CSPG4-Specific CAR T Cells for High-Risk Childhood B Cell Precursor Leukemia



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. KOPN8 Leukemia Cells Express CSPG4
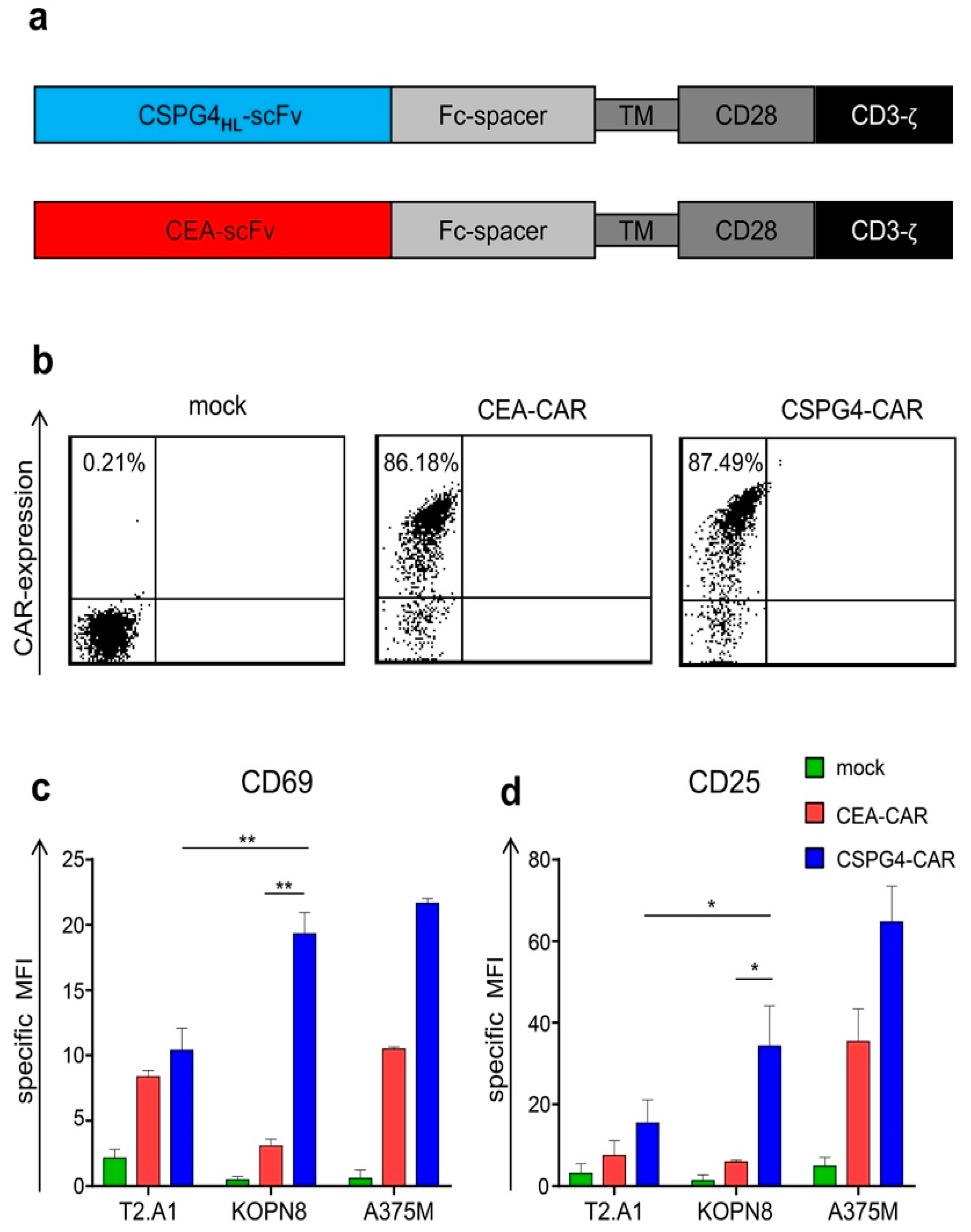
2.2. CSPG4-CAR T Cells are Activated by KOPN8 Leukemia Cells in An Antigen-Specific Fashion
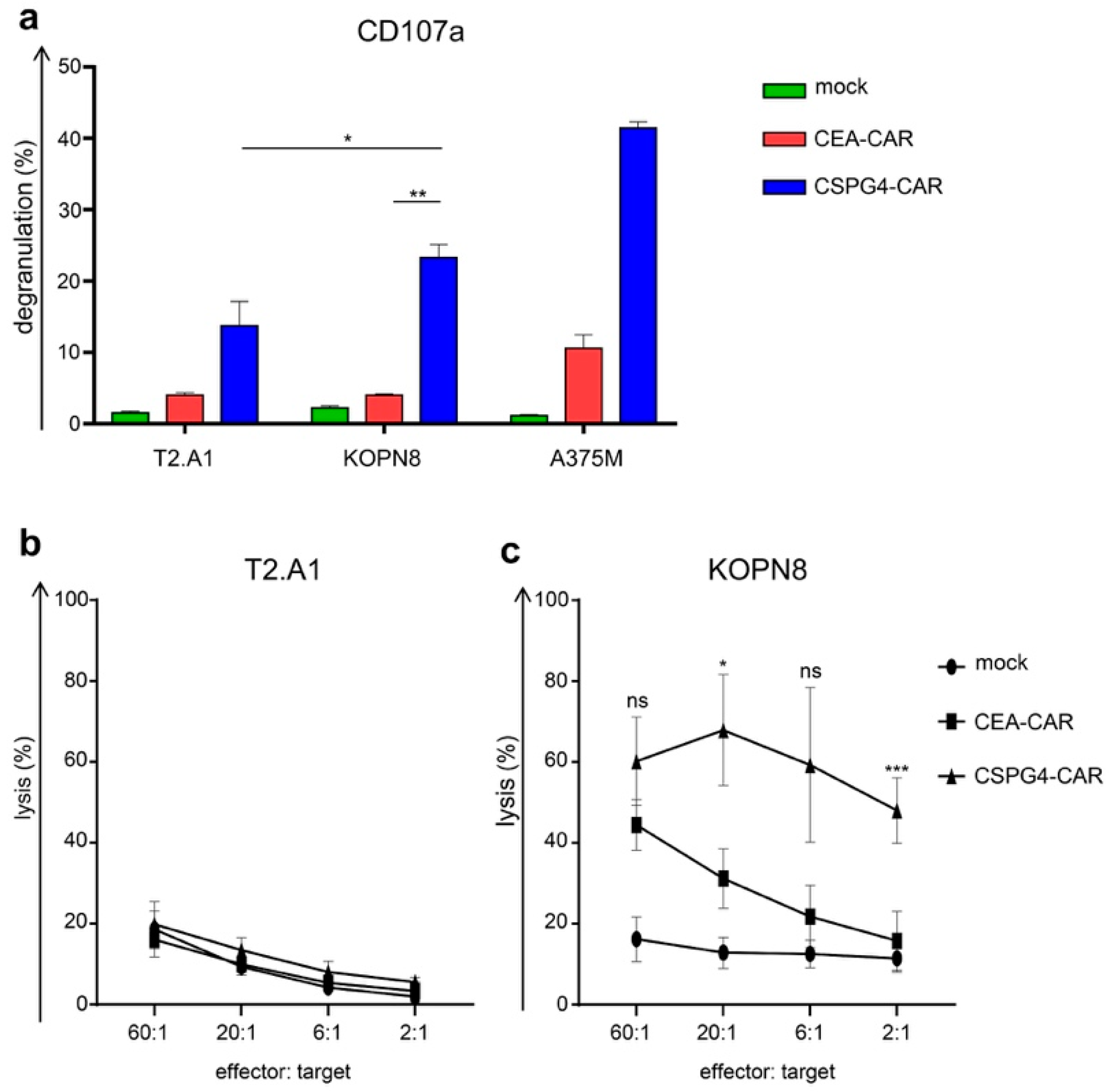
2.3. CSPG4-CAR T Cells Secrete Th1 Cytokines Following Co-Culture with KOPN8 Leukemia Cells
2.4. CSPG4-CAR T Cells Specifically Lyse Leukemia Cells
3. Discussion
4. Materials and Methods
4.1. Cells and Reagents
4.2. T Cell Expansion
4.3. In Vitro Transcription of mRNA
4.4. RNA Electroporation
4.5. Flow Cytometry
4.6. Cytokine Secretion
4.7. Degranulation
4.8. Chromium Release Assay
4.9. Figure Preparation and Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CAR | Chimeric antigen receptor |
| CSPG4 | Chondroitin sulfate proteoglycan 4 |
| CEA | Carcinoembryonic antigen |
| PBMC | Peripheral blood mononuclear cells |
| MLL | Mixed-lineage leukemia |
| ALL | Acute lymphoblastic leukemia |
| CAR | Cfigurehimeric antigen receptor |
References
- June, C.H.; Sadelain, M. Chimeric Antigen Receptor Therapy. N. Engl. J. Med. 2018, 379, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef] [PubMed]
- Schuster, S.J.; Bishop, M.R.; Tam, C.S.; Waller, E.K.; Borchmann, P.; McGuirk, J.P.; Jager, U.; Jaglowski, S.; Andreadis, C.; Westin, J.R.; et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2019, 380, 45–56. [Google Scholar] [CrossRef]
- Sadelain, M.; Riviere, I.; Riddell, S. Therapeutic T cell engineering. Nature 2017, 545, 423–431. [Google Scholar] [CrossRef] [PubMed]
- Rafiq, S.; Brentjens, R.J. Tumors evading CARs-the chase is on. Nat. Med. 2018, 24, 1492–1493. [Google Scholar] [CrossRef] [PubMed]
- Majzner, R.G.; Mackall, C.L. Tumor Antigen Escape from CAR T-cell Therapy. Cancer Discov. 2018, 8, 1219–1226. [Google Scholar] [CrossRef] [PubMed]
- Orlando, E.J.; Han, X.; Tribouley, C.; Wood, P.A.; Leary, R.J.; Riester, M.; Levine, J.E.; Qayed, M.; Grupp, S.A.; Boyer, M.; et al. Genetic mechanisms of target antigen loss in CAR19 therapy of acute lymphoblastic leukemia. Nat. Med. 2018, 24, 1504–1506. [Google Scholar] [CrossRef]
- Fry, T.J.; Shah, N.N.; Orentas, R.J.; Stetler-Stevenson, M.; Yuan, C.M.; Ramakrishna, S.; Wolters, P.; Martin, S.; Delbrook, C.; Yates, B.; et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat. Med. 2018, 24, 20–28. [Google Scholar] [CrossRef]
- Haso, W.; Lee, D.W.; Shah, N.N.; Stetler-Stevenson, M.; Yuan, C.M.; Pastan, I.H.; Dimitrov, D.S.; Morgan, R.A.; FitzGerald, D.J.; Barrett, D.M.; et al. Anti-CD22-chimeric antigen receptors targeting B-cell precursor acute lymphoblastic leukemia. Blood 2013, 121, 1165–1174. [Google Scholar] [CrossRef]
- Gardner, R.; Wu, D.; Cherian, S.; Fang, M.; Hanafi, L.A.; Finney, O.; Smithers, H.; Jensen, M.C.; Riddell, S.R.; Maloney, D.G.; et al. Acquisition of a CD19-negative myeloid phenotype allows immune escape of MLL-rearranged B-ALL from CD19 CAR-T-cell therapy. Blood 2016, 127, 2406–2410. [Google Scholar] [CrossRef] [PubMed]
- Perna, F.; Sadelain, M. Myeloid leukemia switch as immune escape from CD19 chimeric antigen receptor (CAR) therapy. Transl. Cancer Res. 2016, 5, S221–S225. [Google Scholar] [CrossRef] [PubMed]
- Ilieva, K.M.; Cheung, A.; Mele, S.; Chiaruttini, G.; Crescioli, S.; Griffin, M.; Nakamura, M.; Spicer, J.F.; Tsoka, S.; Lacy, K.E.; et al. Chondroitin Sulfate Proteoglycan 4 and Its Potential As an Antibody Immunotherapy Target across Different Tumor Types. Front. Immunol. 2017, 8, 1911. [Google Scholar] [CrossRef] [PubMed]
- Campoli, M.R.; Chang, C.C.; Kageshita, T.; Wang, X.; McCarthy, J.B.; Ferrone, S. Human high molecular weight-melanoma-associated antigen (HMW-MAA): A melanoma cell surface chondroitin sulfate proteoglycan (MSCP) with biological and clinical significance. Crit. Rev. Immunol. 2004, 24, 267–296. [Google Scholar] [CrossRef] [PubMed]
- Yadavilli, S.; Hwang, E.I.; Packer, R.J.; Nazarian, J. The Role of NG2 Proteoglycan in Glioma. Transl. Oncol. 2016, 9, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Osada, T.; Wang, Y.; Yu, L.; Sakakura, K.; Katayama, A.; McCarthy, J.B.; Brufsky, A.; Chivukula, M.; Khoury, T.; et al. CSPG4 protein as a new target for the antibody-based immunotherapy of triple-negative breast cancer. J. Natl. Cancer Inst. 2010, 102, 1496–1512. [Google Scholar] [CrossRef] [PubMed]
- Behm, F.G.; Smith, F.O.; Raimondi, S.C.; Pui, C.H.; Bernstein, I.D. Human homologue of the rat chondroitin sulfate proteoglycan, NG2, detected by monoclonal antibody 7.1, identifies childhood acute lymphoblastic leukemias with t(4;11)(q21;q23) or t(11;19)(q23;p13) and MLL gene rearrangements. Blood 1996, 87, 1134–1139. [Google Scholar] [PubMed]
- Hilden, J.M.; Smith, F.O.; Frestedt, J.L.; McGlennen, R.; Howells, W.B.; Sorensen, P.H.; Arthur, D.C.; Woods, W.G.; Buckley, J.; Bernstein, I.D.; et al. MLL gene rearrangement, cytogenetic 11q23 abnormalities, and expression of the NG2 molecule in infant acute myeloid leukemia. Blood 1997, 89, 3801–3805. [Google Scholar] [PubMed]
- Schwartz, S.; Rieder, H.; Schlager, B.; Burmeister, T.; Fischer, L.; Thiel, E. Expression of the human homologue of rat NG2 in adult acute lymphoblastic leukemia: close association with MLL rearrangement and a CD10(-)/CD24(-)/CD65s(+)/CD15(+) B-cell phenotype. Leukemia 2003, 17, 1589–1595. [Google Scholar] [CrossRef] [PubMed]
- Smith, F.O.; Rauch, C.; Williams, D.E.; March, C.J.; Arthur, D.; Hilden, J.; Lampkin, B.C.; Buckley, J.D.; Buckley, C.V.; Woods, W.G.; et al. The human homologue of rat NG2, a chondroitin sulfate proteoglycan, is not expressed on the cell surface of normal hematopoietic cells but is expressed by acute myeloid leukemia blasts from poor-prognosis patients with abnormalities of chromosome band 11q23. Blood 1996, 87, 1123–1133. [Google Scholar] [PubMed]
- Wuchter, C.; Harbott, J.; Schoch, C.; Schnittger, S.; Borkhardt, A.; Karawajew, L.; Ratei, R.; Ruppert, V.; Haferlach, T.; Creutzig, U.; et al. Detection of acute leukemia cells with mixed lineage leukemia (MLL) gene rearrangements by flow cytometry using monoclonal antibody 7.1. Leukemia 2000, 14, 1232–1238. [Google Scholar] [CrossRef]
- Winters, A.C.; Bernt, K.M. MLL-Rearranged Leukemias-An Update on Science and Clinical Approaches. Front. Pediatr. 2017, 5, 4. [Google Scholar] [CrossRef]
- Xu, J.; Li, L.; Xiong, J.; den Dekker, A.; Ye, A.; Karatas, H.; Liu, L.; Wang, H.; Qin, Z.S.; Wang, S.; et al. MLL1 and MLL1 fusion proteins have distinct functions in regulating leukemic transcription program. Cell Discov. 2016, 2, 16008. [Google Scholar] [CrossRef]
- Nicolosi, P.A.; Dallatomasina, A.; Perris, R. Theranostic impact of NG2/CSPG4 proteoglycan in cancer. Theranostics 2015, 5, 530–544. [Google Scholar] [CrossRef]
- Jordaan, S.; Chetty, S.; Mungra, N.; Koopmans, I.; van Bommel, P.E.; Helfrich, W.; Barth, S. CSPG4: A Target for Selective Delivery of Human Cytolytic Fusion Proteins and TRAIL. Biomedicines 2017, 5, 37. [Google Scholar] [CrossRef]
- Harper, J.R.; Reisfeld, R.A. Inhibition of anchorage-independent growth of human melanoma cells by a monoclonal antibody to a chondroitin sulfate proteoglycan. J. Natl. Cancer Inst. 1983, 71, 259–263. [Google Scholar]
- Rivera, Z.; Ferrone, S.; Wang, X.; Jube, S.; Yang, H.; Pass, H.I.; Kanodia, S.; Gaudino, G.; Carbone, M. CSPG4 as a target of antibody-based immunotherapy for malignant mesothelioma. Clin. Cancer Res. 2012, 18, 5352–5363. [Google Scholar] [CrossRef]
- Wang, X.; Katayama, A.; Wang, Y.; Yu, L.; Favoino, E.; Sakakura, K.; Favole, A.; Tsuchikawa, T.; Silver, S.; Watkins, S.C.; et al. Functional characterization of an scFv-Fc antibody that immunotherapeutically targets the common cancer cell surface proteoglycan CSPG4. Cancer Res. 2011, 71, 7410–7422. [Google Scholar] [CrossRef]
- Chang, C.C.; Campoli, M.; Luo, W.; Zhao, W.; Zaenker, K.S.; Ferrone, S. Immunotherapy of melanoma targeting human high molecular weight melanoma-associated antigen: potential role of nonimmunological mechanisms. Ann. N. Y. Acad. Sci. 2004, 1028, 340–350. [Google Scholar] [CrossRef]
- Brehm, H.; Niesen, J.; Mladenov, R.; Stein, C.; Pardo, A.; Fey, G.; Helfrich, W.; Fischer, R.; Gattenlohner, S.; Barth, S. A CSPG4-specific immunotoxin kills rhabdomyosarcoma cells and binds to primary tumor tissues. Cancer Lett. 2014, 352, 228–235. [Google Scholar] [CrossRef]
- Schwenkert, M.; Birkholz, K.; Schwemmlein, M.; Kellner, C.; Kugler, M.; Peipp, M.; Nettelbeck, D.M.; Schuler-Thurner, B.; Schaft, N.; Dorrie, J.; et al. A single chain immunotoxin, targeting the melanoma-associated chondroitin sulfate proteoglycan, is a potent inducer of apoptosis in cultured human melanoma cells. Melanoma Res. 2008, 18, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Amoury, M.; Mladenov, R.; Nachreiner, T.; Pham, A.T.; Hristodorov, D.; Di, F.S.; Helfrich, W.; Pardo, A.; Fey, G.; Schwenkert, M.; et al. A novel approach for targeted elimination of CSPG4-positive triple-negative breast cancer cells using a MAP tau-based fusion protein. Int. J. Cancer 2016, 139, 916–927. [Google Scholar] [CrossRef] [PubMed]
- De Bruyn, M.; Rybczynska, A.A.; Wei, Y.; Schwenkert, M.; Fey, G.H.; Dierckx, R.A.; van Waarde, A.; Helfrich, W.; Bremer, E. Melanoma-associated Chondroitin Sulfate Proteoglycan (MCSP)-targeted delivery of soluble TRAIL potently inhibits melanoma outgrowth in vitro and in vivo. Mol. Cancer 2010, 9, 301. [Google Scholar] [CrossRef] [PubMed]
- Drake, A.S.; Brady, M.T.; Wang, X.H.; Sait, S.J.; Earp, J.C.; Ghoshal, G.S.; Ferrone, S.; Wang, E.S.; Wetzler, M. Targeting 11q23 positive acute leukemia cells with high molecular weight-melanoma associated antigen-specific monoclonal antibodies. Cancer Immunol. Immunother. 2009, 58, 415–427. [Google Scholar] [CrossRef] [PubMed]
- Beard, R.E.; Zheng, Z.; Lagisetty, K.H.; Burns, W.R.; Tran, E.; Hewitt, S.M.; Abate-Daga, D.; Rosati, S.F.; Fine, H.A.; Ferrone, S.; et al. Multiple chimeric antigen receptors successfully target chondroitin sulfate proteoglycan 4 in several different cancer histologies and cancer stem cells. J. Immunother. Cancer 2014, 2, 25. [Google Scholar] [CrossRef]
- Geldres, C.; Savoldo, B.; Hoyos, V.; Caruana, I.; Zhang, M.; Yvon, E.; Del, V.M.; Creighton, C.J.; Ittmann, M.; Ferrone, S.; et al. T lymphocytes redirected against the chondroitin sulfate proteoglycan-4 control the growth of multiple solid tumors both in vitro and in vivo. Clin. Cancer Res. 2014, 20, 962–971. [Google Scholar] [CrossRef] [PubMed]
- Pellegatta, S.; Savoldo, B.; Di, I.N.; Corbetta, C.; Chen, Y.; Patane, M.; Sun, C.; Pollo, B.; Ferrone, S.; DiMeco, F.; et al. Constitutive and TNFalpha-inducible expression of chondroitin sulfate proteoglycan 4 in glioblastoma and neurospheres: Implications for CAR-T cell therapy. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef]
- Ozerdem, U. Targeting pericytes diminishes neovascularization in orthotopic uveal melanoma in nerve/glial antigen 2 proteoglycan knockout mouse. Ophthalmic Res. 2006, 38, 251–254. [Google Scholar] [CrossRef]
- Ozerdem, U. Targeting of pericytes diminishes neovascularization and lymphangiogenesis in prostate cancer. Prostate 2006, 66, 294–304. [Google Scholar] [CrossRef]
- Tordsson, J.M.; Ohlsson, L.G.; Abrahmsen, L.B.; Karlstrom, P.J.; Lando, P.A.; Brodin, T.N. Phage-selected primate antibodies fused to superantigens for immunotherapy of malignant melanoma. Cancer Immunol. Immunother. 2000, 48, 691–702. [Google Scholar] [CrossRef]
- Harrer, D.C.; Simon, B.; Fujii, S.I.; Shimizu, K.; Uslu, U.; Schuler, G.; Gerer, K.F.; Hoyer, S.; Dorrie, J.; Schaft, N. RNA-transfection of gamma/delta T cells with a chimeric antigen receptor or an alpha/beta T-cell receptor: A safer alternative to genetically engineered alpha/beta T cells for the immunotherapy of melanoma. BMC Cancer 2017, 17, 551. [Google Scholar] [CrossRef] [PubMed]
- Krug, C.; Birkholz, K.; Paulus, A.; Schwenkert, M.; Schmidt, P.; Hoffmann, N.; Hombach, A.; Fey, G.; Abken, H.; Schuler, G.; et al. Stability and activity of MCSP-specific chimeric antigen receptors (CARs) depend on the scFv antigen-binding domain and the protein backbone. Cancer Immunol. Immunother. 2015, 64, 1623–1635. [Google Scholar] [CrossRef] [PubMed]
- Drexler, H.G.; Quentmeier, H.; Macleod, R.A. Malignant hematopoietic cell lines: In vitro models for the study of MLL gene alterations. Leukemia 2004, 18, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Selby, W.L.; Nance, K.V.; Park, H.K. CEA immunoreactivity in metastatic malignant melanoma. Mod. Pathol. 1992, 5, 415–419. [Google Scholar] [PubMed]
- Sadelain, M. CAR therapy: the CD19 paradigm. J. Clin. Investig. 2015, 125, 3392–3400. [Google Scholar] [CrossRef] [PubMed]
- Bagashev, A.; Sotillo, E.; Tang, C.A.; Black, K.L.; Perazzelli, J.; Seeholzer, S.H.; Argon, Y.; Barrett, D.M.; Grupp, S.A.; Hu, C.A.; et al. CD19 alterations emerging after CD19-directed immunotherapy cause retention of the misfolded protein in the endoplasmic reticulum. Mol. Cell Biol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Dreyer, Z.E.; Dinndorf, P.A.; Camitta, B.; Sather, H.; La, M.K.; Devidas, M.; Hilden, J.M.; Heerema, N.A.; Sanders, J.E.; McGlennen, R.; et al. Analysis of the role of hematopoietic stem-cell transplantation in infants with acute lymphoblastic leukemia in first remission and MLL gene rearrangements: a report from the Children’s Oncology Group. J. Clin. Oncol. 2011, 29, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Wang, X.; Kageshita, T.; Wakasugi, S.; Karpf, A.R.; Ferrone, S. Regulation of high molecular weight-melanoma associated antigen (HMW-MAA) gene expression by promoter DNA methylation in human melanoma cells. Oncogene 2006, 25, 2873–2884. [Google Scholar] [CrossRef][Green Version]
- Rosenberg, S.A. IL-2: The first effective immunotherapy for human cancer. J. Immunol. 2014, 192, 5451–5458. [Google Scholar] [CrossRef] [PubMed]
- Golumba-Nagy, V.; Kuehle, J.; Hombach, A.A.; Abken, H. CD28-zeta CAR T Cells Resist TGF-beta Repression through IL-2 Signaling, Which Can Be Mimicked by an Engineered IL-7 Autocrine Loop. Mol. Ther. 2018, 26, 2218–2230. [Google Scholar] [CrossRef] [PubMed]
- Chinen, T.; Kannan, A.K.; Levine, A.G.; Fan, X.; Klein, U.; Zheng, Y.; Gasteiger, G.; Feng, Y.; Fontenot, J.D.; Rudensky, A.Y. An essential role for the IL-2 receptor in Treg cell function. Nat. Immunol. 2016, 17, 1322–1333. [Google Scholar] [CrossRef] [PubMed]
- Kofler, D.M.; Chmielewski, M.; Rappl, G.; Hombach, A.; Riet, T.; Schmidt, A.; Hombach, A.A.; Wendtner, C.M.; Abken, H. CD28 costimulation Impairs the efficacy of a redirected t-cell antitumor attack in the presence of regulatory t cells which can be overcome by preventing Lck activation. Mol. Ther. 2011, 19, 760–767. [Google Scholar] [CrossRef] [PubMed]
- Sander, F.E.; Nilsson, M.; Rydstrom, A.; Aurelius, J.; Riise, R.E.; Movitz, C.; Bernson, E.; Kiffin, R.; Stahlberg, A.; Brune, M.; et al. Role of regulatory T cells in acute myeloid leukemia patients undergoing relapse-preventive immunotherapy. Cancer Immunol. Immunother. 2017, 66, 1473–1484. [Google Scholar] [CrossRef] [PubMed]
- Lamers, C.H.; Sleijfer, S.; van, S.S.; van, E.P.; van, K.B.; Groot, C.; Vulto, A.; den, B.M.; Oosterwijk, E.; Debets, R.; et al. Treatment of metastatic renal cell carcinoma with CAIX CAR-engineered T cells: Clinical evaluation and management of on-target toxicity. Mol. Ther. 2013, 21, 904–912. [Google Scholar] [CrossRef] [PubMed]
- Brudno, J.N.; Kochenderfer, J.N. Toxicities of chimeric antigen receptor T cells: Recognition and management. Blood 2016, 127, 3321–3330. [Google Scholar] [CrossRef]
- Krug, C.; Wiesinger, M.; Abken, H.; Schuler-Thurner, B.; Schuler, G.; Dorrie, J.; Schaft, N. A GMP-compliant protocol to expand and transfect cancer patient T cells with mRNA encoding a tumor-specific chimeric antigen receptor. Cancer Immunol. Immunother. 2014, 63, 999–1008. [Google Scholar] [CrossRef]
- Hombach, A.; Wieczarkowiecz, A.; Marquardt, T.; Heuser, C.; Usai, L.; Pohl, C.; Seliger, B.; Abken, H. Tumor-specific T cell activation by recombinant immunoreceptors: CD3 zeta signaling and CD28 costimulation are simultaneously required for efficient IL-2 secretion and can be integrated into one combined CD28/CD3 zeta signaling receptor molecule. J. Immunol. 2001, 167, 6123–6131. [Google Scholar] [CrossRef]
- Schaft, N.; Dorrie, J.; Muller, I.; Beck, V.; Baumann, S.; Schunder, T.; Kampgen, E.; Schuler, G. A new way to generate cytolytic tumor-specific T cells: Electroporation of RNA coding for a T cell receptor into T lymphocytes. Cancer Immunol. Immunother. 2006, 55, 1132–1141. [Google Scholar] [CrossRef]
- Hofflin, S.; Prommersberger, S.; Uslu, U.; Schuler, G.; Schmidt, C.W.; Lennerz, V.; Dorrie, J.; Schaft, N. Generation of CD8(+) T cells expressing two additional T-cell receptors (TETARs) for personalised melanoma therapy. Cancer Biol. Ther. 2015, 16, 1323–1331. [Google Scholar] [CrossRef]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Harrer, D.C.; Schuler, G.; Dörrie, J.; Schaft, N. CSPG4-Specific CAR T Cells for High-Risk Childhood B Cell Precursor Leukemia. Int. J. Mol. Sci. 2019, 20, 2764. https://doi.org/10.3390/ijms20112764
Harrer DC, Schuler G, Dörrie J, Schaft N. CSPG4-Specific CAR T Cells for High-Risk Childhood B Cell Precursor Leukemia. International Journal of Molecular Sciences. 2019; 20(11):2764. https://doi.org/10.3390/ijms20112764
Chicago/Turabian StyleHarrer, Dennis C., Gerold Schuler, Jan Dörrie, and Niels Schaft. 2019. "CSPG4-Specific CAR T Cells for High-Risk Childhood B Cell Precursor Leukemia" International Journal of Molecular Sciences 20, no. 11: 2764. https://doi.org/10.3390/ijms20112764
APA StyleHarrer, D. C., Schuler, G., Dörrie, J., & Schaft, N. (2019). CSPG4-Specific CAR T Cells for High-Risk Childhood B Cell Precursor Leukemia. International Journal of Molecular Sciences, 20(11), 2764. https://doi.org/10.3390/ijms20112764