Role of Phagocytosis in the Pro-Inflammatory Response in LDL-Induced Foam Cell Formation; a Transcriptome Analysis

 ,
, 
 , , ,
, , ,  ,
,
Abstract
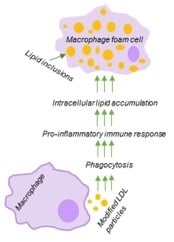
1. Introduction
2. Results
2.1. LDL Accumulation in Human Monocyte-Derived Primary Macrophages
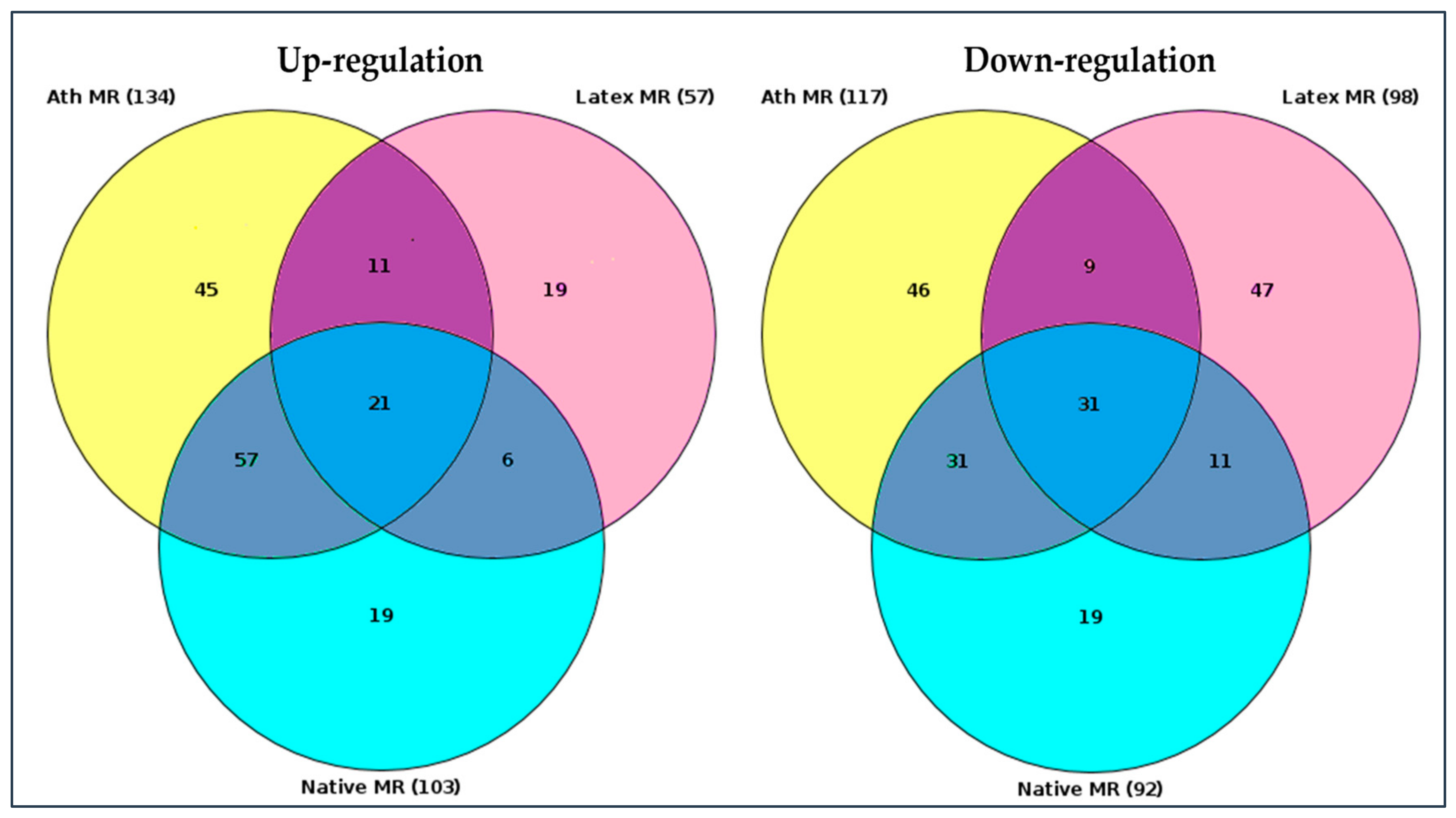
2.2. Search for Master Regulators
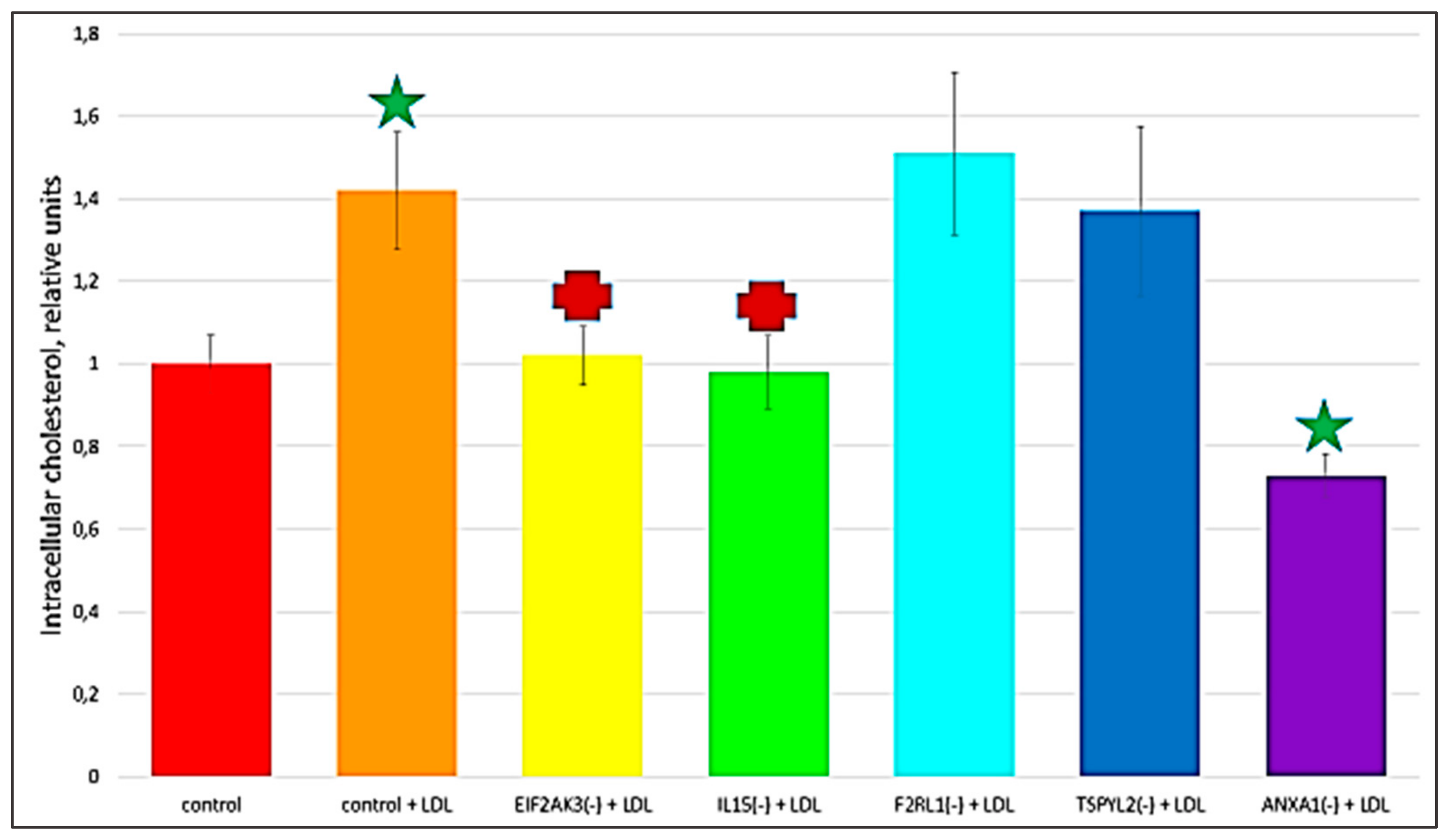
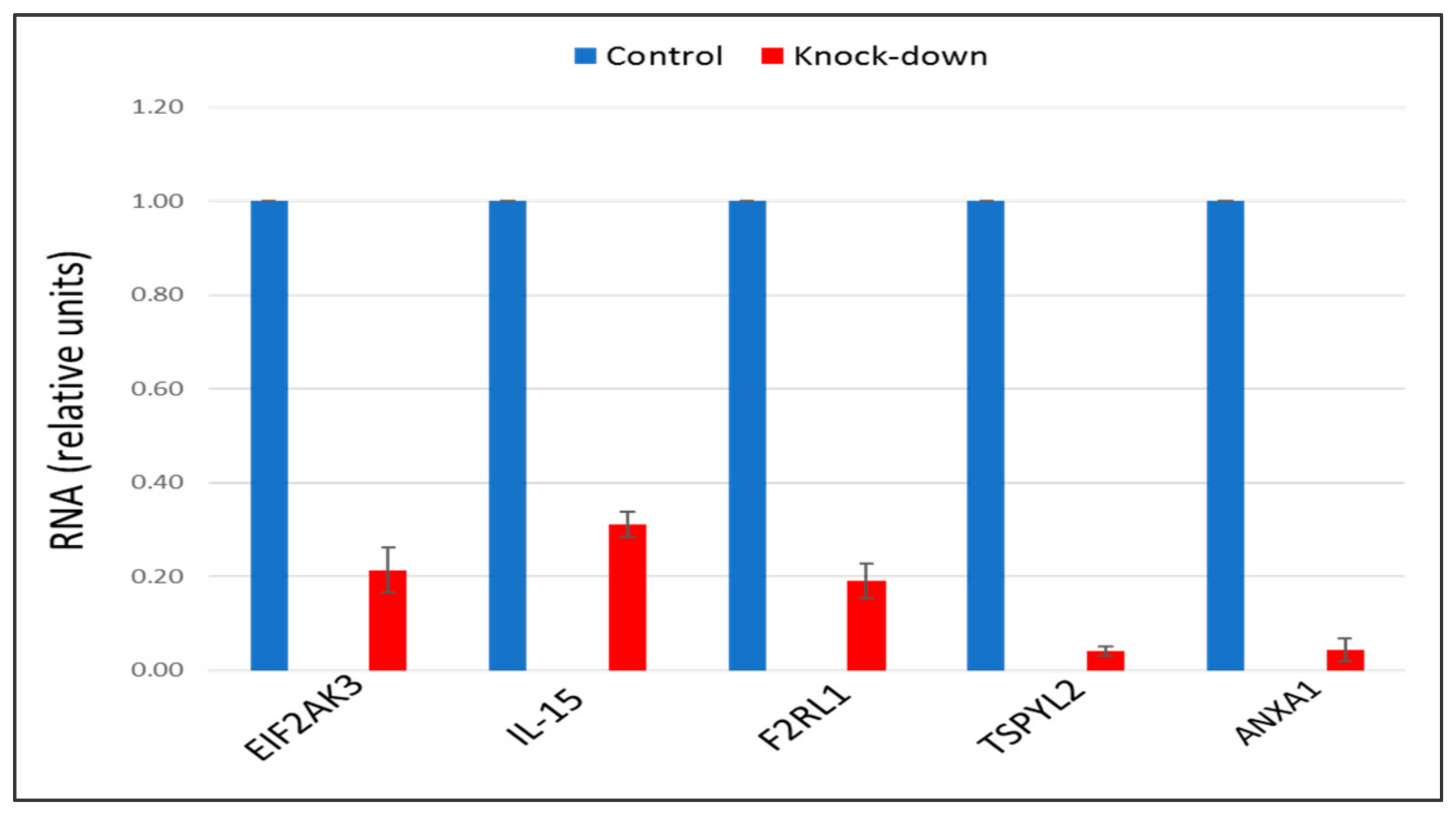
2.3. The Knock-Down of Key Genes
2.4. Identification of the Relevant Signaling Pathways
3. Discussion
4. Materials and Methods
4.1. Lipoproteins and Latex Beads
4.2. Monocyte-derived Macrophages
4.3. Intracellular Cholesterol Measurement
4.4. TRANSFAC and TRANSPATH Databases
4.5. Analysis of Enriched Transcription Factor Binding Sites in Promoters of Differentially Expressed Genes
4.6. Identification of Master Regulators in the Signal Transduction Network
4.7. RNA Sequencing
4.8. Gene Expression Knock-Down by Small Interference RNA (siRNA)
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Li, S.; Gao, G.; Wu, F.; Liu, D.; Zhao, H.; Ke, J.; Liu, Y.; Li, F.; Li, J.; Chen, Z.; et al. Programmed cell death protein 4 deficiency suppresses foam cell formation by activating autophagy in advanced glycation end-product low-density lipoprotein-induced macrophages. J. Cell Biochem. 2018. [Google Scholar] [CrossRef] [PubMed]
- Novikova, O.A.; Laktionov, P.P.; Karpenko, A.A. Mechanisms Underlying Atheroma Induction: The Roles of Mechanotransduction, Vascular Wall Cells, and Blood Cells. Ann. Vasc. Surg. 2018, 53, 224–233. [Google Scholar] [CrossRef] [PubMed]
- Orekhov, A.N. LDL and foam cell formation as the basis of atherogenesis. Curr. Opin. Lipidol. 2018, 29, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Khosravi, M.; Hosseini-Fard, R.; Najafi, M. Circulating low density lipoprotein (LDL). Horm. Mol. Biol. Clin. Investig. 2018, 35. pii: /j/hmbci.2018.35.issue-2/hmbci-2018-0024/hmbci-2018-0024.xml. [Google Scholar] [CrossRef]
- Orsó, E.; Grandl, M.; Schmitz, G. Oxidized LDL-induced endolysosomal phospholipidosis and enzymatically modified LDL-induced foam cell formation determine specific lipid species modulation in human macrophages. Chem. Phys. Lipids. 2011, 164, 479–487. [Google Scholar] [CrossRef]
- Orekhov, A.N.; Ivanova, E.A. Introduction of the special issue “Atherosclerosis and Related Diseases”. Vessel. Plus. 2017, 1, 163–165. [Google Scholar] [CrossRef]
- Tertov, V.V.; Kaplun, V.V.; Sobenin, I.A.; Orekhov, A.N. Low-density lipoprotein modification occurring in human plasma possible mechanism of in vivo lipoprotein desialylation as a primary step of atherogenic modification. Atherosclerosis 1998, 138, 183–195. [Google Scholar] [CrossRef]
- Bhakdi, S.; Dorweiler, B.; Kirchmann, R.; Torzewski, J.; Weise, E.; Tranum-Jensen, J.; Walev, I.; Wieland, E. On the pathogenesis of atherosclerosis: Enzymatic transformation of human low-density lipoprotein to an atherogenic moiety. J. Exp. Med. 1995, 182, 1959–1971. [Google Scholar] [CrossRef]
- Tertov, V.V.; Sobenin, I.A.; Orekhov, A.N. Similarity between naturally occurring modified desialylated, electronegative and aortic low-density lipoprotein. Free Radic. Res. 1996, 25, 313–319. [Google Scholar] [CrossRef]
- Tertov, V.V.; Sobenin, I.A.; Orekhov, A.N.; Jaakkola, O.; Solakivi, T.; Nikkari, T. Characteristics of low- density lipoprotein isolated from circulating immune complexes. Atherosclerosis 1996, 122, 191–199. [Google Scholar] [CrossRef]
- Jaakkola, O.; Solakivi, T.; Tertov, V.V.; Orekhov, A.N.; Miettinen, T.A.; Nikkari, T. Characteristics of low-density lipoprotein subfractions from patients with coronary artery disease. Coron. Artery Dis. 1993, 4, 379–385. [Google Scholar] [CrossRef] [PubMed]
- Tertov, V.V.; Sobenin, I.A.; Gabbasov, Z.A.; Popov, E.G.; Orekhov, A.N. Lipoprotein aggregation as an essential condition of intracellular lipid accumulation caused by modified low density lipoproteins. Biochem. Biophys. Res. Commun. 1989, 163, 489–494. [Google Scholar] [CrossRef]
- Tertov, V.V.; Sobenin, I.A.; Gabbasov, Z.A.; Popov, E.G.; Yaroslavov, A.A.; Jauhiainen, M.; Ehnholm, C.; Smirnov, V.N.; Orekhov, A.N. Three types of naturally occurring modified; lipoproteins induce intracellular lipid accumulation in human aortic intimal cells-the role of lipoprotein aggregation. Eur. J. Clin. Chem. Clin. Biochem. 1992, 30, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Tertov, V.V.; Orekhov, A.N.; Sobenin, I.A.; Gabbasov, Z.A.; Popov, E.G.; Yaroslavov, A.A.; Smirnov, V.N. Three types of naturally occurring modified lipoproteins induce intracellular lipid accumulation due to lipoprotein aggregation. Circ. Res. 1992, 71, 218–228. [Google Scholar] [CrossRef] [PubMed]
- Neele, A.E.; Prange, K.H.; Hoeksema, M.A.; van der Velden, S.; Lucas, T.; Dimmeler, S.; Lutgens, E.; Van den Bossche, J.; de Winther, M.P. Macrophage Kdm6b controls the pro-fibrotic transcriptome signature of foam cells. Epigenomics 2017, 9, 383–391. [Google Scholar] [CrossRef]
- Ho, M.M.; Fraser, D.A. Transcriptome data and gene ontology analysis in human macrophages ingesting modified lipoproteins in the presence or absence of complement protein C1q. Data Brief. 2016, 9, 362–367. [Google Scholar] [CrossRef]
- Shaik-Dasthagirisaheb, Y.B.; Mekasha, S.; He, X.; Gibson, F.C., 3rd; Ingalls, R.R., 3rd; Ingalls, R.R. Signaling events in pathogen-induced macrophage foam cell formation. Pathog. Dis. 2016, 74, ftw074. [Google Scholar] [CrossRef]
- Hu, Y.W.; Zhao, J.Y.; Li, S.F.; Wang, Q.; Zheng, L. Genome-wide profiling to analyze the effects of Ox-LDL induced THP-1 macrophage-derived foam cells on gene expression. Genom. Data 2014, 2, 328–331. [Google Scholar] [CrossRef][Green Version]
- Orekhov, A.N.; Oishi, Y.; Nikiforov, N.G.; Zhelankin, A.V.; Dubrovsky, L.; Sobenin, I.A.; Kel, A.; Stelmashenko, D.; Makeev, V.J.; Foxx, K.; et al. Modified LDL Particles Activate Inflammatory Pathways in Monocyte-derived Macrophages: Transcriptome Analysis. Curr. Pharm. Des. 2018, 24, 3143–3151. [Google Scholar] [CrossRef]
- Ku, G.; Thomas, C.E.; Akeson, A.L.; Jackson, R.L. Induction of interleukin 1 beta expression from human peripheral blood monocyte-derived macrophages by 9-hydroxyoctadecadienoic acid. J. Biol. Chem. 1992, 267, 14183–14188. [Google Scholar]
- Miller, Y.I.; Viriyakosol, S.; Worrall, D.S.; Boullier, A.; Butler, S.; Witztum, J.L. Toll-like receptor 4-dependent and -independent cytokine secretion induced by minimally oxidized low-density lipoprotein in macrophages. Arterioscler Thromb. Vasc. Biol. 2005, 25, 1213–1219. [Google Scholar] [CrossRef]
- Wiesner, P.; Choi, S.H.; Almazan, F.; Benner, C.; Huang, W.; Diehl, C.J.; Gonen, A.; Butler, S.; Witztum, J.L.; Glass, C.K.; et al. Low doses of lipopolysaccharide and minimally oxidized low-density lipoprotein cooperatively activate macrophages via nuclear factor kappa B and activator protein-1: Possible mechanism for acceleration of atherosclerosis by subclinical endotoxemia. Circ. Res. 2010, 107, 56–65. [Google Scholar] [CrossRef]
- Jiang, Y.; Wang, M.; Huang, K.; Zhang, Z.; Shao, N.; Zhang, Y.; Wang, W.; Wang, S. Oxidized low-density lipoprotein induces secretion of interleukin-1β by macrophages via reactive oxygen species-dependent NLRP3 inflammasome activation. Biochem. Biophys. Res. Commun. 2012, 425, 121–126. [Google Scholar] [CrossRef]
- Bekkering, S.; Quintin, J.; Joosten, L.A.; van der Meer, J.W.; Netea, M.G.; Riksen, N.P. Oxidized low-density lipoprotein induces long-term proinflammatory cytokine production and foam cell formation via epigenetic reprogramming of monocytes. Arterioscler Thromb. Vasc. Biol. 2014, 34, 1731–1738. [Google Scholar] [CrossRef]
- Yang, K.; Zhang, X.J.; Cao, L.J.; Liu, X.H.; Liu, Z.H.; Wang, X.Q.; Chen, Q.J.; Lu, L.; Shen, W.F.; Liu, Y. Toll-like receptor 4 mediates inflammatory cytokine secretion in smooth muscle cells induced by oxidized low-density lipoprotein. PLoS ONE 2014, 9, e95935. [Google Scholar] [CrossRef]
- Orekhov, A.N.; Nikiforov, N.G.; Elizova, N.V.; Korobov, G.A.; Aladinskaya, A.V.; Sobenin, I.A.; Bobryshev, Y.V. Tumor Necrosis Factor-α and C-C Motif Chemokine Ligand 18 Associate with Atherosclerotic Lipid Accumulation In situ and In vitro. Curr. Pharm. Des. 2018, 24, 2883–2889. [Google Scholar] [CrossRef]
- Tall, A.R.; Yvan-Charvet, L. Cholesterol, inflammation and innate immunity. Nat. Rev. Immunol. 2015, 15, 104–116. [Google Scholar] [CrossRef]
- Brown, M.S.; Goldstein, J.L. Receptor-mediated endocytosis: Insights from the lipoprotein receptor system. Proc. Natl. Acad. Sci. USA 1979, 76, 3330–3337. [Google Scholar] [CrossRef]
- Goldstein, J.L.; Brown, M.S. A century of cholesterol and coronaries: From plaques to genes to statins. Cell 2015, 161, 161–172. [Google Scholar] [CrossRef]
- Goldstein, J.L.; Brown, M.S. The LDL receptor. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 431–438. [Google Scholar] [CrossRef]
- Goldstein, J.L.; Brown, M.S. Lipoprotein receptors and the control of plasma LDL cholesterol levels. Eur. Heart J. 1992, 13, 34–36. [Google Scholar] [CrossRef]
- Lu, M.; Gursky, O. Aggregation and fusion of low-density lipoproteins in vivo and in vitro. Biomol. Concepts 2013, 4, 501–518. [Google Scholar] [CrossRef]
- Sánchez-Quesada, J.L.; Villegas, S.; Ordóñez-Llanos, J. Electronegative low-density lipoprotein. A link between apolipoprotein B misfolding, lipoprotein aggregation and proteoglycan binding. Curr. Opin. Lipidol. 2012, 23, 479–486. [Google Scholar] [CrossRef]
- Harangi, M.; Szentpéteri, A.; Nádró, B.; Lőrincz, H.; Seres, I.; Páll, D.; Paragh, G. HDL subfraction distribution and HDL function in untreated dyslipidemic patients. Vessel. Plus. 2017, 1, 166–173. [Google Scholar] [CrossRef]
- Nathan, C. Secretory products of macrophages: Twenty-five years on. J. Clin. Investig. 2012, 122, 1189–1190. [Google Scholar] [CrossRef]
- Rekhter, M.D.; Tertov, V.V.; Andreeva, E.R.; Kolpakov, V.A.; Mironov, A.A.; Orekhov, A.N. Lipid accumulation in the subendothelial cells of human aortic intima impairs cell-to-cell contacts: A comparative study in situ and in vitro. Cardiovasc. Pathol. 1993, 2, 53–62. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Kel, A.E.; Stegmaier, P.; Valeev, T.; Koschmann, J.; Poroikov, V.; Kel-Margoulis, O.V.; Wingender, E. Multi-omics “upstream analysis” of regulatory genomic regions helps identifying targets against methotrexate resistance of colon cancer. EuPA Open Proteom. 2016, 13, 1–13. [Google Scholar] [CrossRef]
- Kel, A. Data on master regulators and transcription factor binding sites found by upstream analysis of multi-omics data on methotrexate resistance of colon cancer. Data Brief. 2016, 10, 499–504. [Google Scholar] [CrossRef]
- Orekhov, A.N.; Sobenin, I.A. Modified and Dysfunctional Lipoproteins in Atherosclerosis: Effectors or Biomarkers? Curr. Med. Chem. 2019, 26, 1512–1524. [Google Scholar] [CrossRef]
- Kzhyshkowska, J.; Neyen, C.; Gordon, S. Role of macrophage scavenger receptors in atherosclerosis. Immunobiology 2012, 217, 492–502. [Google Scholar] [CrossRef] [PubMed]
- PrabhuDas, M.R.; Baldwin, C.L.; Bollyky, P.L.; Bowdish, D.M.; Drickamer, K.; Febbraio, M.; Herz, J.; Kobzik, L.; Krieger, M.; Loike, J.; et al. A consensus definitive classification of scavenger receptors and their roles in health and disease. J. Immunol. 2017, 198, 3775–3789. [Google Scholar] [CrossRef] [PubMed]
- Chellan, B.; Rojas, E.; Zhang, C.; Hofmann Bowman, M.A. Enzyme-modified non-oxidized LDL (ELDL) induces human coronary artery smooth muscle cell transformation to a migratory and osteoblast-like phenotype. Sci. Rep. 2018, 8, 11954. [Google Scholar] [CrossRef] [PubMed]
- Pasquin, S.; Laplante, V.; Kouadri, S.; Milasan, A.; Mayer, G.; Tormo, A.J.; Savin, V.; Sharma, M.; Martel, C.; Gauchat, J.F. Cardiotrophin-like Cytokine Increases Macrophage-Foam Cell Transition. J. Immunol. 2018, 201, 2462–2471. [Google Scholar] [CrossRef]
- Salvatore, G.; Bernoud-Hubac, N.; Bissay, N.; Debard, C.; Daira, P.; Meugnier, E.; Proamer, F.; Hanau, D.; Vidal, H.; Aricò, M.; et al. Human monocyte-derived dendritic cells turn into foamy dendritic cells with IL-17A. J. Lipid Res. 2015, 56, 1110–1122. [Google Scholar] [CrossRef]
- Yin, Y.W.; Liao, S.Q.; Zhang, M.J.; Liu, Y.; Li, B.H.; Zhou, Y.; Chen, L.; Gao, C.Y.; Li, J.C.; Zhang, L.L. TLR4-mediated inflammation promotes foam cell formation of vascular smooth muscle cell by upregulating ACAT1 expression. Cell Death Dis. 2014, 18, e1574. [Google Scholar] [CrossRef]
- Alipov, V.I.; Sukhorukov, V.N.; Karagodin, V.P.; Grechko, A.V.; Orekhov, A.N. Chemical composition of circulating native and desialylated low density lipoprotein: What is the difference? Vessel. Plus. 2017, 1, 107–115. [Google Scholar] [CrossRef]
- Hara, A.; Radin, N.S. Lipid extraction of tissues with a low-toxicity solvent. Anal. Biochem. 1978, 90, 420–426. [Google Scholar] [CrossRef]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar]
- Matys, V.; Kel-Margoulis, O.V.; Fricke, E.; Liebich, I.; Land, S.; Barre-Dirrie, A.; Reuter, I.; Chekmenev, D.; Krull, M.; Hornischer, K.; et al. TRANSFAC and its module TRANSCompel: Transcriptional gene regulation in eukaryotes. Nucleic Acids Res. 2006, 34, D108–D110. [Google Scholar] [CrossRef]
- Krull, M.; Pistor, S.; Voss, N.; Kel, A.; Reuter, I.; Kronenberg, D.; Michael, H.; Schwarzer, K.; Potapov, A.; Choi, C.; et al. TRANSPATH: An information resource for storing and visualizing signaling pathways and their pathological aberrations. Nucleic Acids Res. 2006, 34, D546–D551. [Google Scholar] [CrossRef] [PubMed]
- Kel, A.E.; Gössling, E.; Reuter, I.; Cheremushkin, E.; Kel-Margoulis, O.V.; Wingender, E. MATCH: A tool for searching transcription factor binding sites in DNA sequences. Nucleic Acids Res. 2003, 31, 3576–3579. [Google Scholar] [CrossRef] [PubMed]
- Waleev, T.; Shtokalo, D.; Konovalova, T.; Voss, N.; Cheremushkin, E.; Stegmaier, P.; Kel-Margoulis, O.; Wingender, E.; Kel, A. Composite Module Analyst: Identification of transcription factor binding site combinations using genetic algorithm. Nucleic Acids Res. 2006, 34, W541–W545. [Google Scholar] [CrossRef] [PubMed]
- Koschmann, J.; Bhar, A.; Stegmaier, P.; Kel, A.E.; Wingender, E. “Upstream Analysis”: An integrated promoter-pathway analysis approach to causal interpretation of microarray data. Microarrays 2015, 4, 270–280. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No | Treatment | Total Cholesterol (nmol/mg Protein) | |
|---|---|---|---|
| Mean ± SEM | p-Value (Vs. Control) | ||
| 1 | Control | 57 ± 9 | - |
| 2 | Native LDL | 56 ± 5 | NS |
| 3 | Oxidized LDL | 98 ± 11 | 0.014 |
| 4 | Acetylated LDL | 80 ± 6 | 0.039 |
| 5 | Desialylated LDL | 102 ± 8 | 0.004 |
| 6 | Atherogenic LDL | 80 ± 7 | 0.045 |
| 7 | Latex | 51 ± 11 | NS |
| No | Comparison | Number of Differentially Expressed Genes | |
|---|---|---|---|
| Up-Regulated | Down-Regulated | ||
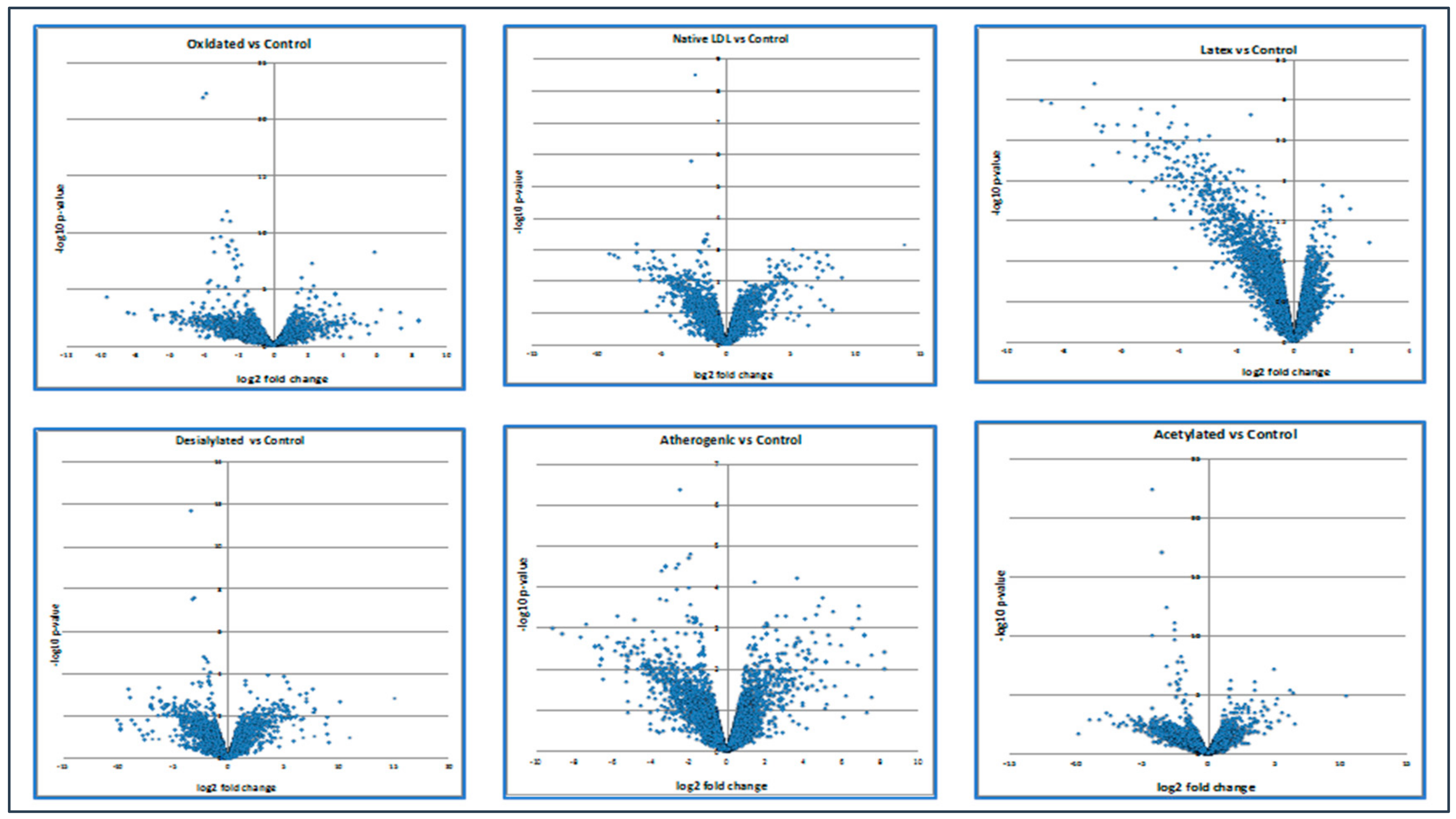
| 2 | Native LDL vs. Control | 177 | 270 |
| 3 | Oxidized LDL vs. Control | 247 | 457 |
| 4 | Acetylated LDL vs. Control | 292 | 452 |
| 5 | Desialylated LDL vs. Control | 241 | 322 |
| 6 | Atherogenic LDL vs. Control | 249 | 366 |
| 7 | Latex vs. Control | 15 | 351 |
| Qty | Gene Symbol | Gene Name | ||
|---|---|---|---|---|
| naturally occurring LDL | up | 11 | EP300 | E1A binding protein p300 |
| IQGAP1 | IQ motif containing GTPase activating protein 1 | |||
| MAP3K14 | mitogen-activated protein kinase kinase kinase 14 | |||
| MAP3K3 | mitogen-activated protein kinase kinase kinase 3 | |||
| NCF2 | neutrophil cytosolic factor 2 | |||
| PIK3R5 | phosphoinositide-3-kinase regulatory subunit 5 | |||
| PRKCD | protein kinase C, delta | |||
| PTGES | prostaglandin E synthase | |||
| RAD23A | RAD23 homolog A, nucleotide excision repair protein | |||
| RIPK2 | receptor interacting serine/threonine kinase 2 | |||
| TAB1 | TGF-beta activated kinase 1/MAP3K7 binding protein 1 | |||
| dn | 9 | AKT1 | v-akt murine thymoma viral oncogene homolog 1 | |
| CDC42 | cell division cycle 42 | |||
| DUSP7 | dual specificity phosphatase 7 | |||
| GSK3B | glycogen synthase kinase 3 beta | |||
| HGF | hepatocyte growth factor (hepapoietin A; scatter factor) | |||
| NCF1 | neutrophil cytosolic factor 1 | |||
| TAOK1 | TAO kinase 1 | |||
| TGFB2 | transforming growth factor beta 2 | |||
| TRAF6 | TNF receptor associated factor 6 | |||
| desialylated LDL | up | 10 | EP300 | E1A binding protein p300 |
| CASP2 | caspase 2 | |||
| DUSP5 | dual specificity phosphatase 5 | |||
| IQGAP1 | IQ motif containing GTPase activating protein 1 | |||
| PIK3R5 | phosphoinositide-3-kinase regulatory subunit 5 | |||
| PTGES | prostaglandin E synthase | |||
| PRKCD | protein kinase C, delta | |||
| PRKCB | protein kinase C, beta | |||
| RAD23A | RAD23 homolog A, nucleotide excision repair protein | |||
| RIPK2 | receptor interacting serine/threonine kinase 2 | |||
| dn | 9 | DUSP7 | dual specificity phosphatase 7 | |
| HGF | hepatocyte growth factor (hepapoietin A; scatter factor) | |||
| MAP2K5 | mitogen-activated protein kinase kinase 5 | |||
| MAP2K7 | mitogen-activated protein kinase kinase 7 | |||
| NCF1 | neutrophil cytosolic factor 1 | |||
| TGFB2 | transforming growth factor beta 2 | |||
| TGFB3 | transforming growth factor beta 3 | |||
| TRADD | TNFRSF1A-associated via death domain | |||
| TRAF6 | TNF receptor associated factor 6 | |||
| acetylated LDL | up | 4 | NCF2 | neutrophil cytosolic factor 2 |
| PRKCD | protein kinase C, delta | |||
| PTGES | prostaglandin E synthase | |||
| SGK1 | serum/glucocorticoid regulated kinase 1 | |||
| dn | 14 | AKT1 | v-akt murine thymoma viral oncogene homolog 1 | |
| CASP8 | caspase 8 | |||
| DUSP1 | dual specificity phosphatase 1 | |||
| DUSP16 | dual specificity phosphatase 16 | |||
| GSK3B | glycogen synthase kinase 3 beta | |||
| HDAC3 | histone deacetylase 3 | |||
| IL1A | interleukin 1 alpha | |||
| MAP2K1 | mitogen-activated protein kinase kinase 1 | |||
| MAP3K1 | mitogen-activated protein kinase kinase kinase 1, E3 ubiquitin protein ligase | |||
| MAP4K2 | mitogen-activated protein kinase kinase kinase kinase 2 | |||
| OTUB1 | OTU deubiquitinase, ubiquitin aldehyde binding 1 | |||
| TAOK1 | TAO kinase 1 | |||
| TRADD | TNFRSF1A-associated via death domain | |||
| TRAF6 | TNF receptor-associated factor 6 | |||
| oxidized LDL | up | 7 | CASP2 | caspase 2 |
| IQGAP1 | IQ motif containing GTPase activating protein 1 | |||
| MAP3K14 | mitogen-activated protein kinase kinase kinase 14 | |||
| NCF2 | neutrophil cytosolic factor 2 | |||
| PRKCB | protein kinase C, beta | |||
| RAD23A | RAD23 homolog A, nucleotide excision repair protein | |||
| SGK1 | serum/glucocorticoid regulated kinase 1 | |||
| dn | 13 | AKT1 | v-akt murine thymoma viral oncogene homolog 1 | |
| CDC42 | cell division cycle 42 | |||
| DUSP1 | dual specificity phosphatase 1 | |||
| DUSP16 | dual specificity phosphatase 16 | |||
| GSK3B | glycogen synthase kinase 3 beta | |||
| HDAC3 | histone deacetylase 3 | |||
| KRAS | Kirsten rat sarcoma viral oncogene homolog | |||
| MAP3K1 | mitogen-activated protein kinase kinase kinase 1, E3 ubiquitin protein ligase | |||
| MAP4K2 | mitogen-activated protein kinase kinase kinase kinase 2 | |||
| MAPK8IP1 | mitogen-activated protein kinase 8 interacting protein 1 | |||
| NCF1 | neutrophil cytosolic factor 1 | |||
| TAOK1 | TAO kinase 1 | |||
| TRAF6 | TNF receptor associated factor 6 |
| Gene Symbol | Functions of Genes (According to NCBI Gene Database) |
|---|---|
| EP300 | The adenovirus E1A-associated cellular p300 transcriptional co-activator protein is encoded by this gene. It acts as histone acetyltransferase regulating transcription through chromatin remodeling that is significant in the cellular proliferation and differentiation. It mediates cAMP-gene regulation by specific binding to phosphorylated cAMP response element-binding (CREB) protein. It plays a role in the stimulation of hypoxia-induced genes such as VEGF, a co-activator of hypoxia-inducible factor 1 alpha (HIF1A). Mutations in this gene cause Rubinstein-Taybi syndrome and may also play a role in epithelial cancer. |
| IQGAP1 | IQ Motif Containing GTPase Activating Protein 1 is a member of the IQGAP protein family encoded by this gene. It regulates cellular morphology and motility by interacting with components of the cytoskeleton, cell adhesion molecules, and several signaling molecules. The gene amplification-upregulated expression of this protein is attributed to two gastric cancer cell lines. |
| MAP3K14 | The mitogen-activated protein kinase kinase kinase 14, a serine/threonine protein-kinase is encoded by this protein. It binds to TRAF2 and activates NF-kappa B. It is involved in an NF-kappa B-inducing signaling cascade that is mutual to receptors of the tumour-necrosis/nerve-growth factor (TNF/NGF) family and to the interleukin-1 type-I receptor. It has sequence similarity with some other MAPKK kinases. |
| MAP3K3 | It is a product of 626-amino acid polypeptide with 96.5% identity to mouse Mekk3. Its catalytic domain is interrelated to those of some other kinases, including mouse Mekk2, tobacco NPK, and yeast Ste11. A 4.6-kb transcript of this gene showing ubiquitous expression was revealed by Northern blot analysis. By activating SEK and MEK1/2, it directly regulates the stress-activated protein kinase (SAPK) and extracellular signal-regulated protein kinase (ERK) pathways, respectively. This protein does not regulate p38 pathway. As demonstrated by co-transfection assays, it can improve a transcription from a nuclear factor kappa-B (NFKB)-dependent reporter gene playing a role in the SAPK pathway. |
| NCF2 | The neutrophil cytosolic factor 2, the 67-kilodalton cytosolic subunit of the multi-protein NADPH oxidase complex found in neutrophils is encoded by this gene. A burst of superoxide delivered to the lumen of the neutrophil phagosome is produced by this oxidase. Mutations in this gene, as well as in other NADPH oxidase subunits, can result in chronic granulomatous disease, a disease that causes recurrent infections by catalase-positive organisms. |
| PIK3R5 | Phosphatidylinositol 3-kinases (PI3Ks) phosphorylate the inositol ring of phosphatidylinositol at the 3-prime position and play important roles in cell growth, proliferation, differentiation, motility, survival, and intracellular trafficking. There are three classes of PI3Ks: I, II and III, and only the class I of PI3Ks is implicated in oncogenesis. The 101 kD regulatory subunit of the class I PI3K gamma complex is a dimeric enzyme that consists of a 110 kD catalytic subunit gamma and a regulatory subunit of either 55, 87 or 101 kD is encoded by this protein. By high-affinity interaction with G-beta-gamma proteins, it recruits the catalytic subunit from the cytosol to the plasma membrane. |
| PRKCD | This gene encodes a member of the protein kinase C family of serine- and threonine-specific protein kinases. Upon activation by diacylglycerol, this protein can act as both a tumor suppressor and a positive regulator of cell cycle progression. The positive or negative regulation of apoptosis is also attributed to this protein. Mutations in this gene cause the autoimmune lymphoproliferative syndrome. |
| PTGES | A glutathione-dependent prostaglandin E synthase is encoded by this gene. The expression of this gene was shown to be induced by proinflammatory cytokine interleukin 1 beta (IL1B). This gene expression can also be induced by tumor suppressor protein TP53 and it may be involved in TP53 induced apoptosis. Knockout studies in mice suggested that this gene may contribute to the pathogenesis of collagen-induced arthritis and mediate acute pain in inflammatory responses. |
| RAD23A | The protein, such as one of two human homologs of Saccharomyces cerevisiae Rad23 is encoded by this gene. The family of Rad23 proteins possess a modular domain structure that consists of a ubiquitin-like domain (UbL), ubiquitin-associated domain 1 (UbA1), UbA2, and XPC-binding domain. Rad23A plays a crucial role in nucleotide excision repair and also in the proteasome delivery of polyubiquitinated proteins. |
| RIPK2 | A member of the receptor-interacting protein (RIP) family of serine/threonine protein kinases is encoded by this gene. The encoded protein incorporates a C-terminal caspase activation and recruitment domain (CARD). Also, it is a component of signaling complexes in both the innate and adaptive immune pathways. It is a powerful activator of NF-kappa B and can induce apoptosis in response to various stimuli. |
| TAB1 | A regulator of the MAP kinase kinase kinase MAP3K7/TAK1, a mediator of various intracellular signaling pathways, including those that induced by TGF beta, interleukin 1, and WNT-1 is encoded by this gene. This protein interacts with TAK1 kinase, thus, activating it. It was demonstrated that its C-terminal portion is sufficient for TAK1 binding and activation, while the N-terminus portion acts as a dominant-negative inhibitor of TGF beta, suggesting that this protein may function as a regulator of TGF beta receptors and TAK1. It also can interact with and activate the mitogen-activated protein kinase 14 (MAPK14/p38alpha) representing an alternative activation pathway contributing to the biological responses of MAPK14 to various stimuli, in addition to the MAPKK pathways. |
| AKT1 | This gene encodes a serine-threonine protein kinase that is catalytically inactive in serum-starved primary and immortalized fibroblasts. AKT1 along with the related AKT2 are activated by platelet-derived growth factor. This gene is a critical mediator of growth factor-induced neuronal survival in the developing nervous system. Factors of survival can suppress apoptosis via activation of the serine/threonine kinase AKT1, which then phosphorylates and inactivates components of the apoptotic machinery. These gene mutations were found to be associated with Proteus syndrome. |
| CDC42 | This gene encodes a small GTPase of the Rho-subfamily, which regulates signaling pathways controlling different functions of cells, including cell morphology, migration, endocytosis, and progression of the cell cycle. This protein shares some similarities with Saccharomyces cerevisiae Cdc 42 being able to complement the yeast cdc42-1 mutant. This protein can regulate actin polymerization by direct binding to Neural Wiskott–Aldrich syndrome protein (N-WASP), which, in turn, activates the actin-related protein-2/3 (Arp2/3) complex. |
| DUSP7 | This gene encodes dual-specificity phosphatase 7 (DUSP7) belonging to a broad heterogeneous subgroup of the type I cysteine-based protein-tyrosine phosphatase superfamily that is able to dephosphorylate both tyrosine and serine/threonine residues. In particular, DUSP7 belongs to a class of DUSPs, designated MKPs, that dephosphorylate the following: MAPK (mitogen-activated protein kinase) proteins ERK (see MIM 601795), JNK (see MIM 601158), and p38 (see MIM 600289) with specificity different from that of individual MKP proteins. MKPs consist of a highly conserved C-terminal catalytic domain and an N-terminal Cdc25 (see MIM 116947)-like (CH2) domain. MAPK activation cascades mediate various cellular physiologic processes, such as proliferation, apoptosis, differentiation, and stress responses. |
| GSK3B | A serine-threonine kinase belonging to the glycogen synthase kinase subfamily is encoded by this gene. It is a negative regulator of glucose homeostasis and is involved in energy metabolism, inflammation, ER-stress, mitochondrial dysfunction, and apoptotic pathways. Defects in this gene were associated with Parkinson’s disease and Alzheimer’s disease. |
| HGF | A binding protein of hepatocyte growth factor receptor that regulates cell growth, cell motility and morphogenesis in numerous cell and tissue types are encoded by this gene. Alternative splicing results in multiple transcript variants, at least one of which encodes a preproprotein that is proteolytically processed to generate alpha and beta chains, which form the mature heterodimer. This protein is secreted by mesenchymal cells and acts as a multi-functional cytokine on cells of mainly epithelial origin. This protein also plays a role in angiogenesis, tumorigenesis, and tissue regeneration. Despite belonging to the encoded protein to the peptidase S1 family of serine proteases, it lacks peptidase activity. These gene mutations can cause non-syndromic hearing loss. |
| NCF1 | This gene encodes a 47 kDa cytosolic subunit of neutrophil NADPH oxidase, which is a multicomponent enzyme that upon activation generates superoxide anion. These gene mutations can cause chronic granulomatous disease. |
| TAOK1 | Thousand and one kinase 1 (TAOK1) protein is encoded by this gene. It is a negative regulator of IL-17-mediated signal transduction and inflammation. The knock-down of TAOK1 promotes IL-17-induced expression of cytokines and chemokines, as well as the activation of mitogen-activated protein kinases and nuclear factor-κB. Independently of its kinase activity, it interacts with IL-17 receptor A (IL-17RA), and prevents the formation of the IL-17R-Act1 (nuclear factor activator 1, also known as tumor necrosis factor receptor-associated factor 3 interacting protein 2) complex in a dose-dependent manner. |
| TGFB2 | This gene encodes a secreted ligand of the TGF-beta (transforming growth factor-beta) superfamily of proteins. The ligands of this family bind various TGF-beta receptors that lead to the recruitment and activation of gene expression regulating transcription factors of the SMAD family. Proteolytic processing of this encoded preproprotein generates a latency-associated peptide (LAP) and a mature peptide, thus, it is found in either a latent form encompassing a mature peptide homodimer, a LAP homodimer, and a latent TGF-beta binding protein, or in an active form encompassing exclusively the mature peptide homodimer. The mature protein may also form heterodimers with other members of the TGF-beta family. Disruption of the TGF-beta/SMAD pathway is associated with a variety of human cancers. A chromosomal translocation of this gene was found to be associated with Peters’ anomaly, a congenital defect of the anterior eye chamber. These gene mutations also can be associated with Loeys-Dietz syndrome. |
| TRAF6 | TNF receptor-associated factor 6 (TRAF6) protein, a member of the TNF receptor-associated factor (TRAF) protein family is encoded by this protein. This protein consists of an amino terminal RING domain followed by four zinc-finger motifs, a central coiled-coil region, and the TRAF-C domain, a highly conserved carboxyl terminal domain. TRAF proteins were found to be associated with and mediate signal transduction from the TNF receptor superfamily members. TRAF6 protein mediates signaling from the TNF receptor superfamily members, as well as members of the Toll/IL-1 family. Signals from receptors, including CD40, TNFSF11/RANCE, and IL-1 were demonstrated to be mediated by this protein. This protein is also able to interact with several protein kinases, including IRAK1/IRAK, SRC, and PKC zeta providing a link between distinct signaling pathways. TRAF6 protein can act as a signal transducer in the NF-kappa B pathway that, in response to proinflammatory cytokines, activates I kappa B kinase (IKK). The interaction of this protein with ubiquitin-conjugating enzymes catalyzing the formation of polyubiquitin chains, such as UBE2N/UBC13, and UBE2V1/UEV1A, was found to be necessary for IKK activation. This protein can also interact with the transforming growth factor (TGF) beta receptor complex and is essential for the activation of SMAD-independent JNK and p38 kinases. |
| Gene Symbol | Innate Immunity | Lipid Metabolism | Phagocytosis |
|---|---|---|---|
| EP300 | + | + | - |
| IQGAP1 | - | ? | + |
| MAP3K14 | + | ? | ? |
| MAP3K3 | + | + | ? |
| NCF2 | + | - | + |
| PIK3R5 | - | - | ? |
| PRKCD | + | + | + |
| PTGES | + | - | - |
| RAD23A | - | - | - |
| RIPK2 | + | ? | + |
| TAB1 | + | ? | ? |
| AKT1 | + | + | + |
| CDC42 | + | + | + |
| DUSP7 | - | ? | ? |
| GSK3B | + | + | + |
| HGF | + | + | + |
| NCF1 | + | + | + |
| TAOK1 | - | ? | ? |
| TGFB2 | + | + | ? |
| TRAF6 | + | ? | + |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Orekhov, A.N.; Nikiforov, N.G.; Sukhorukov, V.N.; Kubekina, M.V.; Sobenin, I.A.; Wu, W.-K.; Foxx, K.K.; Pintus, S.; Stegmaier, P.; Stelmashenko, D.; et al. Role of Phagocytosis in the Pro-Inflammatory Response in LDL-Induced Foam Cell Formation; a Transcriptome Analysis. Int. J. Mol. Sci. 2020, 21, 817. https://doi.org/10.3390/ijms21030817
Orekhov AN, Nikiforov NG, Sukhorukov VN, Kubekina MV, Sobenin IA, Wu W-K, Foxx KK, Pintus S, Stegmaier P, Stelmashenko D, et al. Role of Phagocytosis in the Pro-Inflammatory Response in LDL-Induced Foam Cell Formation; a Transcriptome Analysis. International Journal of Molecular Sciences. 2020; 21(3):817. https://doi.org/10.3390/ijms21030817
Chicago/Turabian StyleOrekhov, Alexander N., Nikita G. Nikiforov, Vasily N. Sukhorukov, Marina V. Kubekina, Igor A. Sobenin, Wei-Kai Wu, Kathy K. Foxx, Sergey Pintus, Philip Stegmaier, Daria Stelmashenko, and et al. 2020. "Role of Phagocytosis in the Pro-Inflammatory Response in LDL-Induced Foam Cell Formation; a Transcriptome Analysis" International Journal of Molecular Sciences 21, no. 3: 817. https://doi.org/10.3390/ijms21030817
APA StyleOrekhov, A. N., Nikiforov, N. G., Sukhorukov, V. N., Kubekina, M. V., Sobenin, I. A., Wu, W.-K., Foxx, K. K., Pintus, S., Stegmaier, P., Stelmashenko, D., Kel, A., Gratchev, A. N., Melnichenko, A. A., Wetzker, R., Summerhill, V. I., Manabe, I., & Oishi, Y. (2020). Role of Phagocytosis in the Pro-Inflammatory Response in LDL-Induced Foam Cell Formation; a Transcriptome Analysis. International Journal of Molecular Sciences, 21(3), 817. https://doi.org/10.3390/ijms21030817