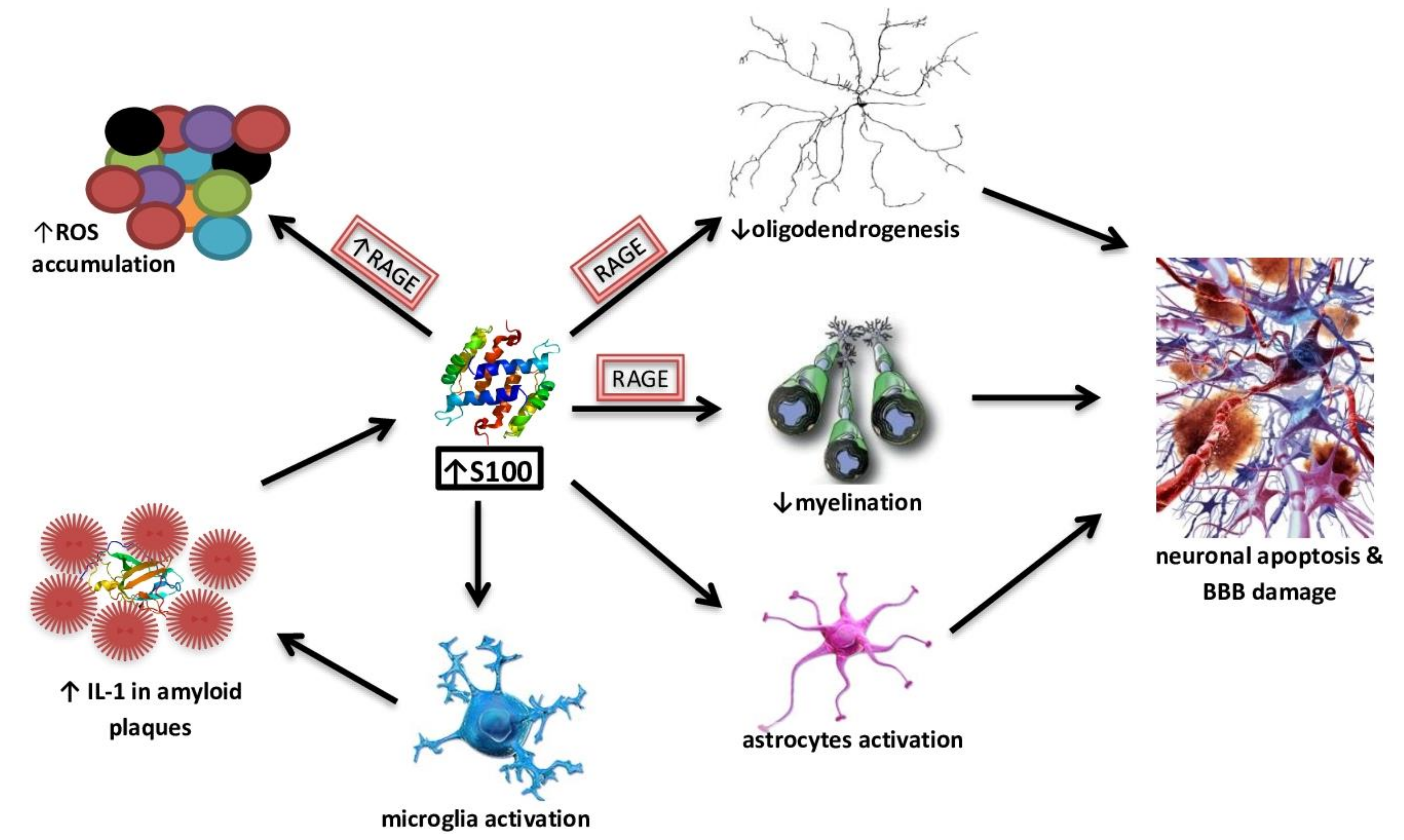
4.2. S100 Protein Family as a Potential Biomarkers of AD
4.3. Diagnostic and Potential AD Biomarkers from Cerebrospinal Fluid
4.4. Diagnostic and Potential Biomarkers from Peripheral Blood
5. Treating AD with Diabetic Medications
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Chatterjee, S.; Mudher, A. Alzheimer’s disease and type 2 diabetes: A critical assessment of the shared pathological traits. Front. Neurosci. 2018. [Google Scholar] [CrossRef] [PubMed]
- Haass, C. Take five—BACE and the γ-secretase quartet conduct Alzheimer’s amyloid β-peptide generation. EMBO J. 2004. [Google Scholar] [CrossRef] [PubMed]
- Alonso, A.D.; Cohen, L.S.; Corbo, C.; Morozova, V.; ElIdrissi, A.; Phillips, G.; Kleiman, F.E. Hyperphosphorylation of Tau Associates With Changes in Its Function Beyond Microtubule Stability. Front. Cell. Neurosci. 2018, 12, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Laßek, M.; Weingarten, J.; Einsfelder, U.; Brendel, P.; Müller, U.; Volknandt, W. Amyloid precursor proteins are constituents of the presynaptic active zone. J. Neurochem. 2013. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, B.; Yang, L.; Guo, Q.; Aithmitti, N.; Songyang, Z.; Zheng, H. Presynaptic and Postsynaptic Interaction of the Amyloid Precursor Protein Promotes Peripheral and Central Synaptogenesis. J. Neurosci. 2009. [Google Scholar] [CrossRef]
- Terry, A.V., Jr.; Buccafusco, J.J. The Cholinergic Hypothesis of Age and Alzheimer’s Disease- Related Cognitive Deficits: Recent Challenges and Their Implications for Novel Drug Development. J. Pharmacol. Exp. Ther. 2003, 306, 821–827. [Google Scholar] [CrossRef]
- Ferreira-vieira, T.H.; Guimaraes, I.M.; Silva, F.R.; Ribeiro, F.M. Alzheimer’s Disease: Targeting the Cholinergic System. Curr. Neuropharmacol. 2016, 14, 101–115. [Google Scholar] [CrossRef]
- Duthie, A.; Chew, D.; Soiza, R.L. Non-psychiatric comorbidity associated with Alzheimer’s disease. QJM 2011. [Google Scholar] [CrossRef]
- Lee, H.J.; Seo, H.I.; Cha, H.Y.; Yang, Y.J.; Kwon, S.H.; Yang, S.J. Diabetes and Alzheimer’s Disease: Mechanisms and Nutritional Aspects. Clin. Nutr. Res. 2018. [Google Scholar] [CrossRef]
- Rad, S.K.; Arya, A.; Karimian, H.; Madhavan, P.; Rizwan, F.; Koshy, S.; Prabhu, G. Mechanism involved in insulin resistance via accumulation of β-amyloid and neurofibrillary tangles: Link between type 2 diabetes and alzheimer’s disease. Drug Des. Devel. Ther. 2018. [Google Scholar] [CrossRef]
- Molofsky, A.V.; Krenick, R.; Ullian, E.; Tsai, H.H.; Deneen, B.; Richardson, W.D.; Barres, B.A.; Rowitch, D.H. Astrocytes and disease: A neurodevelopmental perspective. Genes Dev. 2012. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Chiang, S.H.; Saltiel, A.R. Insulin signaling and the regulation of glucose transport. Mol. Med. 2004. [Google Scholar] [CrossRef] [PubMed]
- Phelps, C.H. Barbiturate-induced glycogen accumulation in brain. An electron microscopic study. Brain Res. 1972. [Google Scholar] [CrossRef]
- Brown, A.M.; Ransom, B.R. Astrocyte glycogen and brain energy metabolism. Glia 2007, 55, 1263–1271. [Google Scholar] [CrossRef]
- Shah, K.; DeSilva, S.; Abbruscato, T. The role of glucose transporters in brain disease: Diabetes and Alzheimer’s disease. Int. J. Mol. Sci. 2012, 13, 12629–12655. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, F.; Iqbal, K.; Grundke-Iqbal, I.; Gong, C.X. Decreased glucose transporters correlate to abnormal hyperphosphorylation of tau in Alzheimer disease. FEBS Lett. 2008. [Google Scholar] [CrossRef]
- Sims-Robinson, C.; Kim, B.; Rosko, A.; Feldman, E.L. How does diabetes accelerate Alzheimer disease pathology? Nat. Rev. Neurol. 2010, 6, 551–559. [Google Scholar] [CrossRef]
- Chen, Z.; Zhong, C. Decoding Alzheimer’s disease from perturbed cerebral glucose metabolism: Implications for diagnostic and therapeutic strategies. Prog. Neurobiol. 2013, 108, 21–43. [Google Scholar] [CrossRef]
- Yao, J.; Irwin, R.W.; Zhao, L.; Nilsen, J.; Hamilton, R.T.; Brinton, R.D. Mitochondrial bioenergetic deficit precedes Alzheimer’s pathology in female mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2009. [Google Scholar] [CrossRef]
- Euser, S.M.; Sattar, N.; Witteman, J.C.M.; Bollen, E.L.E.M.; Sijbrands, E.J.G.; Hofman, A.; Perry, I.J.; Breteler Monique, M.B.; Westendorp Rudi, G.J.; PROSPER and the Rotterdam Study. A prospective analysis of elevated fasting glucose levels and cognitive function in older people: Results from PROSPER and the Rotterdam Study. Diabetes 2010, 59, 1601–1607. [Google Scholar] [CrossRef]
- Mosconi, L.; Tsui, W.H.; De Santi, S.; Li, J.; Rusinek, H.; Convit, A.; Boppana, M.; de Leon, M.J. Reduced hippocampal metabolism in MCI and AD: Automated FDG-PET image analysis. Neurology 2005. [Google Scholar] [CrossRef]
- Pappatà, S.; Salvatore, E.; Postiglione, A. In vivo imaging of neurotransmission and brain receptors in dementia. J. Neuroimaging 2008. [Google Scholar] [CrossRef]
- Cunnane, S.; Nugent, S.; Roy, M.; Courchesne-Loyer, A.; Croteau, E.; Tremblay, S.; Castellano, A.; Pifferi, F.; Bocti, C.; Paquet, N.; et al. Brain fuel metabolism, aging, and Alzheimer’s disease. Nutrition 2011. [Google Scholar] [CrossRef]
- Mosconi, L.; Berti, V.; Glodzik, L.; Pupi, A.; De Santi, S.; De Leon, M.J. Pre-clinical detection of Alzheimer’s disease using FDG-PET, with or without amyloid imaging. J. Alzheimer Dis. 2010. [Google Scholar] [CrossRef]
- Hiltunen, M.; Khandelwal, V.K.; Yaluri, N.; Tiilikainen, T.; Tusa, M.; Koivisto, H.; Krzisch, M.; Vepsäläinen, S.; Mäkinen, P.; Kemppainen, S.; et al. Contribution of genetic and dietary insulin resistance to Alzheimer phenotype in APP/PS1 transgenic mice. J. Cell Mol. Med. 2012, 16, 1206–1222. [Google Scholar] [CrossRef]
- Reiman, E.M.; Caselli, R.J.; Yun, L.S.; Chen, K.; Bandy, D.; Minoshima, S.; Thibodeau, S.N.; Osborne, D. Preclinical evidence of Alzheimer’s disease in persons homozygous for the epsilon 4 allele for apolipoprotein E [see comments]. N. Engl. J. Med. 1996, 334, 752–758. [Google Scholar] [CrossRef]
- Action, B.I. Insulin Regulates Brain Function, but How Does It Get There ? Diabetes 2014, 63, 3992–3997. [Google Scholar] [CrossRef]
- Farris, W.; Mansourian, S.; Chang, Y.; Lindsley, L.; Eckman, E.A.; Frosch, M.P.; Eckman, C.B.; Tanzi, R.E.; Selkoe, D.J.; Guénette, S. Insulin-degrading enzyme regulates the levels of insulin, amyloid β-protein, and the β-amyloid precursor protein intracellular domain in vivo. Proc. Natl. Acad. Sci. USA 2003. [Google Scholar] [CrossRef]
- Zhang, X.; Fu, Z.; Meng, L.; He, M.; Zhang, Z. The Early Events That Initiate β-Amyloid Aggregation in Alzheimer’s Disease. Front. Aging Neurosci. 2018, 10, 1–13. [Google Scholar] [CrossRef]
- Gasparini, L.; Gouras, G.K.; Wang, R.; Gross, R.S.; Beal, M.F.; Greengard, P.; Xu, H. Stimulation of beta-amyloid precursor protein trafficking by insulin reduces intraneuronal beta-amyloid and requires mitogen-activated protein kinase signaling. J. Neurosci. 2001, 21, 2561–2570. [Google Scholar] [CrossRef]
- Matos, M.; Augusto, E.; Oliveira, C.R.; Agostinho, P. Amyloid-beta peptide decreases glutamate uptake in cultured astrocytes: Involvement of oxidative stress and mitogen-activated protein kinase cascades. Neuroscience 2008. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, M.W.; Sanz-blasco, S.; Dolatabadi, N.; Parker, J.; Chon, K.; Lee, M.S.; Soussou, W.; McKercher, S.R.; Ambasudhan, R.; Nakamura, T.; et al. Elevated glucose and oligomeric b-amyloid disrupt synapses via a common pathway of aberrant protein S-nitrosylation. Nat. Commun. 2016, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Tharp, W.G.; Gupta, D.; Smith, J.; Jones, K.P.; Jones, A.M.; Pratley, R.E. Effects of Glucose and Insulin on Secretion of Amyloid- b by Human Adipose Tissue Cells. Obesity 2016, 24, 1471–1479. [Google Scholar] [CrossRef] [PubMed]
- Tarkowski, E.; Andreasen, N.; Tarkowski, A.; Blennow, K. Intrathecal inflammation precedes development of Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 2003. [Google Scholar] [CrossRef]
- An, Y.; Varma, V.R.; Varma, S.; Casanova, R.; Dammer, E.; Pletnikova, O.; Chia, C.W.; Egan, J.M.; Ferrucci, L.; Troncoso, J.; et al. Evidence for brain glucose dysregulation in Alzheimer’s disease. Alzheimer Dement. 2017. [Google Scholar] [CrossRef]
- Janson, J.; Laedtke, T.; Parisi, J.E.; O’Brien, P.; Petersen, R.C.; Butler, P.C. Increased Risk of Type 2 Diabetes in Alzheimer Disease. Diabetes 2004, 53, 474–481. [Google Scholar] [CrossRef]
- Verdile, G.; Keane, K.N.; Cruzat, V.F.; Medic, S.; Sabale, M.; Rowles, J.; Wijesekara, N.; Martins, R.N.; Fraser, P.E.; Newsholme, P. Inflammation and Oxidative Stress: The Molecular Connectivity between Insulin Resistance, Obesity, and Alzheimer’s Disease. Mediat. Inflamm. 2015. [Google Scholar] [CrossRef]
- Ciudin, A. Diabetes mellitus and Alzheimer’s disease: An unforgettable relation. Endocrinol. Nutr. 2016, 63, 191. [Google Scholar] [CrossRef]
- Dos Santos Matioli, M.N.P.; Suemoto, C.K.; Rodriguez, R.D.; Farias, D.S.; Da Silva, M.M.; Leite, R.E.P.; Ferretti-Rebustini, R.E.L.; Farfel, J.M.; Pasqualucci, C.A.; Filho, W.J.; et al. Diabetes is Not Associated with Alzheimer’s Disease Neuropathology. J. Alzheimer Dis. 2017. [Google Scholar] [CrossRef]
- Helzner, E.P.; Scarmeas, N.; Cosentino, S.; Tang, M.X.; Schupf, N.; Stern, Y. Survival in Alzheimer disease: A multiethnic, population-based study of incident cases. Neurology 2008. [Google Scholar] [CrossRef]
- Yu, J.H.; Han, K.; Park, S.; Cho, H.; Lee, D.Y.; Kim, J.W.; Seo, J.A.; Kim, S.G.; Baik, S.H.; Park, Y.G.; et al. Incidence and risk factors for dementia in type 2 diabetes mellitus: A nationwide population-based study in Korea. Diabetes Metab. J. 2020. [Google Scholar] [CrossRef]
- Kerti, L.; Witte, A.V.; Winkler, A.; Grittner, U.; Rujescu, D.; Flöel, A. Higher glucose levels associated with lower memory and reduced hippocampal microstructure. Neurology 2013. [Google Scholar] [CrossRef]
- Macauley, S.L.; Stanley, M.; Caesar, E.E.; Yamada, S.A.; Raichle, M.E.; Perez, R.; Mahan, T.E.; Sutphen, C.L.; Holtzman, D.M. Hyperglycemia modulates extracellular amyloid-β concentrations and neuronal activity in vivo. J. Clin. Investig. 2015, 125, 2463–2467. [Google Scholar] [CrossRef]
- Ho, L.; Qin, W.; Pompl, P.N.; Xiang, Z.; Wang, J.; Zhao, Z.; Peng, Y.; Cambareri, G.; Rocher, A.; Mobbs, C.V.; et al. Diet-induced insulin resistance promotes amyloidosis in a transgenic mouse model of Alzheimer’s disease. FASEB J. 2004, 18, 902–904. [Google Scholar] [CrossRef]
- Yang, Y.; Wu, Y.; Zhang, S.; Song, W. High Glucose Promotes Aβ Production by Inhibiting APP Degradation. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Li, X.H.; Du, L.L.; Cheng, X.S.; Jiang, X.; Zhang, Y.; Lv, B.L.; Liu, R.; Wang, J.-Z.; Zhou, X.-W. Glycation exacerbates the neuronal toxicity of β-amyloid. Cell Death Dis. 2013. [Google Scholar] [CrossRef]
- Solito, E.; Sastre, M. Microglia function in Alzheimer’s disease. Front. Pharmacol. 2012. [Google Scholar] [CrossRef]
- Arancio, O.; Zhang, H.P.; Chen, X.; Lin, C.; Trinchese, F.; Puzzo, D.; Liu, S.m.; Hegde, A.; Yan, S.F.; Stern, A.; et al. RAGE potentiates Aβ-induced perturbation of neuronal function in transgenic mice. EMBO J. 2004. [Google Scholar] [CrossRef]
- Sato, N.; Morishita, R. The roles of lipid and glucose metabolism in modulation of β-amyloid, tau, and neurodegeneration in the pathogenesis of Alzheimer disease. Front. Aging Neurosci. 2015. [Google Scholar] [CrossRef]
- Diehl, T.; Mullins, R.; Kapogiannis, D. Insulin resistance in Alzheimer’s disease. Transl. Res. 2017, 183, 26–40. [Google Scholar] [CrossRef]
- Kleinridders, A.; Ferris, H.A.; Cai, W.; Kahn, C.R. Insulin action in brain regulates systemic metabolism and brain function. Diabetes 2014, 63, 2232–2243. [Google Scholar] [CrossRef]
- Chiu, S.L.; Chen, C.M.; Cline, H.T. Insulin Receptor Signaling Regulates Synapse Number, Dendritic Plasticity, and Circuit Function In Vivo. Neuron 2008. [Google Scholar] [CrossRef]
- Biessels, G.J.; Strachan, M.W.J.; Visseren, F.L.J.; Kappelle, L.J.; Whitmer, R.A. Dementia and cognitive decline in type 2 diabetes and prediabetic stages: towards targeted interventions. Lancet Diabetes Endocrinol. 2014, 2, 246–255. [Google Scholar] [CrossRef]
- Hölscher, C. New drug treatments show neuroprotective effects in Alzheimer’s and Parkinson’s diseases. Neural. Regen. Res. 2014. [Google Scholar] [CrossRef]
- Tumminia, A.; Vinciguerra, F.; Parisi, M.; Frittitta, L. Type 2 diabetes mellitus and alzheimer’s disease: Role of insulin signalling and therapeutic implications. Int. J. Mol. Sci. 2018, 19, 3306. [Google Scholar] [CrossRef]
- Vandal, M.; White, P.J.; Tremblay, C.; St-Amour, I.; Chevrier, G.; Emond, V.; Lefrançois, D.; Virgili, J.; Planel, E.; Giguere, Y.; et al. Insulin reverses the high-fat diet-induced increase in brain Aβ and improves memory in an animal model of Alzheimer disease. Diabetes 2014. [Google Scholar] [CrossRef]
- Devi, L.; Alldred, M.J.; Ginsberg, S.D.; Ohno, M. Mechanisms underlying insulin deficiency-induced acceleration of β-amyloidosis in a mouse model of Alzheimer’s disease. PLoS ONE 2012, 7. [Google Scholar] [CrossRef]
- Son, S.M.; Song, H.; Byun, J.; Park, K.S.; Jang, H.C.; Park, Y.J.; Mook-Jung, I. Accumulation of autophagosomes contributes to enhanced amyloidogenic APP processing under insulin-resistant conditions. Autophagy 2012. [Google Scholar] [CrossRef]
- Ling, X.; Martins, R.N.; Racchi, M.; Craft, S.; Helmerhorst, E. Amyloid beta antagonizes insulin promoted secretion of the amyloid beta protein precursor. J. Alzheimer Dis. 2002. [Google Scholar] [CrossRef]
- De Felice, F.G.; Benedict, C. A key role of insulin receptors in memory. Diabetes 2015. [Google Scholar] [CrossRef]
- Zhao, W.Q.; Townsend, M. Insulin resistance and amyloidogenesis as common molecular foundation for type 2 diabetes and Alzheimer’s disease. Biochim. Biophys. Acta Mol. Basis Dis. 2009. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, F.; Grundke-Iqbal, I.; Iqbal, K.; Gong, C.X. Brain glucose transporters, O-GlcNAcylation and phosphorylation of tau in diabetes and Alzheimer’s disease. J. Neurochem. 2009. [Google Scholar] [CrossRef]
- Sun, Y.; Xiao, Q.; Luo, C.; Zhao, Y.; Pu, D.; Zhao, K.; Chen, J.; Wang, M.; Liao, Z. High-glucose induces tau hyperphosphorylation through activation of TLR9-P38MAPK pathway. Exp. Cell Res. 2017, 359, 312–318. [Google Scholar] [CrossRef]
- Silver, A.C.; Arjona, A.; Walker, W.E.; Fikrig, E. Article The Circadian Clock Controls Toll-like Receptor 9-Mediated Innate and Adaptive Immunity. Immunity 2012, 36, 251–261. [Google Scholar] [CrossRef]
- Shintani, Y.; Kapoor, A.; Kaneko, M.; Smolenski, R.T.; D’Acquisto, F.; Coppen, S.R.; Harada-Shoji, N.; Lee, H.J.; Thiemermann, C.; Takashima, S.; et al. TLR9 mediates cellular protection by modulating energy metabolism in cardiomyocytes and neurons. Proc. Natl. Acad. Sci. USA 2013. [Google Scholar] [CrossRef]
- Mairet-coello, G.; Courchet, J.; Pieraut, S.; Courchet, V.; Maximov, A.; Polleux, F. Article The CAMKK2-AMPK Kinase Pathway Mediates the Synaptotoxic Effects of A b Oligomers through Tau Phosphorylation. Neuron 2013, 78, 94–108. [Google Scholar] [CrossRef]
- Kickstein, E.; Krauss, S.; Thornhill, P.; Rutschow, D.; Zeller, R.; Sharkey, J.; Williamson, R.; Fuchs, M.; Köhler, A.; Glossmann, H.; et al. Biguanide metformin acts on tau phosphorylation via mTOR/protein phosphatase 2A (PP2A) signaling. Proc. Natl. Acad. Sci. USA 2010. [Google Scholar] [CrossRef]
- Cohen, T.J.; Guo, J.L.; Hurtado, D.E.; Kwong, L.K.; Mills, I.P.; Trojanowski, J.Q.; Lee, V.M.Y. The acetylation of tau inhibits its function and promotes pathological tau aggregation. Nat. Commun. 2011. [Google Scholar] [CrossRef]
- Zhu, Y.; Shan, X.; Yuzwa, S.A.; Vocadlo, D.J. The emerging link between O-GlcNAc and Alzheimer disease. J. Biol. Chem. 2014. [Google Scholar] [CrossRef]
- Smet-Nocca, C.; Broncel, M.; Wieruszeski, J.M.; Tokarski, C.; Hanoulle, X.; Leroy, A.; Landrieu, I.; Rolando, C.; Lippens, G.; Hackenberger, C.P. Identification of O-GlcNAc sites within peptides of the Tau protein and their impact on phosphorylation. Mol. Biosyst. 2011. [Google Scholar] [CrossRef]
- Robertson, L.A.; Moya, K.L.; Breen, K.C. The potential role of tau protein O-glycosylation in Alzheimer’s disease. J. Alzheimer Dis. 2004. [Google Scholar] [CrossRef]
- Bhat, R.; Xue, Y.; Berg, S.; Hellberg, S.; Ormö, M.; Nilsson, Y.; Radesäter, A.-C.; Jerning, E.; Markgren, P.-O.; Borgegård, T.; et al. Structural Insights and Biological Effects of Glycogen Synthase Kinase 3-specific Inhibitor AR-A014418. J. Biol. Chem. 2003. [Google Scholar] [CrossRef]
- Cheng, C.M.; Tseng, V.; Wang, J.; Wang, D.; Matyakhina, L.; Bondy, C.A. Tau is hyperphosphorylated in the insulin-like growth factor-I null brain. Endocrinology 2005. [Google Scholar] [CrossRef]
- Schubert, M.; Gautam, D.; Surjo, D.; Ueki, K.; Baudler, S.; Schubert, D.; Kondo, T.; Alber, J.; Galldiks, N.; Küstermann, E.; et al. Role for neuronal insulin resistance in neurodegenerative diseases. Proc. Natl. Acad. Sci. USA 2004, 101, 3100–3105. [Google Scholar] [CrossRef]
- Planel, E.; Tatebayashi, Y.; Miyasaka, T.; Liu, L.; Wang, L.; Herman, M.; Yu, W.H.; Luchsinger, J.A.; Wadzinski, B.; Duff, K.E.; et al. Insulin dysfunction induces in vivo tau hyperphosphorylation through distinct mechanisms. J. Neurosci. 2007. [Google Scholar] [CrossRef]
- Rodriguez-Rodriguez, P.; Sandebring-Matton, A.; Merino-Serrais, P.; Parrado-Fernandez, C.; Rabano, A.; Winblad, B.; Ávila, J.; Ferrer, I.; Cedazo-Minguez, A. Tau hyperphosphorylation induces oligomeric insulin accumulation and insulin resistance in neurons. Brain 2017. [Google Scholar] [CrossRef]
- Babić, M.; Štrac, D.Š.; Mück-Šeler, D.; Pivac, N.; Stanić, G.; Hof, P.R.; Simić, G. Update on the core and developing cerebrospinal fluid biomarkers for Alzheimer disease. Croat. Med. J. 2014. [Google Scholar] [CrossRef]
- Sabbagh, M.N.; Hendrix, S.; Harrison, J.E. FDA position statement “Early Alzheimer’s disease: Developing drugs for treatment, Guidance for Industry”. Alzheimer Dement. Transl. Res. Clin. Interv. 2019. [Google Scholar] [CrossRef]
- EMA. Guideline on the clinical investigation of medicines for the treatment of Alzheimer’s disease. CPMP/EWP/553/95 Rev2. Lekarz POZ 2015, 4, 370–374. [Google Scholar]
- De La Monte, S.M. Contributions of brain insulin resistance and deficiency in amyloid-related neurodegeneration in alzheimers disease. Drugs 2012, 72, 49–66. [Google Scholar] [CrossRef]
- Nobili, F.; Morbelli, S. [18F]FDG-PET as a Biomarker for Early Alzheimer’s Disease. Open Nucl. Med. J. 2014, 2, 46–52. [Google Scholar] [CrossRef]
- Kavanagh, K.; Day, S.M.; Pait, M.C.; Mortiz, W.R.; Newgard, C.B.; Ilkayeva, O.; Mcclain, D.A.; Macauley, S.L. Type-2-Diabetes Alters CSF but Not Plasma Metabolomic and AD Risk Profiles in Vervet Monkeys. Front. Neurosci. 2019. [Google Scholar] [CrossRef]
- Gupta, A.; Singh, A.; Deka, R.C.; Gupta, R.; Jha, R. To Investigate Role of Glycosylated Hemoglobin (Hba1c) as a Biomarker for Prediction of Dementia and Cognitive Dysfunction in Type 2 Diabetic Patients. J. Alzheimer Dis. Park. 2018. [Google Scholar] [CrossRef]
- Luciano, R.; Barraco, G.M.; Muraca, M.; Ottino, S.; Spreghini, M.R.; Sforza, R.W.; Rustico, C.; Morino, G.S.; Manco, M. Biomarkers of Alzheimer disease, insulin resistance, and obesity in childhood. Pediatrics 2015. [Google Scholar] [CrossRef]
- Chetram Deochand, M.T. CSF and Brain Indices of Insulin Resistance, Oxidative Stress and Neuro-Inflammation in Early versus Late Alzheimer’s Disease. J. Alzheimer Dis. Park. 2013. [Google Scholar] [CrossRef]
- Matsuzaki, T.; Sasaki, K.; Tanizaki, Y.; Hata, J.; Fujimi, K.; Matsui, Y.; Sekita, A.; Suzuki, S.O.; Kanba, S.; Kiyohara, Y.; et al. Insulin resistance is associated with the pathology of Alzheimer disease: The Hisayama study. Neurology 2010, 75, 764–770. [Google Scholar] [CrossRef]
- Sedaghat, F.; Notopoulos, A. S100 protein family and its application in clinical practice. Hippokratia 2008, 12, 198. [Google Scholar]
- Steiner, J.; Bogerts, B.; Schroeter, M.L.; Bernstein, H.G. S100B protein in neurodegenerative disorders. Clin. Chem. Lab Med. 2011. [Google Scholar] [CrossRef]
- Chazin, W.J. Relating form and function of EF-hand calcium binding proteins. Acc. Chem. Res. 2011. [Google Scholar] [CrossRef]
- Michetti, F.; Corvino, V.; Geloso, M.C.; Lattanzi, W.; Bernardini, C.; Serpero, L.; Gazzolo, D. The S100B protein in biological fluids: More than a lifelong biomarker of brain distress. J. Neurochem. 2012. [Google Scholar] [CrossRef]
- Austermann, J.; Spiekermann, C.; Roth, J. S100 proteins in rheumatic diseases. Nat. Rev. Rheumatol. 2018. [Google Scholar] [CrossRef]
- Pruenster, M.; Vogl, T.; Roth, J.; Sperandio, M. S100A8/A9: From basic science to clinical application. Pharmacol. Ther. 2016. [Google Scholar] [CrossRef]
- Murao, S.; Collart, F.R.; Huberman, E. A protein containing the cystic fibrosis antigen is an inhibitor of protein kinases. J. Biol. Chem. 1989, 264, 8356–8360. [Google Scholar]
- Pruenster, M.; Kurz, A.R.M.; Chung, K.J.; Cao-Ehlker, X.; Bieber, S.; Nussbaum, C.F.; Bierschenk, S.; Eggersmann, T.K.; Rohwedder, I.; Heinig, K.; et al. Extracellular MRP8/14 is a regulator of β22 integrin-dependent neutrophil slow rolling and adhesion. Nat. Commun. 2015. [Google Scholar] [CrossRef]
- Cheng, P.; Corzo, C.A.; Luetteke, N.; Yu, B.; Nagaraj, S.; Bui, M.M.; Ortiz, M.; Nacken, W.; Sorg, C.; Vogl, T.; et al. Inhibition of dendritic cell differentiation and accumulation of myeloid-derived suppressor cells in cancer is regulated by S100A9 protein. J. Exp. Med. 2008. [Google Scholar] [CrossRef]
- Turovskaya, O.; Foell, D.; Sinha, P.; Vogl, T.; Newlin, R.; Nayak, J.; Nguyen, M.; Olsson, A.; Nawroth, P.P.; Bierhaus, A.; et al. RAGE, carboxylated glycans and S100A8/A9 play essential roles in colitis-associated carcinogenesis. Carcinogenesis 2008. [Google Scholar] [CrossRef]
- Björk, P.; Björk, A.; Vogl, T.; Stenström, M.; Liberg, D.; Olsson, A.; Roth, J.; Ivars, F.; Leanderson, T. Identification of human S100A9 as a novel target for treatment of autoimmune disease via binding to quinoline-3-carboxamides. PLoS Biol. 2009. [Google Scholar] [CrossRef]
- Mrak, R.E.; Griffin, W.S.T. The role of activated astrocytes and of the neurotrophic cytokine S100B in the pathogenesis of Alzheimer’s disease. Neurobiol. Aging 2001. [Google Scholar] [CrossRef]
- Marshak, D.R.; Pesce, S.A.; Stanley, L.C.; Griffin, W.S.T. Increased S100β neurotrophic activity in Alzheimer’s disease temporal lobe. Neurobiol. Aging 1992. [Google Scholar] [CrossRef]
- Petzold, A.; Jenkins, R.; Watt, H.C.; Green, A.J.E.; Thompson, E.J.; Keir, G.; Fox, N.C.; Rossor, M.N. Cerebrospinal fluid S100B correlates with brain atrophy in Alzheimer’s disease. Neurosci. Lett. 2003. [Google Scholar] [CrossRef]
- Businaro, R.; Leone, S.; Fabrizi, C.; Sorci, G.; Donato, R.; Lauro, G.M.; Fumagalli, L. S100B protects LAN-5 neuroblastoma cells against Aβ amyloid-induced neurotoxicity via RAGE engagement at low doses but increases Aβ amyloid neurotoxicity at high doses. J. Neurosci. Res. 2006. [Google Scholar] [CrossRef]
- Mori, T.; Koyama, N.; Arendash, G.W.; Horikoshi-Sakuraba, Y.; Tan, J.; Town, T. Overexpression of human S100B exacerbates cerebral amyloidosis and gliosis in the Tg2576 mouse model of Alzheimer’s disease. Glia 2010. [Google Scholar] [CrossRef]
- Brozzi, F.; Arcuri, C.; Giambanco, I.; Donato, R. S100B protein regulates astrocyte shape and migration via interaction with Src kinase: Implications for astrocyte development, activation, and tumor growth. J. Biol. Chem. 2009. [Google Scholar] [CrossRef]
- Wang, C.; Iashchishyn, I.A.; Pansieri, J.; Nyström, S.; Klementieva, O.; Kara, J.; Horváth, I.; Moskalenko, R.; Rofougaran, R.; Gouras, G.; et al. S100A9-Driven Amyloid-Neuroinflammatory Cascade in Traumatic Brain Injury as a Precursor State for Alzheimer’s Disease. Sci. Rep. 2018, 8, 1–16. [Google Scholar] [CrossRef]
- Rothermundt, M.; Ohrmann, P.; Abel, S.; Siegmund, A.; Pedersen, A.; Ponath, G.; Suslow, T.; Peters, M.; Kaestner, F.; Heindel, W.; et al. Glial cell activation in a subgroup of patients with schizophrenia indicated by increased S100B serum concentrations and elevated myo-inositol. Prog. Neuro Psychopharmacol. Biol. Psychiatry 2007. [Google Scholar] [CrossRef]
- Chaves, M.L.; Camozzato, A.L.; Ferreira, E.D.; Piazenski, I.; Kochhann, R.; Dall’Igna, O.; Mazzini, G.S.; Souza, D.O.; Portela, L.V. Serum levels of S100B and NSE proteins in Alzheimer’s disease patients. J. Neuroinflamm. 2010. [Google Scholar] [CrossRef]
- Peskind, E.R.; Griffin, W.S.T.; Akama, K.T.; Raskind, M.A.; Van Eldik, L.J. Cerebrospinal fluid S100B is elevated in the earlier stages of Alzheimer’s disease. Neurochem. Int. 2001. [Google Scholar] [CrossRef]
- Nooijen, P.T.G.A.; Schoonderwaldt, H.C.; Wevers, R.A.; Hommes, O.R.; Lamers, K.J.B. Neuron-specific enolase, S-100 protein, myelin basic protein and lactate in CSF in Dementia. Dement. Geriatr. Cogn. Disord. 1997. [Google Scholar] [CrossRef]
- Lam, V.; Albrecht, M.A.; Takechi, R.; Giles, C.; James, A.P.; Foster, J.K.; Mamo, J.C.L. The serum concentration of the calcium binding protein S100B is positively associated with cognitive performance in older adults. Front. Aging Neurosci. 2013. [Google Scholar] [CrossRef]
- Kheirouri, S.; Ebrahimi, E.; Alizadeh, M. Association of S100B Serum Levels with Metabolic Syndrome and its Components. Act Med. Port. 2018, 31, 201–206. [Google Scholar] [CrossRef]
- Guo, C.; Zhang, S.; Li, J.Y.; Ding, C.; Yang, Z.H.; Chai, R.; Wang, X.; Wang, Z.-Y. Chronic hyperglycemia induced via the heterozygous knockout of Pdx1 worsens neuropathological lesion in an Alzheimer mouse model. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef]
- Hameed, I.; Masoodi, S.R.; Mir, S.A.; Nabi, M.; Ghazanfar, K.; Ganai, B.A. Type 2 diabetes mellitus: From a metabolic disorder to an inflammatory condition. World J. Diabetes 2015. [Google Scholar] [CrossRef]
- Gerlach, R.; Demel, G.; König, H.G.; Gross, U.; Prehn, J.H.M.; Raabe, A.; Seifert, V.; Kögel, D. Active secretion of S100B from astrocytes during metabolic stress. Neuroscience 2006. [Google Scholar] [CrossRef]
- Olsson, B.; Lautner, R.; Andreasson, U.; Öhrfelt, A.; Portelius, E.; Bjerke, M.; Hölttä, M.; Rosén, C.; Olsson, C.; Strobel, G.; et al. CSF and blood biomarkers for the diagnosis of Alzheimer’s disease: a systematic review and meta-analysis. Lancet Neurol. 2016. [Google Scholar] [CrossRef]
- Molinuevo, J.L.; Ayton, S.; Batrla, R.; Bednar, M.M.; Bittner, T.; Cummings, J.; Fagan, A.M.; Hampel, H.; Mielke, M.M.; Mikulskis, A.; et al. Current state of Alzheimer’s fluid biomarkers. Acta Neuropathol. 2018. [Google Scholar] [CrossRef]
- Marszałek, M. Choroba Alzheimera a produkty degradacji białka APP. Formowanie i różnorodność form fibrylujacych peptydów—Wybrane aspekty. Postepy Hig. Med. Dosw. 2016. [Google Scholar] [CrossRef]
- Blennow, K. A Review of Fluid Biomarkers for Alzheimer’s Disease: Moving from CSF to Blood. Neurol. Ther. 2017. [Google Scholar] [CrossRef]
- Sunderland, T.; Mirza, N.; Putnam, K.T.; Linker, G.; Bhupali, D.; Durham, R.; Soares, H.; Kimmel, L.; Friedman, D.; Bergeson, J.; et al. Cerebrospinal fluid beta-amyloid1-42 and tau in control subjects at risk for Alzheimer’s disease: The effect of APOE epsilon4 allele. Biol. Psychiatry 2004, 56, 670–676. [Google Scholar] [CrossRef]
- Sjögren, M.; Vanderstichele, H.; Ågren, H.; Zachrisson, O.; Edsbagge, M.; Wilkkelsø, C.; Skoog, I.; Wallin, A.; Wahlund, L.O.; Marcusson, J.; et al. Tau and Aβ42 in cerebrospinal fluid from healthy adults 21-93 years of age: Establishment of reference values. Clin. Chem. 2001, 47, 1776–1781. [Google Scholar] [CrossRef]
- Kimura, T.; Sharma, G.; Ishiguro, K.; Hisanaga, S.I. Phospho-tau bar code: Analysis of phosphoisotypes of tau and its application to tauopathy. Front. Neurosci. 2018. [Google Scholar] [CrossRef]
- Agency, E.M. Qualification opinion of Alzheimer’s disease novel methodologies/biomarkers for the use of CSF AB 1-42 and t-tau and/or PET-amyloid imaging ( positive/negative ) as biomarkers for enrichment, for use in regulatory clinical trials in mild and mode. Available online: https://www.ema.europa.eu/en/documents/regulatory-procedural-guideline/qualification-opinion-alzheimers-disease-novel-methodologies/biomarkers-use-cerebrospinal-fluid-amyloid-beta-1-42-t-tau/positron-emission-tomography-amyloid-imaging-positive/negative_en.pdf (accessed on 22 January 2020).
- Decourt, B.; Sabbagh, M.N. BACE1 as a potential biomarker for alzheimer’s disease. J. Alzheimer Dis. 2011. [Google Scholar] [CrossRef]
- Desikan, R.S.; Thompson, W.K.; Holland, D.; Hess, C.P.; Brewer, J.B.; Zetterberg, H.; Blennow, K.; Andreassen, O.A.; McEvoy, L.K.; Hyman, B.T.; et al. Heart fatty acid binding protein and Aβ-associated Alzheimer’s neurodegeneration. Mol. Neurodegener. 2013. [Google Scholar] [CrossRef]
- Lee, W.J.; Liao, Y.C.; Wang, Y.F.; Lin, I.F.; Wang, S.J.; Fuh, J.L. Plasma MCP-1 and cognitive decline in patients with Alzheimer’s disease and mild cognitive impairment: A two-year follow-up study. Sci. Rep. 2018. [Google Scholar] [CrossRef]
- Janelidze, S.; Stomrud, E.; Palmqvist, S.; Zetterberg, H.; Van Westen, D.; Jeromin, A.; Song, L.; Hanlon, D.; Tan Hehir, C.A.; Baker, D.; et al. Plasma β-amyloid in Alzheimer’s disease and vascular disease. Sci. Rep. 2016. [Google Scholar] [CrossRef]
- Nakamura, A.; Kaneko, N.; Villemagne, V.L.; Kato, T.; Doecke, J.; Doré, V.; Fowler, C.; Li, Q.X.; Martins, R.; Rowe, C.; et al. High performance plasma amyloid-β biomarkers for Alzheimer’s disease. Nature 2018. [Google Scholar] [CrossRef]
- Pérez-Grijalba, V.; Pesini, P.; Allué, J.A.; Sarasa, L.; Montañés, M.; Lacosta, A.M.; Diego, C.; Itziar, S.-J.; Mercè, B.; Lluis, T.; et al. Aβ1-17 is a major amyloid-β fragment isoform in cerebrospinal fluid and blood with possible diagnostic value in Alzheimer’s disease. J. Alzheimer Dis. 2014. [Google Scholar] [CrossRef]
- Gisslén, M.; Price, R.W.; Andreasson, U.; Norgren, N.; Nilsson, S.; Hagberg, L.; Fuchs, D.; Spudich, S.; Blennow, K.; Zetterberg, H. Plasma Concentration of the Neurofilament Light Protein (NFL) is a Biomarker of CNS Injury in HIV Infection: A Cross-Sectional Study. EBioMedicine 2016. [Google Scholar] [CrossRef]
- Mattsson, N.; Zetterberg, H.; Janelidze, S.; Insel, P.S.; Andreasson, U.; Stomrud, E.; Palmqvist, S.; Baker, D.; Tan Hehir, C.A.; Jeromin, A. Plasma tau in Alzheimer disease. Neurology 2016. [Google Scholar] [CrossRef]
- Ray, S.; Britschgi, M.; Herbert, C.; Takeda-Uchimura, Y.; Boxer, A.; Blennow, K.; Friedman, L.F.; Galasko, D.R.; Jutel, M.; Karydas, A.; et al. Classification and prediction of clinical Alzheimer’s diagnosis based on plasma signaling proteins. Nat. Med. 2007. [Google Scholar] [CrossRef]
- O’Bryant, S.E.; Xiao, G.; Barber, R.; Reisch, J.; Hall, J.; Cullum, C.M.; Doody, R.; Fairchild, T.; Adams, P.; Wilhelmsen, K.; et al. A blood-based algorithm for the detection of Alzheimer’s disease. Dement. Geriatr. Cogn. Disord. 2011. [Google Scholar] [CrossRef]
- Palmqvist, S.; Janelidze, S.; Stomrud, E.; Zetterberg, H.; Karl, J.; Zink, K.; Bittner, T.; Mattsson, N.; Eichenlaub, U.; Blennow, K.; et al. Performance of Fully Automated Plasma Assays as Screening Tests for Alzheimer Disease-Related β-Amyloid Status. JAMA Neurol. 2019. [Google Scholar] [CrossRef]
- Lewczuk, P.; Ermann, N.; Andreasson, U.; Schultheis, C.; Podhorna, J.; Spitzer, P.; Maler, J.M.; Kornhuber, J.; Blennow, K.; Zetterbergm, H. Plasma neurofilament light as a potential biomarker of neurodegeneration in Alzheimer’s disease. Alzheimer Res. Ther. 2018. [Google Scholar] [CrossRef]
- Schmidt, C.; Wolff, M.; Weitz, M.; Bartlau, T.; Korth, C.; Zerr, I. Rapidly progressive alzheimer disease. Arch. Neurol. 2011. [Google Scholar] [CrossRef]
- Rubio-Perez, C.; Guney, E.; Aguilar, D.; Piñero, J.; Garcia-Garcia, J.; Iadarola, B.; Sanz, F.; Fernandez-Fuentes, N.; Furlong, L.I.; Oliva, B. Genetic and functional characterization of disease associations explains comorbidity. Sci. Rep. 2017. [Google Scholar] [CrossRef]
- de Anda-Jáuregui, G.; Guo, K.; McGregor, B.A.; Hur, J. Exploration of the anti-inflammatory drug space through network pharmacology: Applications for drug repurposing. Front. Physiol. 2018. [Google Scholar] [CrossRef]
- Aguirre-Plans, J.; Piñero, J.; Menche, J.; Sanz, F.; Furlong, L.I.; Schmidt, H.H.H.W.; Oliva, B.; Guney, E. Proximal pathway enrichment analysis for targeting comorbid diseases via network endopharmacology. Pharmaceuticals 2018. [Google Scholar] [CrossRef]
- Yarchoan, M.; Arnold, S.E. Repurposing diabetes drugs for brain insulin resistance in Alzheimer disease. Diabetes 2014. [Google Scholar] [CrossRef]
- Kern, W.; Peters, A.; Fruehwald-Schultes, B.; Deininger, E.; Born, J.; Fehm, H.L. Improving influence of insulin on cognitive functions in humans. Neuroendocrinology 2001. [Google Scholar] [CrossRef]
- Craft, S.; Asthana, S.; Newcomer, J.W.; Wilkinson, C.W.; Tio Matos, I.; Baker, L.D.; Cherrier, M.; Lofgreen, C.; Latendresse, S.; Petrova, A.; et al. Enhancement of memory in Alzheimer disease with insulin and somatostatin, but not glucose. Arch. Gen. Psychiatry 1999. [Google Scholar] [CrossRef]
- Craft, S.; Baker, L.D.; Montine, T.J.; Minoshima, S.; Watson, G.S.; Claxton, A.; Arbuckle, M.; Callaghan, M.; Tsai, E.; Plymate, S.R.; et al. Intranasal insulin therapy for Alzheimer disease and amnestic mild cognitive impairment: A pilot clinical trial. Arch. Neurol. 2012. [Google Scholar] [CrossRef]
- Watson, G.S.; Cholerton, B.A.; Reger, M.A.; Baker, L.D.; Plymate, S.R.; Asthana, S.; Fishel, M.A.; Kulstad, J.J.; Green, P.S.; Cook, D.G.; et al. Preserved cognition in patients with early Alzheimer disease and amnestic mild cognitive impairment during treatment with rosiglitazone: A preliminary study. Am. J. Geriatr. Psychiatry 2005. [Google Scholar] [CrossRef]
- McIntosh, E.C.; Nation, D.A. Importance of treatment status in links between type 2 diabetes and Alzheimer’s disease. Diabetes Care 2019, 42, 972–979. [Google Scholar] [CrossRef]
- Plastino, M.; Fava, A.; Pirritano, D.; Cotronei, P.; Sacco, N.; Sperlì, T.; Spanò, A.; Gallo, D.; Mungari, P.; Consoli, D.; et al. Effects of insulinic therapy on cognitive impairment in patients with Alzheimer disease and Diabetes Mellitus type-2. J. Neurol. Sci. 2010. [Google Scholar] [CrossRef]
- Alagiakrishnan, K.; Senior, P. Antidiabetic drugs and their potential role in treating mild cognitive impairment and Alzheimer’s disease. Discov. Med. 2013, 16, 277–286. [Google Scholar]
- Sharma, G.; Parihar, A.; Talaiya, T.; Dubey, K.; Porwal, B.; Parihar, M.S. Cognitive impairments in type 2 diabetes, risk factors and preventive strategies. J. Basic Clin. Physiol. Pharmacol. 2020, 1–14. [Google Scholar] [CrossRef]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kubis-Kubiak, A.; Dyba, A.; Piwowar, A. The Interplay between Diabetes and Alzheimer’s Disease—In the Hunt for Biomarkers. Int. J. Mol. Sci. 2020, 21, 2744. https://doi.org/10.3390/ijms21082744
Kubis-Kubiak A, Dyba A, Piwowar A. The Interplay between Diabetes and Alzheimer’s Disease—In the Hunt for Biomarkers. International Journal of Molecular Sciences. 2020; 21(8):2744. https://doi.org/10.3390/ijms21082744
Chicago/Turabian StyleKubis-Kubiak, Adriana, Aleksandra Dyba, and Agnieszka Piwowar. 2020. "The Interplay between Diabetes and Alzheimer’s Disease—In the Hunt for Biomarkers" International Journal of Molecular Sciences 21, no. 8: 2744. https://doi.org/10.3390/ijms21082744
APA StyleKubis-Kubiak, A., Dyba, A., & Piwowar, A. (2020). The Interplay between Diabetes and Alzheimer’s Disease—In the Hunt for Biomarkers. International Journal of Molecular Sciences, 21(8), 2744. https://doi.org/10.3390/ijms21082744