
Arabidopsis thaliana Roots Exposed to Extracellular Self-DNA: Evidence of Epigenetic Effects

 , ,
, ,  , ,
, ,  , ,
, ,  and
and
Abstract
1. Introduction
2. Results
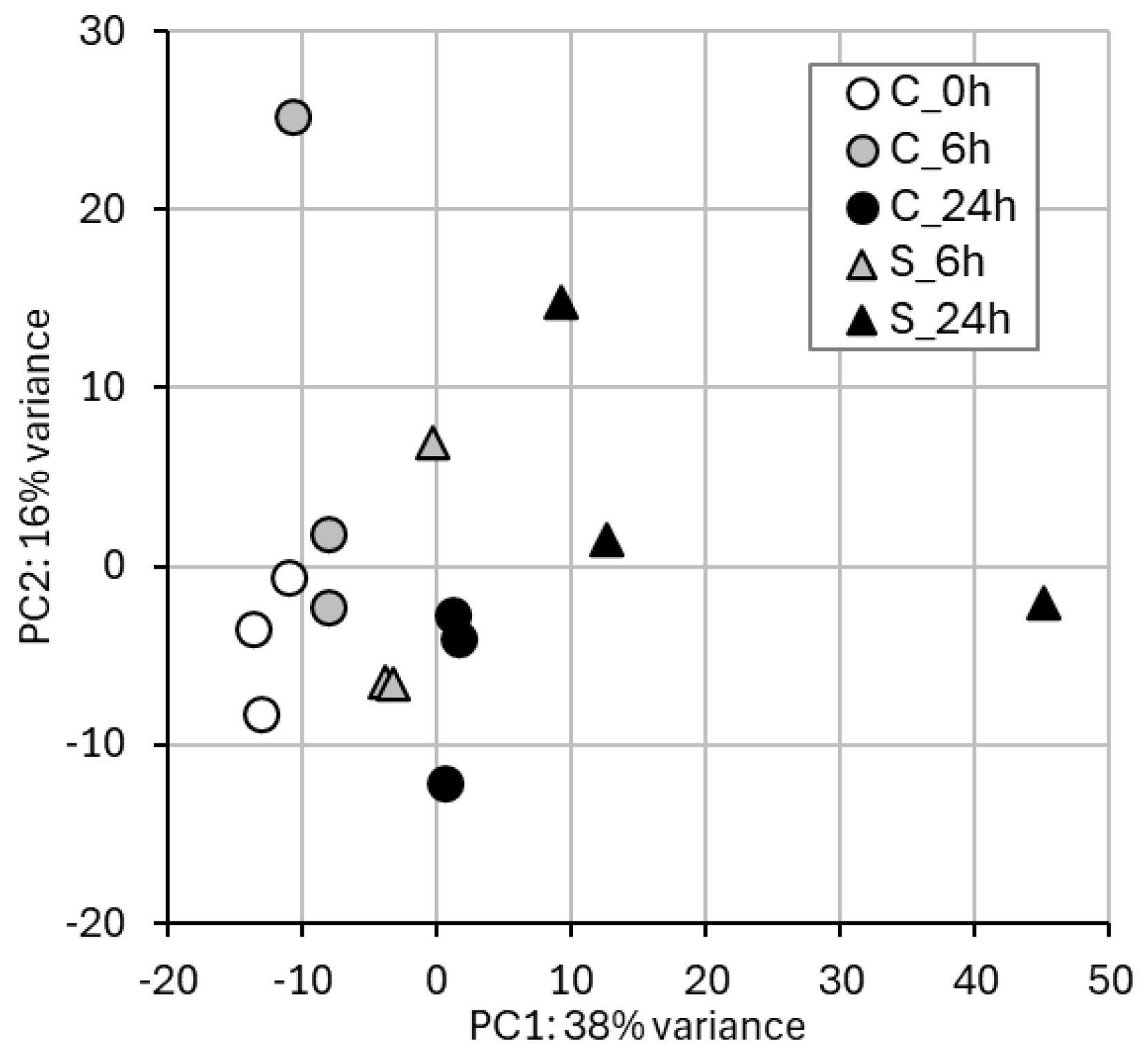
2.1. RNA-Seq and Gene Expression in Treated vs. Control Samples
2.2. Gene Ontology Enrichment
2.3. Differentially Expressed Genes (DEGs)
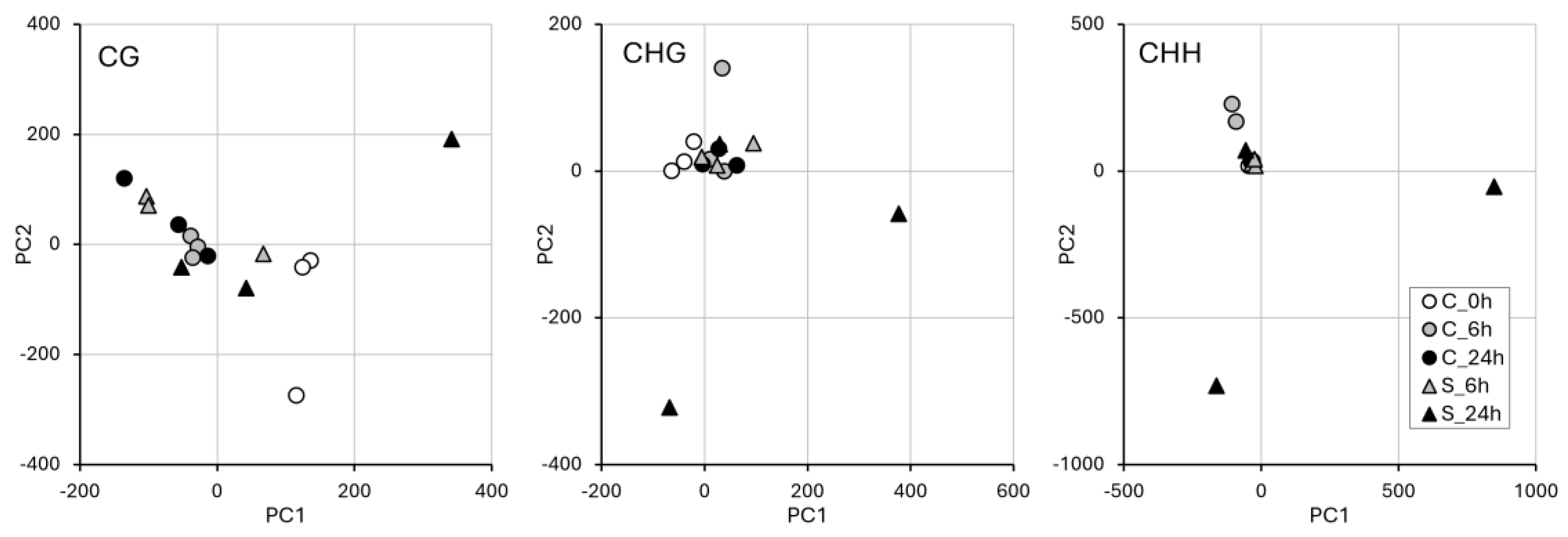
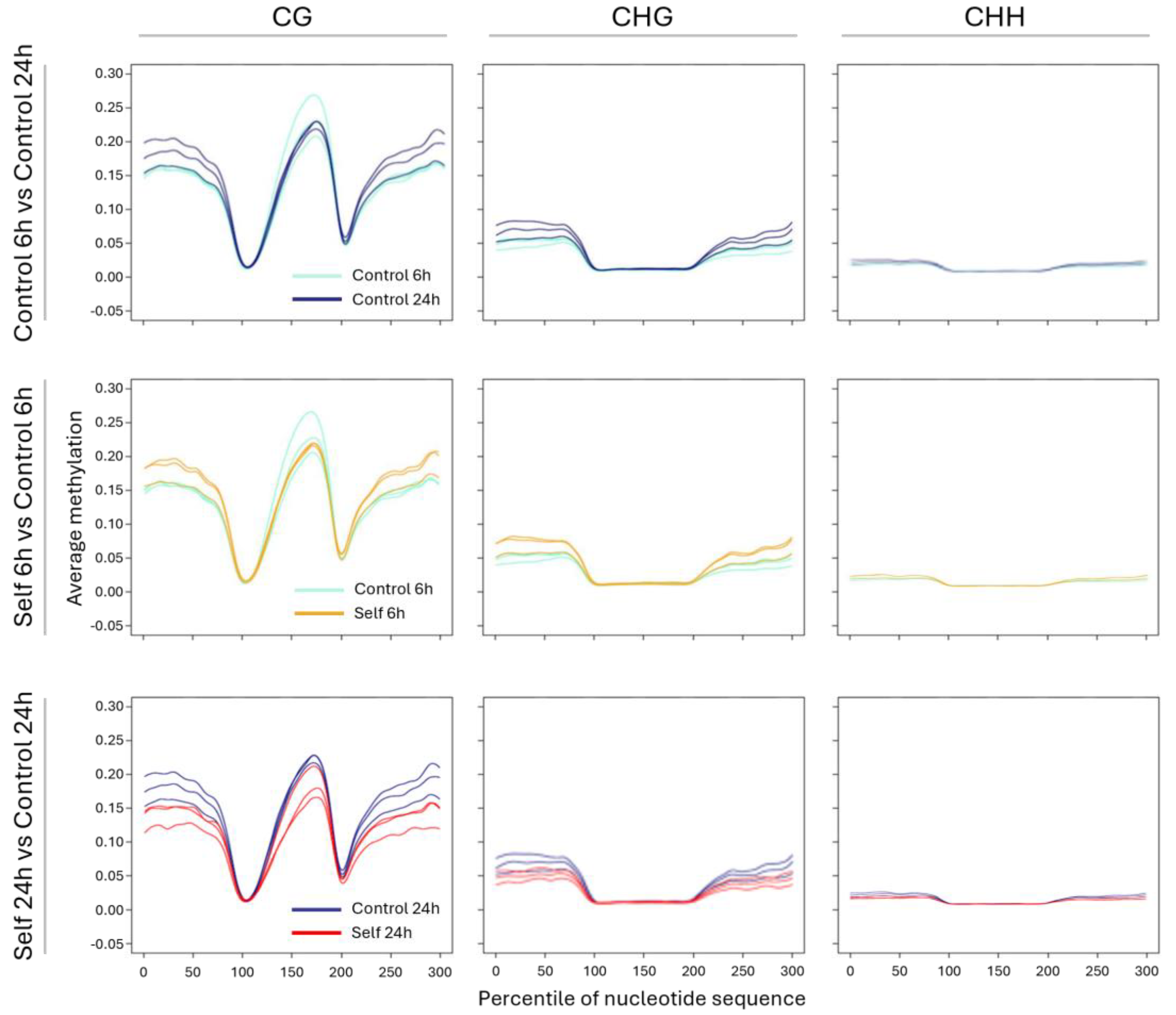
2.4. WGBS and Methylation Level in Treated vs. Control Samples
2.5. Association Between DMR Occurrence and Differential Gene Expression
3. Discussion
3.1. Differentially Expressed Genes at 6 h Exposure
3.2. Differentially Expressed Genes at 24 h Exposure
3.3. Epigenetic Response to Self-DNA Exposure
4. Conclusions
5. Materials and Methods
5.1. Seed Sterilization and Germination
5.2. Growth Medium Slices and Control Samples Preparation
5.3. Sample Collection and Preparation
5.4. Self-DNA Solution Preparation
5.5. Root Exposure and Sampling
5.6. RNA Extraction and mRNA Sequencing
5.7. DNA Extraction and Whole Genome Bisulfite Sequencing (WGBS)
5.8. Bioinformatics Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Comfort, N. From controlling elements to transposons: Barbara McClintock and the Nobel Prize1. Trends Genet. 2001, 17, 475–478. [Google Scholar] [CrossRef] [PubMed]
- Miryeganeh, M.; Saze, H. Epigenetic inheritance and plant evolution. Popul. Ecol. 2020, 62, 17–27. [Google Scholar] [CrossRef]
- Slotkin, R.K.; Martienssen, R. Transposable elements and the epigenetic regulation of the genome. Nat. Rev. Genet. 2007, 8, 272–285. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Shiu, S.; Cal, A.; Borevitz, J.O. Global Analysis of Genetic, Epigenetic and Transcriptional Polymorphisms in Arabidopsis thaliana Using Whole Genome Tiling Arrays. PLoS Genet. 2008, 4, e1000032. [Google Scholar] [CrossRef]
- Jähner, D.; Jaenisch, R. Retrovirus-induced de novo methylation of flanking host sequences correlates with gene inactivity. Nature 1985, 315, 594–597. [Google Scholar] [CrossRef]
- Lippman, Z.; Gendrel, A.-V.; Black, M.; Vaughn, M.W.; Dedhia, N.; Richard McCombie, W.; Lavine, K.; Mittal, V.; May, B.; Kasschau, K.D.; et al. Role of transposable elements in heterochromatin and epigenetic control. Nature 2004, 430, 471–476. [Google Scholar] [CrossRef]
- Parrilla-Doblas, J.T.; Roldán-Arjona, T.; Ariza, R.R.; Córdoba-Cañero, D. Active DNA Demethylation in Plants. Int. J. Mol. Sci. 2019, 20, 4683. [Google Scholar] [CrossRef]
- Belyayev, A. Bursts of transposable elements as an evolutionary driving force. J. Evol. Biol. 2014, 27, 2573–2584. [Google Scholar] [CrossRef]
- Daccord, N.; Celton, J.-M.; Linsmith, G.; Becker, C.; Choisne, N.; Schijlen, E.; Van De Geest, H.; Bianco, L.; Micheletti, D.; Velasco, R.; et al. High-quality de novo assembly of the apple genome and methylome dynamics of early fruit development. Nat. Genet. 2017, 49, 1099–1106. [Google Scholar] [CrossRef]
- Kenchanmane Raju, S.K.; Ritter, E.J.; Niederhuth, C.E. Establishment, maintenance, and biological roles of non-CG methylation in plants. Essays Biochem. 2019, 63, 743–755. [Google Scholar] [CrossRef]
- Gent, J.I.; Dong, Y.; Jiang, J.; Dawe, R.K. Strong epigenetic similarity between maize centromeric and pericentromeric regions at the level of small RNAs, DNA methylation and H3 chromatin modifications. Nucleic Acids Res. 2012, 40, 1550–1560. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Gent, J.I.; Zynda, G.; Song, J.; Makarevitch, I.; Hirsch, C.D.; Hirsch, C.N.; Dawe, R.K.; Madzima, T.F.; McGinnis, K.M.; et al. RNA-directed DNA methylation enforces boundaries between heterochromatin and euchromatin in the maize genome. Proc. Natl. Acad. Sci. USA 2015, 112, 14728–14733. [Google Scholar] [CrossRef] [PubMed]
- Zilberman, D.; Gehring, M.; Tran, R.K.; Ballinger, T.; Henikoff, S. Genome-wide analysis of Arabidopsis thaliana DNA methylation uncovers an interdependence between methylation and transcription. Nat. Genet. 2007, 39, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [CrossRef]
- Muyle, A.M.; Seymour, D.K.; Lv, Y.; Huettel, B.; Gaut, B.S. Gene Body Methylation in Plants: Mechanisms, Functions, and Important Implications for Understanding Evolutionary Processes. Genome Biol. Evol. 2022, 14, evac038. [Google Scholar] [CrossRef]
- Calarco, J.P.; Borges, F.; Donoghue, M.T.A.; Van Ex, F.; Jullien, P.E.; Lopes, T.; Gardner, R.; Berger, F.; Feijó, J.A.; Becker, J.D.; et al. Reprogramming of DNA Methylation in Pollen Guides Epigenetic Inheritance via Small RNA. Cell 2012, 151, 194–205. [Google Scholar] [CrossRef]
- Van Der Graaf, A.; Wardenaar, R.; Neumann, D.A.; Taudt, A.; Shaw, R.G.; Jansen, R.C.; Schmitz, R.J.; Colomé-Tatché, M.; Johannes, F. Rate, spectrum, and evolutionary dynamics of spontaneous epimutations. Proc. Natl. Acad. Sci. USA 2015, 112, 6676–6681. [Google Scholar] [CrossRef]
- Kashkush, K.; Feldman, M.; Levy, A.A. Transcriptional activation of retrotransposons alters the expression of adjacent genes in wheat. Nat. Genet. 2003, 33, 102–106. [Google Scholar] [CrossRef]
- Madlung, A.; Tyagi, A.P.; Watson, B.; Jiang, H.; Kagochi, T.; Doerge, R.W.; Martienssen, R.; Comai, L. Genomic changes in synthetic Arabidopsis polyploids: Genomic changes in Arabidopsis polyploids. Plant J. 2004, 41, 221–230. [Google Scholar] [CrossRef]
- Schmitz, R.J.; He, Y.; Valdés-López, O.; Khan, S.M.; Joshi, T.; Urich, M.A.; Nery, J.R.; Diers, B.; Xu, D.; Stacey, G.; et al. Epigenome-wide inheritance of cytosine methylation variants in a recombinant inbred population. Genome Res. 2013, 23, 1663–1674. [Google Scholar] [CrossRef]
- Eichten, S.R.; Schmitz, R.J.; Springer, N.M. Epigenetics: Beyond Chromatin Modifications and Complex Genetic Regulation. Plant Physiol. 2014, 165, 933–947. [Google Scholar] [CrossRef] [PubMed]
- Fulneček, J.; Matyášek, R.; Kovařík, A. Distribution of 5-methylcytosine residues in 5S rRNA genes in Arabidopsis thaliana and Secale cereale. Mol. Genet. Genom. 2002, 268, 510–517. [Google Scholar] [CrossRef] [PubMed]
- Robertson, K.D.; Wolffe, A.P. DNA methylation in health and disease. Nat. Rev. Genet. 2000, 1, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Secco, D.; Wang, C.; Shou, H.; Schultz, M.D.; Chiarenza, S.; Nussaume, L.; Ecker, J.R.; Whelan, J.; Lister, R. Stress induced gene expression drives transient DNA methylation changes at adjacent repetitive elements. eLife 2015, 4, e09343. [Google Scholar] [CrossRef]
- Bewick, A.J.; Schmitz, R.J. Gene body DNA methylation in plants. Curr. Opin. Plant Biol. 2017, 36, 103–110. [Google Scholar] [CrossRef]
- Herrel, A.; Joly, D.; Danchin, E. Epigenetics in ecology and evolution. Funct. Ecol. 2020, 34, 381–384. [Google Scholar] [CrossRef]
- Skinner, M.K.; Nilsson, E.E. Role of environmentally induced epigenetic transgenerational inheritance in evolutionary biology: Unified Evolution Theory. Environ. Epigenetics 2021, 7, dvab012. [Google Scholar] [CrossRef]
- Gallego-Bartolomé, J. DNA methylation in plants: Mechanisms and tools for targeted manipulation. New Phytol. 2020, 227, 38–44. [Google Scholar] [CrossRef]
- Kumar, S.; Mohapatra, T. Dynamics of DNA Methylation and Its Functions in Plant Growth and Development. Front. Plant Sci. 2021, 12, 596236. [Google Scholar] [CrossRef]
- Dubin, M.J.; Zhang, P.; Meng, D.; Remigereau, M.-S.; Osborne, E.J.; Paolo Casale, F.; Drewe, P.; Kahles, A.; Jean, G.; Vilhjálmsson, B.; et al. DNA methylation in Arabidopsis has a genetic basis and shows evidence of local adaptation. eLife 2015, 4, e05255. [Google Scholar] [CrossRef]
- Galanti, D.; Ramos-Cruz, D.; Nunn, A.; Rodríguez-Arévalo, I.; Scheepens, J.F.; Becker, C.; Bossdorf, O. Genetic and environmental drivers of large-scale epigenetic variation in Thlaspi arvense. PLoS Genet. 2022, 18, e1010452. [Google Scholar] [CrossRef] [PubMed]
- Peña-Ponton, C.; Diez-Rodriguez, B.; Perez-Bello, P.; Becker, C.; McIntyre, L.M.; Van Der Putten, W.; De Paoli, E.; Heer, K.; Opgenoorth, L.; Verhoeven, K.J.F. High-resolution methylome analysis in the clonal Populus nigra cv. ‘Italica’ reveals environmentally sensitive hotspots and drought-responsive TE superfamilies. bioRxiv 2022. [Google Scholar] [CrossRef]
- Arora, H.; Singh, R.K.; Sharma, S.; Sharma, N.; Panchal, A.; Das, T.; Prasad, A.; Prasad, M. DNA methylation dynamics in response to abiotic and pathogen stress in plants. Plant Cell Rep. 2022, 41, 1931–1944. [Google Scholar] [CrossRef]
- Vega-Muñoz, I.; Feregrino-Pérez, A.A.; Torres-Pacheco, I.; Guevara-González, R.G. Exogenous fragmented DNA acts as a damage-associated molecular pattern (DAMP) inducing changes in CpG DNA methylation and defence-related responses in Lactuca sativa. Funct. Plant Biol. 2018, 45, 1065. [Google Scholar] [CrossRef] [PubMed]
- Ferrusquía-Jiménez, N.I.; Chandrakasan, G.; Torres-Pacheco, I.; Rico-Garcia, E.; Feregrino-Perez, A.A.; Guevara-González, R.G. Extracellular DNA: A Relevant Plant Damage-Associated Molecular Pattern (DAMP) for Crop Protection Against Pests—A Review. J. Plant Growth Regul. 2021, 40, 451–463. [Google Scholar] [CrossRef]
- Barbero, F.; Guglielmotto, M.; Capuzzo, A.; Maffei, M. Extracellular Self-DNA (eself-DNA), but Not Heterologous Plant or Insect DNA (etDNA), Induces Plasma Membrane Depolarization and Calcium Signaling in Lima Bean (Phaseolus lunatus) and Maize (Zea mays). Int. J. Mol. Sci. 2016, 17, 1659. [Google Scholar] [CrossRef]
- Duran-Flores, D.; Heil, M. Sources of specificity in plant damaged-self recognition. Curr. Opin. Plant Biol. 2016, 32, 77–87. [Google Scholar] [CrossRef]
- Rassizadeh, L.; Cervero, R.; Flors, V.; Gamir, J. Extracellular DNA as an elicitor of broad-spectrum resistance in Arabidopsis thaliana. Plant Sci. 2021, 312, 111036. [Google Scholar] [CrossRef]
- Ronchi, A.; Foscari, A.; Zaina, G.; De Paoli, E.; Incerti, G. Self-DNA Early Exposure in Cultivated and Weedy Setaria Triggers ROS Degradation Signaling Pathways and Root Growth Inhibition. Plants 2023, 12, 1288. [Google Scholar] [CrossRef]
- Zhou, X.; Gao, H.; Zhang, X.; Khashi u Rahman, M.; Mazzoleni, S.; Du, M.; Wu, F. Plant extracellular self-DNA inhibits growth and induces immunity via the jasmonate signaling pathway. Plant Physiol. 2023, 192, 2475–2491. [Google Scholar] [CrossRef]
- Mazzoleni, S.; Bonanomi, G.; Incerti, G.; Chiusano, M.L.; Termolino, P.; Mingo, A.; Senatore, M.; Giannino, F.; Cartenì, F.; Rietkerk, M.; et al. Inhibitory and toxic effects of extracellular self-DNA in litter: A mechanism for negative plant–soil feedbacks? New Phytol. 2015, 205, 1195–1210. [Google Scholar] [CrossRef] [PubMed]
- Mazzoleni, S.; Cartenì, F.; Bonanomi, G.; Senatore, M.; Termolino, P.; Giannino, F.; Incerti, G.; Rietkerk, M.; Lanzotti, V.; Chiusano, M.L. Inhibitory effects of extracellular self-DNA: A general biological process? New Phytol. 2015, 206, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Chiusano, M.L.; Incerti, G.; Colantuono, C.; Termolino, P.; Palomba, E.; Monticolo, F.; Benvenuto, G.; Foscari, A.; Esposito, A.; Marti, L.; et al. Arabidopsis thaliana Response to Extracellular DNA: Self Versus Nonself Exposure. Plants 2021, 10, 1744. [Google Scholar] [CrossRef] [PubMed]
- Germoglio, M.; Adamo, A.; Incerti, G.; Cartenì, F.; Gigliotti, S.; Storlazzi, A.; Mazzoleni, S. Self-DNA Exposure Induces Developmental Defects and Germline DNA Damage Response in Caenorhabditis elegans. Biology 2022, 11, 262. [Google Scholar] [CrossRef]
- Colombo, M.; Grauso, L.; Lanzotti, V.; Incerti, G.; Adamo, A.; Storlazzi, A.; Gigliotti, S.; Mazzoleni, S. Self-DNA Inhibition in Drosophila melanogaster Development: Metabolomic Evidence of the Molecular Determinants. Biology 2023, 12, 1378. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- de Alteriis, E.; Incerti, G.; Cartenì, F.; Chiusano, M.L.; Colantuono, C.; Palomba, E.; Termolino, P.; Monticolo, F.; Esposito, A.; Bonanomi, G.; et al. Extracellular DNA secreted in yeast cultures is metabolism-specific and inhibits cell proliferation. Microb. Cell 2023, 10, 292–295. [Google Scholar] [CrossRef]
- Anzano, A.; Bonanomi, G.; Mazzoleni, S.; Lanzotti, V. Plant metabolomics in biotic and abiotic stress: A critical overview. Phytochem. Rev. 2022, 21, 503–524. [Google Scholar] [CrossRef]
- Lanzotti, V.; Grauso, L.; Mangoni, A.; Termolino, P.; Palomba, E.; Anzano, A.; Incerti, G.; Mazzoleni, S. Metabolomics and molecular networking analyses in Arabidopsis thaliana show that extracellular self-DNA affects nucleoside/nucleotide cycles with accumulation of cAMP, cGMP and N6-methyl-AMP. Phytochemistry 2022, 204, 113453. [Google Scholar] [CrossRef]
- Palomba, E.; Chiusano, M.L.; Monticolo, F.; Langella, M.C.; Sanchez, M.; Tirelli, V.; de Alteriis, E.; Iannaccone, M.; Termolino, P.; Capparelli, R.; et al. Extracellular Self-DNA Effects on Yeast Cell Cycle and Transcriptome During Batch Growth. Biomolecules 2024, 14, 663. [Google Scholar] [CrossRef]
- Sun, M.; Yang, Z.; Liu, L.; Duan, L. DNA Methylation in Plant Responses and Adaption to Abiotic Stresses. Int. J. Mol. Sci. 2022, 23, 6910. [Google Scholar] [CrossRef]
- Chan, S.W.-L.; Henderson, I.R.; Jacobsen, S.E. Gardening the genome: DNA methylation in Arabidopsis thaliana. Nat. Rev. Genet. 2005, 6, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Mazzoleni, S.; Grauso, L.; de Falco, B.; Mangoni, A.; Termolino, P.; Palomba, E.; Carteni, F.; Lanzotti, V. Metabolomic changes in Arabidopsis thaliana exposed to extracellular self-and nonself-DNA: A reversible effect. Env. Exp. Bot. 2025, 106149. [Google Scholar] [CrossRef]
- Vargas-Hernandez, M.; Macias-Bobadilla, I.; Guevara-Gonzalez, R.G.; Romero-Gomez, S.D.J.; Rico-Garcia, E.; Ocampo-Velazquez, R.V.; Alvarez-Arquieta, L.; Torres-Pacheco, I. Plant hormesis management with biostimulants of biotic origin in agriculture. Front. Plant Sci. 2017, 8, 1762. [Google Scholar] [CrossRef] [PubMed]
- Gallucci, S.; Maffei, M.E. DNA Sensing Across the Tree of Life. Trends Immunol. 2017, 38, 719–732. [Google Scholar] [CrossRef]
- Paludan, S.R.; Bowie, A.G. Immune Sensing of DNA. Immunity 2013, 38, 870–880. [Google Scholar] [CrossRef]
- Krueger, F.; James, F.; Ewels, P.; Afyounian, E.; Weinstein, M.; Schuster-Boeckler, B.; Hulselmans, G. TrimGalore; v0.6.10; The Brabaham Institute: Brabaham, UK, 2023. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 1 March 2023).
- Bock, C. Analysing and interpreting DNA methylation data. Nat. Rev. Genet. 2012, 13, 705–719. [Google Scholar] [CrossRef]
- Krueger, F.; Andrews, S.R. Bismark: A flexible aligner and methylation caller for BisulfiteSeq applications. Bioinformatics 2011, 27, 1571–1572. [Google Scholar] [CrossRef]
- Akalin, A.; Kormaksson, M.; Li, S.; Garrett-Bakelman, F.E.; Figueroa, M.E.; Melnick, A.; Mason, C.E. methylKit: A comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol. 2012, 13, R87. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Love, M.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis. Springer-Verlag New York. Available online: https://ggplot2.tidyverse.org (accessed on 1 April 2023).




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group of DEGs | Down | Up | |||||
|---|---|---|---|---|---|---|---|
| 6 h | 6 and 24 h | 24 h | 6 h | 6 and 24 h | 24 h | ||
| 1. Stress and Defense | |||||||
| Stress Response/Defense: Detoxification/ROS | - | - | 2 | 1 | - | - | |
| Stress Response/Defense: Others | - | - | 2 | ||||
| Stress Response: Cysteine Proteinases | - | - | 2 | ||||
| Stress Response/Defense: Detoxification/Glutathione | - | - | 2 | ||||
| Early Response to Dehydration | 1 | - | - | ||||
| Stress Response and Protein Quality Control | 4 | 1 | - | ||||
| Heat Shock Proteins | 7 | 5 | - | ||||
| Defense and Pathogen Response | 4 | 1 | - | ||||
| Reactive Oxygen Species (ROSs) Response and Detoxification | - | - | 3 | ||||
| Detoxification | 3 | - | - | ||||
| Direct ROS Management | - | - | 5 | ||||
| Indirect ROS Management | - | - | 4 | ||||
| Defense Proteins | - | - | 7 | ||||
| 2. DNA/Protein Processing and Folding | |||||||
| Protein Modification and Degradation | 3 | 1 | - | ||||
| Nucleotide Metabolism | 2 | - | - | ||||
| Protein Folding and Degradation | 1 | 1 | 4 | ||||
| 3. Growth and Development | |||||||
| Cell Wall and Growth Regulation | 6 | - | - | 4 | 1 | - | |
| Embryo Development and Regulation | - | - | 4 | ||||
| Root Hair Development | - | - | 2 | ||||
| Cell Wall Synthesis and Modification | - | - | 4 | ||||
| Cell Cycle and Division (Cyclins) | - | - | 1 | ||||
| Embryo and Seed Development | - | 1 | 4 | ||||
| Chlorophyll/Photosynthesis-related | - | - | 1 | ||||
| Cell Wall-Modifying Enzymes | - | - | 2 | ||||
| 4. Transport, Signaling, and Homeostasis | |||||||
| Ion Transport and Signaling | 5 | - | - | 2 | 3 | - | |
| Signal Transduction and Membrane Functions | 5 | 1 | - | ||||
| Transport and Membrane Trafficking | 6 | 1 | - | ||||
| Cellular Homeostasis and Protection | 3 | - | - | ||||
| Nutrient and Ion Transport | - | - | 3 | ||||
| Hormone and Signaling: Auxin Signaling | - | - | 1 | ||||
| Hormone and Signaling: Auxin and Cytokinin Signaling | - | - | 1 | ||||
| Hormone and Signaling: Terpenoid Synthesis | - | - | 1 | ||||
| Cytochrome P450s | - | - | 3 | ||||
| 5. Energy, Metabolism, and Biosynthesis | |||||||
| Metabolism and Biosynthesis | 9 | 2 | - | - | - | 9 | |
| DNA/RNA Processing | - | - | 1 | ||||
| ATP Production and Regulation | - | 1 | 1 | ||||
| Nucleotide Metabolism | 2 | - | - | ||||
| Redox Balance and Electron Transport | - | - | 4 | ||||
| Metabolism and Transport | - | 2 | 9 | ||||
| Lipid Metabolism (Acyl-CoA) | - | - | 8 | ||||
| Sugar Metabolism and Transport | - | - | 4 | ||||
| 6. Gene Expression and Transcription Regulation | |||||||
| Transcription Factors (TFs) | - | - | 1 | - | - | 7 | |
| NAC Domain TFs | - | - | 5 | ||||
| 7. Uncharacterized/Unknown Proteins/Miscellaneous Roles | 2 | - | 8 | 2 | 1 | 4 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ronchi, A.; Incerti, G.; De Paoli, E.; Panico, S.C.; Sciabbarrasi, G.L.; Termolino, P.; Cartenì, F.; Langella, M.; Chiusano, M.L.; Mazzoleni, S. Arabidopsis thaliana Roots Exposed to Extracellular Self-DNA: Evidence of Epigenetic Effects. Epigenomes 2025, 9, 13. https://doi.org/10.3390/epigenomes9020013
Ronchi A, Incerti G, De Paoli E, Panico SC, Sciabbarrasi GL, Termolino P, Cartenì F, Langella M, Chiusano ML, Mazzoleni S. Arabidopsis thaliana Roots Exposed to Extracellular Self-DNA: Evidence of Epigenetic Effects. Epigenomes. 2025; 9(2):13. https://doi.org/10.3390/epigenomes9020013
Chicago/Turabian StyleRonchi, Alessia, Guido Incerti, Emanuele De Paoli, Speranza Claudia Panico, Giovanni Luca Sciabbarrasi, Pasquale Termolino, Fabrizio Cartenì, Mariachiara Langella, Maria Luisa Chiusano, and Stefano Mazzoleni. 2025. "Arabidopsis thaliana Roots Exposed to Extracellular Self-DNA: Evidence of Epigenetic Effects" Epigenomes 9, no. 2: 13. https://doi.org/10.3390/epigenomes9020013
APA StyleRonchi, A., Incerti, G., De Paoli, E., Panico, S. C., Sciabbarrasi, G. L., Termolino, P., Cartenì, F., Langella, M., Chiusano, M. L., & Mazzoleni, S. (2025). Arabidopsis thaliana Roots Exposed to Extracellular Self-DNA: Evidence of Epigenetic Effects. Epigenomes, 9(2), 13. https://doi.org/10.3390/epigenomes9020013