Identification of Novel Somatic TP53 Mutations in Patients with High-Grade Serous Ovarian Cancer (HGSOC) Using Next-Generation Sequencing (NGS)

 , , , , ,
, , , , ,  and
and
Abstract
:
1. Introduction
2. Results
2.1. Patient Characteristics
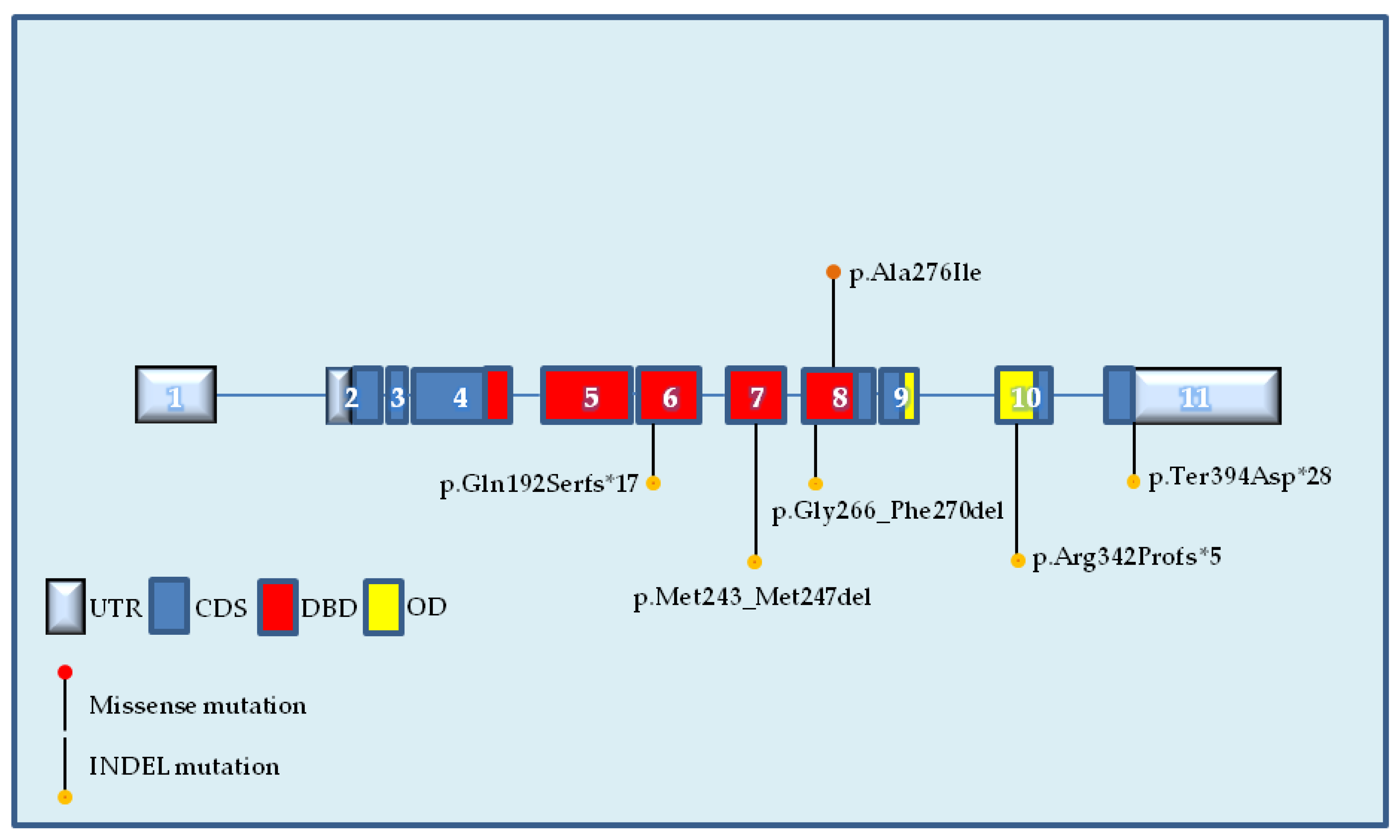
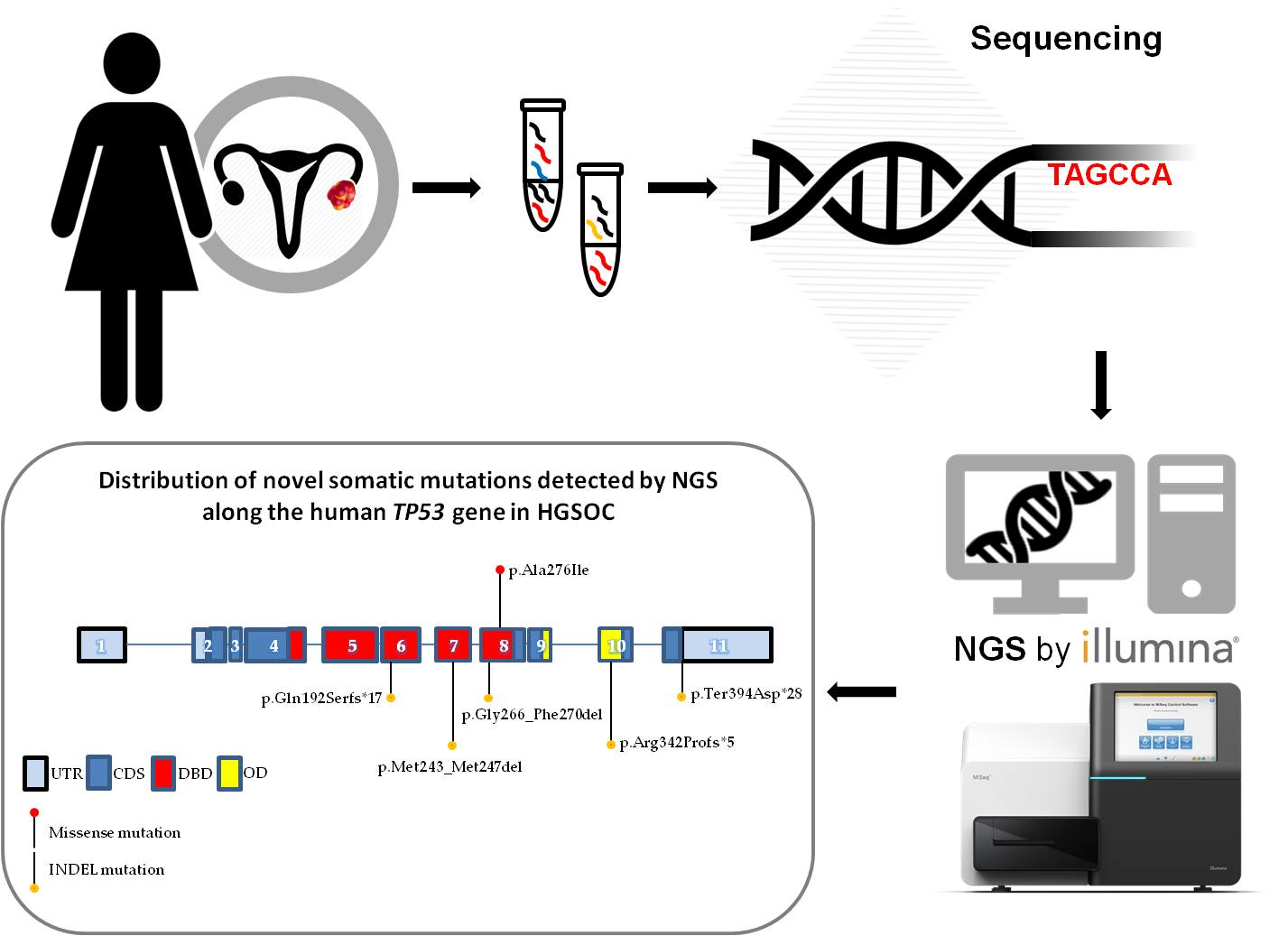
2.2. Identification of Novel TP53 Somatic Mutations by NGS
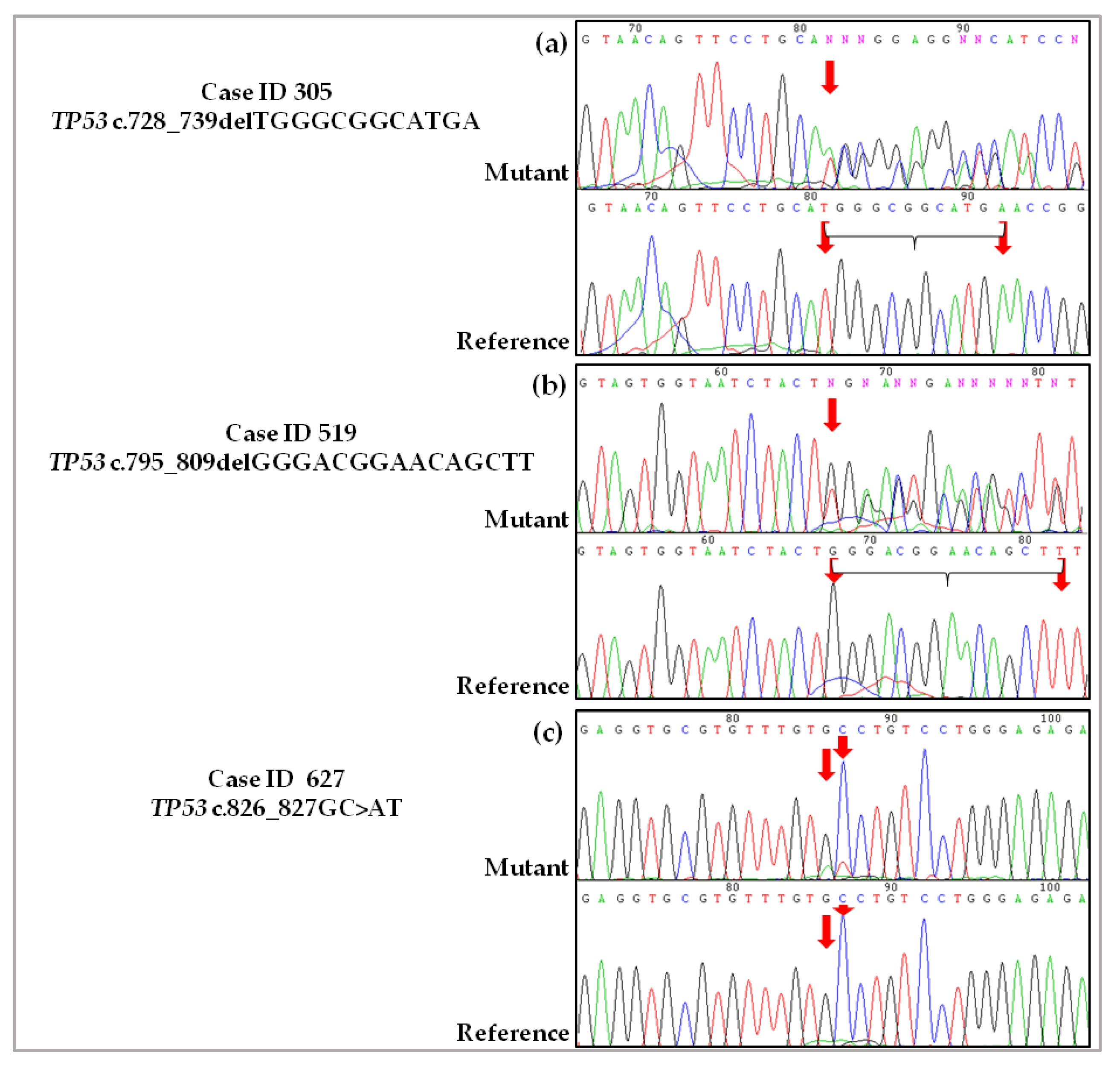
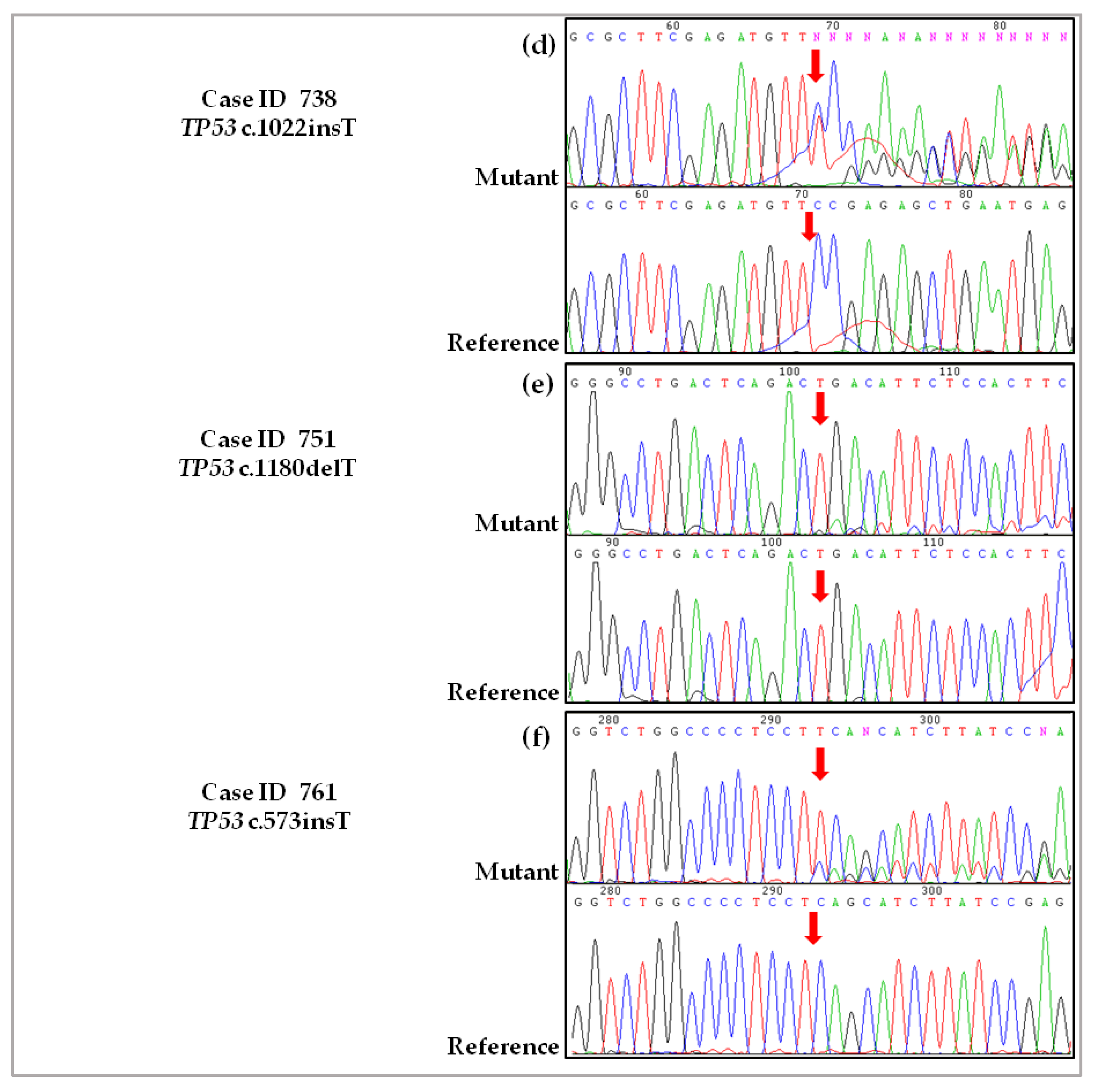
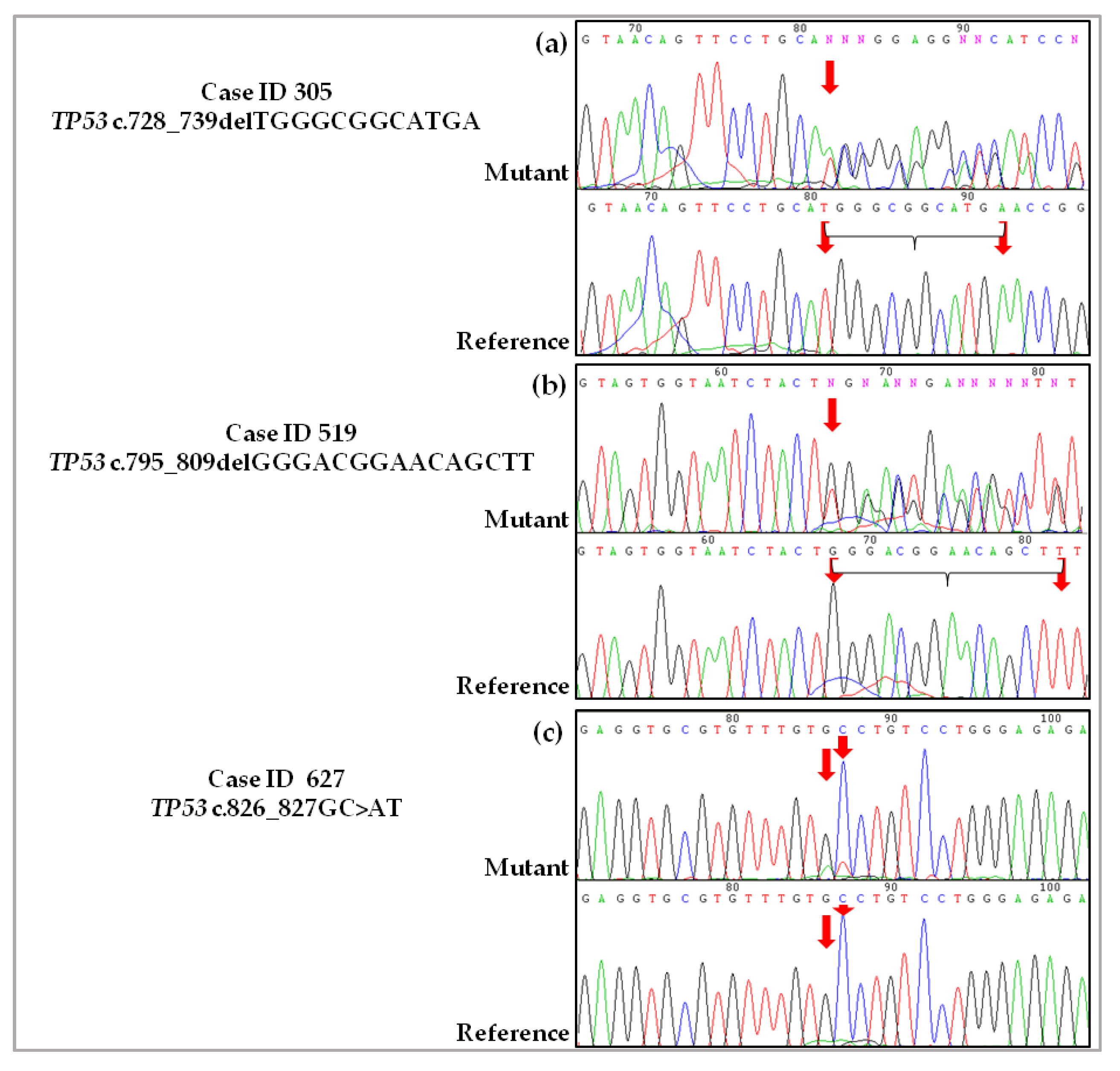
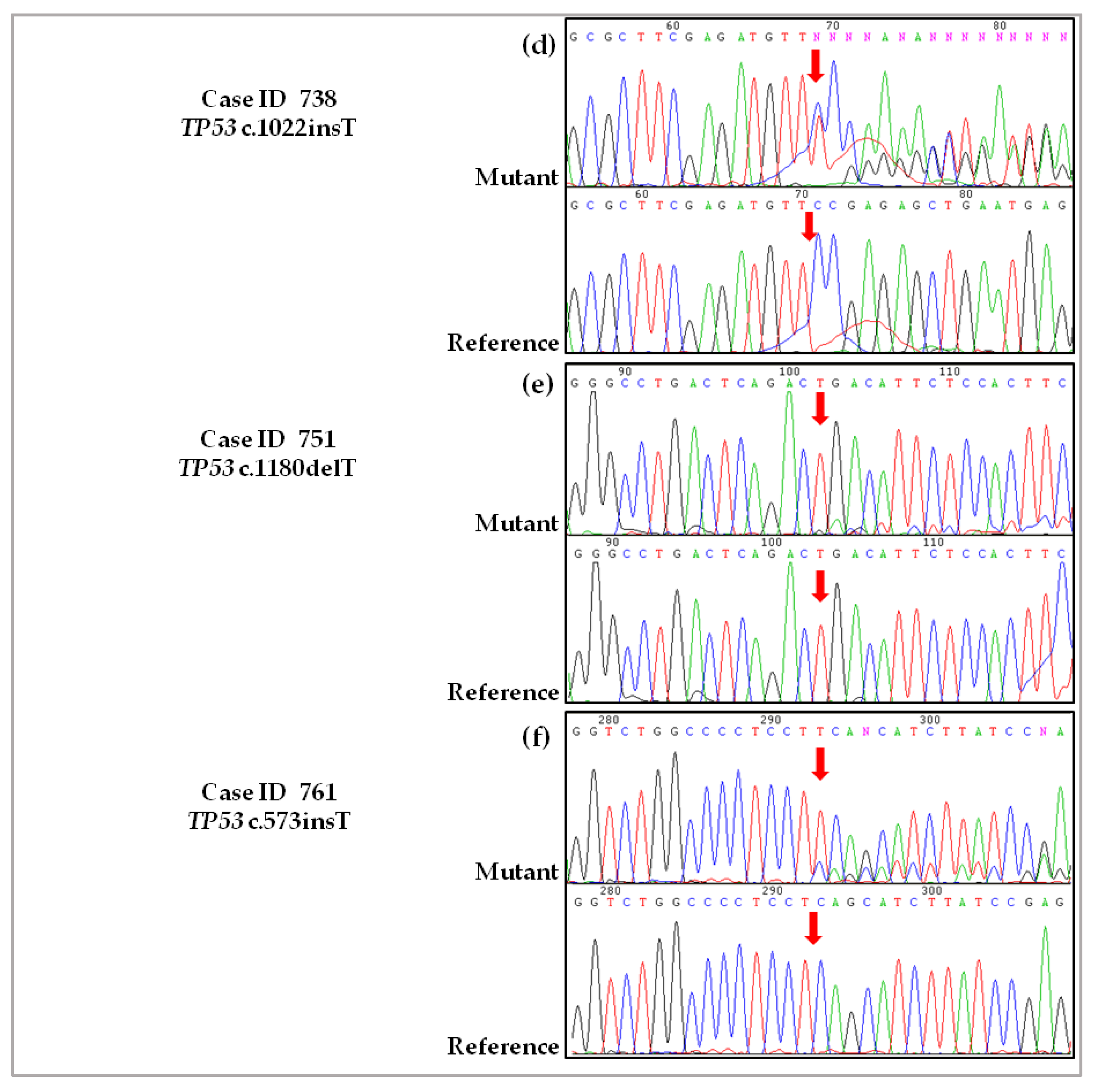
2.3. Validation of De Novo TP53 Somatic Mutations in HGSOC
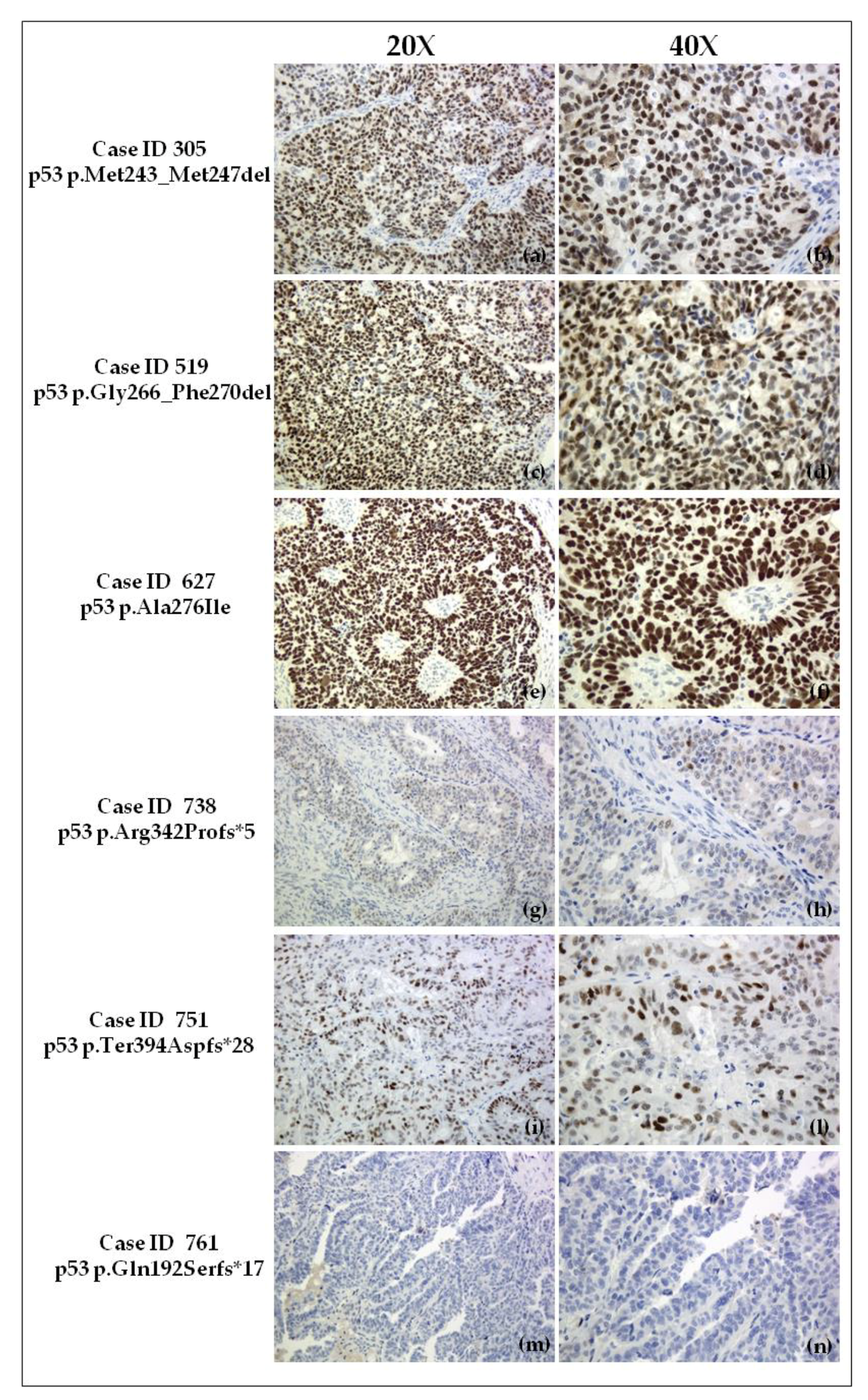
2.4. Immunohistochemical Evaluation of p53 Expression in Tumor Samples
2.5. In Silico Prediction and Structural Analysis
3. Discussion
4. Materials and Methods
4.1. Patients and Human Ethics
4.2. Case History
4.2.1. Case ID 305
4.2.2. Case ID 519
4.2.3. Case ID 627
4.2.4. Case ID 738
4.2.5. Case ID 751
4.2.6. Case ID 761
4.3. Next Generation Sequencing Analysis
4.4. Sanger Sequencing and Pyrosequencing to Validate Novel Somatic Mutations
4.5. p53 Immunohistochemistry
4.6. In Silico Analysis
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Hennessy, B.T.; Coleman, R.L.; Markman, M. Ovarian cancer. Lancet 2009, 374, 1371–1382. [Google Scholar] [CrossRef]
- Ledermann, J.A.; Raja, F.A.; Fotopoulou, C.; Gonzales-Martin, A.; Colombo, N.; Sessa, C.; ESMO Guidelines Working Group. Newly diagnosed and relapsed epithelial ovarian carcinoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2013, 24, vi24–vi32. [Google Scholar] [CrossRef] [PubMed]
- Melamed, A.; Manning-Geist, B.; Bregar, A.J.; Diver, E.J.; Goodman, A.; del Carmen, M.G.; Schorge, J.O.; Rauh-Hain, J.A. Associations between residual disease and survival in epithelial ovarian cancer by histologic type. Gynecol. Oncol. 2017, 147, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Mackay, H.J.; Brady, M.F.; Oza, A.M.; Reuss, A.; Pujade-Lauraine, E.; Swart, A.M.; Siddiqui, N.; Colombo, N.; Bookman, M.A.; Pfisterer, J.; et al. Prognostic relevance of uncommon ovarian histology in women with stage III/IV epithelial ovarian cancer. Int. J. Gynecol. Cancer 2010, 20, 945–952. [Google Scholar] [CrossRef] [PubMed]
- Kurman, R.J.; Shih, I.-M. The Dualistic Model of Ovarian Carcinogenesis: Revisited, Revised, and Expanded. Am. J. Pathol. 2016, 186, 733–747. [Google Scholar] [CrossRef] [PubMed]
- Gockley, A.; Melamed, A.; Bregar, A.J.; Clemmer, J.T.; Birrer, M.; Schorge, J.O.; del Carmen, M.G.; Rauh-Hain, J.A. Outcomes of Women with High-Grade and Low-Grade Advanced-Stage Serous Epithelial Ovarian Cancer. Obstet. Gynecol. 2017, 129, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Bowtell, D.D. The genesis and evolution of high-grade serous ovarian cancer. Nat. Rev. Cancer. 2010, 10, 803–808. [Google Scholar] [CrossRef] [PubMed]
- Silwal-Pandit, L.; Langerød, A.; Børresen-Dale, A.-L. TP53 Mutations in Breast and Ovarian Cancer. Cold Spring Harb. Perspect. Med. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Leitao, M.M.; Soslow, R.A.; Baergen, R.N.; Olvera, N.; Arroyo, C.; Boyd, J. Mutation and expression of the TP53 gene in early stage epithelial ovarian carcinoma. Gynecol Oncol. 2004, 93, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474. [Google Scholar] [CrossRef] [Green Version]
- Freed-Pastor, W.A.; Prives, C. Mutant p53: One name, many proteins. Genes Dev. 2012, 26, 1268–1286. [Google Scholar] [CrossRef] [PubMed]
- Bykov, V.J.N.; Eriksson, S.E.; Bianchi, J.; Wiman, K.G. Targeting mutant p53 for efficient cancer therapy. Nat. Rev. Cancer 2018, 18, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Duffy, M.J.; Synnott, N.C.; Crown, J. Mutant p53 as a target for cancer treatment. Eur. J. Cancer 2017, 83, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Brachova, P.; Thiel, K.W.; Leslie, K.K. The consequence of oncomorphic TP53 mutations in ovarian cancer. Int. J. Mol. Sci. 2013, 14, 19257–19275. [Google Scholar] [CrossRef] [PubMed]
- Brachova, P.; Mueting, S.R.; Carlson, M.J.; Goodheart, M.J.; Button, A.M.; Mott, S.L.; Dai, D.; Thiel, K.W.; Devor, E.J.; Leslie, K.K. TP53 oncomorphic mutations predict resistance to platinum- and taxane-based standard chemotherapy in patients diagnosed with advanced serous ovarian carcinoma. Int. J. Oncol. 2015, 46, 607–618. [Google Scholar] [CrossRef] [PubMed]
- Soussi, T.; Wiman, K.G. TP53: An oncogene in disguise. Cell Death Differ. 2015, 22, 1239–1249. [Google Scholar] [CrossRef] [PubMed]
- Oren, M.; Rotter, V. Mutant p53 gain-of-function in cancer. Cold Spring Harb. Perspect. Biol. 2010, 2, a001107. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.A.; Vousden, K.H. Mutant p53 in cancer: New functions and therapeutic opportunities. Cancer Cell 2014, 25, 304–317. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Reumers, J.; Couceiro, J.R.; De Smet, F.; Gallardo, R.; Rudyak, S.; Cornelis, A.; Zwolinska, A.; Marine, J.C.; Lambrechts, D.; et al. Gain of function of mutant p53 by coaggregation with multiple tumor suppressors. Nat. Chem. Biol. 2011, 7, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Kamps, R.; Brandão, R.D.; Bosch, B.J.; Paulussen, A.D.; Xanthoulea, S.; Blok, M.J.; Romano, A. Next-Generation Sequencing in Oncology: Genetic Diagnosis, Risk Prediction and Cancer Classification. Int. J. Mol. Sci. 2017, 18, 308. [Google Scholar] [CrossRef] [PubMed]
- Su, D.; Zhang, D.; Chen, K.; Lu, J.; Wu, J.; Cao, X.; Ying, L.; Jin, Q.; Ye, Y.; Xie, Z.; et al. High performance of targeted next generation sequencing on variance detection in clinical tumor specimens in comparison with current conventional methods. J. Exp. Clin. Cancer Res. 2017, 36. [Google Scholar] [CrossRef] [PubMed]
- Sakai, K.; Ukita, M.; Schmidt, J.; Wu, L.; de Velasco, M.A.; Roter, A.; Jevons, L.; Nishio, K.; Mandai, M. Clonal composition of human ovarian cancer based on copy number analysis reveals a reciprocal relation with oncogenic mutation status. Cancer Lett. 2017, 405, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Ab Mutalib, N.S.; Syafruddin, S.E.; Md Zain, R.R.; Mohd Dali, A.Z.; Mohd Yunos, R.I.; Saidin, S.; Jamal, R.; Mokhtar, N.M. Molecular characterization of serous ovarian carcinoma using a multigene next generation sequencing cancer panel approach. BMC Res. Notes 2014, 7, 805. [Google Scholar] [CrossRef] [PubMed]
- Khagi, Y.; Goodman, A.M.; Daniels, G.A.; Patel, S.P.; Sacco, A.G.; Randall, J.M.; Bazhenova, L.A.; Kurzrock, R. Hypermutated Circulating Tumor DNA: Correlation with Response to Checkpoint Inhibitor-Based Immunotherapy. Clin. Cancer Res. 2017, 23, 5729–5736. [Google Scholar] [CrossRef] [PubMed]
- Parkinson, C.A.; Gale, D.; Piskorz, A.M.; Biggs, H.; Hodgkin, C.; Addley, H.; Freeman, S.; Moyle, P.; Sala, E.; Sayal, K.; et al. Exploratory Analysis of TP53 Mutations in Circulating Tumour DNA as Biomarkers of Treatment Response for Patients with Relapsed High-Grade Serous Ovarian Carcinoma: A Retrospective Study. PLoS Med. 2016, 13, e1002198. [Google Scholar] [CrossRef] [PubMed]
- Lukman, S.; Lane, D.P.; Verma, C.S. Mapping the structural and dynamical features of multiple p53 DNA binding domains: Insights into loop 1 intrinsic dynamics. PLoS ONE 2013, 8, e80221. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.Y.; Zhou, N.E.; Kay, C.M.; Hodges, R.S. Packing and hydrophobicity effects on protein folding and stability: Effects of β-branched amino acids, valine and isoleucine, on the formation and stability of two-stranded α-helical coiled coils/leucine zippers. Protein Sci. 1993, 2, 383–394. [Google Scholar] [CrossRef] [PubMed]
- Joerger, A.C.; Fersht, A.R. The tumor suppressor p53: From structures to drug discovery. Cold Spring Harb. Perspect. Biol. 2010, 2, a000919. [Google Scholar] [CrossRef] [PubMed]
- Garziera, M.; Canzonieri, V.; Cannizzaro, R.; Geremia, S.; Caggiari, L.; De Zorzi, M.; Maiero, S.; Orzes, E.; Perin, T.; Zanussi, S.; et al. Identification and characterization of CDH1 germline variants in sporadic gastric cancer patients and in individuals at risk of gastric cancer. PLoS ONE 2013, 8, e77035. [Google Scholar] [CrossRef] [PubMed]
- Cole, A.J.; Dwight, T.; Gill, A.J.; Dickson, K.A.; Zhu, Y.; Clarkson, A.; Gard, G.B.; Maidens, J.; Valmadre, S.; Clifton-Bligh, R.; et al. Assessing mutant p53 in primary high-grade serous ovarian cancer using immunohistochemistry and massively parallel sequencing. Sci. Rep. 2016, 6, 26191. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.T.; Mosier, S.L.; Thiess, M.; Beierl, K.F.; Debeljak, M.; Tseng, L.H.; Chen, G.; Yegnasubramanian, S.; Ho, H.; Cope, L.; et al. Clinical validation of KRAS, BRAF, and EGFR mutation detection using next-generation sequencing. Am. J. Clin. Pathol. 2014, 141, 856–866. [Google Scholar] [CrossRef] [PubMed]
- Khoury, M.P.; Bourdon, J.C. The isoforms of the p53 protein. Cold Spring Harb. Perspect. Biol. 2010, 2. [Google Scholar] [CrossRef] [PubMed]
- Chène, P. The role of tetramerization in p53 function. Oncogene 2001, 20, 2611–2617. [Google Scholar] [CrossRef] [PubMed]
- Testa, U.; Petrucci, E.; Pasquini, L.; Castelli, G.; Pelosi, E. Ovarian Cancers: Genetic Abnormalities, Tumor Heterogeneity and Progression, Clonal Evolution and Cancer Stem Cells. Medicines 2018, 5, E16. [Google Scholar] [CrossRef] [PubMed]
- Nunziato, M.; Starnone, F.; Lombardo, B.; Pensabene, M.; Condello, C.; Verdesca, F.; Carlomagno, C.; de Placido, S.; Pastore, L.; Salvatore, F.; et al. Fast Detection of a BRCA2 Large Genomic Duplication by Next Generation Sequencing as a Single Procedure: A Case Report. Int. J. Mol. Sci. 2017, 18, 2487. [Google Scholar] [CrossRef] [PubMed]
- Maru, Y.; Tanaka, N.; Ohira, M.; Itami, M.; Hippo, Y.; Nagase, H. Identification of novel mutations in Japanese ovarian clear cell carcinoma patients using optimized targeted NGS for clinical diagnosis. Gynecol. Oncol. 2017, 144, 377–383. [Google Scholar] [CrossRef] [PubMed]
- Brachova, P.; Mueting, S.R.; Devor, E.J.; Leslie, K.K. Oncomorphic TP53 Mutations in Gynecologic Cancers Lose the Normal Protein:Protein Interactions with the microRNA Microprocessing Complex. J. Cancer Ther. 2014, 5, 506–516. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Nie, L.; Wiederschain, D.; Yuan, Z.M. Identification of p53 sequence elements that are required for MDM2-mediated nuclear export. Mol. Cell. Biol. 2001, 21, 8533–8546. [Google Scholar] [CrossRef] [PubMed]
- Nie, L.; Sasaki, M.; Maki, C.G. Regulation of p53 nuclear export through sequential changes in conformation and ubiquitination. J. Biol. Chem. 2007, 282, 14616–14625. [Google Scholar] [CrossRef] [PubMed]
- Olivier, M.; Hollstein, M.; Hainaut, P. TP53 mutations in human cancers: Origins, consequences, and clinical use. Cold Spring Harb. Perspect. Biol. 2010, 2, a001008. [Google Scholar] [CrossRef] [PubMed]
- Petrovich, M.; Veprintsev, D.B. Effects of CpG methylation on recognition of DNA by the tumour suppressor p53. J. Mol. Biol. 2009, 386, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Yue, X.; Zhao, Y.; Xu, Y.; Zheng, M.; Feng, Z.; Hu, W. Mutant p53 in Cancer: Accumulation, Gain-of-Function, and Therapy. J. Mol. Biol. 2017, 429, 1595–1606. [Google Scholar] [CrossRef] [PubMed]
- Goh, A.M.; Coffill, C.R.; Lane, D.P. The role of mutant p53 in human cancer. J. Pathol. 2011, 223, 116–126. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, E.; Kurman, R.J.; Vang, R.; Sehdev, A.S.; Han, G.; Soslow, R.; Wang, T.L.; Shih, I.-M. TP53 mutations in serous tubal intraepithelial carcinoma and concurrent pelvic high-grade serous carcinoma—Evidence supporting the clonal relationship of the two lesions. J. Pathol. 2012, 226, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.J.; Chun, S.M.; Kim, K.R.; Sohn, I.; Sung, C.O. Clinical relevance of gain-of-function mutations of p53 in high-grade serous ovarian carcinoma. PLoS ONE 2013, 8, e72609. [Google Scholar] [CrossRef] [PubMed]
- Petitjean, A.; Achatz, M.I.; Borresen-Dale, A.L.; Hainaut, P.; Olivier, M. TP53 mutations in human cancers: Functional selection and impact on cancer prognosis and outcomes. Oncogene 2007, 26, 2157–2165. [Google Scholar] [CrossRef] [PubMed]
- Rzepecka, I.K.; Szafron, L.M.; Stys, A.; Felisiak-Golabek, A.; Podgorska, A.; Timorek, A.; Sobiczewski, P.; Pienkowska-Grela, B.; El-Bahrawy, M.; Kupryjanczyk, J. Prognosis of patients with BRCA1-associated ovarian carcinomas depends on TP53 accumulation status in tumor cells. Gynecol. Oncol. 2017, 144, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Leijen, S.; van Geel, R.M.; Sonke, G.S.; de Jong, D.; Rosenberg, E.H.; Marchetti, S.; Pluim, D.; van Werkhoven, E.; Rose, S.; Lee, M.A.; et al. Phase II Study of WEE1 Inhibitor AZD1775 Plus Carboplatin in Patients With TP53-Mutated Ovarian Cancer Refractory or Resistant to First-Line Therapy Within 3 Months. J. Clin. Oncol. 2016, 34, 4354–4361. [Google Scholar] [CrossRef] [PubMed]
- Said, R.; Hong, D.S.; Warneke, C.L.; Lee, J.J.; Wheler, J.J.; Janku, F.; Naing, A.; Falchook, G.S.; Fu, S.; Piha-Paul, S.; et al. P53 mutations in advanced cancers: Clinical characteristics, outcomes, and correlation between progression-free survival and bevacizumab-containing therapy. Oncotarget 2013, 4, 705–714. [Google Scholar] [CrossRef] [PubMed]
- Labbé, C.; Cabanero, M.; Korpanty, G.J.; Tomasini, P.; Doherty, M.K.; Mascaux, C.; Jao, K.; Pitcher, B.; Wang, R.; Pintilie, M.; et al. Prognostic and predictive effects of TP53 co-mutation in patients with EGFR-mutated non-small cell lung cancer (NSCLC). Lung Cancer 2017, 111, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Schwaederlé, M.; Lazar, V.; Validire, P.; Hansson, J.; Lacroix, L.; Soria, J.C.; Pawitan, Y.; Kurzrock, R. VEGF-A Expression Correlates with TP53 Mutations in Non-Small Cell Lung Cancer: Implications for Antiangiogenesis Therapy. Cancer Res. 2015, 75, 1187–1190. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, A.A.; Etemadmoghadam, D.; Temple, J.; Lynch, A.G.; Riad, M.; Sharma, R.; Stewart, C.; Fereday, S.; Caldas, C.; Defazio, A.; et al. Driver mutations in TP53 are ubiquitous in high grade serous carcinoma of the ovary. J. Pathol. 2010, 221, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Hoppenot, C.; Eckert, M.A.; Tienda, S.M.; Lengyel, E. Who are the long-term survivors of high grade serous ovarian cancer? Gynecol. Oncol. 2018, 148, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Konecny, G.E.; Wang, C.; Hamidi, H.; Winterhoff, B.; Kalli, K.R.; Dering, J.; Ginther, C.; Chen, H.W.; Dowdy, S.; Cliby, W.; et al. Prognostic and therapeutic relevance of molecular subtypes in high-grade serous ovarian cancer. J. Natl. Cancer Inst. 2014, 106, dju249. [Google Scholar] [CrossRef] [PubMed]
- Falcone, F.; Scambia, G.; Benedetti Panici, P.; Signorelli, M.; Cormio, G.; Giorda, G.; Bogliolo, S.; Marinaccio, M.; Ghezzi, F.; Rabaiotti, E.; et al. Tertiary cytoreductive surgery in recurrent epithelial ovarian cancer: A multicentre MITO retrospective study. Gynecol. Oncol. 2017, 147, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.J., Jr.; Armstrong, D.K.; Alvarez, R.D.; Bakkum-Gamez, J.N.; Behbakht, K.; Chen, L.M.; Copeland, L.; Crispens, M.A.; DeRosa, M.; Dorigo, O.; et al. Ovarian Cancer, Version 1.2016, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2016, 14, 1134–1163. [Google Scholar] [CrossRef]
- Mylavarapu, S.; Das, A.; Roy, M. Role of BRCA Mutations in the Modulation of Response to Platinum Therapy. Front. Oncol. 2018, 8, 16. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Case ID | Age a | TNM | FIGO | Histology | Grade | R | CT Setting | First Line Treatment | PFI | PFS | OS | Actual Status |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| (year) | (G) | (cm) | (m) | (m) | (m) | |||||||
| 305 | 58 | pT3cN1M0 | IIIC | Serous | G3 | 0 | I line | Carbotaxol | 38 | 43 | 43 | Dead |
| 519 | 52 | pT3cN1M1 | IV | Serous | G3 | 0 | I line | Carbotaxol | 80 | 85 | 98 | Alive |
| 627 | 68 | ypT2aN0M0 | IIA | Serous | G3 | 0 | Neo-adj | Carbotaxol | 64 | 71 | 77 | Alive |
| 738 | 79 | pT2cNxM0 | IIC | Serous | G3 | >1 | NA | NA | NA | NA | 9 | Dead |
| 751 | 71 | pT2cN0 | IIC | Serous | G3 | 0 | I line | Carbotaxol | 29 | 34 | 60 | Alive |
| 761 | 56 | pT3cNxM0 | IIIC | Serous | G3 | 0 | I line | Carbotaxol | 14 | 20 | 58 | Alive |
| Case ID | Exon | Genomic Coordinate | Alt | Ref Read Depth | Alt Read Depth | cDNA Nucleotide Change | AA Change | Mutation Type |
|---|---|---|---|---|---|---|---|---|
| (%) a | ||||||||
| 305 | 7 | 17:7,577,541 | 58.43 | 15,734 | 9194 | c.728_739delTGGGCGGCATGA | p.Met243_Met247del | In-frame |
| INDEL | ||||||||
| 519 | 8 | 17:7,577,128 | 47.84 | 6377 | 3051 | c.795_809delGGGACGGAACAGCTT | p.Gly266_Phe270del | In-frame |
| INDEL | ||||||||
| 627 | 8 | 17:7,577,111 | 22.34 | 4692 | 1048 | c.826_827GC>AT | p.Ala276Ile | Missense |
| 738 | 10 | 17:7,574,004 | 53.41 | 4606 | 2460 | c.1022insT | p.Arg342Profs*5 | Frameshift |
| INDEL | ||||||||
| 751 | 11 | 17:7,572,928 | 8.32 | 2994 | 249 | c.1180delT | p.Ter394Aspfs*28 | Frameshift |
| INDEL | ||||||||
| 761 | 6 | 17:7,578,275 | 73.38 | 7205 | 5287 | c.573insT | p.Gln192Serfs*17 | Frameshift |
| INDEL |
| Case ID | Novel Mutp53 | p53 Domain | p53 IHC a | Predicted Damaging Effect b | Phylogenetic Conservation c | Predicted Splicing Effect | Structural Consequence | Loss of CpG Site d | Platinum Status | OS | Classification of Novel Mutp53 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 305 | p.Met243_Met247del | DBD | ~100% (+++) | Yes | Met243 100% | Yes | Probable strong rearrangement in DNA minor groove binding surface | Yes | Sensitive | 3 ≤ years < 4 † | Uncl |
| Gly244 100% | |||||||||||
| Gly245 100% | |||||||||||
| Met246 100% | |||||||||||
| 519 | p.Gly266_Phe270del | DBD | ~80% (++) | Yes | Gly266 100% | Yes | Probable change in affinity of p53 for target sequence | Yes | Sensitive | >5 years | Uncl |
| Arg267 100% | |||||||||||
| Asn268 60% | |||||||||||
| Ser269 90% | |||||||||||
| Phe270 100% | |||||||||||
| 627 | p.Ala276Ile | DBD | ~80% | Yes | Ala276 100% | No | Probable perturbation in proximity of DNA major groove binding surface | No | Sensitive | >5 years | Uncl |
| 738 | p.Arg342Profs*5 | OD | <5% (−/+) | Yes | Arg342 80% * | Yes | Partial loss of OD | No | NA | <1 year † | LOF |
| Loss of C’-terminal | |||||||||||
| 751 | p.Ter394Aspfs*28 | C′-terminal ** | ~60% (30% +; 30% ++) | No | ** | No | Abnormal protein elongation | No | Sensitive | ≥5 years | Uncl |
| 761 | p.Gln192Serfs*17 | DBD | - | Yes | Gln192 90% * | Yes | Probable strong rearrangement in DNA minor groove binding surface | No | Sensitive | 4 ≤ years < 5 | LOF |
| Massive loss of DBD. | |||||||||||
| Loss of OD and C′-terminal. |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garziera, M.; Cecchin, E.; Canzonieri, V.; Sorio, R.; Giorda, G.; Scalone, S.; De Mattia, E.; Roncato, R.; Gagno, S.; Poletto, E.; et al. Identification of Novel Somatic TP53 Mutations in Patients with High-Grade Serous Ovarian Cancer (HGSOC) Using Next-Generation Sequencing (NGS). Int. J. Mol. Sci. 2018, 19, 1510. https://doi.org/10.3390/ijms19051510
Garziera M, Cecchin E, Canzonieri V, Sorio R, Giorda G, Scalone S, De Mattia E, Roncato R, Gagno S, Poletto E, et al. Identification of Novel Somatic TP53 Mutations in Patients with High-Grade Serous Ovarian Cancer (HGSOC) Using Next-Generation Sequencing (NGS). International Journal of Molecular Sciences. 2018; 19(5):1510. https://doi.org/10.3390/ijms19051510
Chicago/Turabian StyleGarziera, Marica, Erika Cecchin, Vincenzo Canzonieri, Roberto Sorio, Giorgio Giorda, Simona Scalone, Elena De Mattia, Rossana Roncato, Sara Gagno, Elena Poletto, and et al. 2018. "Identification of Novel Somatic TP53 Mutations in Patients with High-Grade Serous Ovarian Cancer (HGSOC) Using Next-Generation Sequencing (NGS)" International Journal of Molecular Sciences 19, no. 5: 1510. https://doi.org/10.3390/ijms19051510