Ischemia/Reperfusion Injury Revisited: An Overview of the Latest Pharmacological Strategies

 ,
,
Abstract
:
1. Introduction
2. Pathophysiology of IRI
2.1. Ischemic Period: Hypoxic Injury and Preparation for Reperfusion
2.2. Reperfusion Injury: the Superlative Damage
2.3. The Ever-Evolving Role of Mitochondria in IRI
2.4. G-Proteins and Endothelium Interplay in IRI
3. Systemic Susceptibility to IRI
4. Therapeutics: the Pharmacological Approach
4.1. Representative Pharmacological Targets
4.2. Pharmacological Targeting of the RISK Pathway
4.3. Pharmacological Targeting of the SAFE Pathway
4.4. Pharmacological Targeting of the cGMP/PKG Pathway
4.5. Pharmacological Targeting of the Mitochondria
4.6. Engaging Multiple Pathways
4.7. Alternative Mechanisms of Protection
4.8. Hyperbaric Oxygen Therapy
5. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Carden, D.L.; Granger, D.N. Pathophysiology of ischaemia-reperfusion injury. J. Pathol. 2000, 190, 255–266. [Google Scholar] [CrossRef]
- Evora, P.R.; Pearson, P.J.; Schaff, H.V. Endothelial function and coronary vasospasm after heart surgery. Arq. Bras. Cardiol. 1993, 61, 119–125. [Google Scholar] [PubMed]
- Evora, P.R.; Pearson, P.J.; Seccombe, J.F.; Schaff, H.V. Ischemia-reperfusion lesion. Physiopathologic aspects and the importance of the endothelial function. Arq. Bras. Cardiol. 1996, 66, 239–245. [Google Scholar] [PubMed]
- Evora, P.R.; Pearson, P.J.; Schaff, H.V. Impaired endothelium-dependent relaxation after coronary reperfusion injury: Evidence for G-protein dysfunction. Ann. Thorac. Surg. 1994, 57, 1550–1556. [Google Scholar] [CrossRef]
- Ibanez, B.; Heusch, G.; Ovize, M.; Van De Werf, F. Evolving Therapies for Myocardial Ischemia/Reperfusion Injury. J. Am. Coll. Cardiol. 2015, 65, 1454–1471. [Google Scholar] [CrossRef] [Green Version]
- Pantazi, E.; Zaouali, M.A.; Bejaoui, M.; Folch-Puy, E.; Ben Abdennebi, H.; Roselló-Catafau, J. Role of sirtuins in ischemia-reperfusion injury. World J. Gastroenterol. 2013, 19, 7594–7602. [Google Scholar] [CrossRef] [PubMed]
- Nastos, C.; Kalimeris, K.; Papoutsidakis, N.; Tasoulis, M.-K.; Lykoudis, P.M.; Theodoraki, K.; Nastou, D.; Smyrniotis, V.; Arkadopoulos, N. Global Consequences of Liver Ischemia/Reperfusion Injury. Oxidative Med. Cell. Longev. 2014, 2014, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Hentia, C.; Rizzato, A.; Camporesi, E.; Yang, Z.; Muntean, D.M.; Săndesc, D.; Bosco, G. An overview of protective strategies against ischemia/reperfusion injury: The role of hyperbaric oxygen preconditioning. Brain Behav. 2018, 8, e00959. [Google Scholar] [CrossRef]
- Kumar, V.; Abbas, A.K.; Aster, J.C. Robbins and Cotran Pathologic Basis of Disease, 9th ed.; Elsevier: Amsterdam, The Netherlands, 2015. [Google Scholar]
- Swärdh, A.; Sjöquist, P.; Wang, Q. Relationship between ischaemic time and ischaemia/reperfusion injury in isolated Langendorff-perfused mouse hearts. Acta Physiol. Scand. 2001, 171, 123–128. [Google Scholar]
- Hasche, E.T.; Fernandes, C.; Ben Freedman, S.; Jeremy, R.W.; Freedman, S.B. Relation between Ischemia Time, Infarct Size, and Left Ventricular Function in Humans. Circulation 1995, 92, 710–719. [Google Scholar] [CrossRef]
- Reimer, K.A.; Lowe, J.E.; Rasmussen, M.M.; Jennings, R.B. The wavefront phenomenon of ischemic cell death. 1. Myocardial infarct size vs. duration of coronary occlusion in dogs. Circulation 1977, 56, 786–794. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.-H.; Huang, C.-C.; Chang, C.-P.; Lin, M.-T.; Niu, K.-C.; Tian, Y.-F. Heat Shock Protein 70 (HSP70) Reduces Hepatic Inflammatory and Oxidative Damage in a Rat Model of Liver Ischemia/Reperfusion Injury with Hyperbaric Oxygen Preconditioning. Med. Sci. Monit. 2018, 24, 8096–8104. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.J.; Choi, A.M.K. Pathways of cell signaling in hyperoxia. Free Radic. Boil. Med. 2003, 35, 341–350. [Google Scholar] [CrossRef]
- Lin, Y.W.; Chen, T.Y.; Hung, C.Y.; Tai, S.H.; Huang, S.Y.; Chang, C.C.; Hung, H.Y.; Lee, E.J. Melatonin protects brain against ischemia/reperfusion injury by attenuating endoplasmic reticulum stress. Int. J. Mol. Med. 2018, 42, 182–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, H.; Zhu, J.; Yue, S.; Lu, L.; Busuttil, R.W.; Kupiec-Weglinski, J.W.; Wang, X.; Zhai, Y.; Zhou, H. The Dichotomy of Endoplasmic Reticulum Stress Response in Liver Ischemia-Reperfusion Injury. Transplantation 2016, 100, 365–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakka, V.P.; Gusain, A.; Raghubir, R. Endoplasmic reticulum stress plays critical role in brain damage after cerebral ischemia/reperfusion in rats. Neurotox. Res. 2010, 17, 189–202. [Google Scholar] [CrossRef]
- De Almeida, T.N.; Victorino, J.P.; Bistafa Liu, J.; Tofoli Queiroz Campos, D.; Graf, C.; Jordani, M.C.; Carneiro d’Albuquerque, L.A.; Mendes, K.D.S.; Castro, E.S.O. Effect of Hepatic Preconditioning with the Use of Methylene Blue on the Liver of Wistar Rats Submitted to Ischemia and Reperfusion. Transplant. Proc. 2018, 50, 841–847. [Google Scholar] [CrossRef]
- Peralta, C.; Gracia-Sancho, J.; Casillas-Ramírez, A.; Gracia‑Sancho, J.; Casillas‑Ramírez, A.; Gracia-Sancho, J.; Casillas-Ramirez, A. Molecular pathways in protecting the liver from ischaemia/reperfusion injury: A 2015 update. Clin. Sci. 2015, 129, 345–362. [Google Scholar]
- Zoratti, M.; Szabò, I. The mitochondrial permeability transition. Biochim. Biophys. Acta BBA Rev. Biomembr. 1995, 1241, 139–176. [Google Scholar] [CrossRef]
- Teoh, N.C.; Farrell, G.C. Hepatic ischemia reperfusion injury: Pathogenic mechanisms and basis for hepatoprotection. J. Gastroenterol. Hepatol. 2003, 18, 891–902. [Google Scholar] [CrossRef]
- Guan, L.-Y.; Fu, P.-Y.; Li, P.-D.; Li, Z.-N.; Liu, H.-Y.; Xin, M.-G.; Li, W. Mechanisms of hepatic ischemia-reperfusion injury and protective effects of nitric oxide. World J. Gastrointest. Surg. 2014, 6, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Datta, G.; Fuller, B.J.; Davidson, B.R. Molecular mechanisms of liver ischemia reperfusion injury: Insights from transgenic knockout models. World J. Gastroenterol. 2013, 19, 1683–1698. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Zhou, H.; Ni, M.; Wang, X.; Busuttil, R.; Kupiec-Weglinski, J.; Zhai, Y. Innate Immune Regulations and Liver Ischemia-Reperfusion Injury. Transplantation 2016, 100, 2601–2610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, X.; Xiao, W.; Xiao, L.; Liu, M. Molecular mechanisms of autophagy in cardiac ischemia/reperfusion injury (Review). Mol. Med. Rep. 2018, 18, 675–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, S.; Wang, Y.; Chen, Y.; Cao, F. The role of the autophagy in myocardial ischemia/reperfusion injury. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2015, 1852, 271–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aghaei, M.; Motallebnezhad, M.; Ghorghanlu, S.; Jabbari, A.; Enayati, A.; Rajaei, M.; Pourabouk, M.; Moradi, A.; Alizadeh, A.M.; Khori, V. Targeting autophagy in cardiac ischemia/reperfusion injury: A novel therapeutic strategy. J. Cell. Physiol. 2019, 234, 16768–16778. [Google Scholar] [CrossRef]
- Zhang, X.; Yan, H.; Yuan, Y.; Gao, J.; Shen, Z.; Cheng, Y.; Shen, Y.; Wang, R.-R.; Wang, X.; Hu, W.-W.; et al. Cerebral ischemia-reperfusion-induced autophagy protects against neuronal injury by mitochondrial clearance. Autophagy 2013, 9, 1321–1333. [Google Scholar] [CrossRef] [Green Version]
- Chouchani, E.T.; Pell, V.R.; James, A.M.; Work, L.M.; Saeb-Parsy, K.; Frezza, C.; Krieg, T.; Murphy, M.P. A Unifying Mechanism for Mitochondrial Superoxide Production during Ischemia-Reperfusion Injury. Cell Metab. 2016, 23, 254–263. [Google Scholar] [CrossRef] [Green Version]
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijević, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord, E.N.J.; Smith, A.C.; et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014, 515, 431–435. [Google Scholar] [CrossRef] [Green Version]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef]
- Pell, V.R.; Chouchani, E.T.; Frezza, C.; Murphy, M.P.; Krieg, T. Succinate metabolism: A new therapeutic target for myocardial reperfusion injury. Cardiovasc. Res. 2016, 111, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Hollander, J.M.; Thapa, D.; Shepherd, D.L. Physiological and structural differences in spatially distinct subpopulations of cardiac mitochondria: Influence of cardiac pathologies. Am. J. Physiol. Circ. Physiol. 2014, 307, H1–H14. [Google Scholar] [CrossRef] [PubMed]
- Javadov, S.; Jang, S.; Parodi-Rullan, R.; Khuchua, Z.; Kuznetsov, A.V. Mitochondrial permeability transition in cardiac ischemia-reperfusion: Whether cyclophilin D is a viable target for cardioprotection? Cell. Mol. Life Sci. CMLS 2017, 74, 2795–2813. [Google Scholar] [CrossRef] [PubMed]
- Fels, J.A.; Manfredi, G. Sex Differences in Ischemia/Reperfusion Injury: The Role of Mitochondrial Permeability Transition. Neurochem. Res. 2019, 44, 2336–2345. [Google Scholar] [CrossRef] [PubMed]
- Casin, K.M.; Fallica, J.; Mackowski, N.; Veenema, R.J.; Chan, A.; Paul, A.S.; Zhu, G.; Bedja, D.; Biswal, S.; Kohr, M.J. S-Nitrosoglutathione Reductase Is Essential for Protecting the Female Heart From Ischemia-Reperfusion Injury. Circ. Res. 2018, 123, 1232–1243. [Google Scholar] [CrossRef] [PubMed]
- Laslett, L.J.; Alagona, P., Jr.; Clark, B.A., 3rd; Drozda, J.P., Jr.; Saldivar, F.; Wilson, S.R.; Poe, C.; Hart, M. The worldwide environment of cardiovascular disease: Prevalence, diagnosis, therapy, and policy issues: A report from the American College of Cardiology. J. Am. Coll. Cardiol. 2012, 60 (Suppl. 25), S1–S49. [Google Scholar] [CrossRef]
- Cohen, M.V.; Yang, X.-M.; Downey, J.M. Smaller infarct after preconditioning does not predict extent of early functional improvement of reperfused heart. Am. J. Physiol. Circ. Physiol. 1999, 277, H1754–H1761. [Google Scholar] [CrossRef] [PubMed]
- Eisen, A.; Fisman, E.Z.; Rubenfire, M.; Freimark, D.; McKechnie, R.; Tenenbaum, A.; Motro, M.; Adler, Y. Ischemic preconditioning: Nearly two decades of research. A comprehensive review. Atherosclerosis 2004, 172, 201–210. [Google Scholar] [CrossRef]
- Schultz, J.E.J.; Hsu, A.K.; Barbieri, J.T.; Li, P.-L.; Gross, G.J. Pertussis toxin abolishes the cardioprotective effect of ischemic preconditioning in intact rat heart. Am. J. Physiol. Circ. Physiol. 1998, 275, H495–H500. [Google Scholar] [CrossRef]
- Waterson, R.E.; Thompson, C.G.; Mabe, N.W.; Kaur, K.; Talbot, J.N.; Neubig, R.R.; Rorabaugh, B.R. Galpha(i2)-mediated protection from ischaemic injury is modulated by endogenous RGS proteins in the mouse heart. Cardiovasc. Res. 2011, 91, 45–52. [Google Scholar] [CrossRef]
- Kohler, D.; Devanathan, V.; Bernardo de Oliveira Franz, C.; Eldh, T.; Novakovic, A.; Roth, J.M.; Granja, T.; Birnbaumer, L.; Rosenberger, P.; Beer-Hammer, S.; et al. Galphai2- and Galphai3-deficient mice display opposite severity of myocardial ischemia reperfusion injury. PLoS ONE 2014, 9, e98325. [Google Scholar] [CrossRef] [PubMed]
- Parra, S.; Huang, X.Y.; Charbeneau, R.A.; Wade, S.M.; Kaur, K.; Rorabaugh, B.R.; Neubig, R.R. Conditional disruption of interactions between Ga-i2 and regulator of G protein signaling (RGS) proteins protects the heart from ischemic injury. BMC Pharmacol. Toxicol. 2014, 15, 29. [Google Scholar] [CrossRef] [PubMed]
- Stewart, A.; Huang, J.; Fisher, R.A. RGS Proteins in Heart: Brakes on the Vagus. Front. Physiol. 2012, 3, 95. [Google Scholar] [CrossRef]
- Yang, J.; Maity, B.; Huang, J.; Gao, Z.; Stewart, A.; Weiss, R.M.; Anderson, M.E.; Fisher, R.A. G-protein inactivator RGS6 mediates myocardial cell apoptosis and cardiomyopathy caused by doxorubicin. Cancer Res. 2013, 73, 1662–1667. [Google Scholar] [CrossRef] [PubMed]
- Rorabaugh, B.R.; Chakravarti, B.; Mabe, N.W.; Seeley, S.L.; Bui, A.D.; Yang, J.Q.; Watts, S.W.; Neubig, R.R.; Fisher, R.A. Regulator of G Protein Signaling 6 Protects the Heart from Ischemic Injury. J. Pharmacol. Exp. Ther. 2017, 360, 409–416. [Google Scholar] [CrossRef]
- Evora, P.R.B. G-Proteins Agonists and NO/cGMP Blockers: Unexplored Frontiers in the Pharmaceutical Industry. Arq. Bras. Cardiol. 2017, 109, 275–276. [Google Scholar] [CrossRef] [PubMed]
- Evora, P.R.B.; Nobre, F. The role of G-proteins in the pathophysiology of the cardiovascular diseases. Arq. Bras. Cardiol. 1999, 72, 209–229. [Google Scholar] [CrossRef]
- Vilalva, K.H.; Mumic, F.T.; Silveira, M.R.G.; Mente, E.D.; Evora, P.R.B.; Castro, E.S.O. Use of Methylene Blue to Treat Hypovolemic Shock Followed by Ischemia-Reperfusion Injury in the Postoperative Orthotopic Liver Transplant Patient: A Case Report. Exp. Clin. Transplant. 2018, 16, 511–514. [Google Scholar]
- Chies, A.B.; Nakazato, P.C.G.; Spadella, M.A.; Zorzi, P.; Gomes, M.C.J.; D’Albuquerque, L.A.C.; Castro-E-Silva, O. Rivastigmine prevents injury induced by ischemia and reperfusion in rat liver. Acta Cir. Bras. 2018, 33, 775–784. [Google Scholar] [CrossRef]
- Nakazato, P.C.G.; Victorino, J.P.; Fina, C.F.; Mendes, K.D.S.; Gomes, M.C.J.; Evora, P.R.B.; D’Albuquerque, L.A.C.; Castro-E-Silva, O. Liver ischemia and reperfusion injury. Pathophysiology and new horizons in preconditioning and therapy. Acta Cir. Bras. 2018, 33, 723–735. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Chen, G.; Wyburn, K.R.; Yin, J.; Bertolino, P.; Eris, J.M.; Alexander, S.I.; Sharland, A.F.; Chadban, S.J. TLR4 activation mediates kidney ischemia/reperfusion injury. J. Clin. Investig. 2007, 117, 2847–2859. [Google Scholar] [CrossRef] [PubMed]
- Koutsogiannidis, C.P.; Johnson, E.O. Pharmacological Neuroprotection in Cardiac Surgery: Effectiveness of Pharmacologic-Preconditioning with Erythromycin. Curr. Vasc. Pharmacol. 2018, 16, 329–335. [Google Scholar] [CrossRef] [PubMed]
- De Perrot, M.; Liu, M.; Waddell, T.K.; Keshavjee, S. Ischemia–Reperfusion–induced Lung Injury. Am. J. Respir. Crit. Care Med. 2003, 167, 490–511. [Google Scholar] [CrossRef] [PubMed]
- Eppinger, M.J.; Deeb, G.M.; Bolling, S.F.; Ward, P.A. Mediators of ischemia-reperfusion injury of rat lung. Am. J. Pathol. 1997, 150, 1773–1784. [Google Scholar] [PubMed]
- Weiser, M.R. Reperfusion injury of ischemic skeletal muscle is mediated by natural antibody and complement. J. Exp. Med. 1996, 183, 2343–2348. [Google Scholar] [CrossRef] [PubMed]
- Menger, M.D.; Pelikan, S.; Steiner, D.; Messmer, K. Microvascular ischemia-reperfusion injury in striated muscle: Significance of “reflow paradox”. Am. J. Physiol. Circ. Physiol. 1992, 263, H1901–H1906. [Google Scholar] [CrossRef] [PubMed]
- Junk, A.K.; Mammis, A.; Savitz, S.I.; Singh, M.; Roth, S.; Malhotra, S.; Rosenbaum, P.S.; Cerami, A.; Brines, M.; Rosenbaum, D.M. Erythropoietin administration protects retinal neurons from acute ischemia-reperfusion injury. Proc. Natl. Acad. Sci. USA 2002, 99, 10659–10664. [Google Scholar] [CrossRef] [Green Version]
- Oharazawa, H.; Igarashi, T.; Yokota, T.; Fujii, H.; Suzuki, H.; Machide, M.; Takahashi, H.; Ohta, S.; Ohsawa, I. Protection of the Retina by Rapid Diffusion of Hydrogen: Administration of Hydrogen-Loaded Eye Drops in Retinal Ischemia-Reperfusion Injury. Investig. Ophthalmol. Vis. Sci. 2010, 51, 487–492. [Google Scholar] [CrossRef]
- Oliver, C.N.; Starke-Reed, P.E.; Stadtman, E.R.; Liu, G.J.; Carney, J.M.; Floyd, R.A. Oxidative damage to brain proteins, loss of glutamine synthetase activity, and production of free radicals during ischemia/reperfusion-induced injury to gerbil brain. Proc. Natl. Acad. Sci. USA 1990, 87, 5144–5147. [Google Scholar] [CrossRef]
- Santos, M.R.; Celotto, A.C.; Capellini, V.K.; Evora, P.R.B.; Piccinato, C.E.; Joviliano, E.E. The protective effect of cilostazol on isolated rabbit femoral arteries under conditions of ischemia and reperfusion: The role of the nitric oxide pathway. Clinics 2012, 67, 171–178. [Google Scholar] [CrossRef]
- Ciscato, J.G.; Capellini, V.K.; Celotto, A.C.; Baldo, C.F.; Joviliano, E.E.; Evora, P.R.; Dalio, M.B.; Piccinato, C.E. Vascular relaxation of canine visceral arteries after ischemia by means of supraceliac aortic cross-clamping followed by reperfusion. Scand. J. Trauma Resusc. Emerg. Med. 2010, 18, 41. [Google Scholar] [CrossRef] [PubMed]
- Cerqueira, N.F.; Hussni, C.A.; Yoshida, W.B. Pathophysiology of mesenteric ischemia/reperfusion: A review. Acta Cir. Bras. 2005, 20, 336–343. [Google Scholar] [CrossRef] [PubMed]
- Hochman, J.S. Cardiogenic shock complicating acute myocardial infarction: Expanding the paradigm. Circulation 2003, 107, 2998–3002. [Google Scholar] [CrossRef] [PubMed]
- Cotter, G.; Kaluski, E.; Milo, O.; Blatt, A.; Salah, A.; Hendler, A.; Krakover, R.; Golick, A.; Vered, Z. LINCS: L-NAME (a NO synthase inhibitor) in the treatment of refractory cardiogenic shock: A prospective randomized study. Eur. Heart J. 2003, 24, 1287–1295. [Google Scholar] [CrossRef]
- De Castro-e-Silva, O.; Centurion, S.; Pacheco, E.G.; Brisotti, J.L.; Oliveira, A.F.; Sasso, K.D. Basics aspects of the ischemia reperfusion injury and of the ischemic preconditioning. Acta Cir. Bras. 2002, 17. [Google Scholar] [CrossRef]
- Salaris, S.C.; Babbs, C.F.; Voorhees, W.D. Methylene blue as an inhibitor of superoxide generation by xanthine oxidase. A potential new drug for the attenuation of ischemia/reperfusion injury. Biochem. Pharmacol. 1991, 42, 499–506. [Google Scholar] [CrossRef]
- Stirpe, F.; Della Corte, E. The regulation of rat liver xanthine oxidase. Conversion in vitro of the enzyme activity from dehydrogenase (type D) to oxidase (type O). J. Boil. Chem. 1969, 244, 3855–3863. [Google Scholar]
- Garcea, G.; Gescher, A.; Steward, W.; Dennison, A.; Berry, D. Oxidative stress in humans following the Pringle manoeuvre. Hepatobiliary Pancreat. Dis. Int. 2006, 5, 210–214. [Google Scholar]
- Vollmar, B.; Glasz, J.; Leiderer, R.; Post, S.; Menger, M.D. Hepatic microcirculatory perfusion failure is a determinant of liver dysfunction in warm ischemia-reperfusion. Am. J. Pathol. 1994, 145, 1421–1431. [Google Scholar]
- Hou, J.; Xia, Y.; Jiang, R.; Chen, D.; Xu, J.; Deng, L.; Huang, X.; Wang, X.; Sun, B. PTPRO plays a dual role in hepatic ischemia reperfusion injury through feedback activation of NF-kappaB. J. Hepatol. 2014, 60, 306–312. [Google Scholar] [CrossRef]
- Li, J.D.; Peng, Y.; Peng, X.Y.; Li, Q.L.; Li, Q. Suppression of nuclear factor-kappaB activity in Kupffer cells protects rat liver graft from ischemia-reperfusion injury. Transplant. Proc. 2010, 42, 1582–1586. [Google Scholar] [CrossRef]
- Braz, M.M.; Elias-Miró, M.; Jiménez-Castro, M.B.; Casillas-Ramírez, A.; Ramalho, F.S.; Peralta, C. The Current State of Knowledge of Hepatic Ischemia-Reperfusion Injury Based on Its Study in Experimental Models. J. Biomed. Biotechnol. 2012, 2012, 1–20. [Google Scholar] [CrossRef]
- Marzi, I.; Rucker, M.; Walcher, F.; Takei, Y. Endothelin-1 is involved in hepatic sinusoidal vasoconstriction after ischemia and reperfusion. Transpl. Int. 1994, 7 (Suppl. 1), S503–S506. [Google Scholar] [CrossRef]
- Pelsers, M.M.; Namiot, Z.; Kisielewski, W.; Namiot, A.; Januszkiewicz, M.; Hermens, W.T.; Glatz, J.F. Intestinal-type and liver-type fatty acid-binding protein in the intestine. Tissue distribution and clinical utility. Clin. Biochem. 2003, 36, 529–535. [Google Scholar] [CrossRef]
- Van Nieuwenhoven, F. Release of Proteins from Isolated Neonatal Rat Cardiomyocytes Subjected to Simulated Ischemia or Metabolic Inhibition is Independent of Molecular Mass. J. Mol. Cell. Cardiol. 1996, 28, 1429–1434. [Google Scholar] [CrossRef] [PubMed]
- Çakir, Ö.Ö.; Toker, A.; Ataseven, H.; Demir, A.; Polat, H. The Importance of Liver-Fatty Acid Binding Protein in Diagnosis of Liver Damage in Patients with Acute Hepatitis. J. Clin. Diagn. Res. 2017, 11, OC17–OC21. [Google Scholar] [PubMed]
- He, N.; Jia, J.-J.; Li, J.-H.; Zhou, Y.-F.; Lin, B.-Y.; Peng, Y.-F.; Chen, J.-J.; Chen, T.-C.; Tong, R.-L.; Jiang, L.; et al. Remote ischemic perconditioning prevents liver transplantation-induced ischemia/reperfusion injury in rats: Role of ROS/RNS and eNOS. World J. Gastroenterol. 2017, 23, 830–841. [Google Scholar] [CrossRef]
- Dufour, D.R.; Lott, J.A.; Nolte, F.S.; Gretch, D.R.; Koff, R.S.; Seeff, L.B. Diagnosis and monitoring of hepatic injury. I. Performance characteristics of laboratory tests. Clin. Chem. 2000, 46, 2027–2049. [Google Scholar] [PubMed]
- Trull, A.K. The clinical validation of novel strategies for monitoring transplant recipients. Clin. Biochem. 2001, 34, 3–7. [Google Scholar] [CrossRef]
- Monbaliu, D.; De Vries, B.; Crabbé, T.; Van Heurn, E.; Verwaest, C.; Roskams, T.; Fevery, J.; Pirenne, J.; Buurman, W. Liver fatty acid-binding protein: An early and sensitive plasma marker of hepatocellular damage and a reliable predictor of graft viability after liver transplantation from non-heart-beating donors. Transplant. Proc. 2005, 37, 413–416. [Google Scholar] [CrossRef]
- Wang, G.; Bonkovsky, H.L.; De Lemos, A.; Burczynski, F.J. Recent insights into the biological functions of liver fatty acid binding protein 1. J. Lipid Res. 2015, 56, 2238–2247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, C.L.; Gonwa, T.A.; Wilkinson, A.H. Pathophysiology of renal disease associated with liver disorders: Implications for liver transplantation. Part I. Liver Transplant. 2002, 8, 91–109. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Maher, J.M.; Chen, C.; Klaassen, C.D. Hepatic ischemia-reperfusion induces renal heme oxygenase-1 via NF-E2-related factor 2 in rats and mice. Mol. Pharmacol. 2007, 71, 817–825. [Google Scholar] [CrossRef] [PubMed]
- Sutton, T.A.; Mang, H.E.; Campos, S.B.; Sandoval, R.M.; Yoder, M.C.; Molitoris, B.A. Injury of the renal microvascular endothelium alters barrier function after ischemia. Am. J. Physiol. Physiol. 2003, 285, F191–F198. [Google Scholar] [CrossRef] [Green Version]
- Molitoris, B.A. Actin cytoskeleton in ischemic acute renal failure. Kidney Int. 2004, 66, 871–883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.T.; Park, S.W.; Kim, M.; D’Agati, V.D. Acute kidney injury after hepatic ischemia and reperfusion injury in mice. Lab. Investig. 2009, 89, 196–208. [Google Scholar] [CrossRef] [PubMed]
- Kadkhodaee, M.; Mikaeili, S.; Zahmatkesh, M.; Golab, F.; Seifi, B.; Arab, H.-A.; Shams, S.; Mahdavi-Mazdeh, M. Alteration of renal functional, oxidative stress and inflammatory indices following hepatic ischemia-reperfusion. Gen. Physiol. Biophys. 2012, 31, 195–202. [Google Scholar] [CrossRef] [Green Version]
- Polat, C.; Tokyol, C.; Kahraman, A.; Sabuncuoğlu, B.; Yilmaz, S. The effects of desferrioxamine and quercetin on hepatic ischemia–reperfusion induced renal disturbance. Prostaglandins Leukot. Essent. Fat. Acids 2006, 74, 379–383. [Google Scholar] [CrossRef]
- Park, S.W.; Chen, S.W.; Kim, M.; D’Agati, V.D.; Lee, H.T. Human activated protein C attenuates both hepatic and renal injury caused by hepatic ischemia and reperfusion injury in mice. Kidney Int. 2009, 76, 739–750. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, S.; Serizawa, A.; Sakaguchi, T.; Tsuchiya, Y.; Kojima, Y.; Okamoto, K.; Kurachi, K.; Konno, H.; Fujise, Y.; Baba, S.; et al. The roles of platelet-activating factor and endothelin-1 in renal damage after total hepatic ischemia and reperfusion. Transplantation 2000, 69, 2267–2273. [Google Scholar] [CrossRef]
- Tucker, D.; Lu, Y.; Zhang, Q. From Mitochondrial Function to Neuroprotection-an Emerging Role for Methylene Blue. Mol. Neurobiol. 2018, 55, 5137–5153. [Google Scholar] [CrossRef] [PubMed]
- Dziedzic, T. Systemic inflammation as a therapeutic target in acute ischemic stroke. Expert Rev. Neurother. 2015, 15, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Jin, R.; Liu, L.; Zhang, S.; Nanda, A.; Li, G. Role of inflammation and its mediators in acute ischemic stroke. J. Cardiovasc. Transl. Res. 2013, 6, 834–851. [Google Scholar] [CrossRef] [PubMed]
- Licata, G.; Tuttolomondo, A.; Corrao, S.; Di Raimondo, D.; Fernandez, P.; Caruso, C.; Avellone, G.; Pinto, A. Immunoinflammatory Activation during the Acute Phase of Lacunar and Non-Lacunar Ischemic Stroke: Association with Time of Onset and Diabetic State. Int. J. Immunopathol. Pharmacol. 2006, 19, 639–646. [Google Scholar] [CrossRef] [PubMed]
- Schauer, R.J.; Kalmuk, S.; Gerbes, A.L.; Leiderer, R.; Meissner, H.; Schildberg, F.W.; Messmer, K.; Bilzer, M. Intravenous administration of glutathione protects parenchymal and non-parenchymal liver cells against reperfusion injury following rat liver transplantation. World J. Gastroenterol. 2004, 10, 864–870. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Ma, Q.; Zhu, P.; Ren, J.; Reiter, R.J.; Chen, Y. Protective role of melatonin in cardiac ischemia-reperfusion injury: From pathogenesis to targeted therapy. J. Pineal Res. 2018, 64, e12471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerksick, C.; Willoughby, D. The Antioxidant Role of Glutathione and N-Acetyl-Cysteine Supplements and Exercise-Induced Oxidative Stress. J. Int. Soc. Sports Nutr. 2005, 2, 38–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kantor, P.F.; Lucien, A.; Kozak, R.; Lopaschuk, G.D. The Antianginal Drug Trimetazidine Shifts Cardiac Energy Metabolism from Fatty Acid Oxidation to Glucose Oxidation by Inhibiting Mitochondrial Long-Chain 3-Ketoacyl Coenzyme a Thiolase. Circ. Res. 2000, 86, 580–588. [Google Scholar] [CrossRef] [PubMed]
- Lopaschuk, G.D. Metabolic Modulators in Heart Disease: Past, Present, and Future. Can. J. Cardiol. 2017, 33, 838–849. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Chen, J.M.; Huang, H.; Kuznicki, M.; Zheng, S.; Sun, W.; Quan, N.; Wang, L.; Yang, H.; Guo, H.M.; et al. The protective effect of trimetazidine on myocardial ischemia/reperfusion injury through activating AMPK and ERK signaling pathway. Metabolism 2016, 65, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Rossello, X.; Yellon, D.M. The RISK pathway and beyond. Basic Res. Cardiol. 2018, 113, 2. [Google Scholar] [CrossRef] [PubMed]
- Eltzschig, H.K.; Eckle, T. Ischemia and reperfusion--from mechanism to translation. Nat. Med. 2011, 17, 1391–1401. [Google Scholar] [CrossRef] [PubMed]
- Burley, D.S.; Ferdinandy, P.; Baxter, G.F. Cyclic GMP and protein kinase-G in myocardial ischaemia-reperfusion: Opportunities and obstacles for survival signaling. Br. J. Pharmacol. 2007, 152, 855–869. [Google Scholar] [CrossRef] [PubMed]
- Cadenas, S. ROS and redox signaling in myocardial ischemia-reperfusion injury and cardioprotection. Free Radic. Boil. Med. 2018, 117, 76–89. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Li, T.; Ji, T.; Yi, W.; Yang, Z.; Wang, S.; Yang, Y.; Gu, C. AMPK: Potential Therapeutic Target for Ischemic Stroke. Theranostics 2018, 8, 4535–4551. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.-Y.; Yiang, G.-T.; Liao, W.-T.; Tsai, A.P.-Y.; Cheng, Y.-L.; Cheng, P.-W.; Li, C.-Y.; Li, C.-J. Current Mechanistic Concepts in Ischemia and Reperfusion Injury. Cell. Physiol. Biochem. 2018, 46, 1650–1667. [Google Scholar] [CrossRef] [PubMed]
- Hausenloy, D.J.; Lecour, S.; Yellon, D.M. Reperfusion Injury Salvage Kinase and Survivor Activating Factor Enhancement Prosurvival Signaling Pathways in Ischemic Postconditioning: Two Sides of the Same Coin. Antioxid. Redox Signal. 2011, 14, 893–907. [Google Scholar] [CrossRef] [Green Version]
- Yellon, D.M.; Hausenloy, D.J. New directions for protecting the heart against ischaemia–reperfusion injury: Targeting the Reperfusion Injury Salvage Kinase (RISK)-pathway. Cardiovasc. Res. 2004, 61, 448–460. [Google Scholar]
- Ghaboura, N.; Tamareille, S.; Ducluzeau, P.H.; Grimaud, L.; Loufrani, L.; Croue, A.; Tourmen, Y.; Henrion, D.; Furber, A.; Prunier, F. Diabetes mellitus abrogates erythropoietin-induced cardioprotection against ischemic-reperfusion injury by alteration of the RISK/GSK-3beta signaling. Basic Res. Cardiol. 2011, 106, 147–162. [Google Scholar] [CrossRef]
- Schulman, D.; Latchman, D.S.; Yellon, D.M. Urocortin protects the heart from reperfusion injury via upregulation of p42/p44 MAPK signaling pathway. Am. J. Physiol. Circ. Physiol. 2002, 283, H1481–H1488. [Google Scholar] [CrossRef] [Green Version]
- Yellon, D.M.; Hausenloy, D.J. Mechanisms of disease: Myocardial reperfusion injury. N. Engl. J. Med. 2007, 357, 1121–1135. [Google Scholar] [CrossRef]
- Murphy, E.; Steenbergen, C. Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury. Physiol. Rev. 2008, 88, 581–609. [Google Scholar] [CrossRef] [PubMed]
- Walters, A.M.; Porter, G.A., Jr.; Brookes, P.S. Mitochondria as a drug target in ischemic heart disease and cardiomyopathy. Circ. Res. 2012, 111, 1222–1236. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lv, F.; Jin, L.; Peng, W.; Song, R.; Ma, J.; Cao, C.-M.; Xiao, R.-P. MG53 participates in ischaemic postconditioning through the RISK signalling pathway. Cardiovasc. Res. 2011, 91, 108–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hausenloy, D.J.; Yellon, D.M. Reperfusion injury salvage kinase signalling: Taking a RISK for cardioprotection. Heart Fail. Rev. 2007, 12, 217–234. [Google Scholar] [CrossRef] [PubMed]
- Lecour, S. Activation of the protective Survivor Activating Factor Enhancement (SAFE) pathway against reperfusion injury: Does it go beyond the RISK pathway? J. Mol. Cell. Cardiol. 2009, 47, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Tartaglia, L.A.; Weber, R.F.; Figari, I.S.; Reynolds, C.; Palladino, M.A.; Goeddel, D.V. The two different receptors for tumor necrosis factor mediate distinct cellular responses. Proc. Natl. Acad. Sci. USA 1991, 88, 9292–9296. [Google Scholar] [CrossRef] [PubMed]
- Lecour, S.; James, R.W. When are pro-inflammatory cytokines SAFE in heart failure? Eur. Heart J. 2011, 32, 680–685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amit-Vazina, M.; Shishodia, S.; Harris, D.; Van, Q.; Wang, M.; Weber, D.; Alexanian, R.; Talpaz, M.; Aggarwal, B.B.; Estrov, Z. Atiprimod blocks STAT3 phosphorylation and induces apoptosis in multiple myeloma cells. Br. J. Cancer 2005, 93, 70–80. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Huang, Q.; Shi, B.; Eksarko, P.; Temkin, V.; Pope, R.M. Regulation of Mcl-1 expression in rheumatoid arthritis synovial macrophages. Arthritis Rheum. 2006, 54, 3174–3181. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, J.; Wei, H.; Tian, Z. STAT3-decoy oligodeoxynucleotide inhibits the growth of human lung cancer via down-regulating its target genes. Oncol. Rep. 2007, 17, 1377–1382. [Google Scholar] [CrossRef] [PubMed]
- Torbenson, M.; Yang, S.Q.; Liu, H.Z.; Huang, J.; Gage, W.; Diehl, A.M. STAT-3 Overexpression and p21 Up-Regulation Accompany Impaired Regeneration of Fatty Livers. Am. J. Pathol. 2002, 161, 155–161. [Google Scholar] [CrossRef] [Green Version]
- Ikezoe, T.; Saito, T.; Bandobashi, K.; Yang, Y.; Koeffler, H.P.; Taguchi, H. HIV-1 protease inhibitor induces growth arrest and apoptosis of human multiple myeloma cells via inactivation of signal transducer and activator of transcription 3 and extracellular signal-regulated kinase 1/2. Mol. Cancer Ther. 2004, 3, 473–479. [Google Scholar] [PubMed]
- Marqués, J.M.; Belza, I.; Holtmann, B.; Pennica, D.; Prieto, J.; Bustos, M. Cardiotrophin-1 is an essential factor in the natural defense of the liver against apoptosis. Hepatology 2007, 45, 639–648. [Google Scholar] [CrossRef] [PubMed]
- Pavo, N.; Lukovic, D.; Zlabinger, K.; Zimba, A.; Lorant, D.; Goliasch, G.; Winkler, J.; Pils, D.; Auer, K.; Ankersmit, H.J.; et al. Sequential activation of different pathway networks in ischemia-affected and non-affected myocardium, inducing intrinsic remote conditioning to prevent left ventricular remodeling. Sci. Rep. 2017, 7, 43958. [Google Scholar] [CrossRef] [PubMed]
- Feyzizadeh, S.; Badalzadeh, R. Application of ischemic postconditioning’s algorithms in tissues protection: Response to methodological gaps in preclinical and clinical studies. J. Cell. Mol. Med. 2017, 21, 2257–2267. [Google Scholar] [CrossRef] [PubMed]
- Xin, W.K.; Yang, X.L.; Rich, T.C.; Krieg, T.; Barrington, R.; Cohen, M.V.; Downey, J.M. All Preconditioning-Related G Protein-Coupled Receptors Can Be Demonstrated in the Rabbit Cardiomyocyte. J. Cardiovasc. Pharmacol. Ther. 2012, 17, 190–198. [Google Scholar] [CrossRef] [PubMed]
- Costa, M.A.; Elesgaray, R.; Balaszczuk, A.M.; Arranz, C. Role of NPR-C natriuretic receptor in nitric oxide system activation induced by atrial natriuretic peptide. Regul. Pept. 2006, 135, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Shiva, S.; Sack, M.N.; Greer, J.J.; Duranski, M.; Ringwood, L.A.; Burwell, L.; Wang, X.; MacArthur, P.H.; Shoja, A.; Raghavachari, N.; et al. Nitrite augments tolerance to ischemia/reperfusion injury via the modulation of mitochondrial electron transfer. J. Exp. Med. 2007, 204, 2089–2102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duranski, M.R.; Greer, J.J.; Dejam, A.; Jaganmohan, S.; Hogg, N.; Langston, W.; Patel, R.P.; Yet, S.-F.; Wang, X.; Kevil, C.G.; et al. Cytoprotective effects of nitrite during in vivo ischemia-reperfusion of the heart and liver. J. Clin. Investig. 2005, 115, 1232–1240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, M.V.; Yang, X.-M.; Liu, Y.; Solenkova, N.V.; Downey, J.M. Cardioprotective PKG-independent NO signaling at reperfusion. Am. J. Physiol. Circ. Physiol. 2010, 299, H2028–H2036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inserte, J.; Garcia-Dorado, D. The cGMP/PKG pathway as a common mediator of cardioprotection: Translatability and mechanism. Br. J. Pharmacol. 2015, 172, 1996–2009. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.-B.; Cadete, V.J.; Sawicka, J.; Wozniak, M.; Sawicki, G. Effect of the myosin light chain kinase inhibitor ML-7 on the proteome of hearts subjected to ischemia–reperfusion injury. J. Proteom. 2012, 75, 5386–5395. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.S.; Liu, B.; Luo, X.J.; Zhang, J.J.; Li, N.S.; Ma, Q.L.; Jiang, J.L.; Li, Y.J.; Li, Q.; Peng, J. A novel function of nuclear nonmuscle myosin regulatory light chain in promotion of xanthine oxidase transcription after myocardial ischemia/reperfusion. Free Radic. Biol. Med. 2015, 83, 115–128. [Google Scholar] [CrossRef] [PubMed]
- Evora, P.R.; Alves Junior, L.; Ferreira, C.A.; Menardi, A.C.; Bassetto, S.; Rodrigues, A.J.; Scorzoni Filho, A.; Vicente, W.V. Twenty years of vasoplegic syndrome treatment in heart surgery. Methylene blue revised. Rev. Bras. Cir. Cardiovasc. 2015, 30, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, J.M.; Filho, A.P.; Rodrigues, A.J.; Vicente, W.V.D.A.; Evora, P.R.B. Methylene blue for clinical anaphylaxis treatment: A case report. Sao Paulo Med. J. 2007, 125, 60–62. [Google Scholar] [CrossRef] [PubMed]
- Rosique, R.G.; Rosique, M.; Rosique, I.; Tirapelli, L.; Silva, O.C.E.; Dos Santos, J.; Evora, P. Effect of Methylene Blue on the Hemodynamic Instability Resulting from Liver Ischemia and Reperfusion in Rabbits. Transplant. Proc. 2011, 43, 3643–3651. [Google Scholar] [CrossRef]
- Tian, W.F.; Zeng, S.; Sheng, Q.; Chen, J.L.; Weng, P.; Zhang, X.T.; Yuan, J.J.; Pang, Q.F.; Wang, Z.Q. Methylene Blue Protects the Isolated Rat Lungs from Ischemia-Reperfusion Injury by Attenuating Mitochondrial Oxidative Damage. Lung 2018, 196, 73–82. [Google Scholar] [CrossRef]
- Wang, L.; Chen, B.; Lin, B.; Ye, Y.; Bao, C.; Zhao, X.; Jin, L.; Xiong, X. Methylene Blue Attenuates Lung Injury Induced by Hindlimb Ischemia Reperfusion in Rats. Mediat. Inflamm. 2018, 2018, 1–8. [Google Scholar] [CrossRef]
- Sarac, F.; Kilincaslan, H.; Kilic, E.; Koldaş, M.; Terzi, E.H.; Aydogdu, I.; Kılıncaslan, H.; Kılıc, E. Methylene blue attenuates renal ischemia–reperfusion injury in rats. J. Pediatr. Surg. 2015, 50, 1067–1071. [Google Scholar] [CrossRef]
- Liu, P.-G.; He, S.-Q.; Zhang, Y.-H.; Wu, J. Protective effects of apocynin and allopurinol on ischemia/reperfusion-induced liver injury in mice. World J. Gastroenterol. 2008, 14, 2832–2837. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.-Q.; Qiu, T.; Zhang, L.; Chen, Z.-B.; Wang, Z.-S.; Ma, X.-X.; Li, D. Allopurinol preconditioning attenuates renal ischemia/reperfusion injury by inhibiting HMGB1 expression in a rat model. Acta Cir. Bras. 2016, 31, 176–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, J.; Kim, D.; Kim, K.; Ryu, S.; Han, S.; Shin, B.; Kim, G.; Gwak, M.; Ko, J. Ischemic Preconditioning Produces Comparable Protection Against Hepatic Ischemia/Reperfusion Injury Under Isoflurane and Sevoflurane Anesthesia in Rats. Transplant. Proc. 2017, 49, 2188–2193. [Google Scholar] [CrossRef] [PubMed]
- Abu-Amara, M.; Gurusamy, K.; Hori, S.; Glantzounis, G.; Fuller, B.; Davidson, B.R. Systematic review of randomized controlled trials of pharmacological interventions to reduce ischaemia-reperfusion injury in elective liver resection with vascular occlusion. HPB 2010, 12, 4–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Su, L.; Zhang, X.; Zhang, C.; Wang, L.; Li, Y.; Zhang, Y.; He, T.; Zhu, X.; Cui, L. Ulinastatin downregulates TLR4 and NF-kB expression and protects mouse brains against ischemia/reperfusion injury. Neurol. Res. 2017, 39, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Li, P.; Li, Z.; Liu, H.; Guan, L.; Zhang, L. Effects of Ulinastatin on Liver Warm Ischemia Reperfusion Injury in Mice. Transplantation 2018, 102, S705. [Google Scholar] [CrossRef]
- Xu, Z.; Yu, J.; Wu, J.; Qi, F.; Wang, H.; Wang, Z.; Wang, Z. The Effects of Two Anesthetics, Propofol and Sevoflurane, on Liver Ischemia/Reperfusion Injury. Cell. Physiol. Biochem. 2016, 38, 1631–1642. [Google Scholar] [CrossRef]
- Yang, S.; Chou, W.-P.; Pei, L. Effects of propofol on renal ischemia/reperfusion injury in rats. Exp. Ther. Med. 2013, 6, 1177–1183. [Google Scholar] [CrossRef] [Green Version]
- Malek, M.; Nematbakhsh, M. Renal ischemia/reperfusion injury; from pathophysiology to treatment. J. Ren. Inj. Prev. 2015, 4, 20–27. [Google Scholar]
- Li, P.D.; Fu, P.Y.; Li, Z.N.; Ye, Y.S.; Wang, Z.P.; Liu, H.Y.; Li, W.; Guan, L.Y. Effects of Ulinastatin on liver warm ischemia reperfusion injury in mice. Transplantation 2016, 100, S705. [Google Scholar] [CrossRef]
- Katayama, Y.; Inaba, T.; Nito, C.; Ueda, M.; Katsura, K. Neuroprotective effects of erythromycin on cerebral ischemia reperfusion-injury and cell viability after oxygen-glucose deprivation in cultured neuronal cells. Brain Res. 2014, 1588, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Soliman, S.; Ishrat, T.; Fouda, A.Y.; Patel, A.; Pillai, B.; Fagan, S.C. Sequential Therapy with Minocycline and Candesartan Improves Long-Term Recovery After Experimental Stroke. Transl. Stroke Res. 2015, 6, 309–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozak, A.; Ergul, A.; El-Remessy, A.B.; Johnson, M.H.; Machado, L.S.; Elewa, H.F.; Abdelsaid, M.; Wiley, D.C.; Fagan, S.C. Candesartan augments ischemia-induced proangiogenic state and results in sustained improvement after stroke. Stroke 2009, 40, 1870–1876. [Google Scholar] [CrossRef] [PubMed]
- Price, N.L.; Gomes, A.P.; Ling, A.J.; Duarte, F.V.; Martin-Montalvo, A.; North, B.J.; Agarwal, B.; Ye, L.; Ramadori, G.; Teodoro, J.S.; et al. SIRT1 is required for AMPK activation and the beneficial effects of resveratrol on mitochondrial function. Cell Metab. 2012, 15, 675–690. [Google Scholar] [CrossRef] [PubMed]
- Athar, M.; Back, J.H.; Kopelovich, L.; Bickers, D.R.; Kim, A.L. Multiple molecular Targets of Resveratrol: Anti-carcinogenic Mechanisms. Arch. Biochem. Biophys. 2009, 486, 95–102. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, M.R.; Nabavi, S.F.; Manayi, A.; Daglia, M.; Hajheydari, Z.; Nabavi, S.M. Resveratrol and the mitochondria: From triggering the intrinsic apoptotic pathway to inducing mitochondrial biogenesis, a mechanistic view. Biochim. Biophys. Acta BBA Gen. Subj. 2016, 1860, 727–745. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, S.S.; Cantó, C. The molecular targets of resveratrol. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2015, 1852, 1114–1123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sweitzer, N.K.; Airhart, S. New Therapeutic Target in Heart Failure: Achieving and Maintaining Normokalemia. Circulation 2018, 137, 1331–1333. [Google Scholar] [CrossRef] [PubMed]
- Cannavo, A.; Komici, K.; Bencivenga, L.; D’Amico, M.L.; Gambino, G.; Liccardo, D.; Ferrara, N.; Rengo, G. GRK2 as a therapeutic target for heart failure. Expert Opin. Ther. Targets 2018, 22, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Lin, P.; Li, P.; Feng, L.; Ren, Q.; Xie, X.; Xu, J. Ghrelin protects the heart against ischemia/reperfusion injury via inhibition of TLR4/NLRP3 inflammasome pathway. Life Sci. 2017, 186, 50–58. [Google Scholar] [CrossRef]
- Bakker, P.J.; Butter, L.M.; Claessen, N.; Teske, G.J.; Sutterwala, F.S.; Florquin, S.; Leemans, J.C. A tissue-specific role for Nlrp3 in tubular epithelial repair after renal ischemia/reperfusion. Am. J. Pathol. 2014, 184, 2013–2022. [Google Scholar] [CrossRef] [PubMed]
- Kojima, M.; Kangawa, K. Drug Insight: The functions of ghrelin and its potential as a multitherapeutic hormone. Nat. Clin. Pract. Endocrinol. Metab. 2006, 2, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Soeki, T.; Kishimoto, I.; Schwenke, D.O.; Tokudome, T.; Horio, T.; Yoshida, M.; Hosoda, H.; Kangawa, K. Ghrelin suppresses cardiac sympathetic activity and prevents early left ventricular remodeling in rats with myocardial infarction. Am. J. Physiol. Circ. Physiol. 2008, 294, H426–H432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Budas, G.R.; Disatnik, M.-H.; Mochly-Rosen, D. Aldehyde dehydrogenase 2 in cardiac protection: A new therapeutic target? Trends Cardiovasc. Med. 2009, 19, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-H.; Budas, G.R.; Churchill, E.N.; Disatnik, M.-H.; Hurley, T.D.; Mochly-Rosen, D. Activation of aldehyde dehydrogenase-2 reduces ischemic damage to the heart. Science 2008, 321, 1493–1495. [Google Scholar] [CrossRef] [PubMed]
- Doser, T.A.; Turdi, S.; Thomas, D.P.; Epstein, P.N.; Li, S.-Y.; Ren, J. Transgenic overexpression of aldehyde dehydrogenase-2 rescues chronic alcohol intake-induced myocardial hypertrophy and contractile dysfunction. Circulation 2009, 119, 1941–1949. [Google Scholar] [CrossRef]
- Ma, H.; Guo, R.; Yu, L.; Zhang, Y.; Ren, J. Aldehyde dehydrogenase 2 (ALDH2) rescues myocardial ischaemia/reperfusion injury: Role of autophagy paradox and toxic aldehyde. Eur. Heart J. 2011, 32, 1025–1038. [Google Scholar] [CrossRef]
- Panisello-Roselló, A.; Lopez, A.; Folch-Puy, E.; Carbonell, T.; Rolo, A.; Palmeira, C.; Adam, R.; Net, M.; Roselló-Catafau, J. Role of aldehyde dehydrogenase 2 in ischemia reperfusion injury: An update. World J. Gastroenterol. 2018, 24, 2984–2994. [Google Scholar] [CrossRef]
- Panisello-Rosello, A.; Alva, N.; Flores, M.; Lopez, A.; Castro Benitez, C.; Folch-Puy, E.; Rolo, A.; Palmeira, C.; Adam, R.; Carbonell, T.; et al. Aldehyde Dehydrogenase 2 (ALDH2) in Rat Fatty Liver Cold Ischemia Injury. Int. J. Mol. Sci. 2018, 19, 2479. [Google Scholar] [CrossRef]
- Shimizu, M.; Wang, Q.-D.; Sjöquist, P.-O.; Rydén, L. Angiotensin II Type 1 Receptor Blockade with Candesartan Protects the Porcine Myocardium from Reperfusion-Induced Injury. J. Cardiovasc. Pharmacol. 1998, 32, 231–238. [Google Scholar] [CrossRef]
- Venkat, P.; Shen, Y.; Chopp, M.; Chen, J. Cell-based and pharmacological neurorestorative therapies for ischemic stroke. Neuropharmacology 2018, 134, 310–322. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Zhang, Y.; Hu, S.; Shi, C.; Zhu, P.; Ma, Q.; Jin, Q.; Cao, F.; Tian, F.; Chen, Y. Melatonin protects cardiac microvasculature against ischemia/reperfusion injury via suppression of mitochondrial fission-VDAC1-HK2-mPTP-mitophagy axis. J. Pineal Res. 2017, 63, e12413. [Google Scholar] [CrossRef] [PubMed]
- Zhai, M.; Li, B.; Duan, W.; Jing, L.; Zhang, B.; Zhang, M.; Yu, L.; Liu, Z.; Yu, B.; Ren, K.; et al. Melatonin ameliorates myocardial ischemia reperfusion injury through SIRT3-dependent regulation of oxidative stress and apoptosis. J. Pineal Res. 2017, 63, e12419. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Li, D.; Zhu, P.; Hu, S.; Hu, N.; Ma, S.; Zhang, Y.; Han, T.; Ren, J.; Cao, F.; et al. Melatonin suppresses platelet activation and function against cardiac ischemia/reperfusion injury via PPARgamma/FUNDC1/mitophagy pathways. J. Pineal Res. 2017, 63. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Zhao, Y.; Xu, L.; Gao, L.; Su, Y.; Lin, N.; Pu, J. The nuclear melatonin receptor ROR α is a novel endogenous defender against myocardial ischemia/reperfusion injury. J. Pineal Res. 2016, 60, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.M.; Gong, B.; Duan, W.X.; Fan, C.X.; Zhang, J.; Li, Z.; Xue, X.D.; Xu, Y.L.; Meng, D.D.; Li, B.Y.; et al. Melatonin ameliorates myocardial ischemia/reperfusion injury in type 1 diabetic rats by preserving mitochondrial function: Role of AMPK-PGC-1 alpha-SIRT3 signaling. Sci. Rep. 2017, 7, 41337. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Ozasa, H.; Horikawa, S. Edaravone protects against lung injury induced by intestinal ischemia/reperfusion in rat. Free Radic. Boil. Med. 2005, 38, 369–374. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.M.; Flentjar, N.J.; De Haan, J.; Hertzog, P.; Kola, I.; Crack, P.J.; Iannello, R.C. Increased infarct size and exacerbated apoptosis in the glutathione peroxidase-1 (Gpx-1) knockout mouse brain in response to ischemia/reperfusion injury. J. Neurochem. 2001, 78, 1389–1399. [Google Scholar]
- Suyavaran, A.; Ramamurthy, C.; Mareeswaran, R.; Subastri, A.; Lokeswara Rao, P.; Thirunavukkarasu, C. TNF-alpha suppression by glutathione preconditioning attenuates hepatic ischemia reperfusion injury in young and aged rats. Inflamm. Res. 2015, 64, 71–81. [Google Scholar] [CrossRef]
- Whillier, S.; Raftos, J.E.; Chapman, B.; Kuchel, P.W. Role ofN-acetylcysteine and cystine in glutathione synthesis in human erythrocytes. Redox Rep. 2009, 14, 115–124. [Google Scholar] [CrossRef]
- Kalimeris, K.; Briassoulis, P.; Ntzouvani, A.; Nomikos, T.; Papaparaskeva, K.; Politi, A.; Batistaki, C.; Kostopanagiotou, G. N-acetylcysteine ameliorates liver injury in a rat model of intestinal ischemia reperfusion. J. Surg. Res. 2016, 206, 263–272. [Google Scholar] [CrossRef]
- Kim, H.Y.; Kim, S.J.; Lee, S.M. Activation of NLRP3 and AIM2 inflammasomes in Kupffer cells in hepatic ischemia/reperfusion. FEBS J. 2015, 282, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Weigand, M.A.; Plachky, J.; Thies, J.C.; Spies-Martin, D.; Otto, G.; Martin, E.; Bardenheuer, H.J. N-acetylcysteine attenuates the increase in alpha-glutathione S-transferase and circulating ICAM-1 and VCAM-1 after reperfusion in humans undergoing liver transplantation. Transplantation 2001, 72, 694–698. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, J.O.G.; Ignarro, L.J.; Lundström, I.; Jynge, P.; Almén, T. Calmangafodipir [Ca4Mn(DPDP)5], mangafodipir (MnDPDP) and MnPLED with special reference to their SOD mimetic and therapeutic properties. Drug Discov. Today 2015, 20, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Ben Mosbah, I.; Mouchel, Y.; Pajaud, J.; Ribault, C.; Lucas, C.; Laurent, A.; Boudjema, K.; Morel, F.; Corlu, A.; Compagnon, P. Pretreatment with Mangafodipir Improves Liver Graft Tolerance to Ischemia/Reperfusion Injury in Rat. PLoS ONE 2012, 7, e50235. [Google Scholar] [CrossRef] [PubMed]
- El-Saadi, W.; Puskar, W.; Skogvard, P.; Engvall, J.E.; Andersson, R.G.; Karlsson, J.-E.; Ali, M.; Maret, E.; Jynge, P. Mangafodipir as a cardioprotective adjunct to reperfusion therapy: A feasibility study in patients with ST-segment elevation myocardial infarction. Eur. Hear. J. Cardiovasc. Pharmacother. 2015, 1, 39–45. [Google Scholar]
- Beckman, J.S.; Koppenol, W.H. Nitric oxide, superoxide, and peroxynitrite: The good, the bad, and the ugly. Am. J. Physiol. Cell Physiol. 1996, 271, C1424–C1437. [Google Scholar] [CrossRef]
- Marcelino, P.; Marinho, H.S.; Campos, M.C.; Neves, A.R.; Real, C.; Fontes, F.S.; Carvalho, A.; Feio, G.; Martins, M.B.F.; Corvo, M.L. Therapeutic activity of superoxide dismutase-containing enzymosomes on rat liver ischaemia-reperfusion injury followed by magnetic resonance microscopy. Eur. J. Pharm. Sci. 2017, 109, 464–471. [Google Scholar] [CrossRef]
- Carillon, J.; Rouanet, J.-M.; Cristol, J.-P.; Brion, R. Superoxide Dismutase Administration, a Potential Therapy Against Oxidative Stress Related Diseases: Several Routes of Supplementation and Proposal of an Original Mechanism of Action. Pharm. Res. 2013, 30, 2718–2728. [Google Scholar] [CrossRef]
- Chen, C.; Lu, W.; Wu, G.; Lv, L.; Chen, W.; Huang, L.; Wu, X.; Xu, N.; Wu, Y. Cardioprotective effects of combined therapy with diltiazem and superoxide dismutase on myocardial ischemia-reperfusion injury in rats. Life Sci. 2017, 183, 50–59. [Google Scholar] [CrossRef]
- Pinto, A.; Immohr, M.B.; Jahn, A.; Jenke, A.; Boeken, U.; Lichtenberg, A.; Akhyari, P. The extracellular isoform of superoxide dismutase has a significant impact on cardiovascular ischaemia and reperfusion injury during cardiopulmonary bypass. Eur. J. Cardio-Thorac. Surg. 2016, 50, 1035–1044. [Google Scholar] [CrossRef] [Green Version]
- Wieland, E.; Schütz, E.; Armstrong, V.W.; Küthe, F.; Heller, C.; Oellerich, M. Idebenone protects hepatic microsomes against oxygen radical-mediated damage in organ preservation solutions. Transplantation 1995, 60, 444–450. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.P.; Lei, D.W.; Shao, Z.B.; Zhou, X.R. Synergistic neuroprotective effect of rasagiline and idebenone against retinal ischemia-reperfusion injury via the Lin28-let-7-Dicer pathway. Oncotarget 2018, 9, 12137–12153. [Google Scholar] [Green Version]
- Obici, S.; Feng, Z.; Arduini, A.; Conti, R.; Rossetti, L. Inhibition of hypothalamic carnitine palmitoyltransferase-1 decreases food intake and glucose production. Nat. Med. 2003, 9, 756–761. [Google Scholar] [CrossRef] [PubMed]
- Bremer, J. The Effect of Fasting on the Activity of Liver Carnitine Palmitoyltransferase and Its Inhibition by Malonyl-Coa. Biochim. Biophys. Acta 1981, 665, 628–631. [Google Scholar] [CrossRef]
- Chavez, P.N.; Stanley, W.C.; McElfresh, T.A.; Huang, H.; Sterk, J.P.; Chandler, M.P. Effect of hyperglycemia and fatty acid oxidation inhibition during aerobic conditions and demand-induced ischemia. Am. J. Physiol. Circ. Physiol. 2003, 284, H1521–H1527. [Google Scholar] [CrossRef] [Green Version]
- Linke, A.; Chandler, M.P.; Young, M.E.; Penn, M.S.; Gupte, S.; D’Agostino, C.; Hintze, T.H.; Lionetti, V.; Stanley, W.C.; Recchia, F.A. Carnitine palmitoyl transferase-I inhibition prevents ventricular remodeling and delays decompensation in pacing-induced heart failure. Cardiovasc. Res. 2005, 66, 454–461. [Google Scholar]
- Chang, K.-C.; Liang, J.-T.; Wu, M.-S.; Chang, K.-C.; Tseng, C.-D.; Lu, S.-C.; Liang, J.-T.; Wu, M.-S.; Tsai, M.-S.; Hsu, K.-L.; et al. Effects of acetyl-L-carnitine and oxfenicine on aorta stiffness in diabetic rats. Eur. J. Clin. Investig. 2010, 40, 1002–1010. [Google Scholar] [CrossRef] [PubMed]
- Maarman, G.; Marais, E.; Lochner, A.; Du Toit, E.F.; Toit, E.F. Effect of Chronic CPT-1 Inhibition on Myocardial Ischemia-Reperfusion Injury (I/R) in a Model of Diet-Induced Obesity. Cardiovasc. Drugs Ther. 2012, 26, 205–216. [Google Scholar] [CrossRef] [PubMed]
- Lopaschuk, G.D.; Wall, S.R.; Olley, P.M.; Davies, N.J. Etomoxir, a carnitine palmitoyltransferase I inhibitor, protects hearts from fatty acid-induced ischemic injury independent of changes in long chain acylcarnitine. Circ. Res. 1988, 63, 1036–1043. [Google Scholar] [CrossRef]
- O’Connor, R.S.; Guo, L.; Ghassemi, S.; Snyder, N.W.; Worth, A.J.; Weng, L.; Kam, Y.; Philipson, B.; Trefely, S.; Nunez-Cruz, S.; et al. The CPT1a inhibitor, etomoxir induces severe oxidative stress at commonly used concentrations. Sci. Rep. 2018, 8, 6289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, L.; Campbell, R.; Scheuermann-Freestone, M.; Taylor, R.; Gunaruwan, P.; Williams, L.; Ashrafian, H.; Horowitz, J.; Fraser, A.G.; Clarke, K.; et al. Metabolic modulation with perhexiline in chronic heart failure—A randomized, controlled trial of short-term use of a novel treatment. Circulation 2005, 112, 3280–3288. [Google Scholar] [CrossRef] [PubMed]
- Holubarsch, C.J.F.; Rohrbach, M.; Karrasch, M.; Boehm, E.; Polonski, L.; Ponikowski, P.; Rhein, S. A double-blind randomized multicentre clinical trial to evaluate the efficacy and safety of two doses of etomoxir in comparison with placebo in patients with moderate congestive heart failure: The ERGO (etomoxir for the recovery of glucose oxidation) study. Clin. Sci. 2007, 113, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Abozguia, K.; Elliott, P.; McKenna, W.J.; Phan, T.T.; Nallur-Shivu, G.; Ahmed, I.; Maher, A.R.; Kaur, K.; Taylor, J.; Henning, A.; et al. Metabolic Modulator Perhexiline Corrects Energy Deficiency and Improves Exercise Capacity in Symptomatic Hypertrophic Cardiomyopathy. Circulation 2010, 122, 1562–1569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boros, D.; Thompson, J.; Larson, D.F. Adenosine regulation of the immune response initiated by ischemia reperfusion injury. Perfusion 2016, 31, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Borea, P.A.; Varani, K.; Vincenzi, F.; Baraldi, P.G.; Tabrizi, M.A.; Merighi, S.; Gessi, S. The A3 adenosine receptor: History and perspectives. Pharmacol. Rev. 2015, 67, 74–102. [Google Scholar] [CrossRef] [PubMed]
- Paez, D.T.; Garces, M.; Calabró, V.; Bin, E.P.; D´annunzio, V.; Del Mauro, J.; Marchini, T.; Höcht, C.; Evelson, P.; Gelpi, R.J.; et al. Adenosine A1 receptors and mitochondria: Targets of remote ischemic preconditioning. Am. J. Physiol. Circ. Physiol. 2019, 316, H743–H750. [Google Scholar] [CrossRef]
- Ahmad, A.; Ahmad, S.; Glover, L.; Miller, S.M.; Shannon, J.M.; Guo, X.; Franklin, W.A.; Bridges, J.P.; Schaack, J.B.; Colgan, S.P.; et al. Adenosine A2A receptor is a unique angiogenic target of HIF-2alpha in pulmonary endothelial cells. Proc. Natl. Acad. Sci. USA 2009, 106, 10684–10689. [Google Scholar] [CrossRef]
- Cao, J.; Lin, H.; Li, W.; Dong, Z.; Shi, Y.; Zhang, X.; Xiao, R. Ischemia postconditioning protects dermal microvascular endothelial cells of rabbit epigastric skin flaps against apoptosis via adenosine A2a receptors. J. Plast. Surg. Hand Surg. 2019, 53, 76–82. [Google Scholar] [CrossRef]
- Mehaffey, J.H.; Money, D.; Charles, E.J.; Schubert, S.; Piñeros, A.F.; Wu, D.; Bontha, S.V.; Hawkins, R.; Teman, N.R.; Laubach, V.E.; et al. Adenosine 2A Receptor Activation Attenuates Ischemia Reperfusion Injury During Extracorporeal Cardiopulmonary Resuscitation. Ann. Surg. 2019, 269, 1176–1183. [Google Scholar] [CrossRef]
- Chen, J.F.; Huang, Z.; Ma, J.; Zhu, J.; Moratalla, R.; Standaert, D.; Moskowitz, M.A.; Fink, J.S.; Schwarzschild, M.A. A(2A) adenosine receptor deficiency attenuates brain injury induced by transient focal ischemia in mice. J. Neurosci. 1999, 19, 9192–9200. [Google Scholar] [CrossRef] [PubMed]
- Gui, L.; Duan, W.; Tian, H.; Li, C.; Zhu, J.; Chen, J.-F.; Zheng, J. Adenosine A2A receptor deficiency reduces striatal glutamate outflow and attenuates brain injury induced by transient focal cerebral ischemia in mice. Brain Res. 2009, 1297, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Atef, R.M.; Agha, A.M.; Abdel-Rhaman, A.A.; Nassar, N.N. The Ying and Yang of Adenosine A1 and A2A Receptors on ERK1/2 Activation in a Rat Model of Global Cerebral Ischemia Reperfusion Injury. Mol. Neurobiol. 2018, 55, 1284–1298. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, R.A.; Agha, A.M.; Nassar, N.N. SCH58261 the selective adenosine A(2A) receptor blocker modulates ischemia reperfusion injury following bilateral carotid occlusion: Role of inflammatory mediators. Neurochem. Res. 2012, 37, 538–547. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, R.; Agha, A.; Abdel-Rahman, A.; Nassar, N. Role of adenosine A2A receptor in cerebral ischemia reperfusion injury: Signaling to phosphorylated extracellular signal-regulated protein kinase (pERK1/2). Neuroscience 2016, 314, 145–159. [Google Scholar] [CrossRef]
- Huerter, M.E.; Sharma, A.K.; Zhao, Y.; Charles, E.J.; Kron, I.L.; Laubach, V.E. Attenuation of Pulmonary Ischemia-Reperfusion Injury by Adenosine A2B Receptor Antagonism. Ann. Thorac. Surg. 2016, 102, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Wan, T.C.; Tampo, A.; Kwok, W.-M.; Auchampach, J.A. Ability of CP-532,903 to protect mouse hearts from ischemia/reperfusion injury is dependent on expression of A3 adenosine receptors in cardiomyoyctes. Biochem. Pharmacol. 2019, 163, 21–31. [Google Scholar] [CrossRef]
- Ohana, G.; Cohen, S.; Rath-Wolfson, L.; Fishman, P. A3 adenosine receptor agonist, CF102, protects against hepatic ischemia/reperfusion injury following partial hepatectomy. Mol. Med. Rep. 2016, 14, 4335–4341. [Google Scholar] [CrossRef]
- Montaner, J.; Ramiro, L.; Simats, A.; Hernández-Guillamon, M.; Delgado, P.; Bustamante, A.; Rosell, A. Matrix metalloproteinases and ADAMs in stroke. Cell. Mol. Life Sci. 2019, 76, 3117–3140. [Google Scholar] [CrossRef]
- Bell, R.M.; Kunuthur, S.P.; Hendry, C.; Bruce-Hickman, D.; Davidson, S.; Yellon, D.M. Matrix metalloproteinase inhibition protects CyPD knockout mice independently of RISK/mPTP signalling: A parallel pathway to protection. Basic Res. Cardiol. 2013, 108, 331. [Google Scholar] [CrossRef]
- Daly, M.C.; Atkinson, S.J.; Varisco, B.M.; Klingbeil, L.; Hake, P.; Lahni, P.; Piraino, G.; Wu, D.; Hogan, S.P.; Zingarelli, B.; et al. Role of matrix metalloproteinase-8 as a mediator of injury in intestinal ischemia and reperfusion. FASEB J. 2016, 30, 3453–3460. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.D.; Maretti-Mira, A.C.; Wang, L.; DeLeve, L.D. Liver-Selective MMP-9 Inhibition in the Rat Eliminates Ischemia-Reperfusion Injury and Accelerates Liver Regeneration. Hepatology 2019, 69, 314–328. [Google Scholar] [CrossRef] [PubMed]
- Baghirova, S.; Hughes, B.G.; Poirier, M.; Kondo, M.Y.; Schulz, R. Nuclear matrix metalloproteinase-2 in the cardiomyocyte and the ischemic-reperfused heart. J. Mol. Cell. Cardiol. 2016, 94, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Mukherjee, A.; Das, N.; Swarnakar, S. Protective roles of nanomelatonin in cerebral ischemia-reperfusion of aged brain: Matrixmetalloproteinases as regulators. Exp. Gerontol. 2017, 92, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Cortes, A.L.; Gonsalez, S.R.; Rioja, L.S.; Oliveira, S.S.C.; Santos, A.L.S.; Prieto, M.C.; Melo, P.A.; Lara, L.S. Protective outcomes of low-dose doxycycline on renal function of Wistar rats subjected to acute ischemia/reperfusion injury. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Naoumov, N.V. Cyclophilin inhibition as potential therapy for liver diseases. J. Hepatol. 2014, 61, 1166–1174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernardi, P.; Di Lisa, F. The mitochondrial permeability transition pore: Molecular nature and role as a target in cardioprotection. J. Mol. Cell. Cardiol. 2015, 78, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, G.; Matoba, T.; Nakano, Y.; Nagaoka, K.; Ishikita, A.; Nakano, K.; Funamoto, D.; Sunagawa, K.; Egashira, K. Nanoparticle-Mediated Targeting of Cyclosporine A Enhances Cardioprotection Against Ischemia-Reperfusion Injury Through Inhibition of Mitochondrial Permeability Transition Pore Opening. Sci. Rep. 2016, 6, 20467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piot, C.; Croisille, P.; Staat, P.; Thibault, H.; Rioufol, G.; Mewton, N.; Elbelghiti, R.; Cung, T.T.; Bonnefoy, E.; Angoulvant, D.; et al. Effect of cyclosporine on reperfusion injury in acute myocardial infarction. N. Engl. J. Med. 2008, 359, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.A.; Perry, J.B.; Allen, M.E.; Sabbah, H.N.; Stauffer, B.L.; Shaikh, S.R.; Cleland, J.G.; Colucci, W.S.; Butler, J.; Voors, A.A.; et al. Expert consensus document: Mitochondrial function as a therapeutic target in heart failure. Nat. Rev. Cardiol. 2017, 14, 238–250. [Google Scholar] [CrossRef] [PubMed]
- Ottani, F.; Latini, R.; Staszewsky, L.; La Vecchia, L.; Locuratolo, N.; Sicuro, M.; Masson, S.; Barlera, S.; Milani, V.; Lombardi, M.; et al. Cyclosporine A in Reperfused Myocardial Infarction: The Multicenter, Controlled, Open-Label CYCLE Trial. J. Am. Coll. Cardiol. 2016, 67, 365–374. [Google Scholar] [CrossRef] [PubMed]
- Fakharnia, F.; Khodagholi, F.; Dargahi, L.; Ahmadiani, A. Prevention of Cyclophilin D-Mediated mPTP Opening Using Cyclosporine-A Alleviates the Elevation of Necroptosis, Autophagy and Apoptosis-Related Markers Following Global Cerebral Ischemia-Reperfusion. J. Mol. Neurosci. 2017, 61, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Hausenloy, D.J.; Duchen, M.R.; Yellon, D.M. Inhibiting mitochondrial permeability transition pore opening at reperfusion protects against ischaemia-reperfusion injury. Cardiovasc. Res. 2003, 60, 617–625. [Google Scholar] [CrossRef] [PubMed]
- Paeshuyse, J.; Kaul, A.; De Clercq, E.; Rosenwirth, B.; Dumont, J.M.; Scalfaro, P.; Bartenschlager, R.; Neyts, J. The non-immunosuppressive cyclosporin DEBIO-025 is a potent inhibitor of hepatitis C virus replication in vitro. Hepatology 2006, 43, 761–770. [Google Scholar] [CrossRef] [PubMed]
- Coelmont, L.; Kaptein, S.; Paeshuyse, J.; Vliegen, I.; Dumont, J.M.; Vuagniaux, G.; Neyts, J. Debio 025, a Cyclophilin Binding Molecule, Is Highly Efficient in Clearing Hepatitis C Virus (HCV) Replicon-Containing Cells When Used Alone or in Combination with Specifically Targeted Antiviral Therapy for HCV (STAT-C) Inhibitors. Antimicrob. Agents Chemother. 2009, 53, 967–976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reutenauer, J.; Dorchies, O.M.; Patthey-Vuadens, O.; Vuagniaux, G.; Ruegg, U.T. Investigation of Debio 025, a cyclophilin inhibitor, in the dystrophic mdx mouse, a model for Duchenne muscular dystrophy. Br. J. Pharmacol. 2008, 155, 574–584. [Google Scholar] [CrossRef]
- Gomez, L.; Thibault, H.; Gharib, A.; Dumont, J.-M.; Vuagniaux, G.; Scalfaro, P.; Derumeaux, G.; Ovize, M. Inhibition of mitochondrial permeability transition improves functional recovery and reduces mortality following acute myocardial infarction in mice. Am. J. Physiol. Circ. Physiol. 2007, 293, H1654–H1661. [Google Scholar] [CrossRef] [Green Version]
- Rosenwirth, B.; Billich, A.; Datema, R.; Donatsch, P.; Hammerschmid, F.; Harrison, R.; Hiestand, P.; Jaksche, H.; Mayer, P.; Peichl, P. Inhibition of human immunodeficiency virus type 1 replication by SDZ NIM 811, a nonimmunosuppressive cyclosporine analog. Antimicrob. Agents Chemother. 1994, 38, 1763–1772. [Google Scholar] [CrossRef] [Green Version]
- Argaud, L.; Gateauroesch, O.; Muntean, D.; Chalabreysse, L.; Loufouat, J.; Robert, D.; Ovize, M. Specific inhibition of the mitochondrial permeability transition prevents lethal reperfusion injury. J. Mol. Cell. Cardiol. 2005, 38, 367–374. [Google Scholar] [CrossRef]
- Readnower, R.D.; Pandya, J.D.; McEwen, M.L.; Pauly, J.R.; Springer, J.E.; Sullivan, P.G. Post-Injury Administration of the Mitochondrial Permeability Transition Pore Inhibitor, NIM811, Is Neuroprotective and Improves Cognition after Traumatic Brain Injury in Rats. J. Neurotrauma 2011, 28, 1845–1853. [Google Scholar] [CrossRef]
- Theruvath, T.P.; Zhong, Z.; Pediaditakis, P.; Ramshesh, V.K.; Currin, R.T.; Tikunov, A.; Holmuhamedov, E.; Lemasters, J.J. Minocycline and N-methyl-4-isoleucine cyclosporin (NIM811) mitigate storage/reperfusion injury after rat liver transplantation through suppression of the mitochondrial permeability transition. Hepatology 2008, 47, 236–246. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, S.; Saito, M.; Kinoshita, Y.; Ohmasa, F.; Dimitriadis, F.; Shomori, K.; Hayashi, A.; Satoh, K. Nicorandil ameliorates ischaemia-reperfusion injury in the rat kidney. Br. J. Pharmacol. 2011, 163, 272–282. [Google Scholar] [CrossRef]
- Pithadia, A.B.; Panchal, S.S.; Patel, D.J. Neuroprotective effects of potassium channel openers on cerebral ischemia-reperfusion injury in diabetic rats. Bull. Fac. Pharm. Cairo Univ. 2017, 55, 95–100. [Google Scholar] [CrossRef]
- Coetzee, W.A. Multiplicity of effectors of the cardioprotective agent, diazoxide. Pharmacol. Ther. 2013, 140, 167–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamanaka, K.; Eldeiry, M.; Aftab, M.; Ryan, T.J.; Roda, G.; Meng, X.; Weyant, M.J.; Cleveland, J.C.; Fullerton, D.A.; Reece, T.B. Pretreatment with Diazoxide Attenuates Spinal Cord Ischemia-Reperfusion Injury Through Signaling Transducer and Activator of Transcription 3 Pathway. Ann. Thorac. Surg. 2019, 107, 733–739. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, K.; Eldeiry, M.; Aftab, M.; Mares, J.; Ryan, T.J.; Meng, X.; Weyant, M.J.; Cleveland, J.C.; Fullerton, D.A.; Reece, T.B. Optimized induction of beta common receptor enhances the neuroprotective function of erythropoietin in spinal cord ischemic injury. J. Thorac. Cardiovasc. Surg. 2018, 155, 2505–2516. [Google Scholar] [CrossRef]
- Dourado, S.F.D.M.; Barbeiro, D.F.; Koike, M.K.; Barbeiro, H.V.; Da Silva, F.P.; Machado, M.C.C. Diazoxide reduces local and remote organ damage in a rat model of intestinal ischemia reperfusion. J. Surg. Res. 2018, 225, 118–124. [Google Scholar] [CrossRef]
- Lesnefsky, E.J.; Chen, Q.; Moghaddas, S.; Hassan, M.O.; Tandler, B.; Hoppel, C.L. Blockade of Electron Transport during Ischemia Protects Cardiac Mitochondria. J. Boil. Chem. 2004, 279, 47961–47967. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Hoppel, C.L.; Lesnefsky, E.J. Blockade of electron transport before cardiac ischemia with the reversible inhibitor amobarbital protects rat heart mitochondria. J. Pharmacol. Exp. Ther. 2006, 316, 200–207. [Google Scholar] [CrossRef]
- Chance, B.; Williams, G.R.; Hollunger, G. Inhibition of electron and energy transfer in mitochondria. I. Effects of Amytal, thiopental, rotenone, progesterone, and methylene glycol. J. Boil. Chem. 1963, 238, 418–431. [Google Scholar]
- Zhang, W.; Sha, Y.; Wei, K.; Wu, C.; Ding, D.; Yang, Y.; Zhu, C.; Zhang, Y.; Ding, G.; Zhang, A.; et al. Rotenone ameliorates chronic renal injury caused by acute ischemia/reperfusion. Oncotarget 2018, 9, 24199–24208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boylston, J.A.; Sun, J.; Chen, Y.; Gucek, M.; Sack, M.N.; Murphy, E. Characterization of the cardiac succinylome and its role in ischemia-reperfusion injury. J. Mol. Cell. Cardiol. 2015, 88, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Chandrika, B.B.; Yang, C.; Ou, Y.; Feng, X.K.; Muhoza, D.; Holmes, A.F.; Theus, S.; Deshmukh, S.; Haun, R.S.; Kaushal, G.P. Endoplasmic Reticulum Stress-Induced Autophagy Provides Cytoprotection from Chemical Hypoxia and Oxidant Injury and Ameliorates Renal Ischemia-Reperfusion Injury. PLoS ONE 2015, 10, e0140025. [Google Scholar] [CrossRef] [PubMed]
- Kimura, H.; Shibuya, N.; Kimura, Y. Hydrogen Sulfide Is a Signaling Molecule and a Cytoprotectant. Antioxid. Redox Signal. 2012, 17, 45–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karwi, Q.G.; Bice, J.S.; Baxter, G.F. Pre- and postconditioning the heart with hydrogen sulfide (H2S) against ischemia/reperfusion injury in vivo: A systematic review and meta-analysis. Basic Res. Cardiol. 2018, 113, 6. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Biggs, T.D.; Xian, M. Hydrogen sulfide (H2S) releasing agents: Chemistry and biological applications. Chem. Commun. 2014, 50, 11788–11805. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Whiteman, M.; Guan, Y.Y.; Neo, K.L.; Cheng, Y.; Lee, S.W.; Zhao, Y.; Baskar, R.; Tan, C.-H.; Moore, P.K. Characterization of a Novel, Water-Soluble Hydrogen Sulfide-Releasing Molecule (GYY4137): New Insights Into the Biology of Hydrogen Sulfide. Circulation 2008, 117, 2351–2360. [Google Scholar] [CrossRef] [PubMed]
- Donnarumma, E.; Ali, M.J.; Rushing, A.M.; Scarborough, A.L.; Bradley, J.M.; Organ, C.L.; Islam, K.N.; Polhemus, D.J.; Evangelista, S.; Cirino, G.; et al. Zofenopril Protects Against Myocardial Ischemia–Reperfusion Injury by Increasing Nitric Oxide and Hydrogen Sulfide Bioavailability. J. Am. Heart Assoc. 2016, 5, 7. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Li, Z.; Organ, C.L.; Park, C.-M.; Yang, C.-T.; Pacheco, A.; Wang, D.; Lefer, D.J.; Xian, M. pH-Controlled Hydrogen Sulfide Release for Myocardial Ischemia-Reperfusion Injury. J. Am. Chem. Soc. 2016, 138, 6336–6339. [Google Scholar] [CrossRef]
- Karwi, Q.G.; Bornbaum, J.; Boengler, K.; Torregrossa, R.; Whiteman, M.; Wood, M.E.; Schulz, R.; Baxter, G.F. AP39, a mitochondria-targeting hydrogen sulfide (H2S) donor, protects against myocardial reperfusion injury independently of salvage kinase signalling. Br. J. Pharmacol. 2017, 174, 287–301. [Google Scholar] [CrossRef]
- Ikeda, K.; Marutani, E.; Hirai, S.; Wood, M.E.; Whiteman, M.; Ichinose, F. Mitochondria-targeted hydrogen sulfide donor AP39 improves neurological outcomes after cardiac arrest in mice. Nitric Oxide 2015, 49, 90–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, A.; Oláh, G.; Szczesny, B.; Wood, M.E.; Whiteman, M.; Szabó, C. AP39, A Mitochondrially Targeted Hydrogen Sulfide Donor, Exerts Protective Effects in Renal Epithelial Cells Subjected to Oxidative Stress in Vitro and in Acute Renal Injury in Vivo. Shock 2016, 45, 88–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narayanan, B.A.; Narayanan, N.K.; Re, G.G.; Nixon, D.W. Differential expression of genes induced by resveratrol in LNCaP cells: P53-mediated molecular targets. Int. J. Cancer 2003, 104, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Gross, E.R. Ligand triggers of classical preconditioning and postconditioning. Cardiovasc. Res. 2006, 70, 212–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, L.; Donaldson, C.J.; Smith, G.W.; Vale, W.W. The Structures of the Mouse and Human Urocortin Genes (Ucn and UCN). Genomics 1998, 50, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Baxter, G.F.; Mocanu, M.M.; Brar, B.K.; Latchman, D.S.; Yellon, D.M. Cardioprotective effects of transforming growth factor-beta1 during early reoxygenation or reperfusion are mediated by p42/p44 MAPK. J. Cardiovasc. Pharmacol. 2001, 38, 930–939. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, R.; Valuckaite, V.; Staron, M.L.; Theccanat, T.; D’Souza, K.M.; Alverdy, J.C.; Akhter, S.A. High-molecular-weight polyethylene glycol protects cardiac myocytes from hypoxia- and reoxygenation-induced cell death and preserves ventricular function. Am. J. Physiol. Circ. Physiol. 2011, 300, H1733–H1742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasut, G.; Panisello, A.; Folch-Puy, E.; Lopez, A.; Castro-Benítez, C.; Calvo, M.; Carbonell, T.; García-Gil, A.; Adam, R.; Roselló-Catafau, J. Polyethylene glycols: An effective strategy for limiting liver ischemia reperfusion injury. World J. Gastroenterol. 2016, 22, 6501–6508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bejaoui, M.; Pantazi, E.; Folch-Puy, E.; Panisello, A.; Calvo, M.; Pasut, G.; Rimola, A.; Navasa, M.; Adam, R.; Rosello-Catafau, J. Protective Effect of Intravenous High Molecular Weight Polyethylene Glycol on Fatty Liver Preservation. Biomed. Res. Int. 2015, 2015, 794287. [Google Scholar] [CrossRef]
- Abuchowski, A.; Van Es, T.; Palczuk, N.C.; Davis, F.F. Alteration of immunological properties of bovine serum albumin by covalent attachment of polyethylene glycol. J. Boil. Chem. 1977, 252, 3578–3581. [Google Scholar]
- Lopez, A.; Panisello-Rosello, A.; Castro-Benitez, C.; Adam, R. Glycocalyx Preservation and NO Production in Fatty Livers—The Protective Role of High Molecular Polyethylene Glycol in Cold Ischemia Injury. Int. J. Mol. Sci. 2018, 19, 2375. [Google Scholar] [CrossRef] [PubMed]
- Skyschally, A.; Gres, P.; Hoffmann, S.; Haude, M.; Erbel, R.; Schulz, R.; Heusch, G. Bidirectional role of tumor necrosis factor-alpha in coronary microembolization—Progressive contractile dysfunction versus delayed protection against infarction. Circ. Res. 2007, 100, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Schulz, R.; Heusch, G. Tumor necrosis factor-alpha and its receptors 1 and 2: Yin and Yang in myocardial infarction? Circulation 2009, 119, 1355–1357. [Google Scholar] [CrossRef] [PubMed]
- Ward-Kavanagh, L.K.; Lin, W.W.; Šedý, J.R.; Ware, C.F.; Šedý, J.S. The TNF Receptor Superfamily in Co-stimulating and Co-inhibitory Responses. Immunity 2016, 44, 1005–1019. [Google Scholar] [CrossRef] [PubMed]
- Shohami, E. Dual role of tumor necrosis factor alpha in brain injury. Cytokine Growth Factor Rev. 1999, 10, 119–130. [Google Scholar] [CrossRef]
- Flaherty, M.P.; Guo, Y.; Tiwari, S.; Rezazadeh, A.; Hunt, G.; Sanganalmath, S.K.; Tang, X.-L.; Bolli, R.; Dawn, B. The role of TNF-alpha receptors p55 and p75 in acute myocardial ischemia/reperfusion injury and late preconditioning. J. Mol. Cell. Cardiol. 2008, 45, 735–741. [Google Scholar] [CrossRef] [PubMed]
- Shibata, R.; Sato, K.; Pimentel, D.R.; Takemura, Y.; Kihara, S.; Ohashi, K.; Funahashi, T.; Ouchi, N.; Walsh, K. Adiponectin protects against myocardial ischemia-reperfusion injury through AMPK- and COX-2-dependent mechanisms. Nat. Med. 2005, 11, 1096–1103. [Google Scholar] [CrossRef]
- Bauche, I.B.; El Mkadem, S.A.; Pottier, A.-M.; Senou, M.; Many, M.-C.; Rezsöhazy, R.; Penicaud, L.; Maeda, N.; Funahashi, T.; Brichard, S.M. Overexpression of Adiponectin Targeted to Adipose Tissue in Transgenic Mice: Impaired Adipocyte Differentiation. Endocrinology 2007, 148, 1539–1549. [Google Scholar] [CrossRef] [Green Version]
- Gao, C.; Liu, Y.; Yu, Q.; Yang, Q.; Li, B.; Sun, L.; Yan, W.; Cai, X.; Gao, E.; Xiong, L.; et al. TNF-alpha antagonism ameliorates myocardial ischemia-reperfusion injury in mice by upregulating adiponectin. Am. J. Physiol. Heart Circ. Physiol. 2015, 308, H1583–H1591. [Google Scholar] [CrossRef]
- Tasdemir, C.; Tasdemir, S.; Vardi, N.; Ates, B.; Parlakpinar, H.; Kati, B.; Karaaslan, M.G.; Acet, A. Protective Effect of Infliximab on Ischemia/Reperfusion-Induced Damage in Rat Kidney. Ren. Fail. 2012, 34, 1144–1149. [Google Scholar] [CrossRef]
- Nagata, Y.; Fujimoto, M.; Nakamura, K.; Isoyama, N.; Matsumura, M.; Fujikawa, K.; Uchiyama, K.; Takaki, E.; Takii, R.; Nakai, A.; et al. Anti-TNF-alpha Agent Infliximab and Splenectomy Are Protective Against Renal Ischemia-Reperfusion Injury. Transplantation 2016, 100, 1675–1682. [Google Scholar] [CrossRef] [PubMed]
- Hiroyoshi, T.; Tsuchida, M.; Uchiyama, K.; Fujikawa, K.; Komatsu, T.; Kanaoka, Y.; Matsuyama, H. Splenectomy protects the kidneys against ischemic reperfusion injury in the rat. Transpl. Immunol. 2012, 27, 8–11. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, B.; Dash, R.K.; Stowe, D.F.; Bosnjak, Z.J.; Camara, A.K.S. Isoflurane modulates cardiac mitochondrial bioenergetics by selectively attenuating respiratory complexes. Biochim. Biophys. Acta BBA Bioenerg. 2014, 1837, 354–365. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Kass, D.A. Phosphodiesterases and cardiac cGMP: Evolving roles and controversies. Trends Pharmacol. Sci. 2011, 32, 360–365. [Google Scholar] [CrossRef] [PubMed]
- Salloum, F.N.; Takenoshita, Y.; Ockaili, R.A.; Daoud, V.P.; Chou, E.; Yoshida, K.; Kukreja, R.C. Sildenafil and vardenafil but not nitroglycerin limit myocardial infarction through opening of mitochondrial K(ATP) channels when administered at reperfusion following ischemia in rabbits. J. Mol. Cell. Cardiol. 2007, 42, 453–458. [Google Scholar] [CrossRef] [PubMed]
- Ebner, B.; Ebner, A.; Reetz, A.; Böhme, S.; Schauer, A.; Strasser, R.H.; Weinbrenner, C. Pharmacological postconditioning by bolus injection of phosphodiesterase-5 inhibitors vardenafil and sildenafil. Mol. Cell. Biochem. 2013, 379, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Azevedo, I.M.; da Silva, G.V.; de Medeiros, A.S.V.C.; de Medeiros Dumaresq de Souza, A.S.V.C.; de Macêdo, R.K.A.; Medeiros, A.C. Tadalafil combined with remote ischemic preconditioning in the prevention of renal ischemia/reperfusion injury. J. Surg. Clin. Res. 2018, 9, 11–23. [Google Scholar]
- Khatib, S.Y. The Protective Effects of Phosphodiesterase 5 Inhibitors “Sildenafil & Ordonafil” on Ischemia-Reperfusion Injury. FASEB J. 2019, 33, 833. [Google Scholar]
- Lee, K.H.; Lee, S.-R.; Cho, H.; Woo, J.S.; Kang, J.H.; Jeong, Y.-M.; Cheng, X.W.; Kim, W.-S.; Kim, W. Cardioprotective effects of PKG activation by soluble GC activator, BAY 60-2770, in ischemia-reperfusion-injured rat hearts. PLoS ONE 2017, 12, e0180207. [Google Scholar] [CrossRef]
- Peralta, C.; Hotter, G.; Closa, D.; Gelpi, E.; Bulbena, O.; Roselló-Catafau, J. Protective effect of preconditioning on the injury associated to hepatic ischemia-reperfusion in the rat: Role of nitric oxide and adenosine. Hepatology 1997, 25, 934–937. [Google Scholar] [CrossRef]
- Jakobsen, Ø.; Næsheim, T.; Aas, K.N.; Sørlie, D.; Steensrud, T. Adenosine instead of supranormal potassium in cardioplegia: It is safe, efficient, and reduces the incidence of postoperative atrial fibrillation. A randomized clinical trial. J. Thorac. Cardiovasc. Surg. 2013, 145, 812–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fredholm, B.B. Adenosine, an endogenous distress signal, modulates tissue damage and repair. Cell Death Differ. 2007, 14, 1315–1323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiel, M.; Chambers, J.D.; Chouker, A.; Fischer, S.; Zourelidis, C.; Bardenheuer, H.J.; Arfors, K.E.; Peter, K. Effect of adenosine on the expression of beta(2) integrins and L-selectin of human polymorphonuclear leukocytes in vitro. J. Leukoc. Boil. 1996, 59, 671–682. [Google Scholar] [CrossRef] [PubMed]
- Fredholm, B.B.; IJzerman, A.P.; Jacobson, K.A.; Klotz, K.N.; Linden, J. International Union of Pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacol. Rev. 2001, 53, 527–552. [Google Scholar] [PubMed]
- Evora, P.R.; Roselino, C.H.; Schiaveto, P.M. Methylene blue in anaphylactic shock. Ann. Emerg. Med. 1997, 30, 240. [Google Scholar]
- Evora, P.R.; Rodrigues, A.J. Methylene blue revised. J. Thorac. Cardiovasc. Surg. 2006, 131, 250–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evora, P.R.B.; Ribeiro, P.J.D.F.; Vicente, W.V.D.A.; Dos Reis, C.L.; Rodrigues, A.J.; Menardi, A.C.; Junior, L.A.; Évora, P.M.; Bassetto, S. Methylene blue for vasoplegic syndrome treatment in heart surgery: Fifteen years of questions, answers, doubts and certainties. Braz. J. Cardiovasc. Surg. 2009, 24, 279–288. [Google Scholar] [CrossRef]
- Evora, P.R.B. Methylene Blue Is a Guanylate Cyclase Inhibitor That Does Not Interfere with Nitric Oxide Synthesis. Tex. Heart Inst. J. 2016, 43, 103. [Google Scholar] [CrossRef]
- Castro-E-Silva, O.; D’Albuquerque, L.A.C.; Silveira, M.R.G.; Zorzi, P.; Liu, J.B.; Campos, D.T.Q.; Victorino, J.P.; Jordani, M.C.; Mendes, K.D.S.; Évora, P.R.B. Evaluation of the therapeutic effect of methylene blue on the liver of rats submitted to ischemia and reperfusion. Acta Cir. Bras. 2018, 33, 1043–1051. [Google Scholar] [CrossRef] [Green Version]
- Albuquerque, F.P.; Laureano, E.; Jordani-Gomes, M.C.; Fina, C.F.; Vanni, C.; Mente, E.D.; Vollet Filho, J.D.; Bagnato, V.S.; Dalbuquerque, L.A.C.; Evora, P.R.B.; et al. Prophylactic Use of Laser Light and Methylene Blue on Ischemia and Liver Reperfusion Injury. Transplant. Proc. 2019, 51, 1549–1554. [Google Scholar] [CrossRef]
- Nantais, J.; Dumbarton, T.C.; Farah, N.; Maxan, A.; Zhou, J.; Minor, S.; Lehmann, C. Impact of methylene blue in addition to norepinephrine on the intestinal microcirculation in experimental septic shock. Clin. Hemorheol. Microcirc. 2014, 58, 97–105. [Google Scholar] [PubMed]
- Lévy, B.; Fritz, C.; Tahon, E.; Jacquot, A.; Auchet, T.; Kimmoun, A. Vasoplegia treatments: The past, the present, and the future. Crit. Care 2018, 22, 52. [Google Scholar] [CrossRef] [PubMed]
- Barbosa Evora, P.R. Blocking soluble guanylate cyclase could be the present and future of NO/cGMP inhibition for vasoplegia treatment. Crit. Care 2018, 22, 104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schinzel, A.C.; Takeuchi, O.; Huang, Z.; Fisher, J.K.; Zhou, Z.; Rubens, J.; Hetz, C.; Danial, N.N.; Moskowitz, M.A.; Korsmeyer, S.J. Cyclophilin D is a component of mitochondrial permeability transition and mediates neuronal cell death after focal cerebral ischemia. Proc. Natl. Acad. Sci. USA 2005, 102, 12005–12010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurokawa, T.; Kobayashi, H.; Nonami, T.; Harada, A.; Nakao, A.; Sugiyama, S.; Ozawa, T.; Takagi, H. Beneficial effects of cyclosporine on postischemic liver injury in rats. Transplantation 1992, 53, 308–311. [Google Scholar] [CrossRef]
- Zhang, C.-X.; Cheng, Y.; Liu, D.-Z.; Liu, M.; Cui, H.; Zhang, B.-L.; Mei, Q.-B.; Zhou, S.-Y. Mitochondria-targeted cyclosporin a delivery system to treat myocardial ischemia reperfusion injury of rats. J. Nanobiotechnol. 2019, 17, 18. [Google Scholar] [CrossRef] [PubMed]
- Ghaffari, S.; Kazemi, B.; Toluey, M.; Sepehrvand, N. The Effect of Prethrombolytic Cyclosporine-A Injection on Clinical Outcome of Acute Anterior ST-Elevation Myocardial Infarction. Cardiovasc. Ther. 2013, 31, E34–E39. [Google Scholar] [CrossRef]
- Haddad, E.; McAlister, V.; Renouf, E.; Malthaner, R.; Kjaer, M.S.; Gluud, L.L. Cyclosporin versus tacrolimus for liver transplanted patients. Cochrane Database Syst. Rev. 2006. [Google Scholar] [CrossRef]
- Webster, A.C.; Taylor, R.R.; Chapman, J.R.; Craig, J.C. Tacrolimus versus cyclosporin as primary immunosuppression for kidney transplant recipients. Cochrane Database Syst. Rev. 2005. [Google Scholar] [CrossRef]
- Penninga, L.; Penninga, E.I.; Møller, C.H.; Iversen, M.; Steinbrüchel, D.A.; Gluud, C. Tacrolimus versus cyclosporin as primary immunosuppression for lung transplant recipients. Cochrane Database Syst. Rev. 2013. [Google Scholar] [CrossRef]
- Parodi-Rullan, R.M.; Soto-Prado, J.; Diaz-Cordero, S.I.; Vega-Lugo, J.; Chapa-Dubocq, X.; Javadov, S. Sanglifehrin A Has No Protective Effects in Non-reperfused Myocardial Infarction: The Role of Mitochondria. FASEB J. 2018, 32, 618. [Google Scholar]
- Sloan, R.C.; Moukdar, F.; Frasier, C.R.; Patel, H.D.; Bostian, P.A.; Lust, R.M.; Brown, D.A. Mitochondrial permeability transition in the diabetic heart: Contributions of thiol redox state and mitochondrial calcium to augmented reperfusion injury. J. Mol. Cell. Cardiol. 2012, 52, 1009–1018. [Google Scholar] [CrossRef] [PubMed]
- Bibli, S.I.; Papapetropoulos, A.; Iliodromitis, E.K.; Daiber, A.; Randriamboavonjy, V.; Steven, S.; Brouckaert, P.; Chatzianastasiou, A.; Kypreos, K.E.; Hausenloy, D.J.; et al. Nitroglycerine limits infarct size through S-nitrosation of cyclophilin D: A novel mechanism for an old drug. Cardiovasc. Res. 2019, 115, 625–636. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Sileikyte, J.; Neuenswander, B.; Hedrick, M.P.; Chung, T.D.Y.; Aube, J.; Schoenen, F.J.; Forte, M.A.; Bernardi, P. N-Phenylbenzamides as Potent Inhibitors of the Mitochondrial Permeability Transition Pore. ChemMedChem 2016, 11, 283–288. [Google Scholar] [CrossRef] [PubMed]
- Fancelli, D.; Abate, A.; Amici, R.; Bernardi, P.; Ballarini, M.; Cappa, A.; Carenzi, G.; Colombo, A.; Contursi, C.; Di Lisa, F.; et al. Cinnamic Anilides as New Mitochondrial Permeability Transition Pore Inhibitors Endowed with Ischemia-Reperfusion Injury Protective Effect in Vivo. J. Med. Chem. 2014, 57, 5333–5347. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.J.; Fancelli, D.; Wong, M.; Niedzwiecki, M.; Ballarini, M.; Plyte, S.; Chang, Q. GNX-4728, a novel small molecule drug inhibitor of mitochondrial permeability transition, is therapeutic in a mouse model of amyotrophic lateral sclerosis. Front. Cell. Neurosci. 2014, 8, 433. [Google Scholar] [CrossRef] [PubMed]
- Richardson, A.P.; Halestrap, A.P. Quantification of active mitochondrial permeability transition pores using GNX-4975 inhibitor titrations provides insights into molecular identity. Biochem. J. 2016, 473, 1129–1140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, S.; Šileikytė, J.; Schiavone, M.; Neuenswander, B.; Argenton, F.; Aube, J.; Hedrick, M.P.; Chung, T.D.Y.; Forte, M.A.; Bernardi, P.; et al. Discovery, Synthesis, and Optimization of Diarylisoxazole-3-carboxamides as Potent Inhibitors of the Mitochondrial Permeability Transition Pore. ChemMedChem 2015, 10, 1655–1671. [Google Scholar] [CrossRef] [PubMed]
- Schaller, S.; Paradis, S.; Ngoh, G.A.; Assaly, R.; Buisson, B.; Drouot, C.; Ostuni, M.A.; Lacapere, J.-J.; Bassissi, F.; Bordet, T.; et al. TRO40303, a New Cardioprotective Compound, Inhibits Mitochondrial Permeability Transition. J. Pharmacol. Exp. Ther. 2010, 333, 696–706. [Google Scholar] [CrossRef] [PubMed]
- Atar, D.; Arheden, H.; Berdeaux, A.; Bonnet, J.L.; Carlsson, M.; Clemmensen, P.; Cuvier, V.; Danchin, N.; Dubois-Rande, J.L.; Engblom, H.; et al. Effect of intravenous TRO40303 as an adjunct to primary percutaneous coronary intervention for acute ST-elevation myocardial infarction: MITOCARE study results. Eur. Heart J. 2015, 36, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Butt, N.; Bache-Mathiesen, L.K.; Nordrehaug, J.E.; Tuseth, V.; Munk, P.S.; Bonarjee, V.; Hall, T.S.; Jensen, S.E.; Halvorsen, S.; Firat, H.; et al. Administration of the Mitochondrial Permeability Transition Pore Inhibitor, TRO40303, prior to Primary Percutaneous Coronary Intervention, Does Not Affect the Levels of Pro-Inflammatory Cytokines or Acute-Phase Proteins. Cardiology 2017, 138, 122–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miao, J.X.; Huang, Z.J.; Liu, S.; Li, X.Y.; Jia, P.Y.; Guo, Y.X.; Wu, N.; Jia, D.L. Hydroxytyrosol protects against myocardial ischemia reperfusion injury by inhibiting mitochondrial permeability transition pore opening. Exp. Ther. Med. 2019, 17, 671–678. [Google Scholar] [CrossRef] [PubMed]
- Wojtovich, A.P.; Brookes, P.S. The endogenous mitochondrial complex II inhibitor malonate regulates mitochondrial ATP-sensitive potassium channels: Implications for ischemic preconditioning. Biochim. Biophys. Acta BBA Bioenergy 2008, 1777, 882–889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wojtovich, A.P.; Brookes, P.S. The complex II inhibitor atpenin A5 protects against cardiac ischemia-reperfusion injury via activation of mitochondrial KATP channels. Basic Res. Cardiol. 2009, 104, 121–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, B.; Zhu, X.; Chen, C.L.; Hu, K.; Swartz, H.M.; Chen, Y.R.; He, G. Opening of the mitoKATP channel and decoupling of mitochondrial complex II and III contribute to the suppression of myocardial reperfusion hyperoxygenation. Mol. Cell. Biochem. 2010, 337, 25–38. [Google Scholar] [CrossRef] [PubMed]
- St-Pierre, J.; Buckingham, J.A.; Roebuck, S.J.; Brand, M.D. Topology of Superoxide Production from Different Sites in the Mitochondrial Electron Transport Chain. J. Boil. Chem. 2002, 277, 44784–44790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orr, A.L.; Vargas, L.; Turk, C.N.; Baaten, J.E.; Matzen, J.T.; Dardov, V.J.; Attle, S.J.; Li, J.; Quackenbush, D.C.; Goncalves, R.L.S.; et al. Suppressors of superoxide production from mitochondrial complex III. Nat. Methods 2015, 11, 834–836. [Google Scholar] [CrossRef] [Green Version]
- Scialo, F.; Sriram, A.; Fernandez-Ayala, D.; Gubina, N.; Lohmus, M.; Nelson, G.; Logan, A.; Cooper, H.M.; Navas, P.; Enriquez, J.A.; et al. Mitochondrial ROS Produced via Reverse Electron Transport Extend Animal Lifespan. Cell Metab. 2016, 23, 725–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmeisser, S.; Priebe, S.; Groth, M.; Monajembashi, S.; Hemmerich, P.; Guthke, R.; Platzer, M.; Ristow, M. Neuronal ROS signaling rather than AMPK/sirtuin-mediated energy sensing links dietary restriction to lifespan extension. Mol. Metab. 2013, 2, 92–102. [Google Scholar] [CrossRef] [PubMed]
- De Haes, W.; Frooninckx, L.; Van Assche, R.; Smolders, A.; Depuydt, G.; Billen, J.; Braeckman, B.P.; Schoofs, L.; Temmerman, L. Metformin promotes lifespan through mitohormesis via the peroxiredoxin PRDX-2. Proc. Natl. Acad. Sci. USA 2014, 111, E2501–E2509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dröse, S.; Hanley, P.J.; Brandt, U. Ambivalent effects of diazoxide on mitochondrial ROS production at respiratory chain complexes I and III. Biochim. Biophys. Acta BBA Gen. Subj. 2009, 1790, 558–565. [Google Scholar] [CrossRef] [PubMed]
- Scialò, F.; Fernández-Ayala, D.J.; Sanz, A. Role of Mitochondrial Reverse Electron Transport in ROS Signaling: Potential Roles in Health and Disease. Front. Physiol. 2017, 8, 428. [Google Scholar] [CrossRef] [PubMed]
- Fato, R.; Bergamini, C.; Bortolus, M.; Maniero, A.L.; Leoni, S.; Ohnishi, T.; Lenaz, G. Differential effects of mitochondrial Complex I inhibitors on production of reactive oxygen species. Biochim. Biophys. Acta 2009, 1787, 384–392. [Google Scholar] [CrossRef] [PubMed]
- Korge, P.; Calmettes, G.; Weiss, J.N. Reactive oxygen species production in cardiac mitochondria after complex I inhibition: Modulation by substrate-dependent regulation of the NADH/NAD(+) ratio. Free Radic. Biol. Med. 2016, 96, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Detaille, D.; Pasdois, P.; Sémont, A.; Dos Santos, P.; Diolez, P. An old medicine as a new drug to prevent mitochondrial complex I from producing oxygen radicals. PLoS ONE 2019, 14, e0216385. [Google Scholar] [CrossRef] [PubMed]
- Lambert, A.J.; Brand, M.D. Inhibitors of the Quinone-binding Site Allow Rapid Superoxide Production from Mitochondrial NADH:Ubiquinone Oxidoreductase (Complex I). J. Boil. Chem. 2004, 279, 39414–39420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambert, A.J.; Brand, M.D. Superoxide production by NADH:ubiquinone oxidoreductase (complex I) depends on the pH gradient across the mitochondrial inner membrane. Biochem. J. 2004, 382, 511–517. [Google Scholar] [CrossRef]
- Hirst, J.; King, M.S.; Pryde, K.R. The production of reactive oxygen species by complex I. Biochem. Soc. Trans. 2008, 36, 976–980. [Google Scholar] [CrossRef] [PubMed]
- Pryde, K.R.; Hirst, J. Superoxide Is Produced by the Reduced Flavin in Mitochondrial Complex I a Single, Unified Mechanism That Applies during both Forward and Reverse Electron Transfer. J. Biol. Chem. 2011, 286, 18056–18065. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, H.; Takagi, T.; Uchiyama, K.; Higashihara, H.; Katada, K.; Isozaki, Y.; Naito, Y.; Yoshida, N.; Yoshikawa, T. Rotenone, a mitochondrial electron transport inhibitor, ameliorates ischemia-reperfusion-induced intestinal mucosal damage in rats. Redox Rep. 2004, 9, 313–316. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Zhang, Y.; Zhao, D.; Ding, G.; Huang, S.; Zhang, A.; Jia, Z. Rotenone Remarkably Attenuates Oxidative Stress, Inflammation, and Fibrosis in Chronic Obstructive Uropathy. Mediat. Inflamm. 2014, 2014, 670106. [Google Scholar] [CrossRef] [PubMed]
- Tanaka-Esposito, C.; Chen, Q.; Lesnefsky, E.J. Blockade of electron transport before ischemia protects mitochondria and decreases myocardial injury during reperfusion in aged rat hearts. Transl. Res. 2012, 160, 207–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nadtochiy, S.M.; Burwell, L.S.; Ingraham, C.A.; Spencer, C.M.; Friedman, A.E.; Pinkert, C.A.; Brookes, P.S. In vivo cardioprotection by S-nitroso-2-mercaptopropionyl glycine. J. Mol. Cell. Cardiol. 2009, 46, 960–968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chouchani, E.T.; Methner, C.; Nadtochiy, S.M.; Logan, A.; Pell, V.R.; Ding, S.; James, A.M.; Cocheme, H.M.; Reinhold, J.; Lilley, K.S.; et al. Cardioprotection by S-nitrosation of a cysteine switch on mitochondrial complex I. Nat. Med. 2013, 19, 753–759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohsin, A.A.; Chen, Q.; Quan, N.; Rousselle, T.; Maceyka, M.W.; Samidurai, A.; Thompson, J.; Hu, Y.; Li, J.; Lesnefsky, E.J. Mitochondrial Complex I Inhibition by Metformin Limits Reperfusion Injury. J. Pharmacol. Exp. Ther. 2019, 369, 282–290. [Google Scholar] [CrossRef] [PubMed]
- Gadicherla, A.K.; Stowe, D.F.; Antholine, W.E.; Yang, M.; Camara, A.K. Damage to mitochondrial complex I during cardiac ischemia reperfusion injury is reduced indirectly by anti-anginal drug ranolazine. Biochim. Biophys. Acta 2012, 1817, 419–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brand, M.D.; Goncalves, R.L.; Orr, A.L.; Vargas, L.; Gerencser, A.A.; Borch Jensen, M.; Wang, Y.T.; Melov, S.; Turk, C.N.; Matzen, J.T.; et al. Suppressors of Superoxide-H2O2 Production at Site IQ of Mitochondrial Complex I Protect against Stem Cell Hyperplasia and Ischemia-Reperfusion Injury. Cell Metab. 2016, 24, 582–592. [Google Scholar] [CrossRef]
- Dröse, S.; Bleier, L.; Brandt, U. A Common Mechanism Links Differently Acting Complex II Inhibitors to Cardioprotection: Modulation of Mitochondrial Reactive Oxygen Species Production. Mol. Pharmacol. 2011, 79, 814–822. [Google Scholar] [CrossRef] [Green Version]
- Gutman, M.; Silamn, N. The steady state activity of succinate dehydrogenase in the presence of opposing effectors. 1. The effect of L malate and CoQH2 on the enzymic activity. Mol. Cell. Biochem. 1975, 7, 51–58. [Google Scholar] [CrossRef]
- Zeylemaker, W.; Slater, E. The inhibition of succinate dehydrogenase by oxaloacetate. Biochim. Biophys. Acta BBA Enzym. 1967, 132, 210–212. [Google Scholar] [CrossRef]
- Valls-Lacalle, L.; Barba, I.; Miro-Casas, E.; Alburquerque-Bejar, J.J.; Ruiz-Meana, M.; Fuertes-Agudo, M.; Rodriguez-Sinovas, A.; Garcia-Dorado, D. Succinate dehydrogenase inhibition with malonate during reperfusion reduces infarct size by preventing mitochondrial permeability transition. Cardiovasc. Res. 2016, 109, 374–384. [Google Scholar] [CrossRef] [PubMed]
- Valls-Lacalle, L.; Barba, I.; Miró-Casas, E.; Ruiz-Meana, M.; Rodríguez-Sinovas, A.; Garcia-Dorado, D. Selective Inhibition of Succinate Dehydrogenase in Reperfused Myocardium with Intracoronary Malonate Reduces Infarct Size. Sci. Rep. 2018, 8, 2442. [Google Scholar] [CrossRef] [PubMed]
- Merlen, G.; Raymond, V.A.; Cassim, S.; Lapierre, P.; Bilodeau, M. Oxaloacetate Protects Rat Liver from Experimental Warm Ischemia/Reperfusion Injury by Improving Cellular Energy Metabolism. Liver Transplant. 2019, 25, 627–639. [Google Scholar] [CrossRef] [PubMed]
- Marosi, M.; Fuzik, J.; Nagy, D.; Rákos, G.; Kis, Z.; Vécsei, L.; Toldi, J.; Ruban-Matuzani, A.; Teichberg, V.I.; Farkas, T. Oxaloacetate restores the long-term potentiation impaired in rat hippocampus CA1 region by 2-vessel occlusion. Eur. J. Pharmacol. 2009, 604, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.L.; Shestov, A.A.; Worth, A.J.; Nath, K.; Nelson, D.S.; Leeper, D.B.; Glickson, J.D.; Blair, I.A. Inhibition of Mitochondrial Complex II by the Anticancer Agent Lonidamine. J. Biol. Chem. 2016, 291, 42–57. [Google Scholar] [CrossRef] [Green Version]
- Rudolph, V.; Rudolph, T.K.; Schopfer, F.J.; Bonacci, G.; Woodcock, S.R.; Cole, M.P.; Baker, P.R.; Ramani, R.; Freeman, B.A. Endogenous generation and protective effects of nitro-fatty acids in a murine model of focal cardiac ischaemia and reperfusion. Cardiovasc. Res. 2010, 85, 155–166. [Google Scholar] [CrossRef]
- Koenitzer, J.R.; Bonacci, G.; Woodcock, S.R.; Chen, C.S.; Cantu-Medellin, N.; Kelley, E.E.; Schopfer, F.J. Fatty acid nitroalkenes induce resistance to ischemic cardiac injury by modulating mitochondrial respiration at complex II. Redox Biol. 2016, 8, 1–10. [Google Scholar] [CrossRef]
- Khoo, N.K.H.; Li, L.; Salvatore, S.R.; Schopfer, F.J.; Freeman, B.A. Electrophilic fatty acid nitroalkenes regulate Nrf2 and NF-kappaB signaling: A medicinal chemistry investigation of structure-function relationships. Sci. Rep. 2018, 8, 2295. [Google Scholar] [CrossRef]
- Nadtochiy, S.M.; Zhu, Q.M.; Urciuoli, W.; Rafikov, R.; Black, S.M.; Brookes, P.S. Nitroalkenes confer acute cardioprotection via adenine nucleotide translocase 1. J. Biol. Chem. 2012, 287, 3573–3580. [Google Scholar] [CrossRef]
- Nadtochiy, S.M.; Baker, P.R.; Freeman, B.A.; Brookes, P.S. Mitochondrial nitroalkene formation and mild uncoupling in ischaemic preconditioning: Implications for cardioprotection. Cardiovasc. Res. 2009, 82, 333–340. [Google Scholar] [CrossRef]
- Nadtochiy, S.M.; Madukwe, J.; Hagen, F.K.; Brookes, P.S. Mitochondrially targeted nitro-linoleate: A new tool for the study of cardioprotection. Br. J. Pharmacol. 2014, 171, 2091–2098. [Google Scholar] [CrossRef] [PubMed]
- Hoek, T.L.V.; Becker, L.B.; Shao, Z.; Li, C.; Schumacker, P.T. Reactive Oxygen Species Released from Mitochondria during Brief Hypoxia Induce Preconditioning in Cardiomyocytes. J. Boil. Chem. 1998, 273, 18092–18098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oldenburg, O.; Cohen, M.V.; Downey, J.M. Mitochondrial K(ATP) channels in preconditioning. J. Mol. Cell. Cardiol. 2003, 35, 569–575. [Google Scholar] [CrossRef]
- Bleier, L.; Drose, S. Superoxide generation by complex III: From mechanistic rationales to functional consequences. Biochim. Biophys. Acta 2013, 1827, 1320–1331. [Google Scholar] [CrossRef] [Green Version]
- Halestrap, A.P.; Clarke, S.J.; Khaliulin, I. The role of mitochondria in protection of the heart by preconditioning. Biochim. Biophys. Acta BBA Bioenergy 2007, 1767, 1007–1031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guzy, R.D.; Mack, M.M.; Schumacker, P.T. Mitochondrial Complex III is Required for Hypoxia-Induced ROS Production and Gene Transcription in Yeast. Antioxid. Redox Signal. 2007, 9, 1317–1328. [Google Scholar] [CrossRef]
- Klimova, T.; Chandel, N.S. Mitochondrial complex III regulates hypoxic activation of HIF. Cell Death Differ. 2008, 15, 660–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandel, N.S.; McClintock, D.S.; Feliciano, C.E.; Wood, T.M.; Melendez, J.A.; Rodriguez, A.M.; Schumacker, P.T. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1 alpha during hypoxia—A mechanism of O-2 sensing. J. Biol. Chem. 2000, 275, 25130–25138. [Google Scholar] [CrossRef]
- Stepanova, A.; Konrad, C.; Manfredi, G.; Springett, R.; Ten, V.; Galkin, A. The dependence of brain mitochondria reactive oxygen species production on oxygen level is linear, except when inhibited by antimycin A. J. Neurochem. 2019, 148, 731–745. [Google Scholar] [CrossRef]
- Baur, J.A.; Chen, D.; Chini, E.N.; Chua, K.; Cohen, H.Y.; de Cabo, R.; Deng, C.; Dimmeler, S.; Gius, D.; Guarente, L.P.; et al. Dietary restriction: Standing up for sirtuins. Science 2010, 329, 1012–1013. [Google Scholar] [CrossRef]
- Braidy, N.; Guillemin, G.J.; Mansour, H.; Chan-Ling, T.; Poljak, A.; Grant, R. Age related changes in NAD+ metabolism oxidative stress and Sirt1 activity in wistar rats. PLoS ONE 2011, 6, e19194. [Google Scholar] [CrossRef] [PubMed]
- Porter, G.A.; Urciuoli, W.R.; Brookes, P.S.; Nadtochiy, S.M. SIRT3 deficiency exacerbates ischemia-reperfusion injury: Implication for aged hearts. Am. J. Physiol. Circ. Physiol. 2014, 306, H1602–H1609. [Google Scholar] [CrossRef] [PubMed]
- Banks, A.S.; Kon, N.; Knight, C.; Matsumoto, M.; Gutierrez-Juarez, R.; Rossetti, L.; Gu, W.; Accili, D. SirT1 gain of function increases energy efficiency and prevents diabetes in mice. Cell Metab. 2008, 8, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-H.; Li, S.-A.; Huang, C.-H.; Su, H.-H.; Chen, Y.-H.; Chang, J.T.; Huang, S.-S. Sirt1 Activation by Post-ischemic Treatment with Lumbrokinase Protects Against Myocardial Ischemia-Reperfusion Injury. Front. Pharmacol. 2018, 9, 636. [Google Scholar] [CrossRef]
- Li, R.; Ma, X.; Song, Y.; Zhang, Y.; Xiong, W.; Li, L.; Zhou, L. Anti-colorectal cancer targets of resveratrol and biological molecular mechanism: Analyses of network pharmacology, human and experimental data. J. Cell. Biochem. 2019, 120, 11265–11273. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Tang, C.; Tai, L.W.; Yao, W.; Guo, P.; Hong, J.; Yang, X.; Li, X.; Jin, Z.; Ke, J.; et al. Flurbiprofen axetil attenuates cerebral ischemia/reperfusion injury by reducing inflammation in a rat model of transient global cerebral ischemia/reperfusion. Biosci. Rep. 2018, 38, 4. [Google Scholar] [CrossRef] [PubMed]
- Weng, X.-F.; Li, S.-T.; Song, Q.; Zhu, Q.; Song, D.-D.; Qin, Z.-H.; Xie, Y. Protective Effect of Nicotinamide Adenine Dinucleotide Phosphate on Renal Ischemia-Reperfusion Injury. Kidney Blood Press. Res. 2018, 43, 651–663. [Google Scholar] [CrossRef] [PubMed]
- Weisbrot-Lefkowitz, M.; Reuhl, K.; Perry, B.; Chan, P.H.; Inouye, M.; Mirochnitchenko, O. Overexpression of human glutathione peroxidase protects transgenic mice against focal cerebral ischemia/reperfusion damage. Mol. Brain Res. 1998, 53, 333–338. [Google Scholar] [CrossRef]
- Beadle, R.M.; Williams, L.K.; Kuehl, M.; Bowater, S.; Abozguia, K.; Leyva, F.; Yousef, Z.; Wagenmakers, A.J.; Thies, F.; Horowitz, J.; et al. Improvement in Cardiac Energetics by Perhexiline in Heart Failure due to Dilated Cardiomyopathy. JACC Heart Fail. 2015, 3, 202–211. [Google Scholar] [CrossRef]
- Nadtochiy, S.M.; Wang, Y.T.; Nehrke, K.; Munger, J.; Brookes, P.S. Cardioprotection by nicotinamide mononucleotide (NMN): Involvement of glycolysis and acidic pH. J. Mol. Cell. Cardiol. 2018, 121, 155–162. [Google Scholar] [CrossRef]
- Mc Rosano, G.; Vitale, C. Metabolic Modulation of Cardiac Metabolism in Heart Failure. Card. Fail. Rev. 2018, 4, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Chong, C.-R.; Ong, G.J.; Horowitz, J.D. Emerging drugs for the treatment of angina pectoris. Expert Opin. Emerg. Drugs 2016, 21, 1–12. [Google Scholar] [CrossRef]
- Bonora, M.; Wieckowski, M.R.; Sinclair, D.A.; Kroemer, G.; Pinton, P.; Galluzzi, L. Targeting mitochondria for cardiovascular disorders: Therapeutic potential and obstacles. Nat. Rev. Cardiol. 2019, 16, 33–55. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Patra, A.; Sadowska, G.B.; Stonestreet, B.S. Ischemic-Reperfusion Injury Increases Matrix Metalloproteinases and Tissue Metalloproteinase Inhibitors in Fetal Sheep Brain. Dev. Neurosci. 2018, 40, 234–245. [Google Scholar] [CrossRef]
- Duarte, S.; Baber, J.; Fujii, T.; Coito, A.J. Matrix metalloproteinases in liver injury, repair and fibrosis. Matrix Boil. 2015, 44, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Cerofolini, L.; Fragai, M.; Luchinat, C. Mechanism and Inhibition of Matrix Metalloproteinases. Curr. Med. Chem. 2018, 26, 2609–2633. [Google Scholar] [CrossRef] [PubMed]
- Bao, D.-S.; Wu, Y.-K.; Wang, G.-Y.; Yang, S.-J.; Liang, G.-B.; Xie, Z.-H.; Rong, S.; Fu, S.-J. Hyperbaric Oxygenation Protects Against Ischemia-Reperfusion Injury in Transplanted Rat Kidneys by Triggering Autophagy and Inhibiting Inflammatory Response. Ann. Transplant. 2017, 22, 75–82. [Google Scholar] [CrossRef]
- Huchim, O.; Rivas-Sosa, F.; Rivera-Canul, N.; Mendez-Dominguez, N. 350 años de la medicina hiperbárica: Aspectos históricos, fisiopatogénicos y terapéuticos. Gac. Med. Mex. 2017, 153, 938–945. [Google Scholar] [CrossRef]
- Fife, C.E.; Eckert, K.A.; Carter, M.J. An Update on the Appropriate Role for Hyperbaric Oxygen: Indications and Evidence. Plast. Reconstr. Surg. 2016, 138, 107S–116S. [Google Scholar] [CrossRef]
- Tezcan, O.; Caliskan, A.; Demirtas, S.; Yavuz, C.; Kuyumcu, M.; Nergiz, Y.; Guzel, A.; Karahan, O.; Ari, S.; Soker, S.; et al. Effects of Hyperbaric Oxygen Treatment on Renal System. Iran. J. Kidney Dis. 2017, 11, 18–22. [Google Scholar]
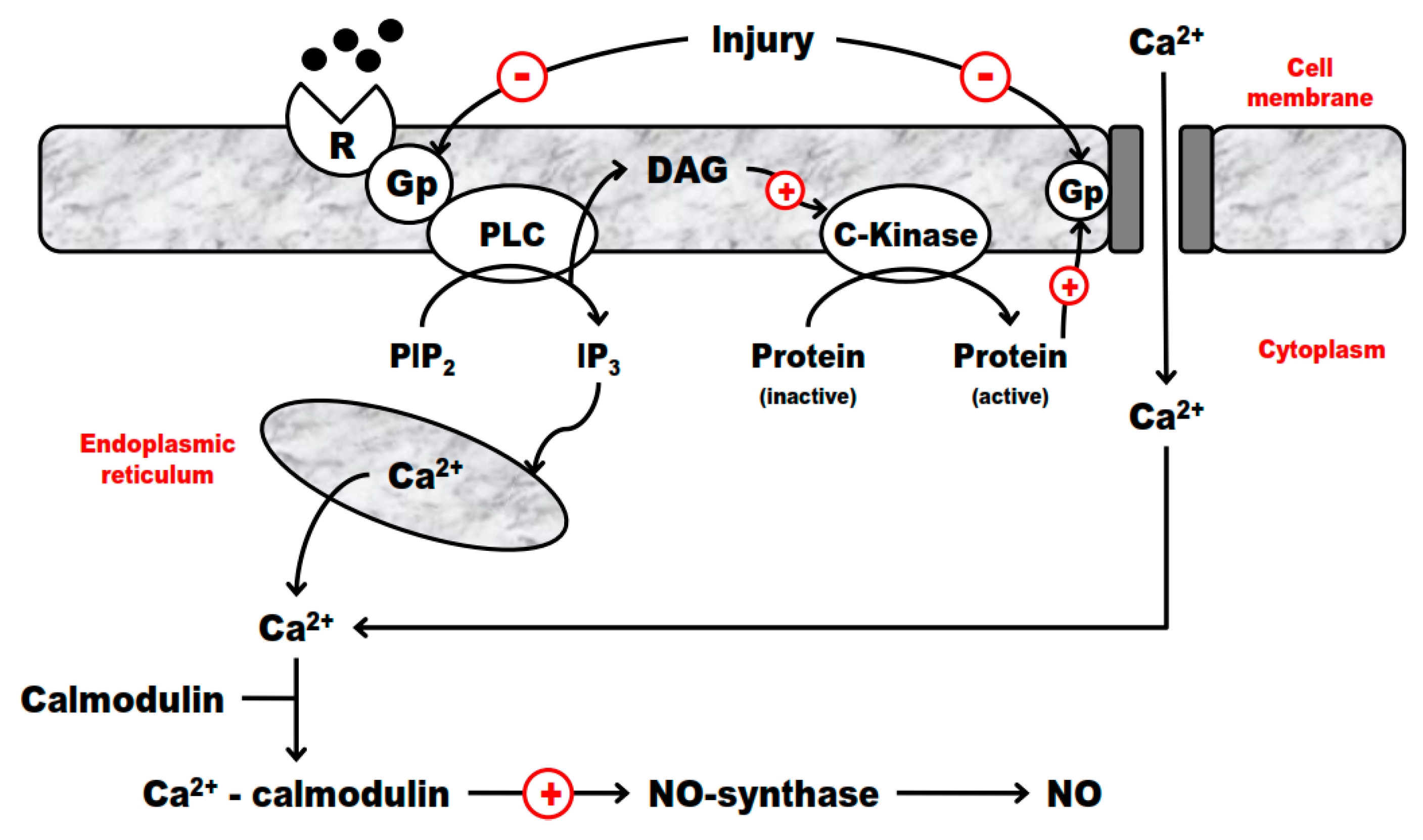
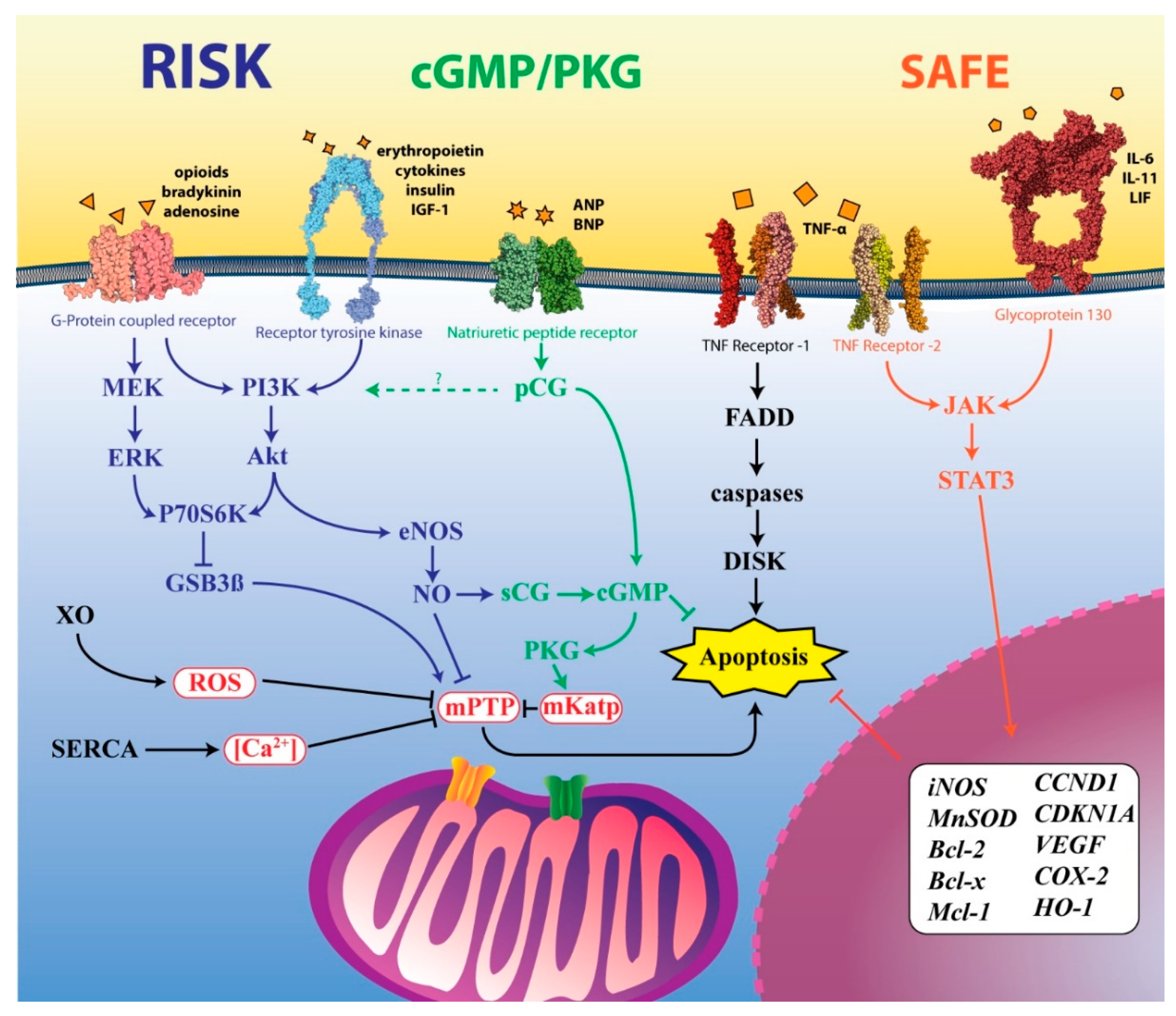

{kind=link}
{kind=link}
{kind=link}
| Ultrastructural Alteration | Reversible Injury | Irreversible Injury |
|---|---|---|
| Plasma membrane | formation of bubbles, reduction, and distortion of microvilli, appearance of myelin figures and loosening of intracellular bonds | evident discontinuity of plasma membranes and organelles |
| Mitochondrial | edema, rarefaction, and appearance of amorphous densities rich in phospholipids | dilatation of mitochondria with formation of amorphous densities, intracytoplasmic myelin figures and cotton-like material (denatured proteins) |
| Nuclear | disintegration of granular and fibrillar elements | karyolysis (DNase activity), pyknosis (shrinking of the nucleus and increased basophilia), karyorrhexis (nuclear fragmentation) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soares, R.O.S.; Losada, D.M.; Jordani, M.C.; Évora, P.; Castro-e-Silva, O. Ischemia/Reperfusion Injury Revisited: An Overview of the Latest Pharmacological Strategies. Int. J. Mol. Sci. 2019, 20, 5034. https://doi.org/10.3390/ijms20205034
Soares ROS, Losada DM, Jordani MC, Évora P, Castro-e-Silva O. Ischemia/Reperfusion Injury Revisited: An Overview of the Latest Pharmacological Strategies. International Journal of Molecular Sciences. 2019; 20(20):5034. https://doi.org/10.3390/ijms20205034
Chicago/Turabian StyleSoares, Ricardo O. S., Daniele M. Losada, Maria C. Jordani, Paulo Évora, and Orlando Castro-e-Silva. 2019. "Ischemia/Reperfusion Injury Revisited: An Overview of the Latest Pharmacological Strategies" International Journal of Molecular Sciences 20, no. 20: 5034. https://doi.org/10.3390/ijms20205034