Methotrexate an Old Drug with New Tricks

 ,
,
Abstract
1. Introduction
2. History
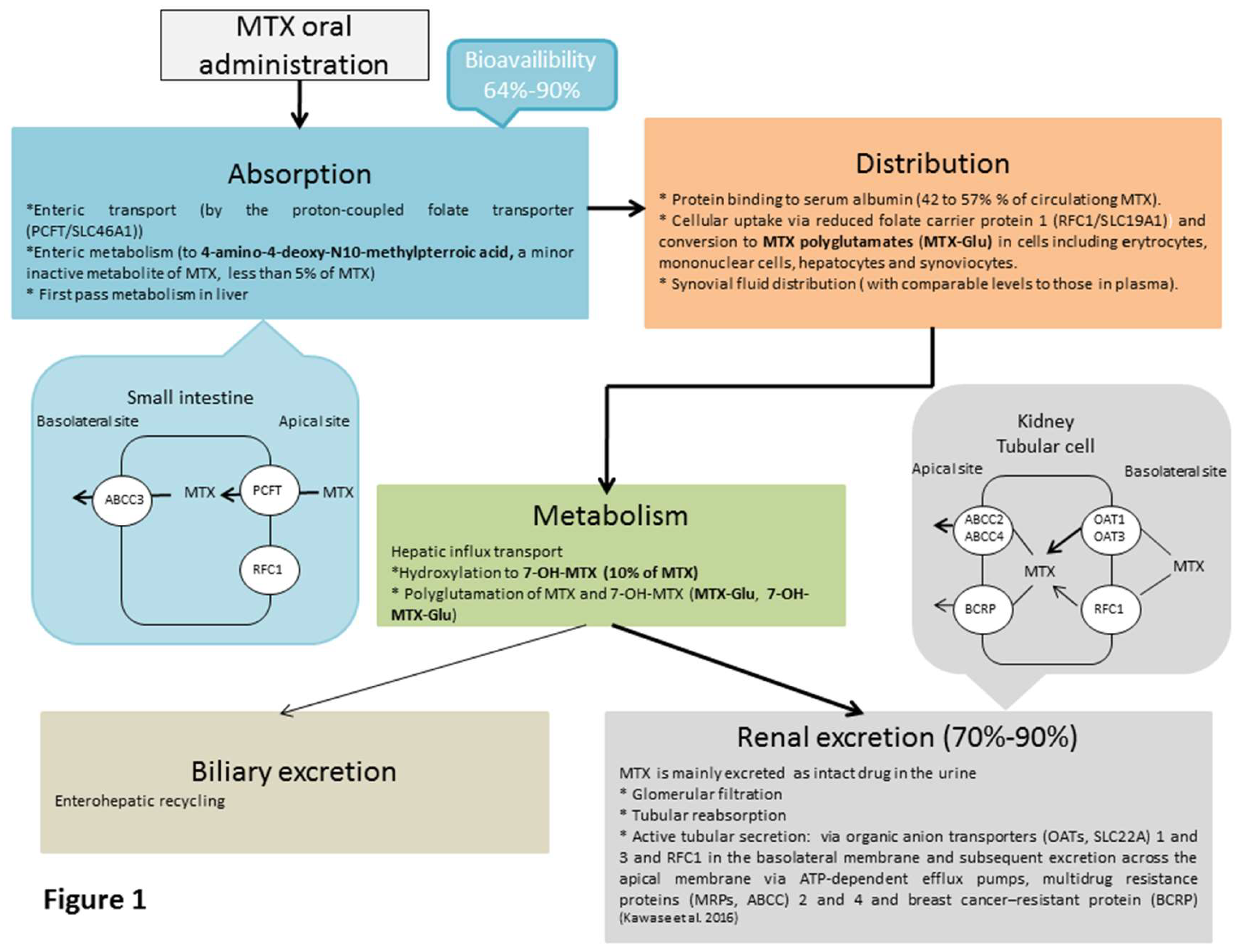
3. Pharmacokinetics of MTX
4. MTX Therapeutic Mechanisms of Action in Inflammatory Settings
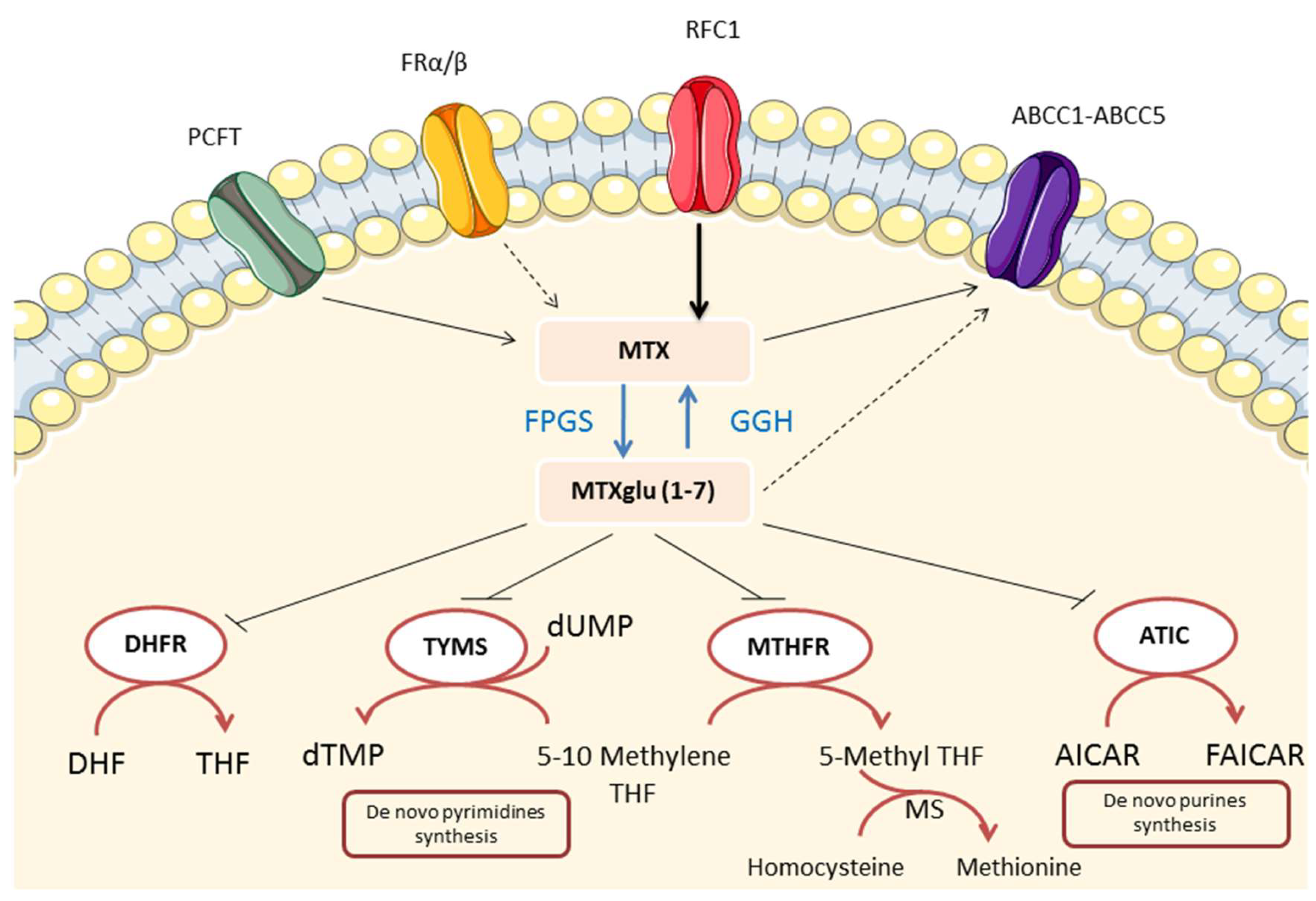
4.1. Folate Antagonism
4.2. Effects on Extracellular Adenosine Accumulation
4.3. Effects on Polyamine Production
4.4. Generation of Reactive Oxygen Species
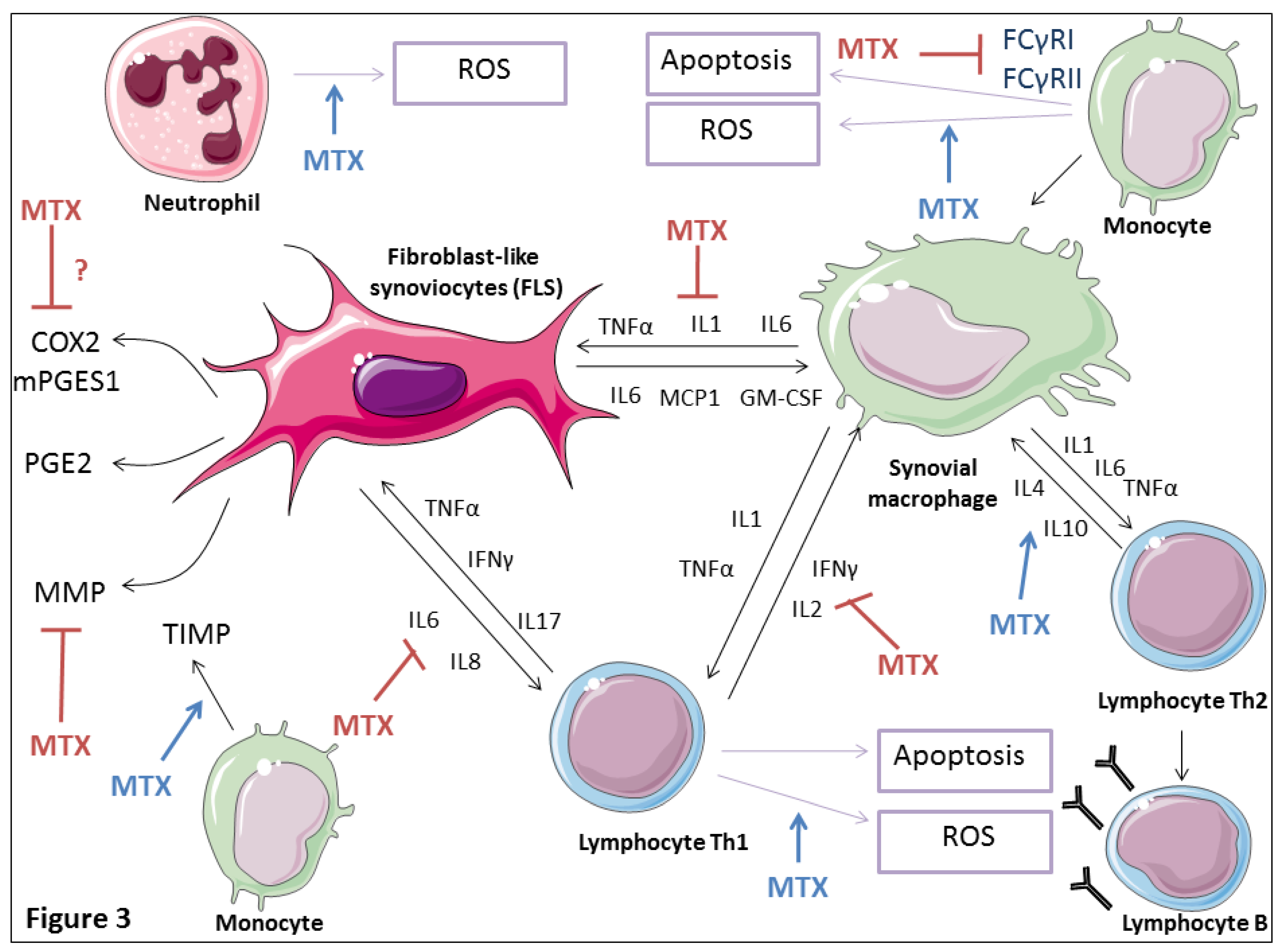
4.5. Effects on Cytokine Production
4.6. Effects on Matrix Metalloproteinases
4.7. Effects on Prostaglandin Production
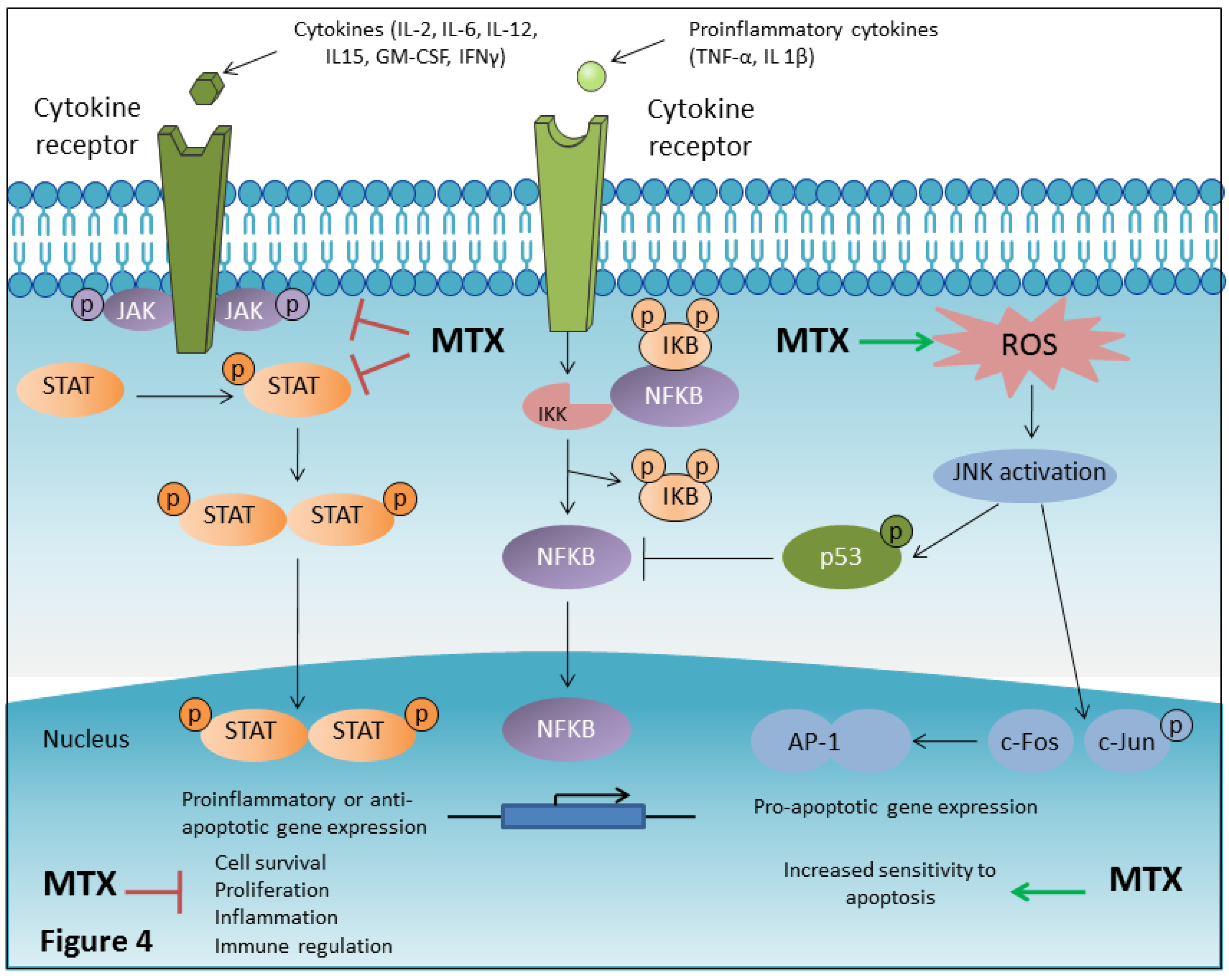
4.8. MTX Inhibits NF-κB Activity
4.9. MTX is a JAK/STAT Pathway Inhibitor
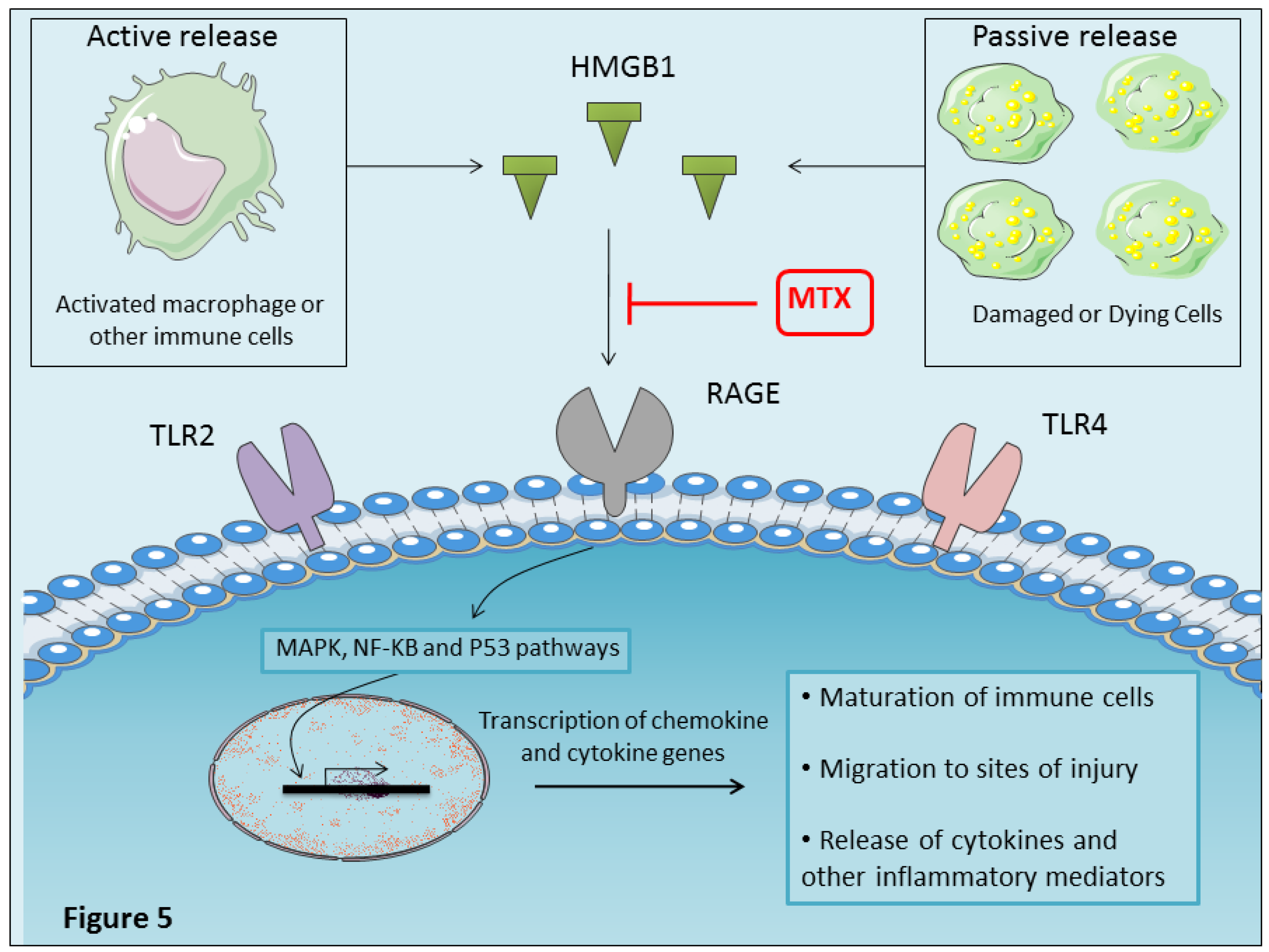
4.10. MTX Inhibits Proinflammatory HMGBI Alarmin Effects
5. MTX Adverse Effects Mechanisms of Action
6. MTX Response Variability
7. MTX and Chronic Viral Arthritis
8. Conclusions and Perspectives
Conflicts of Interest
References
- Chan, E.S.L.; Cronstein, B.N. Mechanisms of action of methotrexate. Bull. NYU Hosp. Jt. Dis. 2013, 71, S5–S8. [Google Scholar]
- Chan, E.S.L.; Cronstein, B.N. Methotrexate—How does it really work? Nat. Rev. Rheumatol. 2010, 6, 175–178. [Google Scholar] [CrossRef] [PubMed]
- Genestier, L.; Paillot, R.; Quemeneur, L.; Izeradjene, K.; Revillard, J.P. Mechanisms of action of methotrexate. Immunopharmacology 2000, 47, 247–257. [Google Scholar] [CrossRef]
- Sutaria, R.B.; Amaral, J.K.; Schoen, R.T. Emergence and treatment of chikungunya arthritis. Curr. Opin. Rheumatol. 2018, 30, 256–263. [Google Scholar] [CrossRef] [PubMed]
- Marks, M.; Marks, J.L. Viral arthritis. Clin. Med. 2016, 16, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Perl, A. Mechanisms of viral pathogenesis in rheumatic disease. Ann. Rheum. Dis. 1999, 58, 454–461. [Google Scholar] [CrossRef]
- Suhrbier, A.; Jaffar-Bandjee, M.C.; Gasque, P. Arthritogenic alphaviruses—An overview. Nat. Rev. Rheumatol. 2012, 8, 420–429. [Google Scholar] [CrossRef] [PubMed]
- Conway, R.; Carey, J.J. Risk of liver disease in methotrexate treated patients. World J. Hepatol. 2017, 9, 1092–1100. [Google Scholar] [CrossRef]
- WHO. WHO Model Lists of Essential Medicines 21th List. Available online: http://www.who.int/medicines/publications/essentialmedicines/en/ (accessed on 22 July 2019).
- Bijlsma, J.W.J.; Jacobs, J.W.G. Methotrexate: Still the anchor drug in RA treatment. Jt. Bone Spine 2009, 76, 452–454. [Google Scholar] [CrossRef]
- Singh, J.A.; Furst, D.E.; Bharat, A.; Curtis, J.R.; Kavanaugh, A.F.; Kremer, J.M.; Moreland, L.; O’Dell, J.; Winthrop, K.; Beukelman, T.; et al. 2012 Update of the 2008 American College of Rheumatology (ACR) Recommendations for the use of Disease-Modifying Anti-Rheumatic Drugs and Biologics in the treatment of Rheumatoid Arthritis (RA). Arthritis Care Res. 2012, 64, 625–639. [Google Scholar] [CrossRef]
- Emery, P.; Sebba, A.; Huizinga, T.W.J. Biologic and oral disease-modifying antirheumatic drug monotherapy in rheumatoid arthritis. Ann. Rheum. Dis. 2013, 72, 1897–1904. [Google Scholar] [CrossRef] [PubMed]
- Cronstein, B.N. Low-dose methotrexate: A mainstay in the treatment of rheumatoid arthritis. Pharmacol. Rev. 2005, 57, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.; Cronstein, B.N. Understanding the mechanisms of action of methotrexate: Implications for the treatment of rheumatoid arthritis. Bull. NYU Hosp. Jt. Dis. 2007, 65, 168–173. [Google Scholar] [PubMed]
- Prey, S.; Paul, C. Effect of folic or folinic acid supplementation on methotrexate-associated safety and efficacy in inflammatory disease: A systematic review. Br. J. Dermatol. 2009, 160, 622–628. [Google Scholar] [CrossRef] [PubMed]
- Schnabel, A.; Gross, W.L. Low-dose methotrexate in rheumatic diseases—Efficacy, side effects, and risk factors for side effects. Semin. Arthritis Rheum. 1994, 23, 310–327. [Google Scholar] [CrossRef]
- Romão, V.C.; Lima, A.; Bernardes, M.; Canhão, H.; Fonseca, J.E. Three decades of low-dose methotrexate in rheumatoid arthritis: Can we predict toxicity? Immunol. Res. 2014, 60, 289–310. [Google Scholar] [CrossRef]
- Farber, S.; Diamond, L.K. Temporary remissions in acute leukemia in children produced by folic acid antagonist, 4-aminopteroyl-glutamic acid. N. Engl. J. Med. 1948, 238, 787–793. [Google Scholar] [CrossRef]
- Seeger, D.R.; Cosulich, D.B.; Smith, J.M.; Hultquist, M.E. Analogs of Pteroylglutamic Acid. III. 4-Amino Derivatives. J. Am. Chem. Soc. 1949, 71, 1753–1758. [Google Scholar] [CrossRef]
- Thiersch, I.B. Bone-marrow changes in man after treatment with aminopterin, amethopterin, and aminoanfol. With special reference to megaloblastosis and tumor remission. Cancer 1949, 2, 877–883. [Google Scholar] [CrossRef]
- Gubner, R.; Cote, L.; Hughes, J.; Oleson, J.J.; Ruegsegger, J.M.; Williams, J.H. Comparative Effects of Aminopterin, Cortisone and Acth in Experimental Formaldehyde Arthritis and Psoriatic Arthritis. J. Investig. Dermatol. 1952, 19, 297–305. [Google Scholar] [CrossRef][Green Version]
- Gubner, R.; August, S.; Ginsberg, V. Therapeutic suppression of tissue reactivity. II. Effect of aminopterin in rheumatoid arthritis and psoriasis. Am. J. Med. Sci. 1951, 221, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Weinblatt, M.E.; Coblyn, J.S.; Fox, D.A.; Fraser, P.A.; Holdsworth, D.E.; Glass, D.N.; Trentham, D.E. Efficacy of Low-Dose Methotrexate in Rheumatoid Arthritis. N. Engl. J. Med. 1985, 312, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Braun, J.; Rau, R. An update on methotrexate. Curr. Opin. Rheumatol. 2009, 21, 216–223. [Google Scholar] [CrossRef] [PubMed]
- Bannwarth, B.; Labat, L.; Moride, Y.; Schaeverbeke, T. Methotrexate in rheumatoid arthritis. An update. Drugs 1994, 47, 25–50. [Google Scholar] [CrossRef] [PubMed]
- Aletaha, D.; Neogi, T.; Silman, A.J.; Funovits, J.; Felson, D.T.; Bingham, C.O.; Birnbaum, N.S.; Burmester, G.R.; Bykerk, V.P.; Cohen, M.D.; et al. 2010 Rheumatoid arthritis classification criteria: An American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum. 2010, 62, 2569–2581. [Google Scholar] [CrossRef] [PubMed]
- Curtis, J.R.; Singh, J.A. The Use of Biologics in Rheumatoid Arthritis: Current and Emerging Paradigms of Care. Clin. Ther. 2011, 33, 679–707. [Google Scholar] [CrossRef] [PubMed]
- Smolen, J.S.; Landewé, R.; Bijlsma, J.; Burmester, G.; Chatzidionysiou, K.; Dougados, M.; Nam, J.; Ramiro, S.; Voshaar, M.; Van Vollenhoven, R.; et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2016 update. Ann. Rheum. Dis. 2017, 76, 960–977. [Google Scholar] [CrossRef]
- Matsuoka, H.; Ohi, N.; Mihara, M.; Suzuki, H.; Miyamoto, K.; Maruyama, N.; Tsuji, K.; Kato, N.; Akimoto, T.; Takeda, Y.; et al. Antirheumatic Agents: Novel Methotrexate Derivatives Bearing a Benzoxazine or Benzothiazine Moiety. J. Med. Chem. 1997, 40, 105–111. [Google Scholar] [CrossRef]
- Kokuryo, Y.; Kawata, K.; Nakatani, T.; Kugimiya, A.; Tamura, Y.; Kawada, K.; Matsumoto, M.; Suzuki, R.; Kuwabara, K.; Hori, Y.; et al. Synthesis and evaluation of novel fluorinated methotrexate derivatives for application to rheumatoid arthritis treatment. J. Med. Chem. 1997, 40, 3280–3291. [Google Scholar] [CrossRef]
- Mello, S.B.V.; Tavares, E.R.; Guido, M.C.; Bonfá, E.; Maranhão, R.C. Anti-inflammatory effects of intravenous methotrexate associated with lipid nanoemulsions on antigen-induced arthritis. Clinics 2016, 71, 54–58. [Google Scholar] [CrossRef]
- Tamura, T.; Higuchi, Y.; Kitamura, H.; Murao, N.; Saitoh, R.; Morikawa, T.; Sato, H. Novel hyaluronic acid-methotrexate conjugate suppresses joint inflammation in the rat knee: Efficacy and safety evaluation in two rat arthritis models. Arthritis Res. Ther. 2016, 18, 79. [Google Scholar]
- Inoue, K.; Yuasa, H. Molecular Basis for Pharmacokinetics and Pharmacodynamics of Methotrexate in Rheumatoid Arthritis Therapy. Drug Metab. Pharmacokinet. 2014, 29, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Visser, K.; van der Heijde, D. Optimal dosage and route of administration of methotrexate in rheumatoid arthritis: A systematic review of the literature. Ann. Rheum. Dis. 2008, 68, 1094–1099. [Google Scholar] [CrossRef] [PubMed]
- Bello, A.E.; Perkins, E.L.; Jay, R.; Efthimiou, P. Recommendations for optimizing methotrexate treatment for patients with rheumatoid arthritis. Open Access Rheumatol. 2017, 9, 67–79. [Google Scholar] [CrossRef] [PubMed]
- Desmoulin, S.K.; Hou, Z.; Gangjee, A.; Matherly, L.H. The human proton-coupled folate transporter: Biology and therapeutic applications to cancer. Cancer Biol. Ther. 2012, 13, 1355–1373. [Google Scholar] [CrossRef]
- Grim, J.; Chládek, J.; Martínková, J. Pharmacokinetics and pharmacodynamics of methotrexate in non-neoplastic diseases. Clin. Pharm. 2003, 42, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Hoekstra, M.; Haagsma, C.; Neef, C.; Proost, J.; Knuif, A.; van de Laar, M. Bioavailability of higher dose methotrexate comparing oral and subcutaneous administration in patients with rheumatoid arthritis. J. Rheumatol. 2004, 31, 645–648. [Google Scholar]
- Schiff, M.H.; Jaffe, J.S.; Freundlich, B. Head-to-head, randomised, crossover study of oral versus subcutaneous methotrexate in patients with rheumatoid arthritis: Drug-exposure limitations of oral methotrexate at doses ≥15 mg may be overcome with subcutaneous administration. Ann. Rheum. Dis. 2014, 73, 1549–1551. [Google Scholar] [CrossRef]
- Hillson, J.L.; Furst, D.E. Pharmacology and pharmacokinetics of methotrexate in rheumatic disease. Practical issues in treatment and design. Rheum. Dis. Clin. N. Am. 1997, 23, 757–778. [Google Scholar] [CrossRef]
- Bianchi, G.; Caporali, R.; Todoerti, M.; Mattana, P. Methotrexate and Rheumatoid Arthritis: Current Evidence Regarding Subcutaneous Versus Oral Routes of Administration. Adv. Ther. 2016, 33, 369–378. [Google Scholar] [CrossRef]
- Paxton, J.W. Protein binding of methotrexate in sera from normal human beings: Effect of drug concentration, pH, temperature, and storage. J. Pharmacol. Methods 1981, 5, 203–213. [Google Scholar] [CrossRef]
- Herman, R.A.; Veng-Pedersen, P.; Hoffman, J.; Koehnke, R.; Furst, D.E. Pharmacokinetics of Low-Dose Methotrexate in Rheumatoid Arthritis Patients. J. Pharm. Sci. 1989, 78, 165–171. [Google Scholar] [CrossRef]
- Tracy, T.S.; Worster, T.; Bradley, J.D.; Greene, P.K.; Brater, D.C. Methotrexate disposition following concomitant administration of ketoprofen, piroxicam and flurbiprofen in patients with rheumatoid arthritis. Br. J. Clin. Pharm. 1994, 37, 453–456. [Google Scholar] [CrossRef] [PubMed]
- Bannwarth, B.; Péhourcq, F.; Schaeverbeke, T.; Dehais, J. Clinical pharmacokinetics of low-dose pulse methotrexate in rheumatoid arthritis. Clin. Pharm. 1996, 30, 194–210. [Google Scholar] [CrossRef] [PubMed]
- Seideman, P.; Beck, O.; Eksborg, S.; Wennberg, M. The pharmacokinetics of methotrexate and its 7-hydroxy metabolite in patients with rheumatoid arthritis. Br. J. Clin. Pharm. 1993, 35, 409–412. [Google Scholar] [CrossRef]
- Nuernberg, B.; Koehnke, R.; Solsky, M.; Hoffman, J.; Furst, D.E. Biliary elimination of low-dose methotrexate in humans. Arthritis Rheum. 1990, 33, 898–902. [Google Scholar] [CrossRef] [PubMed]
- Bressolle, F.; Bologna, C.; Kinowski, J.; Sany, J.; Combe, B. Effects of moderate renal insufficiency on pharmacokinetics of methotrexate in rheumatoid arthritis patients. Ann. Rheum. Dis. 1998, 57, 110–113. [Google Scholar] [CrossRef] [PubMed]
- Olsen, E.A. The pharmacology of methotrexate. J. Am. Acad. Dermatol. 1991, 25, 306–318. [Google Scholar] [CrossRef]
- Fiehn, C. Methotrexate transport mechanisms: The basis for targeted drug delivery and ß-folate-receptor-specific treatment. Clin. Exp. Rheumatol. 2010, 28, S40–S45. [Google Scholar] [PubMed]
- Chabner, B.A.; Allegra, C.J.; Curt, G.A.; Clendeninn, N.J.; Baram, J.; Koizumi, S.; Drake, J.C.; Jolivet, J. Polyglutamation of methotrexate. Is methotrexate a prodrug? J. Clin. Investig. 1985, 76, 907–912. [Google Scholar] [CrossRef] [PubMed]
- Panetta, J.C.; Wall, A.; Pui, C.H.; Relling, M.V.; Evans, W.E. Methotrexate Intracellular Disposition in Acute Lymphoblastic Leukemia: A Mathematical Model of γ-Glutamyl Hydrolase Activity. Clin. Cancer Res. 2002, 8, 2423–2429. [Google Scholar] [PubMed]
- Brown, P.M.; Pratt, A.G.; Isaacs, J.D. Mechanism of action of methotrexate in rheumatoid arthritis, and the search for biomarkers. Nat. Rev. Rheumatol. 2016, 12, 731. [Google Scholar] [CrossRef] [PubMed]
- Murakami, T.; Mori, N. Involvement of Multiple Transporters-mediated Transports in Mizoribine and Methotrexate Pharmacokinetics. Pharmaceuticals 2012, 5, 802–836. [Google Scholar] [CrossRef] [PubMed]
- Stamp, L.K.; O’Donnell, J.L.; Chapman, P.T.; Zhang, M.; Frampton, C.; James, J.; Barclay, M.L. Determinants of red blood cell methotrexate polyglutamate concentrations in rheumatoid arthritis patients receiving long-term methotrexate treatment. Arthritis Rheum. 2009, 60, 2248–2256. [Google Scholar] [CrossRef] [PubMed]
- Dervieux, T.; Furst, D.; Lein, D.O.; Capps, R.; Smith, K.; Caldwell, J.; Kremer, J. Pharmacogenetic and metabolite measurements are associated with clinical status in patients with rheumatoid arthritis treated with methotrexate: Results of a multicentred cross sectional observational study. Ann. Rheum. Dis. 2005, 64, 1180–1185. [Google Scholar] [CrossRef] [PubMed]
- Fairbanks, L.D.; Rückemann, K.; Qiu, Y.; Hawrylowicz, C.M.; Richards, D.F.; Swaminathan, R.; Kirschbaum, B.; Simmonds, H.A. Methotrexate inhibits the first committed step of purine biosynthesis in mitogen-stimulated human T-lymphocytes: A metabolic basis for efficacy in rheumatoid arthritis? Biochem. J. 1999, 342, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Genestier, L.; Paillot, R.; Fournel, S.; Ferraro, C.; Miossec, P.; Revillard, J.P. Immunosuppressive properties of methotrexate: Apoptosis and clonal deletion of activated peripheral T cells. J. Clin. Investig. 1998, 102, 322–328. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, A.; Hakoda, M.; Yamanaka, H.; Kamatani, N.; Kashiwazaki, S. Divergent effects of methotrexate on the clonal growth of T and B lymphocytes and synovial adherent cells from patients with rheumatoid arthritis. Ann. Rheum. Dis. 1996, 55, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Morgan, S.L.; Baggott, J.E.; Vaughn, W.H.; Austin, J.S.; Veitch, T.A.; Lee, J.Y.; Koopman, W.J.; Krumdieck, C.L.; Alarcón, G.S. Supplementation with folic acid during methotrexate therapy for rheumatoid arthritis. A double-blind, placebo-controlled trial. Ann. Intern. Med. 1994, 121, 833–841. [Google Scholar] [CrossRef]
- Cronstein, B.N.; Haskó, G. Regulation of Inflammation by Adenosine. Front. Immunol. 2013, 4, 85. [Google Scholar]
- Haskó, G.; Cronstein, B.N. Adenosine: An endogenous regulator of innate immunity. Trends Immunol. 2004, 25, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Stamp, L.K.; Hazlett, J.; Roberts, R.L.; Frampton, C.; Highton, J.; Hessian, P.A. Adenosine receptor expression in rheumatoid synovium: A basis for methotrexate action. Arthritis Res. Ther. 2012, 14, R138. [Google Scholar] [CrossRef] [PubMed]
- Cronstein, B.N.; Sitkovsky, M. Adenosine and adenosine receptors in the pathogenesis and treatment of rheumatic diseases. Nat. Rev. Rheumatol. 2017, 13, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Forrest, C.M.; Harman, G.; McMillan, R.B.; Stoy, N.; Stone, T.W.; Darlington, L.G. Modulation of cytokine release by purine receptors in patients with rheumatoid arthritis. Clin. Exp. Rheumatol. 2005, 23, 89–92. [Google Scholar]
- Varani, K.; Padovan, M.; Vincenzi, F.; Targa, M.; Trotta, F.; Govoni, M.; Borea, P.A. A2A and A3 adenosine receptor expression in rheumatoid arthritis: Upregulation, inverse correlation with disease activity score and suppression of inflammatory cytokine and metalloproteinase release. Arthritis Res. 2011, 13, R197. [Google Scholar] [CrossRef]
- Baggott, J.E.; Vaughn, W.H.; Hudson, B.B. Inhibition of 5-aminoimidazole-4-carboxamide ribotide transformylase, adenosine deaminase and 5′-adenylate deaminase by polyglutamates of methotrexate and oxidized folates and by 5-aminoimidazole-4-carboxamide riboside and ribotide. Biochem. J. 1986, 236, 193–200. [Google Scholar] [CrossRef]
- Montesinos, M.C.; Takedachi, M.; Thompson, L.F.; Wilder, T.F.; Fernández, P.; Cronstein, B.N. The antiinflammatory mechanism of methotrexate depends on extracellular conversion of adenine nucleotides to adenosine by ecto-5′-nucleotidase: Findings in a study of ecto-5′-nucleotidase gene-deficient mice. Arthritis Rheum. 2007, 56, 1440–1445. [Google Scholar] [CrossRef]
- Cronstein, B.N.; Eberle, M.A.; Gruber, H.E.; Levin, R.I. Methotrexate inhibits neutrophil function by stimulating adenosine release from connective tissue cells. Proc. Natl. Acad. Sci. USA 1991, 88, 2441–2445. [Google Scholar] [CrossRef]
- Cronstein, B.N.; Naime, D.; Ostad, E. The antiinflammatory mechanism of methotrexate. Increased adenosine release at inflamed sites diminishes leukocyte accumulation in an in vivo model of inflammation. J. Clin. Investig. 1993, 92, 2675. [Google Scholar] [CrossRef]
- Montesinos, M.C.; Desai, A.; Delano, D.; Chen, J.F.; Fink, J.S.; Jacobson, M.A.; Cronstein, B.N. Adenosine A2A or A3 receptors are required for inhibition of inflammation by methotrexate and its analog MX-68. Arthritis Rheum. 2003, 48, 240–247. [Google Scholar] [CrossRef]
- Montesinos, M.C.; Desai, A.; Cronstein, B.N. Suppression of inflammation by low-dose methotrexate is mediated by adenosine A2A receptor but not A3 receptor activation in thioglycollate-induced peritonitis. Arthritis Res. 2006, 8, R53. [Google Scholar]
- Riksen, N.P.; Barrera, P.; van den Broek, P.H.H.; van Riel, P.L.C.M.; Smits, P.; Rongen, G.A. Methotrexate modulates the kinetics of adenosine in humans in vivo. Ann. Rheum. Dis. 2006, 65, 465–470. [Google Scholar] [CrossRef] [PubMed]
- Montesinos, M.C.; Yap, J.S.; Desai, A.; Posadas, I.; McCrary, C.T.; Cronstein, B.N. Reversal of the antiinflammatory effects of methotrexate by the nonselective adenosine receptor antagonists theophylline and caffeine: Evidence that the antiinflammatory effects of methotrexate are mediated via multiple adenosine receptors in rat adjuvant arthritis. Arthritis Rheum. 2000, 43, 656–663. [Google Scholar] [PubMed]
- Nesher, G.; Mates, M.; Zevin, S. Effect of caffeine consumption on efficacy of methotrexate in rheumatoid arthritis. Arthritis Rheum. 2003, 48, 571–572. [Google Scholar] [CrossRef] [PubMed]
- Benito-Garcia, E.; Heller, J.E.; Chibnik, L.B.; Maher, N.E.; Matthews, H.M.; Bilics, J.A.; Weinblatt, M.E.; Shadick, N.A. Dietary caffeine intake does not affect methotrexate efficacy in patients with rheumatoid arthritis. J. Rheumatol. 2006, 33, 1275–1281. [Google Scholar] [PubMed]
- Yukioka, K.; Wakitani, S.; Yukioka, M.; Furumitsu, Y.; Shichikawa, K.; Ochi, T.; Goto, H.; Matsui-Yuasa, I.; Otani, S.; Nishizawa, Y. Polyamine levels in synovial tissues and synovial fluids of patients with rheumatoid arthritis. J. Rheumatol. 1992, 19, 689–692. [Google Scholar]
- Nesher, G.; Osborn, T.G.; Moore, T.L. In vitro effects of methotrexate on polyamine levels in lymphocytes from rheumatoid arthritis patients. Clin. Exp. Rheumatol. 1996, 14, 395–399. [Google Scholar] [PubMed]
- Smith, D.M.; Johnson, J.A.; Turner, R.A. Biochemical perturbations of BW 91Y (3-deazaadenosine) on human neutrophil chemotactic potential and lipid metabolism. Int. J. Tissue React. 1991, 13, 1–18. [Google Scholar]
- Herman, S.; Zurgil, N.; Deutsch, M. Low dose methotrexate induces apoptosis with reactive oxygen species involvement in T lymphocytic cell lines to a greater extent than in monocytic lines. Inflamm. Res. 2005, 54, 273–280. [Google Scholar] [CrossRef]
- Phillips, D.C.; Woollard, K.J.; Griffiths, H.R. The anti-inflammatory actions of methotrexate are critically dependent upon the production of reactive oxygen species. Br. J. Pharm. 2003, 138, 501–511. [Google Scholar] [CrossRef]
- Chalupsky, K.; Cai, H. Endothelial dihydrofolate reductase: Critical for nitric oxide bioavailability and role in angiotensin II uncoupling of endothelial nitric oxide synthase. Proc. Natl. Acad. Sci. USA 2005, 102, 9056–9061. [Google Scholar] [CrossRef] [PubMed]
- Crabtree, M.J.; Hale, A.B.; Channon, K.M. Dihydrofolate reductase protects endothelial nitric oxide synthase from uncoupling in tetrahydrobiopterin deficiency. Free Radic. Biol. Med. 2011, 50, 1639–1646. [Google Scholar] [CrossRef] [PubMed]
- Spurlock, C.F.; Gass, H.M.; Bryant, C.J.; Wells, B.C.; Olsen, N.J.; Aune, T.M. Methotrexate-mediated inhibition of nuclear factor κB activation by distinct pathways in T cells and fibroblast-like synoviocytes. Rheumatology 2015, 54, 178–187. [Google Scholar] [CrossRef] [PubMed]
- Sung, J.Y.; Hong, J.H.; Kang, H.S.; Choi, I.; Lim, S.D.; Lee, J.K.; Seok, J.H.; Lee, J.H.; Hur, G.M. Methotrexate suppresses the interleukin-6 induced generation of reactive oxygen species in the synoviocytes of rheumatoid arthritis. Immunopharmacology 2000, 47, 35–44. [Google Scholar] [CrossRef]
- Brennan, F.M. A follow-up to “Anti-cytokine therapy in chronic destructive arthritis” by Wim B van den Berg. Arthritis Res. 2001, 3, 211–213. [Google Scholar] [CrossRef] [PubMed]
- Dolhain, R.J.; Tak, P.P.; Dijkmans, B.A.; De Kuiper, P.; Breedveld, F.C.; Miltenburg, A.M. Methotrexate reduces inflammatory cell numbers, expression of monokines and of adhesion molecules in synovial tissue of patients with rheumatoid arthritis. Br. J. Rheumatol. 1998, 37, 502–508. [Google Scholar] [CrossRef] [PubMed]
- Brody, M.; Böhm, I.; Bauer, R. Mechanism of action of methotrexate: Experimental evidence that methotrexate blocks the binding of interleukin 1 beta to the interleukin 1 receptor on target cells. Eur. J. Clin. Chem. Clin. Biochem. 1993, 31, 667–674. [Google Scholar] [CrossRef]
- Neurath, M.F.; Hildner, K.; Becker, C.; Schlaak, J.F.; Barbulescu, K.; Germann, T.; Schmitt, E.; Schirmacher, P.; Haralambous, S.; Pasparakis, M.; et al. Methotrexate specifically modulates cytokine production by T cells and macrophages in murine collagen-induced arthritis (CIA): A mechanism for methotrexate-mediated immunosuppression. Clin. Exp. Immunol. 1999, 115, 42. [Google Scholar] [CrossRef] [PubMed]
- Hildner, K.; Finotto, S.; Becker, C.; Schlaak, J.; Schirmacher, P.; Galle, P.R.; Märker-Hermann, E.; Neurath, M.F. Tumour necrosis factor (TNF) production by T cell receptor-primed T lymphocytes is a target for low dose methotrexate in rheumatoid arthritis. Clin. Exp. Immunol. 1999, 118, 137–146. [Google Scholar] [CrossRef]
- Rudwaleit, M.; Yin, Z.; Siegert, S.; Grolms, M.; Radbruch, A.; Braun, J.; Sieper, J. Response to methotrexate in early rheumatoid arthritis is associated with a decrease of T cell derived tumour necrosis factor α, increase of interleukin 10, and predicted by the initial concentration of interleukin 4. Ann. Rheum. Dis. 2000, 59, 311–314. [Google Scholar] [CrossRef] [PubMed]
- Gerards, A.H.; de Lathouder, S.; Groot, E.R.; Dijkmans, B.A.C.; Aarden, L.A. Inhibition of cytokine production by methotrexate. Studies in healthy volunteers and patients with rheumatoid arthritis. Rheumatology 2003, 42, 1189–1196. [Google Scholar] [CrossRef] [PubMed]
- Miranda-Carús, M.E.; Balsa, A.; Benito-Miguel, M.; Pérez de Ayala, C.; Martín-Mola, E. IL-15 and the initiation of cell contact-dependent synovial fibroblast-T lymphocyte cross-talk in rheumatoid arthritis: Effect of methotrexate. J. Immunol. 2004, 173, 1463–1476. [Google Scholar] [CrossRef] [PubMed]
- Constantin, A.; Loubet-Lescoulié, P.; Lambert, N.; Yassine-Diab, B.; Abbal, M.; Mazières, B.; Préval, C.; de Cantagrel, A. Antiinflammatory and immunoregulatory action of methotrexate in the treatment of rheumatoid arthritis: Evidence of increased interleukin-4 and interleukin-10 gene expression demonstrated in vitro by competitive reverse transcriptase-polymerase chain reaction. Arthritis Rheum. 1998, 41, 48–57. [Google Scholar] [PubMed]
- Cutolo, M.; Sulli, A.; Pizzorni, C.; Seriolo, B.; Straub, R. Anti-inflammatory mechanisms of methotrexate in rheumatoid arthritis. Ann. Rheum. Dis. 2001, 60, 729–735. [Google Scholar] [CrossRef] [PubMed]
- Segal, R.; Mozes, E.; Yaron, M.; Tartakovsky, B. The effects of methotrexate on the production and activity of interleukin-1. Arthritis Rheum. 1989, 32, 370–377. [Google Scholar] [CrossRef]
- Bergström, B.; Carlsten, H.; Ekwall, A.K.H. Methotrexate inhibits effects of platelet-derived growth factor and interleukin-1β on rheumatoid arthritis fibroblast-like synoviocytes. Arthritis Res. 2018, 20, 49. [Google Scholar]
- Burmester, G.R.; Stuhlmüller, B.; Keyszer, G.; Kinne, R.W. Mononuclear phagocytes and rheumatoid synovitis. Mastermind or workhorse in arthritis? Arthritis Rheum. 1997, 40, 5–18. [Google Scholar] [CrossRef]
- Wijngaarden, S.; van Roon, J.A.G.; van de Winkel, J.G.J.; Bijlsma, J.W.J.; Lafeber, F.P.J.G. Downregulation of activating Fcγ receptors on monocytes of patients with rheumatoid arthritis upon methotrexate treatment. Rheumatology 2005, 44, 729–734. [Google Scholar] [CrossRef]
- Murphy, G.; Knäuper, V.; Atkinson, S.; Butler, G.; English, W.; Hutton, M.; Stracke, J.; Clark, I. Matrix metalloproteinases in arthritic disease. Arthritis Res. Ther. 2002, 4, S39–S49. [Google Scholar] [CrossRef]
- Vincenti, M.P.; Clark, I.M.; Brinckerhoff, C.E. Using inhibitors of metalloproteinases to treat arthritis. Easier said than done? Arthritis Rheum. 1994, 37, 1115–1126. [Google Scholar] [CrossRef]
- Fiedorczyk, M.; Klimiuk, P.A.; Sierakowski, S.; Gindzienska-Sieskiewicz, E.; Chwiecko, J. Serum matrix metalloproteinases and tissue inhibitors of metalloproteinases in patients with early rheumatoid arthritis. J. Rheumatol. 2006, 33, 1523–1529. [Google Scholar]
- Ho, C.Y.; Wong, C.K.; Li, E.K.; Tam, L.S.; Lam, C.W.K. Suppressive effect of combination treatment of leflunomide and methotrexate on chemokine expression in patients with rheumatoid arthritis. Clin. Exp. Immunol. 2003, 133, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Seitz, M.; Dayer, J.M. Enhanced production of tissue inhibitor of metalloproteinases by peripheral blood mononuclear cells of rheumatoid arthritis patients responding to methotrexate treatment. Rheumatology 2000, 39, 637–645. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Firestein, G.S.; Paine, M.M.; Boyle, D.L. Mechanisms of Methotrexate Action in Rheumatoid Arthritis. Arthritis Rheum. 1994, 37, 193–200. [Google Scholar] [CrossRef]
- Zhang, Y.; Jiang, J.; Xie, J.; Xu, C.; Wang, C.; Yin, L.; Yang, L.; Sung, K.-L.P. Combined effects of tumor necrosis factor-α and interleukin-1β on lysyl oxidase and matrix metalloproteinase expression in human knee synovial fibroblasts in vitro. Exp. Med. 2017, 14, 5258–5266. [Google Scholar] [CrossRef] [PubMed]
- Stichtenoth, D.O.; Thorén, S.; Bian, H.; Peters-Golden, M.; Jakobsson, P.J.; Crofford, L.J. Microsomal Prostaglandin E Synthase is Regulated by Proinflammatory Cytokines and Glucocorticoids in Primary Rheumatoid Synovial Cells. J. Immunol. 2001, 167, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Westman, M.; Korotkova, M.; af Klint, E.; Stark, A.; Audoly, L.P.; Klareskog, L.; Ulfgren, A.K.; Jakobsson, P.J. Expression of microsomal prostaglandin E synthase 1 in rheumatoid arthritis synovium. Arthritis Rheum. 2004, 50, 1774–1780. [Google Scholar] [CrossRef] [PubMed]
- Garg, N.K.; Singh, B.; Tyagi, R.K.; Sharma, G.; Katare, O.P. Effective transdermal delivery of methotrexate through nanostructured lipid carriers in an experimentally induced arthritis model. Colloids Surf. B Biointerfaces 2016, 147, 17–24. [Google Scholar] [CrossRef]
- Novaes, G.S.; Mello, S.B.; Laurindo, I.M.; Cossermelli, W. Low dose methotrexate decreases intraarticular prostaglandin and interleukin 1 levels in antigen induced arthritis in rabbits. J. Rheumatol. 1996, 23, 2092–2097. [Google Scholar]
- Williams, A.; Goodfellow, R.; Topley, N.; Amos, N.; Williams, B. The suppression of rat collagen-induced arthritis and inhibition of macrophage derived mediator release by liposomal methotrexate formulations. Inflamm. Res. 2000, 49, 155–161. [Google Scholar] [CrossRef]
- Vergne, P.; Liagre, B.; Bertin, P.; Cook-Moreau, J.; Treves, R.; Beneytout, J.L.; Rigaud, M. Methotrexate and cyclooxygenase metabolism in cultured human rheumatoid synoviocytes. J. Rheumatol. 1998, 25, 433–440. [Google Scholar] [PubMed]
- Mello, S.B.; Barros, D.M.; Silva, A.S.; Laurindo, I.M.; Novaes, G.S. Methotrexate as a preferential cyclooxygenase 2 inhibitor in whole blood of patients with rheumatoid arthritis. Rheumatology 2000, 39, 533–536. [Google Scholar] [CrossRef] [PubMed]
- Furst, D.E.; Kremer, J.M. Methotrexate in rheumatoid arthritis. Arthritis Rheum. 1988, 31, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Gheorghe, K.R.; Sadique, S.; Leclerc, P.; Idborg, H.; Wobst, I.; Catrina, A.I.; Jakobsson, P.-J.; Korotkova, M. Limited effect of anti-rheumatic treatment on 15-prostaglandin dehydrogenase in rheumatoid arthritis synovial tissue. Arthritis Res. 2012, 14, R121. [Google Scholar] [CrossRef] [PubMed]
- Inoue, H.; Takamori, M.; Nagata, N.; Nishikawa, T.; Oda, H.; Yamamoto, S.; Koshihara, Y. An investigation of cell proliferation and soluble mediators induced by interleukin 1β in human synovial fibroblasts: Comparative response in osteoarthritis and rheumatoid arthritis. Inflamm. Res. 2001, 50, 65–72. [Google Scholar] [PubMed]
- Hinz, M.; Arslan, S.Ç.; Scheidereit, C. It takes two to tango: IκBs, the multifunctional partners of NF-κB. Immunol. Rev. 2012, 246, 59–76. [Google Scholar] [CrossRef]
- Atreya, I.; Atreya, R.; Neurath, M.F. NF-κB in inflammatory bowel disease. J. Intern. Med. 2008, 263, 591–596. [Google Scholar] [CrossRef]
- Tak, P.P.; Firestein, G.S. NF-κB: A key role in inflammatory diseases. J. Clin. Investig. 2001, 107, 7–11. [Google Scholar] [CrossRef]
- Makarov, S.S. NF-kappaB in rheumatoid arthritis: A pivotal regulator of inflammation, hyperplasia, and tissue destruction. Arthritis Res. 2001, 3, 200–206. [Google Scholar] [CrossRef]
- Ak, P.; Levine, A.J. p53 and NF-κB: Different strategies for responding to stress lead to a functional antagonism. FASEB J. 2010, 24, 3643–3652. [Google Scholar] [CrossRef]
- Seemayer, C.; Kuchen, S.; Neidhart, M.; Kuenzler, P.; Rihoskova, V.; Neumann, E.; Pruschy, M.; Aicher, W.; Muller-Ladner, U.; Gay, R.; et al. p53 in rheumatoid arthritis synovial fibroblasts at sites of invasion. Ann. Rheum. Dis. 2003, 62, 1139–1144. [Google Scholar] [CrossRef] [PubMed]
- Firestein, G.S. NF-κB: Holy Grail for rheumatoid arthritis? Arthritis Rheum. 2004, 50, 2381–2386. [Google Scholar] [CrossRef] [PubMed]
- Spurlock, C.F.; Tossberg, J.T.; Matlock, B.K.; Olsen, N.J.; Aune, T.M. Methotrexate inhibits NF-κB activity via lincRNA-p21 induction. Arthritis Rheumatol. 2014, 66, 2947–2957. [Google Scholar] [CrossRef] [PubMed]
- O’Shea, J.J.; Plenge, R. JAK and STAT Signaling Molecules in Immunoregulation and Immune-Mediated Disease. Immunity 2012, 36, 542–550. [Google Scholar] [CrossRef] [PubMed]
- Ivashkiv, L.B.; Hu, X. The JAK/STAT pathway in rheumatoid arthritis: Pathogenic or protective? Arthritis Rheum. 2003, 48, 2092–2096. [Google Scholar] [CrossRef] [PubMed]
- Vaddi, K.; Luchi, M. JAK inhibition for the treatment of rheumatoid arthritis: A new era in oral DMARD therapy. Expert Opin. Investig. Drugs 2012, 21, 961–973. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.; Fisher, K.H.; Snowden, J.A.; Danson, S.J.; Brown, S.; Zeidler, M.P. Methotrexate is a JAK/STAT Pathway Inhibitor. PLoS ONE 2015, 10, e0130078. [Google Scholar] [CrossRef]
- Thomas, S.J.; Fisher, K.; Brown, S.; Snowden, J.A.; Danson, S.; Zeidler, M. Methotrexate Is a Suppressor of JAK/STAT Pathway Activation Which Inhibits JAK2V617F Induced Signalling. Blood 2014, 124, 4577. [Google Scholar]
- Klune, J.R.; Dhupar, R.; Cardinal, J.; Billiar, T.R.; Tsung, A. HMGB1: Endogenous Danger Signaling. Mol. Med. 2008, 14, 476–484. [Google Scholar] [CrossRef]
- Castiglioni, A.; Canti, V.; Rovere-Querini, P.; Manfredi, A.A. High-mobility group box 1 (HMGB1) as a master regulator of innate immunity. Cell Tissue Res. 2011, 343, 189–199. [Google Scholar] [CrossRef]
- Scaffidi, P.; Misteli, T.; Bianchi, M.E. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 2002, 418, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.M.; Yan, S.D.; Yan, S.F.; Stern, D.M. The multiligand receptor RAGE as a progression factor amplifying immune and inflammatory responses. J. Clin. Investig. 2001, 108, 949–955. [Google Scholar] [CrossRef] [PubMed]
- Bierhaus, A.; Humpert, P.M.; Stern, D.M.; Arnold, B.; Nawroth, P.P. Advanced glycation end product receptor-mediated cellular dysfunction. Ann. N. Y. Acad. Sci. 2005, 1043, 676–680. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Sun, W.; Gao, R.; Su, Y.; Umehara, H.; Dong, L.; Gong, F. The role of high mobility group box chromosomal protein 1 in rheumatoid arthritis. Rheumatology 2013, 52, 1739–1747. [Google Scholar] [CrossRef][Green Version]
- Kokkola, R.; Sundberg, E.; Ulfgren, A.-K.; Palmblad, K.; Li, J.; Wang, H.; Ulloa, L.; Yang, H.; Yan, X.-J.; Furie, R.; et al. High mobility group box chromosomal protein 1: A novel proinflammatory mediator in synovitis. Arthritis Rheum. 2002, 46, 2598–2603. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, N.; Kawahara, K.; Yone, K.; Hashiguchi, T.; Yamakuchi, M.; Goto, M.; Inoue, K.; Yamada, S.; Ijiri, K.; Matsunaga, S.; et al. High mobility group box chromosomal protein 1 plays a role in the pathogenesis of rheumatoid arthritis as a novel cytokine. Arthritis Rheum. 2003, 48, 971–981. [Google Scholar] [CrossRef] [PubMed]
- Pullerits, R.; Jonsson, I.M.; Verdrengh, M.; Bokarewa, M.; Andersson, U.; Erlandsson-Harris, H.; Tarkowski, A. High mobility group box chromosomal protein 1, a DNA binding cytokine, induces arthritis. Arthritis Rheum. 2003, 48, 1693–1700. [Google Scholar] [CrossRef] [PubMed]
- Kokkola, R.; Li, J.; Sundberg, E.; Aveberger, A.C.; Palmblad, K.; Yang, H.; Tracey, K.J.; Andersson, U.; Harris, H.E. Successful treatment of collagen-induced arthritis in mice and rats by targeting extracellular high mobility group box chromosomal protein 1 activity. Arthritis Rheum. 2003, 48, 2052–2058. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xu, P.; Xu, K.; Cai, Y.-S.; Sun, M.; Yang, L.; Sun, J.; Lu, S. Methotrexate affects HMGB1 expression in rheumatoid arthritis, and the downregulation of HMGB1 prevents rheumatoid arthritis progression. Mol. Cell. Biochem. 2016, 420, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Kuroiwa, Y.; Takakusagi, Y.; Kusayanagi, T.; Kuramochi, K.; Imai, T.; Hirayama, T.; Ito, I.; Yoshida, M.; Sakaguchi, K.; Sugawara, F. Identification and Characterization of the Direct Interaction between Methotrexate (MTX) and High-Mobility Group Box 1 (HMGB1) Protein. PLoS ONE 2013, 8, e63073. [Google Scholar] [CrossRef]
- Braun, J. Optimal administration and dosage of methotrexate. Clin. Exp. Rheumatol. 2010, 28, S46–S51. [Google Scholar] [PubMed]
- van Ede, A.E.; Laan, R.F.J.M.; Blom, H.J.; De Abreu, R.A.; van de Putte, L.B.A. Methotrexate in rheumatoid arthritis: An updatewith focus on mechanisms involved in toxicity. Semin. Arthritis Rheum. 1998, 27, 277–292. [Google Scholar] [CrossRef]
- Abolmaali, S.S.; Tamaddon, A.M.; Dinarvand, R. A review of therapeutic challenges and achievements of methotrexate delivery systems for treatment of cancer and rheumatoid arthritis. Cancer Chemother. Pharm. 2013, 71, 1115–1130. [Google Scholar] [CrossRef] [PubMed]
- Goodman, S.M.; Cronstein, B.N.; Bykerk, V.P. Outcomes Related to Methotrexate Dose and Route of Administration in Patients with Rheumatoid Arthritis: A Systematic Literature Review. Clin. Exp. Rheumatol. 2015, 33, 272–278. [Google Scholar] [PubMed]
- Kromann, C.B.; Lage-Hansen, P.R.; Koefoed, M.; Jemec, G.B.E. Does switching from oral to subcutaneous administration of methotrexate influence on patient reported gastro-intestinal adverse effects? J. Dermatol. Treat. 2015, 26, 188–190. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Yang, Z.; Kang, P.; Xie, X. Subcutaneous administration of methotrexate at high doses makes a better performance in the treatment of rheumatoid arthritis compared with oral administration of methotrexate: A systematic review and meta-analysis. Semin. Arthritis Rheum. 2016, 45, 656–662. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhou, H.; Liu, L. Side effects of methotrexate therapy for rheumatoid arthritis: A systematic review. Eur. J. Med. Chem. 2018, 158, 502–516. [Google Scholar] [CrossRef] [PubMed]
- Paul, M.; Hemshekhar, M.; Thushara, R.M.; Sundaram, M.S.; NaveenKumar, S.K.; Naveen, S.; Devaraja, S.; Somyajit, K.; West, R.; Nayaka, S.C.; et al. Methotrexate Promotes Platelet Apoptosis via JNK-Mediated Mitochondrial Damage: Alleviation by N-Acetylcysteine and N-Acetylcysteine Amide. PLoS ONE 2015, 10, e0127558. [Google Scholar] [CrossRef] [PubMed]
- Kremer, J.M.; Galivan, J.; Streckfuss, A.; Kamen, B. Methotrexate metabolism analysis in blood and liver of rheumatoid arthritis patients. Association with hepatic folate deficiency and formation of polyglutamates. Arthritis Rheum. 1986, 29, 832–835. [Google Scholar] [CrossRef]
- Chan, E.S.L.; Montesinos, M.C.; Fernandez, P.; Desai, A.; Delano, D.L.; Yee, H.; Reiss, A.B.; Pillinger, M.H.; Chen, J.F.; Schwarzschild, M.A.; et al. Adenosine A2A receptors play a role in the pathogenesis of hepatic cirrhosis. Br. J. Pharm. 2006, 148, 1144–1155. [Google Scholar] [CrossRef]
- Che, J.; Chan, E.S.L.; Cronstein, B.N. Adenosine A2A Receptor Occupancy Stimulates Collagen Expression by Hepatic Stellate Cells via Pathways Involving Protein Kinase A, Src, and Extracellular Signal-Regulated Kinases 1/2 Signaling Cascade or p38 Mitogen-Activated Protein Kinase Signaling Pathway. Mol. Pharm. 2007, 72, 1626–1636. [Google Scholar]
- Aithal, G.P. Hepatotoxicity related to antirheumatic drugs. Nat. Rev. Rheumatol. 2011, 7, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Ortega-Alonso, A.; Andrade, R.J. Chronic liver injury induced by drugs and toxins. J. Dig. Dis. 2018, 19, 514–521. [Google Scholar] [CrossRef] [PubMed]
- Vardi, N.; Parlakpinar, H.; Cetin, A.; Erdogan, A.; Cetin Ozturk, I. Protective Effect of β-Carotene on Methotrexate–Induced Oxidative Liver Damage. Toxicol. Pathol. 2010, 38, 592–597. [Google Scholar] [CrossRef]
- Kim, Y.J.; Song, M.; Ryu, J.C. Inflammation in methotrexate-induced pulmonary toxicity occurs via the p38 MAPK pathway. Toxicology 2009, 256, 183–190. [Google Scholar] [CrossRef]
- Lynch, J.P.; McCune, W.J. Immunosuppressive and cytotoxic pharmacotherapy for pulmonary disorders. Am. J. Respir. Crit. Care Med. 1997, 155, 395–420. [Google Scholar] [CrossRef]
- Salaffi, F.; Manganelli, P.; Carotti, M.; Subiaco, S.; Lamanna, G.; Cervini, C. Methotrexate-induced pneumonitis in patients with rheumatoid arthritis and psoriatic arthritis: Report of five cases and review of the literature. Clin. Rheumatol. 1997, 16, 296–304. [Google Scholar] [CrossRef]
- Lateef, O.; Shakoor, N.; Balk, R.A. Methotrexate pulmonary toxicity. Expert Opin. Drug Saf. 2005, 4, 723–730. [Google Scholar] [CrossRef]
- Ohbayashi, M.; Suzuki, M.; Yashiro, Y.; Fukuwaka, S.; Yasuda, M.; Kohyama, N.; Kobayashi, Y.; Yamamoto, T. Induction of pulmonary fibrosis by methotrexate treatment in mice lung in vivo and in vitro. J. Toxicol. Sci. 2010, 35, 653–661. [Google Scholar] [CrossRef]
- Golden, M.R.; Katz, R.S.; Balk, R.A.; Golden, H.E. The relationship of preexisting lung disease to the development of methotrexate pneumonitis in patients with rheumatoid arthritis. J. Rheumatol. 1995, 22, 1043–1047. [Google Scholar]
- Verstappen, S.M.M.; Bakker, M.F.; Heurkens, A.H.M.; Veen, M.J.; van der Kruize, A.A.; Geurts, M.A.W.; Bijlsma, J.W.J.; Jacobs, J.W.G. Adverse events and factors associated with toxicity in patients with early rheumatoid arthritis treated with methotrexate tight control therapy: The CAMERA study. Ann. Rheum. Dis. 2010, 69, 1044–1048. [Google Scholar] [CrossRef] [PubMed]
- Kremer, J.M.; Petrillo, G.F.; Hamilton, R.A. Pharmacokinetics and renal function in patients with rheumatoid arthritis receiving a standard dose of oral weekly methotrexate: Association with significant decreases in creatinine clearance and renal clearance of the drug after 6 months of therapy. J. Rheumatol. 1995, 22, 38–40. [Google Scholar] [PubMed]
- Seideman, P.; Müller-Suur, R.; Ekman, E. Renal effects of low dose methotrexate in rheumatoid arthritis. J. Rheumatol. 1993, 20, 1126–1128. [Google Scholar] [CrossRef] [PubMed]
- Abelson, H.T.; Fosburg, M.T.; Beardsley, G.P.; Goorin, A.M.; Gorka, C.; Link, M.; Link, D. Methotrexate-induced renal impairment: Clinical studies and rescue from systemic toxicity with high-dose leucovorin and thymidine. JCO 1983, 1, 208–216. [Google Scholar] [CrossRef] [PubMed]
- Grönroos, M.; Chen, M.; Jahnukainen, T.; Capitanio, A.; Aizman, R.I.; Celsi, G. Methotrexate induces cell swelling and necrosis in renal tubular cells. Pediatr. Blood Cancer 2006, 46, 624–629. [Google Scholar] [CrossRef] [PubMed]
- Cronstein, B.N. The mechanism of action of methotrexate. Rheum. Dis. Clin. N. Am. 1997, 23, 739–755. [Google Scholar] [CrossRef]
- Li, X.; Abe, E.; Yamakawa, Y.; Yoneda, G.; Fujino, R.; Yamashita, M.; Iida, Y.; Jono, H.; Saito, H. Effect of Administration Duration of Low Dose Methotrexate on Development of Acute Kidney Injury in Rats. J. Kidney 2016. [Google Scholar] [CrossRef]
- Merrill, J.T.; Shen, C.; Schreibman, D.; Coffey, D.; Zakharenko, O.; Fisher, R.; Lahita, R.G.; Salmon, J.; Cronstein, B.N. Adenosine A1 receptor promotion of multinucleated giant cell formation by human monocytes: A mechanism for methotrexate-induced nodulosis in rheumatoid arthritis. Arthritis Rheum. 1997, 40, 1308–1315. [Google Scholar] [PubMed]
- Albrecht, K.; Müller-Ladner, U. Side effects and management of side effects of methotrexate in rheumatoid arthritis. Clin. Exp. Rheumatol. 2010, 28, S95–S101. [Google Scholar]
- Thakkar, M.M.; Engemann, S.C.; Walsh, K.M.; Sahota, P.K. Adenosine and the homeostatic control of sleep: Effects of A1 receptor blockade in the perifornical lateral hypothalamus on sleep-wakefulness. Neuroscience 2008, 153, 875–880. [Google Scholar] [CrossRef]
- Bernini, J.C.; Fort, D.W.; Griener, J.C.; Kane, B.J.; Chappell, W.B.; Kamen, B.A. Aminophylline for methotrexate-induced neurotoxicity. Lancet 1995, 345, 544–547. [Google Scholar] [CrossRef]
- Quinn, C.T.; Griener, J.C.; Bottiglieri, T.; Hyland, K.; Farrow, A.; Kamen, B.A. Elevation of homocysteine and excitatory amino acid neurotransmitters in the CSF of children who receive methotrexate for the treatment of cancer. J. Clin. Oncol. 1997, 15, 2800–2806. [Google Scholar] [CrossRef] [PubMed]
- Millot, F.; Dhondt, J.L.; Mazingue, F.; Mechinaud, F.; Ingrand, P.; Guilhot, F. Changes of Cerebral Biopterin and Biogenic Amine Metabolism in Leukemic Children Receiving 5 g/m2 Intravenous Methotrexate. Pediatric Res. 1995, 37, 151–154. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Buckley, L.M.; Bullaboy, C.A.; Leichtman, L.; Marquez, M. Multiple congenital anomalies associated with weekly low-dose methotrexate treatment of the mother. Arthritis Rheum. 1997, 40, 971–973. [Google Scholar] [CrossRef] [PubMed]
- May, K.P.; West, S.G.; McDermott, M.T.; Huffer, W.E. The effect of low-dose methotrexate on bone metabolism and histomorphometry in rats. Arthritis Rheum. 1994, 37, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Cranney, A.B.; McKendry, R.J.; Wells, G.A.; Ooi, D.S.; Kanigsberg, N.D.; Kraag, G.R.; Smith, C.D. The effect of low dose methotrexate on bone density. J. Rheumatol. 2001, 28, 2395–2399. [Google Scholar]
- Patel, S.; Patel, G.; Johnson, D.; Ogunremi, L.; Barron, J. Effect of low dose weekly methotrexate on bone mineral density and bone turnover. Ann. Rheum. Dis. 2003, 62, 186–187. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, Y.; Suwa, A.; Ikeda, Y.; Hirakata, M. Pneumocystis jiroveci pneumonia associated with low-dose methotrexate treatment for rheumatoid arthritis: Report of two cases and review of the literature. Mod. Rheumatol. 2006, 16, 36–38. [Google Scholar] [CrossRef] [PubMed]
- Veen, M.J.; van der Heide, A.; van der Kruize, A.A.; Bijlsma, J.W. Infection rate and use of antibiotics in patients with rheumatoid arthritis treated with methotrexate. Ann. Rheum. Dis. 1994, 53, 224–228. [Google Scholar] [CrossRef] [PubMed]
- Bernatsky, S.; Hudson, M.; Suissa, S. Anti-rheumatic drug use and risk of serious infections in rheumatoid arthritis. Rheumatology 2007, 46, 1157–1160. [Google Scholar] [CrossRef]
- Doran, M.F.; Crowson, C.S.; Pond, G.R.; O’Fallon, W.M.; Gabriel, S.E. Predictors of infection in rheumatoid arthritis. Arthritis Rheum. 2002, 46, 2294–2300. [Google Scholar] [CrossRef] [PubMed]
- Smitten, A.L.; Choi, H.K.; Hochberg, M.C.; Suissa, S.; Simon, T.A.; Testa, M.A.; Chan, K.A. The risk of hospitalized infection in patients with rheumatoid arthritis. J. Rheumatol. 2008, 35, 387–393. [Google Scholar] [PubMed]
- Salliot, C.; van der Heijde, D. Long-term safety of methotrexate monotherapy in patients with rheumatoid arthritis: A systematic literature research. Ann. Rheum. Dis. 2009, 68, 1100–1104. [Google Scholar] [CrossRef] [PubMed]
- McLean-Tooke, A.; Aldridge, C.; Waugh, S.; Spickett, G.P.; Kay, L. Methotrexate, rheumatoid arthritis and infection risk: What is the evidence? Rheumatology 2009, 48, 867–871. [Google Scholar] [CrossRef]
- Beauparlant, P.; Papp, K.; Haraoui, B. The incidence of cancer associated with the treatmentof rheumatoid arthritis. Semin. Arthritis Rheum. 1999, 29, 148–158. [Google Scholar] [CrossRef]
- Baecklund, E.; Ekbom, A.; Sparén, P.; Feltelius, N.; Klareskog, L. Disease activity and risk of lymphoma in patients with rheumatoid arthritis: Nested case-control study. BMJ 1998, 317, 180–181. [Google Scholar] [CrossRef]
- Kamel, O.W.; van de Rijn, M.; LeBrun, D.P.; Weiss, L.M.; Warnke, R.A.; Dorfman, R.F. Lymphoid neoplasms in patients with rheumatoid arthritis and dermatomyositis: Frequency of Epstein-Barr virus and other features associated with immunosuppression. Hum. Pathol. 1994, 25, 638–643. [Google Scholar] [CrossRef]
- Moder, K.G.; Tefferi, A.; Cohen, M.D.; Menke, D.M.; Luthra, H.S. Hematologic malignancies and the use of methotrexate in rheumatoid arthritis: A retrospective study. Am. J. Med. 1995, 99, 276–281. [Google Scholar] [CrossRef]
- Puig, J.G.; Fox, I.H. Ethanol-induced activation of adenine nucleotide turnover. Evidence for a role of acetate. J. Clin. Investig. 1984, 74, 936–941. [Google Scholar] [CrossRef]
- St Clair, E.W.; Rice, J.R.; Snyderman, R. Pneumonitis complicating low-dose methotrexate therapy in rheumatoid arthritis. Arch. Intern. Med. 1985, 145, 2035–2038. [Google Scholar] [CrossRef]
- Kane, B.J.; Kuhn, J.G.; Roush, M.K. Pentostatin: An Adenosine Deaminase Inhibitor for the Treatment of Hairy Cell Leukemia. Ann. Pharm. 1992, 26, 939–947. [Google Scholar] [CrossRef] [PubMed]
- Motegi, S.; Ishikawa, O. Methotrexate-induced accelerated nodulosis in a patient with rheumatoid arthritis and scleroderma. Acta Derm. Venereol. 2014, 94, 357–358. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Izzedine, H.; Launay-Vacher, V.; Karie, S.; Caramella, C.; de Person, F.; Deray, G. Is low-dose methotrexate nephrotoxic? Case report and review of the literature. Clin. Nephrol. 2005, 64, 315–319. [Google Scholar] [CrossRef] [PubMed]
- Widemann, B.C.; Adamson, P.C. Understanding and Managing Methotrexate Nephrotoxicity. Oncologist 2006, 11, 694–703. [Google Scholar] [CrossRef] [PubMed]
- Saag, K.G.; Teng, G.G.; Patkar, N.M.; Anuntiyo, J.; Finney, C.; Curtis, J.R.; Paulus, H.E.; Mudano, A.; Pisu, M.; Elkins-Melton, M.; et al. American College of Rheumatology 2008 recommendations for the use of nonbiologic and biologic disease-modifying antirheumatic drugs in rheumatoid arthritis. Arthritis Rheum. 2008, 59, 762–784. [Google Scholar] [CrossRef] [PubMed]
- Smolen, J.S.; Aletaha, D. Rheumatoid arthritis therapy reappraisal: Strategies, opportunities and challenges. Nat. Rev. Rheumatol. 2015, 11, 276–289. [Google Scholar] [CrossRef]
- Ranganathan, P. An update on methotrexate pharmacogenetics in rheumatoid arthritis. Pharmacogenomics 2008, 9, 439–451. [Google Scholar] [CrossRef]
- Owen, S.A.; Hider, S.L.; Martin, P.; Bruce, I.N.; Barton, A.; Thomson, W. Genetic polymorphisms in key methotrexate pathway genes are associated with response to treatment in rheumatoid arthritis patients. Pharm. J. 2013, 13, 227–234. [Google Scholar] [CrossRef]
- Qiu, Q.; Huang, J.; Shu, X.; Fan, H.; Zhou, Y.; Xiao, C. Polymorphisms and Pharmacogenomics for the Clinical Efficacy of Methotrexate in Patients with Rheumatoid Arthritis: A Systematic Review and Meta-analysis. Sci. Rep. 2017, 7, 44015. [Google Scholar] [CrossRef]
- Dervieux, T.; Wessels, J.A.M.; van der Straaten, T.; Penrod, N.; Moore, J.H.; Guchelaar, H.-J.; Kremer, J.M. Gene-gene interactions in folate and adenosine biosynthesis pathways affect methotrexate efficacy and tolerability in rheumatoid arthritis. Pharmacogenet. Genom. 2009, 19, 935–944. [Google Scholar] [CrossRef]
- Wessels, J.A.M.; Kooloos, W.M.; Jonge, R.D.; Vries-Bouwstra, J.K.D.; Allaart, C.F.; Linssen, A.; Collee, G.; Sonnaville, P.D.; Lindemans, J.; Huizinga, T.W.J.; et al. Relationship between genetic variants in the adenosine pathway and outcome of methotrexate treatment in patients with recent-onset rheumatoid arthritis. Arthritis Rheum. 2006, 54, 2830–2839. [Google Scholar] [CrossRef] [PubMed]
- Wessels, J.A.M.; van der Kooij, S.M.; le Cessie, S.; Kievit, W.; Barerra, P.; Allaart, C.F.; Huizinga, T.W.J.; Guchelaar, H.J. Pharmacogenetics Collaborative Research Group A clinical pharmacogenetic model to predict the efficacy of methotrexate monotherapy in recent-onset rheumatoid arthritis. Arthritis Rheum. 2007, 56, 1765–1775. [Google Scholar] [CrossRef] [PubMed]
- Pastore, S.; Stocco, G.; Favretto, D.; De Iudicibus, S.; Taddio, A.; d’Adamo, P.; Malusà, N.; Addobbati, R.; Decorti, G.; Lepore, L.; et al. Genetic determinants for methotrexate response in juvenile idiopathic arthritis. Front Pharm. 2015, 6, 52. [Google Scholar] [CrossRef] [PubMed]
- Senapati, S.; Singh, S.; Das, M.; Kumar, A.; Gupta, R.; Kumar, U.; Jain, S.; Juyal, R.; Thelma, B.K. Genome-wide analysis of methotrexate pharmacogenomics in rheumatoid arthritis shows multiple novel risk variants and leads for TYMS regulation. Pharm. Genom. 2014, 24, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Cobb, J.; Cule, E.; Moncrieffe, H.; Hinks, A.; Ursu, S.; Patrick, F.; Kassoumeri, L.; Flynn, E.; Bulatović, M.; Wulffraat, N.; et al. Genome-Wide Data Reveals Novel Genes for Methotrexate Response in a Large Cohort of Juvenile Idiopathic Arthritis Cases. Pharm. J. 2014, 14, 356–364. [Google Scholar] [CrossRef]
- Taylor, J.C.; Bongartz, T.; Massey, J.; Mifsud, B.; Spiliopoulou, A.; Scott, I.C.; Wang, J.; Morgan, M.; Plant, D.; Colombo, M.; et al. Genome-wide association study of response to methotrexate in early rheumatoid arthritis patients. Pharm. J. 2018, 18, 528–538. [Google Scholar] [CrossRef] [PubMed]
- Nakaya, H.I.; Gardner, J.; Poo, Y.S.; Major, L.; Pulendran, B.; Suhrbier, A. Gene Profiling of Chikungunya Virus Arthritis in a Mouse Model Reveals Significant Overlap with Rheumatoid Arthritis. Arthritis Rheum 2012, 64, 3553–3563. [Google Scholar] [CrossRef]
- Chen, W.; Foo, S.S.; Rulli, N.E.; Taylor, A.; Sheng, K.-C.; Herrero, L.J.; Herring, B.L.; Lidbury, B.A.; Li, R.W.; Walsh, N.C.; et al. Arthritogenic alphaviral infection perturbs osteoblast function and triggers pathologic bone loss. Proc. Natl. Acad. Sci. USA 2014, 111, 6040–6045. [Google Scholar] [CrossRef]
- Kudaeva, F.M.; Speechley, M.R.; Pope, J.E. A systematic review of viral exposures as a risk for rheumatoid arthritis. Semin. Arthritis Rheum. 2018, 48, 587–596. [Google Scholar] [CrossRef]
- Amaral, J.K.; Sutaria, R.; Schoen, R.T. Treatment of Chronic Chikungunya Arthritis with Methotrexate: A Systematic Review. Arthritis Care Res. 2018, 70, 1501–1508. [Google Scholar] [CrossRef]
- Pereira, J.K.A.; Schoen, R.T. Management of chikungunya arthritis. Clin. Rheumatol. 2017, 36, 2179–2186. [Google Scholar] [CrossRef] [PubMed]
- Javelle, E.; Ribera, A.; Degasne, I.; Gaüzère, B.A.; Marimoutou, C.; Simon, F. Specific Management of Post-Chikungunya Rheumatic Disorders: A Retrospective Study of 159 Cases in Reunion Island from 2006–2012. PLoS Negl. Trop. Dis. 2015, 9, e0003603. [Google Scholar] [CrossRef] [PubMed]
- Ganu, M.A.; Ganu, A.S. Post-chikungunya chronic arthritis—Our experience with DMARDs over two year follow up. J. Assoc. Phys. India 2011, 59, 83–86. [Google Scholar]
- Ravindran, V.; Alias, G. Efficacy of combination DMARD therapy vs. hydroxychloroquine monotherapy in chronic persistent chikungunya arthritis: A 24-week randomized controlled open label study. Clin. Rheumatol. 2017, 36, 1335–1340. [Google Scholar] [CrossRef] [PubMed]
- Simon, F.; Javelle, E.; Oliver, M.; Leparc-Goffart, I.; Marimoutou, C. Chikungunya Virus Infection. Curr. Infect. Dis. Rep. 2011, 13, 218–228. [Google Scholar] [CrossRef] [PubMed]
- Foissac, M.; Javelle, E.; Ray, S.; Guérin, B.; Simon, F. Post-Chikungunya Rheumatoid Arthritis, Saint Martin. Emerg. Infect. Dis. 2015, 21, 530–532. [Google Scholar] [CrossRef] [PubMed]
- Goupil, B.A.; Mores, C.N. A Review of Chikungunya Virus-induced Arthralgia: Clinical Manifestations, Therapeutics, and Pathogenesis. Open Rheumatol. J. 2016, 10, 129–140. [Google Scholar] [CrossRef]
- Malvy, D.; Ezzedine, K.; Mamani-Matsuda, M.; Autran, B.; Tolou, H.; Receveur, M.C.; Pistone, T.; Rambert, J.; Moynet, D.; Mossalayi, D. Destructive arthritis in a patient with chikungunya virus infection with persistent specific IgM antibodies. BMC Infect. Dis. 2009, 9, 200. [Google Scholar] [CrossRef]
- Taylor, A.; Sheng, K.C.; Herrero, L.J.; Chen, W.; Rulli, N.E.; Mahalingam, S. Methotrexate Treatment Causes Early Onset of Disease in a Mouse Model of Ross River Virus-Induced Inflammatory Disease through Increased Monocyte Production. PLoS ONE 2013, 8, e71146. [Google Scholar] [CrossRef]
- Bedoui, Y.; Giry, C.; Jaffar-Bandjee, M.C.; Selambarom, J.; Guiraud, P.; Gasque, P. Immunomodulatory drug methotrexate used to treat patients with chronic inflammatory rheumatisms post-chikungunya does not impair the synovial antiviral and bone repair responses. PLoS Negl. Trop. Dis. 2018, 12, e0006634. [Google Scholar] [CrossRef]
- Miner, J.J.; Cook, L.E.; Hong, J.P.; Smith, A.M.; Richner, J.M.; Shimak, R.M.; Young, A.R.; Monte, K.; Poddar, S.; Crowe, J.E.; et al. Therapy with CTLA4-Ig and an antiviral monoclonal antibody controls chikungunya virus arthritis. Sci. Transl. Med. 2017, 9, eaah3438. [Google Scholar] [CrossRef] [PubMed]
- Palazzi, C.; D’Angelo, S.; Olivieri, I. Hepatitis C virus-related arthritis. Autoimmun. Rev. 2008, 8, 48–51. [Google Scholar] [CrossRef] [PubMed]
- Nissen, M.J.; Fontanges, E.; Allam, Y.; Zoulim, F.; Trépo, C.; Miossec, P. Rheumatological manifestations of hepatitis C: Incidence in a rheumatology and non-rheumatology setting and the effect of methotrexate and interferon. Rheumatology 2005, 44, 1016–1020. [Google Scholar] [CrossRef] [PubMed]
- Mok, M.Y.; Ng, W.L.; Yuen, M.F.; Wong, R.W.; Lau, C.S. Safety of disease modifying anti-rheumatic agents in rheumatoid arthritis patients with chronic viral hepatitis. Clin. Exp. Rheumatol. 2000, 18, 363–368. [Google Scholar]
- Kujawska, A.; Clements, M.; Wise, C.M.; Roberts, W.N. Hepatitis C and methotrexate. Arthritis Care Res. 2003, 49, 843–845. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.L.; Jing, J.; Mo, Y.Q.; Ma, J.D.; Yang, L.J.; Chen, L.F.; Zhang, X.; Yan, T.; Zheng, D.H.; Pessler, F.; et al. Presence of hepatitis B virus in synovium and its clinical significance in rheumatoid arthritis. Arthritis Res. 2018, 20, 130. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Nakazono, K.; Murasawa, A.; Mita, Y.; Hata, K.; Saito, N.; Kikuchi, M.; Yoshida, K.; Nakano, M.; Gejyo, F. Development of fulminant hepatitis B (precore variant mutant type) after the discontinuation of low-dose methotrexate therapy in a rheumatoid arthritis patient. Arthritis Rheum. 2001, 44, 339–342. [Google Scholar] [CrossRef]
- Ostuni, P.; Botsios, C.; Punzi, L.; Sfriso, P.; Todesco, S. Hepatitis B reactivation in a chronic hepatitis B surface antigen carrier with rheumatoid arthritis treated with infliximab and low dose methotrexate. Ann. Rheum. Dis. 2003, 62, 686–687. [Google Scholar] [CrossRef]
- Laohapand, C.; Arromdee, E.; Tanwandee, T. Long-term use of methotrexate does not result in hepatitis B reactivation in rheumatologic patients. Hepatol. Int. 2015, 9, 202–208. [Google Scholar] [CrossRef]
- Tang, K.T.; Chen, Y.H.; Lin, C.H.; Chen, D.Y. Methotrexate is not associated with increased liver cirrhosis in a population-based cohort of rheumatoid arthritis patients with chronic hepatitis C. Sci. Rep. 2016, 6, 33104. [Google Scholar]
- Tang, K.T.; Hung, W.T.; Chen, Y.H.; Lin, C.H.; Chen, D.Y. Methotrexate is not associated with increased liver cirrhosis in a population-based cohort of rheumatoid arthritis patients with chronic hepatitis B. Sci. Rep. 2016, 6, 22387. [Google Scholar]
- Nguyen, B.Y.; Reveille, J.D. Rheumatic manifestations associated with HIV in the highly active antiretroviral therapy era. Curr. Opin. Rheumatol. 2009, 21, 404–410. [Google Scholar] [CrossRef] [PubMed]
- Almoallim, H.; Jali, I.; Wali, G. Successful use of antitumor necrosis factor-alpha biological therapy in managing human immunodeficiency virus-associated arthritis: Three case studies from Saudi Arabia. Jt. Bone Spine 2013, 80, 426–428. [Google Scholar] [CrossRef] [PubMed]
- Carroll, M.B.; Fields, J.H.; Clerc, P.G. Rheumatoid arthritis in patients with HIV: Management challenges. Open Access Rheumatol. 2016, 8, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Duvic, M.; Johnson, T.M.; Rapini, R.P.; Freese, T.; Brewton, G.; Rios, A. Acquired Immunodeficiency Syndrome—Associated Psoriasis and Reiter’s Syndrome. Arch. Derm. 1987, 123, 1622–1632. [Google Scholar] [CrossRef] [PubMed]
- Menon, K.; Voorhees, A.S.V.; Bebo, B.F.; Gladman, D.D.; Hsu, S.; Kalb, R.E.; Lebwohl, M.G.; Strober, B.E. Psoriasis in patients with HIV infection: From the Medical Board of the National Psoriasis Foundation. J. Am. Acad. Dermatol. 2010, 62, 291–299. [Google Scholar] [CrossRef]
- Maurer, T.A.; Zackheim, H.S.; Tuffanelli, L.; Berger, T.G. The use of methotrexate for treatment of psoriasis in patients with HIV infection. J. Am. Acad. Dermatol. 1994, 31, 372–375. [Google Scholar] [CrossRef]
- Masson, C.; Chennebault, J.M.; Leclech, C. Is HIV infection contraindication to the use of methotrexate in psoriatic arthritis? J. Rheumatol. 1995, 22, 2191. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Organ System | MTX Related Adverse Events | Toxic Mechanism of Action |
|---|---|---|
| Gastrointestinal | Nausea; Vomiting; Diarrhea; Mucositis and stomatitis | Gastrointestinal toxicities and bone marrow suppression seem to be directly related to folate antagonism, because these tissues have high cell turnover with a high requirement for purines, thymidine, and pyrimidine [37,143,144]. Supplementation of folic or folinic acid may diminish toxicity. Gastrointestinal symptoms of nausea and diarrhea may be more frequent with oral MTX [145]. Switching from oral to parental administration was shown to significantly decrease the frequency of adverse gastrointestinal events in patients with RA [146,147], suggesting that other mechanisms may account for MTX induced gastrointestinal toxicity. The pathogenic mechanism underlying gastrointestinal side effects may also be related to the change of plasma homocysteine [148]. |
| Hematological | Anaemia; Leucopenia; Thrombopenia; Pancytopenia | Recently, MTX-induced thrombocytopenia was shown to be mediated by MTX-induced activation of platelet apoptosis via JNK and oxidative stress [149]. |
| Hepatic | Elevated liver enzymes | Long-term MTX administration can cause accumulation of MTX polyglutamates in the liver and decreased folate levels. The depletion of hepatic folate stores by accumulated MTX poly glutamates is one possible toxic effect of MTX on the liver [150]. Folate supplementation has been associated with a reduced incidence of elevated transaminases induced by MTX treatment [15]. |
| Steatosis, fibrosis, cirrhosis | MTX-related hepatic fibrosis may be mediated through an adenosine pathway. MTX was shown to enhance adenosine release from cultured hepatoma (HepG2) cells. Adenosine A2A receptor occupancy stimulates collagen production by hepatic stellate cell lines [151,152]. Unlike wild-type mice, mice deficient for the adenosine A2A receptor or treated with an adenosine A2A receptor antagonist (ZM241385) were protected from developing liver fibrosis when challenged by hepatotoxin (carbon tetrachloride or thiocetamide) [151]. MTX-related liver fibrosis may also be mediated by its capacity to interfere with the generation of methionine from homocysteine. Excess of homocysteine induces endoplasmic reticulum stress promoting fat accumulation in the liver. Homocysteine can also activate hepatic stellate cells and proinflammatory cytokines, leading to liver fibrosis [153,154]. MTX-induced hepatic damage may be related to the generation of reactive oxygen species (ROS). MTX was shown to cause oxidative tissue damage by increasing lipid peroxidation in the liver tissue and decreasing the level of antioxidant enzymes in rats [155]. | |
| Pulmonary | Interstitial pneumonitis; Pneumocystis carinii pneumonia; Pulmonary fibrosis | Pulmonary toxicity has been shown to occur at both high- and low-dose MTX treatment, suggesting an idiosyncratic reaction not linked to folate antagonism [49]. Several mechanisms for the pathogenesis of MTX-induced pneumonitis have been suggested including hypersensitivity, direct drug toxicity to the lung tissue, immunosuppression or altered cytokine expression contributing to the pulmonary inflammatory response and tissue damage [156]. Typical bronchoalveolar lavage and histological findings support the concept that MTX-induced pneumonitis represents a hypersensitivity reaction [157,158,159]. MTX also induces injury to alveolar epithelial walls and pulmonary fibrosis, suggesting a direct drug toxicity route [160]. MTX pulmonary toxicity may be mediated by mitogen-activated protein kinase (MAPK) pathways activation and cytokine release [156]. MTX can compromise the immune response and increase the risk for opportunistic infections due to Pneumocystis carinii [161]. |
| Renal | A decrease in glomerular filtration rate; Renal insufficiency (only in pre-existing, severely impaired renal function) | In contrast to high-dose MTX, which can lead to direct tubulus toxicity and subsequent renal failure, renal toxicities induced by low-dose MTX are rare. Low dose MTX has been associated with decrease in glomerular filtration rate (GFR) [162,163,164]. MTX and its major metabolite 7-OH-MTX are relatively insoluble in acid urine and may act as a direct toxin on the tubular epithelium, or precipitate within the tubular lumen, which can lead to intratubular obstruction resulting in a decrease in GFR (particularly at high doses) [49,165]. Evidence for a direct toxic effect of MTX on renal tubular cells has been demonstrated [166]; Low doses MTX can induce cell swelling and necrosis in renal tubular cells, which may lead to permanent tubular damage [166]. MTX associated renal toxicity may be explained by an increase in plasma adenosine concentration in extracellular fluid and subsequent activation of A1 receptors in renal tissue, reducing renal blood flow and salt and water excretion [167]. Long duration of low dose MTX administration caused severe kidney injury and renal MTX accumulation in a rat model. 4-hydroxynonenal (4-HNE) and malondialdehyde (MDA), which are reliable oxidative stress markers, were significantly increased in Long-MTX treated rats suggesting that MTX-induced kidney injury may be mediated through an increase in oxidative stress [168]. |
| Dermatologic | Nodulosis (rare); Alopecia; Rash; Anaphylactic reactions | MTX-induced nodulosis may be mediated by adenosine through the adenosine A1 receptor [169]. MTX was shown to induce the generation of multinucleated giant cells, as does adenosine A1 receptor occupancy. This effect of MTX was reversed by a specific adenosine A1 receptor antagonist. Alopecia related to MTX treatment seems to be related to folate antagonism. In low dose MTX treatment, alopecia is rare and generally resolves several months after discontinuation [17,170]. |
| Central nervous system (CNS) | Lethargy and fatigue; Headache, vertigo (less frequent) | Neurotoxicity of MTX may be related to MTX induced adenosine release and accumulation in the CNS. By acting at the A1 receptor on the perifornical lateral hypothalamus, adenosine may regulate wakefulness and somnolence and so potentially explaining asthenia and sleepiness experienced by some patients after MTX intake [171,172]. Other possible mechanisms of MTX-induced neurotoxicity are increased homocysteine levels and their excitatory amino acid neurotransmitter metabolites as homocysteic acid and cysteinesulfinic acid [173] and impairment of biopterin regerenating system in the brain, resulting in a reduced monoamine neurotransmitters availability [174]. |
| Urogenital | Teratogenecity; oligospermia; gynecomastia (rare) | Use of MTX should be avoided before or during pregnancy because of its documented embryotoxicity and teratogenicity [175]. |
| Musculoskeletal | Osteopathy; Osteoporosis | The effect of low dose MTX on bone was described in rats. Prolonged administration of low dose MTX in rats caused significant osteopenia with reduced osteoblast activity and increased osteoclast recruitment, which results in increased bone resorption [176]. However, no detrimental impact of MTX on the skeleton has been reported in patients treated with low dose MTX. MTX seems to have no clinically significant effect on bone mineral density (BMD) or on the osteoblast lineage [177,178]. |
| Immunologic | Opportunistic infections | There is a belief amongst rheumatologists that MTX, as an immunosuppressant drug, is asssociated with the development of opportunistic infections. Weekly low-doses MTX can affect T cell activity [58], and cases of Pneumocystis pneumonia, nocardiosis, aspergillum, cryptococcosis, herpes zoster, herpes simplex and listeria-meningitis have been reported [170,179]. Despite some studies suggesting an increased risk of infection with MTX [180,181], several other studies have found that low-dose MTX does not appear to significantly increase the risk of infections in RA patients [182,183,184,185]. This risk appears to be associated with disease activity, comorbidities (diabetes, alcoholism) and the use of glucocorticoids, but not directly with MTX treatment [182]. It is well recognized that RA patients have significant increased risk of infection possibly due to chronic immune activation and inflammation which may impair immune function [185]. An increased risk of infection associated with MTX is possibly offset by the improvement of the immunological function secondary to the control of inflammation [185]. |
| Others | Lymphoproliferative disorders | Lymphoproliferative disorders occur with increased frequency in RA patients compared to the general population, especially in the setting of high disease activity [170,184,186,187]. A relationship between MTX treatment and the occurrence of lymphoproliferative disorders in RA has been suggested. Long-term MTX therapy was associated with Epstein–Barr virus-related lymphoproliferative disorders with spontaneous regression after MTX withdrawal [188]. Despite its association with Epstein–Barr-associated lymphomas, there is currently no clear evidence that MTX provides additional risk of lymphoproliferative disorders to that of RA itself [184,189]. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bedoui, Y.; Guillot, X.; Sélambarom, J.; Guiraud, P.; Giry, C.; Jaffar-Bandjee, M.C.; Ralandison, S.; Gasque, P. Methotrexate an Old Drug with New Tricks. Int. J. Mol. Sci. 2019, 20, 5023. https://doi.org/10.3390/ijms20205023
Bedoui Y, Guillot X, Sélambarom J, Guiraud P, Giry C, Jaffar-Bandjee MC, Ralandison S, Gasque P. Methotrexate an Old Drug with New Tricks. International Journal of Molecular Sciences. 2019; 20(20):5023. https://doi.org/10.3390/ijms20205023
Chicago/Turabian StyleBedoui, Yosra, Xavier Guillot, Jimmy Sélambarom, Pascale Guiraud, Claude Giry, Marie Christine Jaffar-Bandjee, Stéphane Ralandison, and Philippe Gasque. 2019. "Methotrexate an Old Drug with New Tricks" International Journal of Molecular Sciences 20, no. 20: 5023. https://doi.org/10.3390/ijms20205023
APA StyleBedoui, Y., Guillot, X., Sélambarom, J., Guiraud, P., Giry, C., Jaffar-Bandjee, M. C., Ralandison, S., & Gasque, P. (2019). Methotrexate an Old Drug with New Tricks. International Journal of Molecular Sciences, 20(20), 5023. https://doi.org/10.3390/ijms20205023