S-Adenosylmethionine Treatment of Colorectal Cancer Cell Lines Alters DNA Methylation, DNA Repair and Tumor Progression-Related Gene Expression


 , , ,
, , ,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Cultures
2.2. Cell Proliferation Analysis with SRB Assay
2.3. Cell Cycle Analysis by Flow Cytometry
2.4. Whole Genomic Expression Analysis with Microarray Technique
2.5. DNA Isolation from the Treated Cells
2.6. Global DNA Methylation Analysis with LINE-1 Pyrosequencing
2.7. Whole Genomic DNA Methylation Analysis with Illumina Bead Array Technique
2.8. ELISA Assay and Immunocytochemistry for γ-H2AX Detection
2.9. Genomic Stability Detection with Comet Assay
2.10. Micronuclei Scoring and Staining
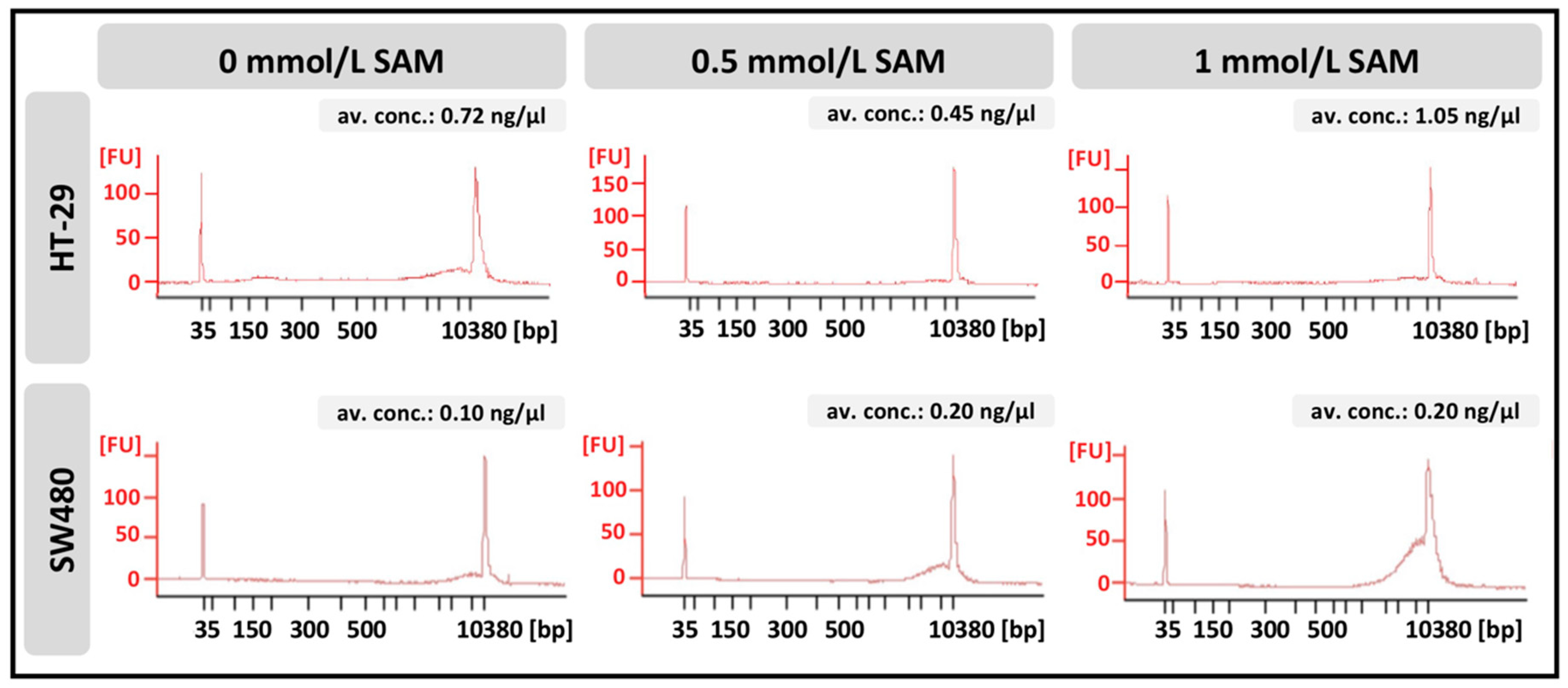
2.11. Quantitative and Qualitative Analysis of Cell-Free DNA with DNA Isolation
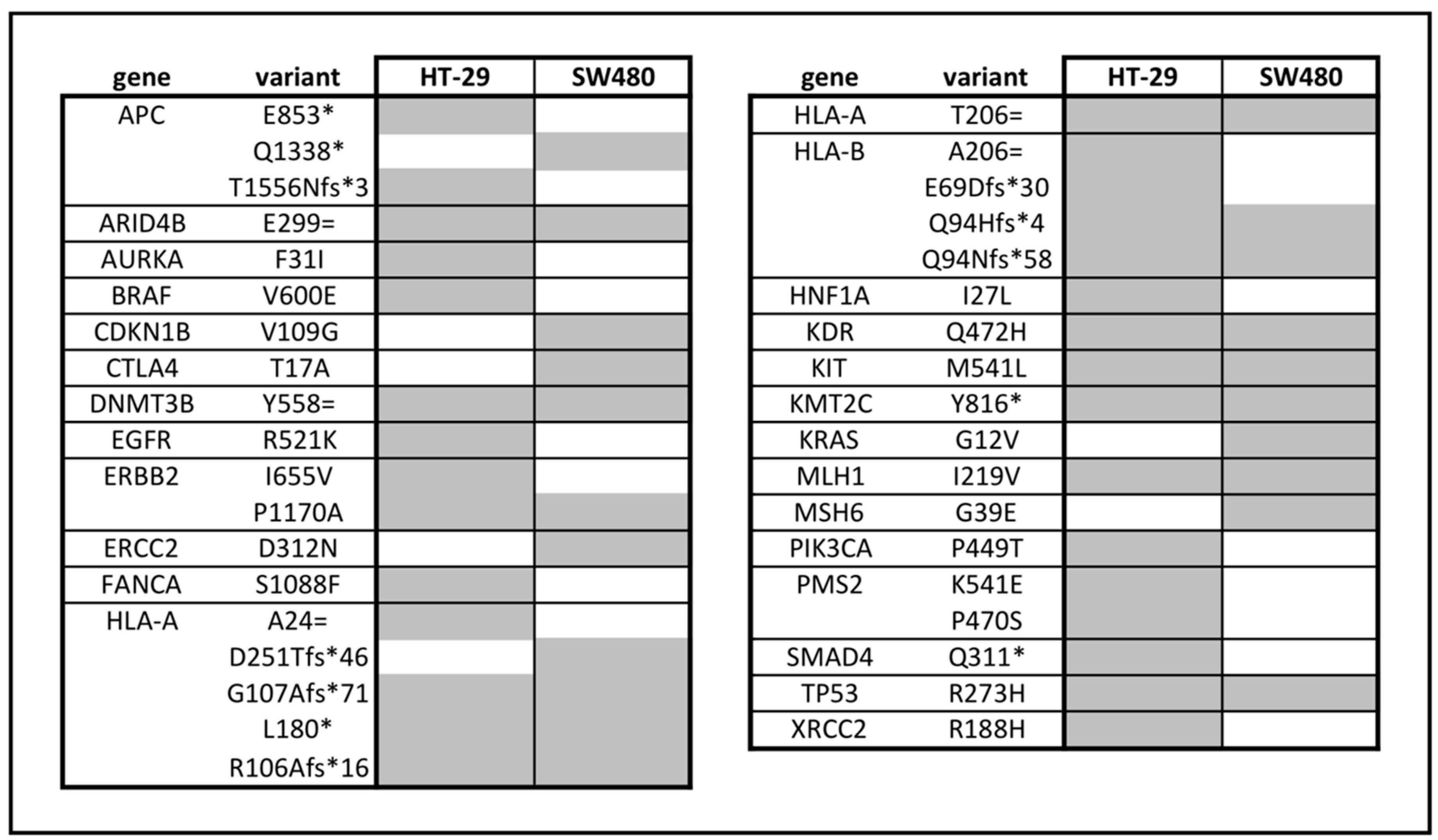
2.12. Whole Exome Sequencing
3. Results
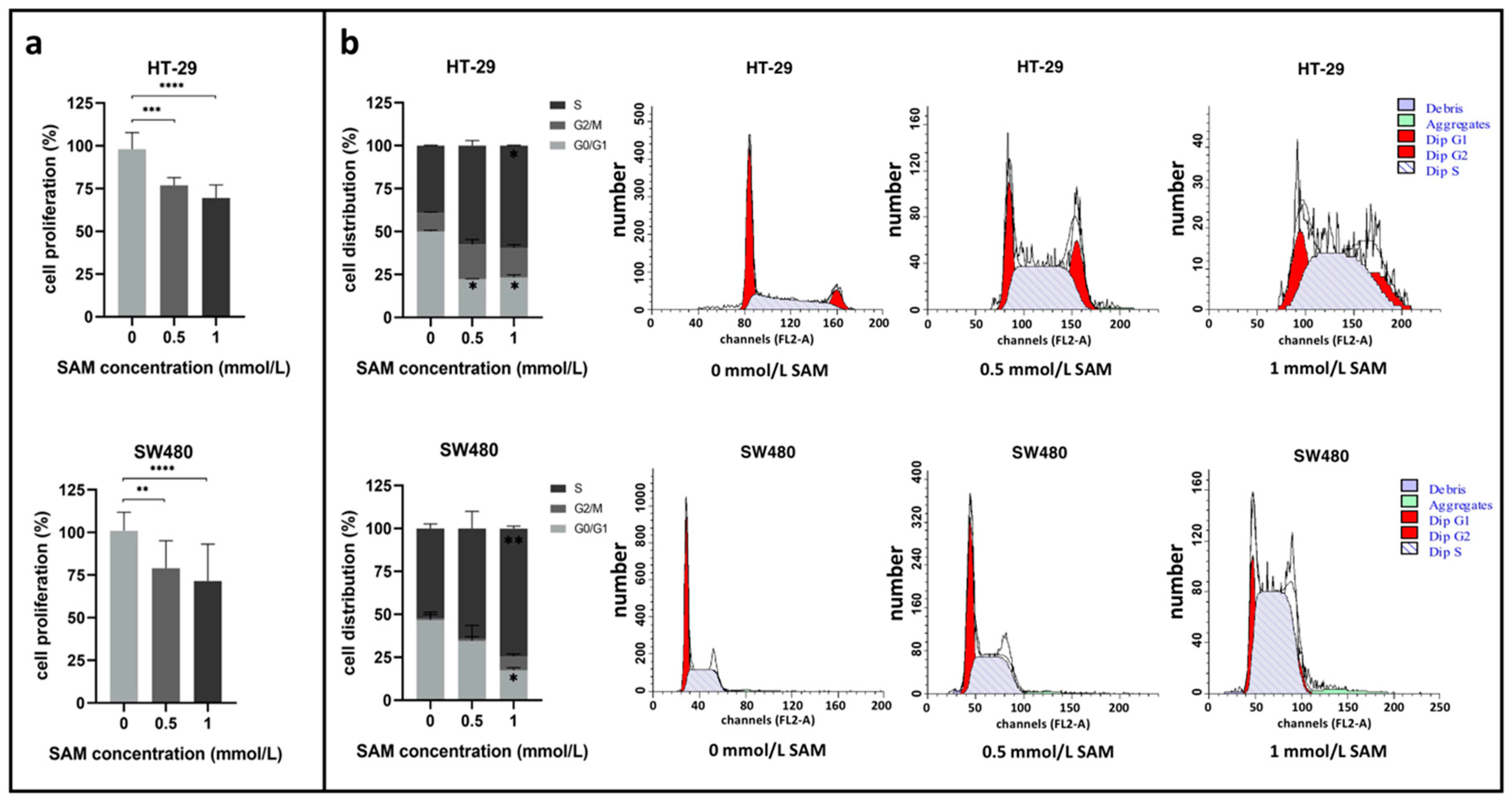
3.1. Effect of SAM Treatment on Cell Proliferation and Cell Cycle
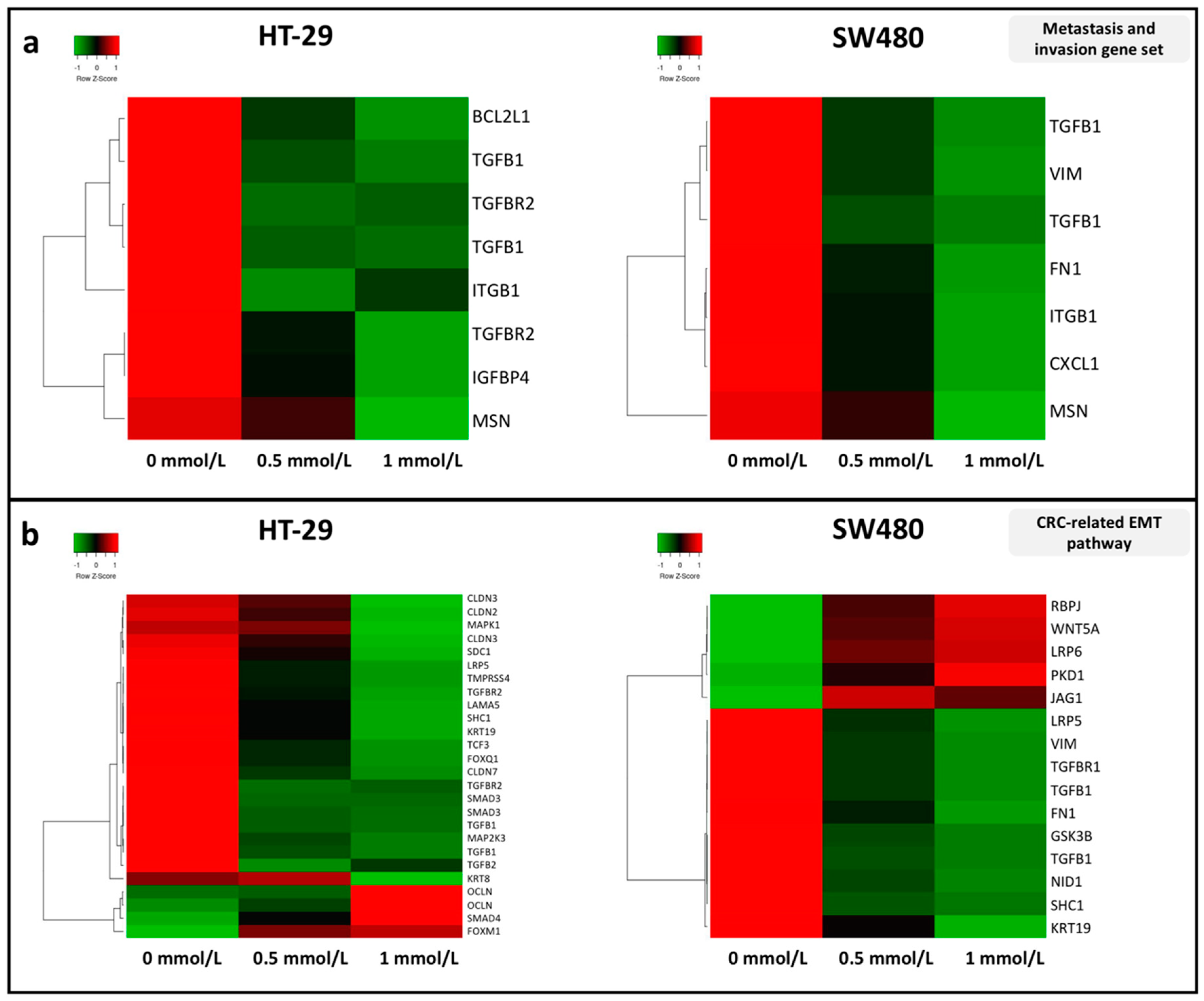
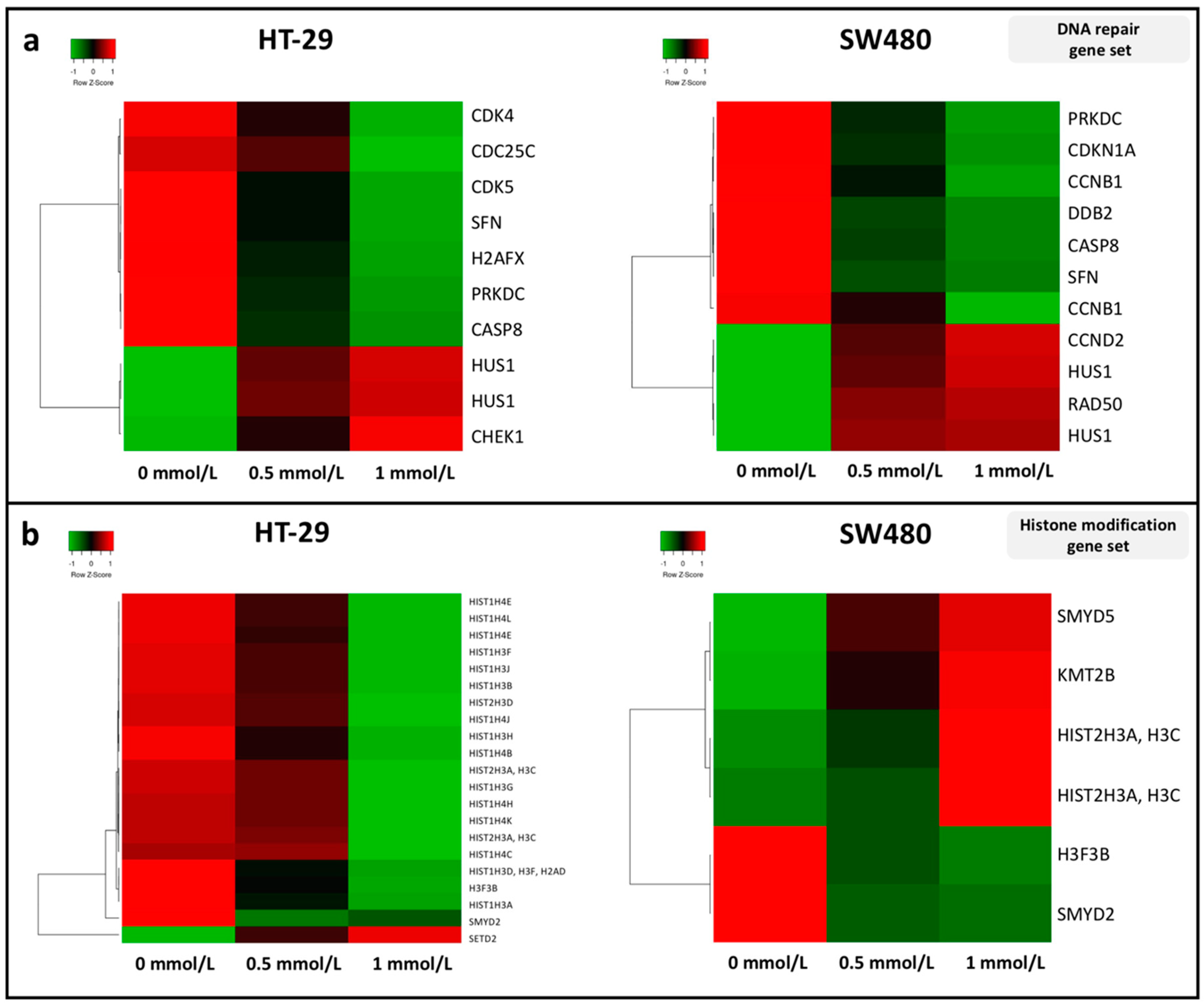
3.2. Effect of SAM Treatment on the Expression of Genes Involved in Tumor Progression
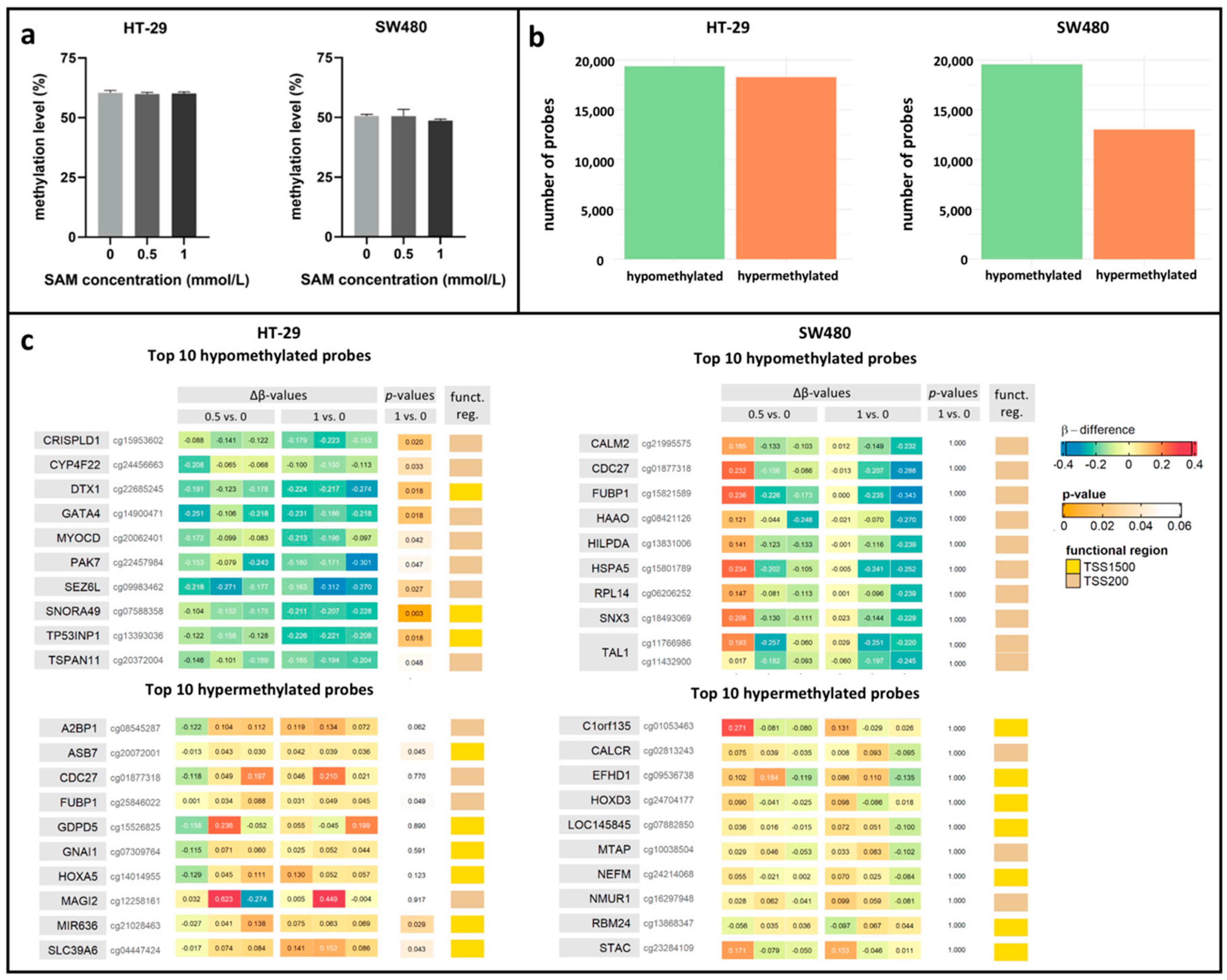
3.3. Effect of SAM Treatment on Global and Promoter-Specific DNA Methylation Level
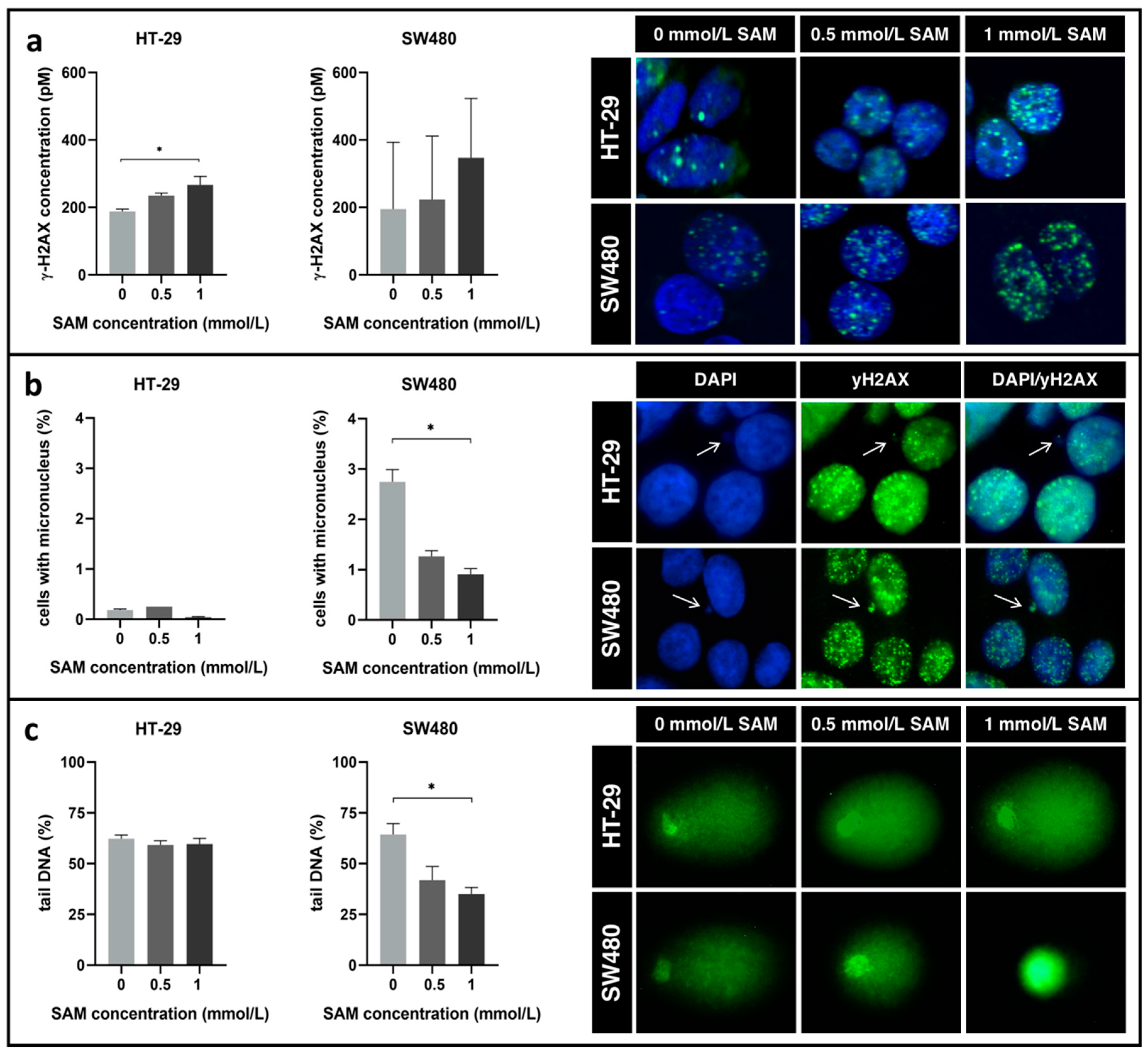
3.4. Effect of SAM Treatment on Genomic Stability
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Dai, W. Genomic Instability and Cancer. J. Carcinog Mutagen 2014, 5, 1000165. [Google Scholar] [PubMed]
- Shen, Z. Genomic instability and cancer: An introduction. J. Mol. Cell Biol. 2011, 3, 1–3. [Google Scholar] [CrossRef]
- Włodarczyk, M.; Włodarczyk, J.; Siwiński, P.; Sobolewska-Włodarczyk, A.; Fichna, J. Genetic Molecular Subtypes in Optimizing Personalized Therapy for Metastatic Colorectal Cancer. Curr. Drug Targets 2018, 19, 1731–1737. [Google Scholar] [CrossRef]
- Smeby, J.; Sveen, A.; Merok, M.A.; Danielsen, S.A.; Eilertsen, I.A.; Guren, M.G.; Dienstmann, R.; Nesbakken, A.; Lothe, R.A. CMS-dependent prognostic impact of KRAS and BRAFV600E mutations in primary colorectal cancer. Ann. Oncol. 2018, 29, 1227–1234. [Google Scholar] [CrossRef]
- Grady, W.M. Genomic instability and colon cancer. Cancer Metastasis Rev. 2004, 23, 11–27. [Google Scholar] [CrossRef]
- Markowitz, S.D.; Bertagnolli, M.M. Molecular Basis of Colorectal Cancer. N. Engl. J. Med 2009, 361, 2449–2460. [Google Scholar] [CrossRef]
- Guinney, J.; Dienstmann, R.; Wang, X.; de Reyniès, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef]
- Kulis, M.; Esteller, M. DNA methylation and cancer. Adv Genet. 2010, 70, 27–56. [Google Scholar] [PubMed]
- Meaney, M.J.; Szyf, M. Environmental programming of stress responses through DNA methylation: Life at the interface between a dynamic environment and a fixed genome. Dialogues Clin. Neurosci. 2005, 7, 103–123. [Google Scholar] [PubMed]
- Das, P.M.; Singal, R. DNA methylation and cancer. J. Clin. Oncol. 2004, 22, 4632–4642. [Google Scholar] [CrossRef] [PubMed]
- Gautrey, H.E.; van Otterdijk, S.D.; Cordell, H.J.; Mathers, J.C.; Strathdee, G. DNA methylation abnormalities at gene promoters are extensive and variable in the elderly and phenocopy cancer cells. FASEB J. 2014, 28, 3261–3272. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Li, Y.N.; Wang, F.; Zhang, W.M.; Geng, X. S-adenosylmethionine inhibits the growth of cancer cells by reversing the hypomethylation status of c-myc and H-ras in human gastric cancer and colon cancer. Int. J. Biol. Sci. 2010, 6, 784–795. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.C. S-Adenosylmethionine. Int. J. Biochem. Cell Biol. 2000, 32, 391–395. [Google Scholar] [CrossRef]
- Gören, J.L.; Stoll, A.L.; Damico, K.E.; Sarmiento, I.A.; Cohen, B.M. Bioavailability and lack of toxicity of S-adenosyl-L-methionine (SAMe) in humans. Pharmacotherapy 2004, 24, 1501–1507. [Google Scholar] [CrossRef] [PubMed]
- Parashar, S.; Cheishvili, D.; Arakelian, A.; Hussain, Z.; Tanvir, I.; Khan, H.A.; Szyf, M.; Rabbani, S.A. S-adenosylmethionine blocks osteosarcoma cells proliferation and invasion in vitro and tumor metastasis in vivo: Therapeutic and diagnostic clinical applications. Cancer Med. 2015, 4, 732–744. [Google Scholar] [CrossRef]
- Stoyanov, E.; Mizrahi, L.; Olam, D.; Schnitzer-Perlman, T.; Galun, E.; Goldenberg, D.S. Tumor-suppressive effect of S-adenosylmethionine supplementation in a murine model of inflammation-mediated hepatocarcinogenesis is dependent on treatment longevity. Oncotarget 2017, 8, 104772–104784. [Google Scholar] [CrossRef]
- Zhao, Y.; Li, J.S.; Guo, M.Z.; Feng, B.S.; Zhang, J.P. Inhibitory effect of S-adenosylmethionine on the growth of human gastric cancer cells in vivo and in vitro. Chin. J. Cancer 2010, 29, 752–760. [Google Scholar] [CrossRef]
- Schmidt, T.; Leha, A.; Salinas-Riester, G. Treatment of prostate cancer cells with S-adenosylmethionine leads to genome-wide alterations in transcription profiles. Gene 2016, 595, 161–167. [Google Scholar] [CrossRef]
- Pakneshan, P.; Szyf, M.; Farias-Eisner, R.; Rabbani, S.A.; Rabbani, S.A. Reversal of the hypomethylation status of urokinase (uPA) promoter blocks breast cancer growth and metastasis. J. Biol. Chem. 2004, 279, 31735–31744. [Google Scholar] [CrossRef]
- Guruswamy, S.; Swamy, M.V.; Choi, C.I.; Steele, V.E.; Rao, C.V. S-adenosyl L-methionine inhibits azoxymethane-induced colonic aberrant crypt foci in F344 rats and suppresses human colon cancer Caco-2 cell growth in 3D culture. Int. J. Cancer 2008, 122, 25–30. [Google Scholar] [CrossRef]
- Loenen, W.A.M. S-Adenosylmethionine: Jack of all trades and master of everything? Biochem. Soc. Trans. 2006, 34, 330–333. [Google Scholar] [CrossRef] [PubMed]
- Berg, K.C.G.; Eide, P.W.; Eilertsen, I.A.; Johannessen, B.; Bruun, J.; Danielsen, S.A.; Bjørnslett, M.; Meza-Zepeda, L.A.; Eknæs, M.; Lind, G.E.; et al. Multi-omics of 34 colorectal cancer cell lines—a resource for biomedical studies. Mol. Cancer 2017, 16, 116. [Google Scholar] [CrossRef] [PubMed]
- Li, T.W.H.; Zhang, Q.; Oh, P.; Xia, M.; Chen, H.; Bemanian, S.; Lastra, N.; Circ, M.; Moyer, M.P.; Mato, J.M.; et al. S-Adenosylmethionine and methylthioadenosine inhibit cellular FLICE inhibitory protein expression and induce apoptosis in colon cancer cells. Mol. Pharmacol. 2009, 76, 192–200. [Google Scholar] [CrossRef]
- Yang, H.; Sadda, M.R.; Li, M.; Zeng, Y.; Chen, L.; Bae, W.; Ou, X.; Runnegar, M.T.; Mato, J.M.; Lu, S.C. S-adenosylmethionine and its metabolite induce apoptosis in HepG2 cells: Role of protein phosphatase 1 and Bcl-x(S). Hepatology 2004, 40, 221–231. [Google Scholar] [CrossRef]
- Kalmár, A.; Nagy, Z.B.; Galamb, O.; Csabai, I.; Bodor, A.; Wichmann, B.; Valcz, G.; Barták, B.K.; Tulassay, Z.; Igaz, P.; et al. Genome-wide expression profiling in colorectal cancer focusing on lncRNAs in the adenoma-carcinoma transition. BMC Cancer 2019, 19, 1059. [Google Scholar] [CrossRef]
- Valcz, G.; Buzás, E.I.; Kittel, Á.; Krenács, T.; Visnovitz, T.; Spisák, S.; Török, G.; Homolya, L.; Zsigrai, S.; Kiszler, G.; et al. En bloc release of MVB-like small extracellular vesicle clusters by colorectal carcinoma cells. J. Extracell Vesicles 2019, 8, 1596668. [Google Scholar] [CrossRef]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ data to high confidence variant calls: The Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinform. 2013, 43, 11.10.1–11.10.33. [Google Scholar]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef] [PubMed]
- Bakhoum, S.F.; Ngo, B.; Laughney, A.M.; Cavallo, J.A.; Murphy, C.J.; Ly, P.; Shah, P.; Sriram, R.K.; Watkins, T.B.K.; Taunk, N.K.; et al. Chromosomal instability drives metastasis through a cytosolic DNA response. Nature 2018, 553, 467–472. [Google Scholar] [CrossRef]
- Gaykalova, D.A.; Zizkova, V.; Guo, T.; Tiscareno, I.; Wei, Y.; Vatapalli, R.; Hennessey, P.T.; Ahn, J.; Danilova, L.; Khan, Z.; et al. Integrative computational analysis of transcriptional and epigenetic alterations implicates DTX1 as a putative tumor suppressor gene in HNSCC. Oncotarget 2017, 8, 15349–15363. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.F.; Fang, F.; Hu, S.Y.; Lu, J.; Cao, L.; Zhao, W.L.; Xiao, P.F.; Li, Z.H.; Wang, N.N.; Xu, L.X.; et al. Hypermethylation of the GATA binding protein 4 (GATA4) promoter in Chinese pediatric acute myeloid leukemia. BMC Cancer 2015, 15, 756. [Google Scholar] [CrossRef]
- Sepulveda, J.L.; Gutierrez-Pajares, J.L.; Luna, A.; Yao, Y.; Tobias, J.W.; Thomas, S.; Woo, Y.; Giorgi, F.; Komissarova, E.V.; Califano, A.; et al. High-definition CpG methylation of novel genes in gastric carcinogenesis identified by next-generation sequencing. Mod. Pathol. 2016, 29, 182–193. [Google Scholar] [CrossRef] [PubMed]
- Weng, W.; Yang, Q.; Huang, M.; Qiao, Y.; Xie, Y.; Yu, Y.; jing, A.; Li, Z. c-Myc inhibits TP53INP1 expression via promoter methylation in esophageal carcinoma. Biochem. Biophys. Res. Commun. 2011, 405, 278–284. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.W.; Luo, J.; Weng, Y.I.; Mutch, D.G.; Goodfellow, P.J.; Miller, D.S.; Huang, T.H.M. Promoter hypermethylation of CIDEA, HAAO and RXFP3 associated with microsatellite instability in endometrial carcinomas. Gynecol. Oncol. 2010, 117, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Musialik, E.; Bujko, M.; Kober, P.; Wypych, A.; Gawle-Krawczyk, K.; Matysiak, M.; Siedlecki, J.A. Promoter methylation and expression levels of selected hematopoietic genes in pediatric B-cell acute lymphoblastic leukemia. Blood Res. 2015, 50, 26–32. [Google Scholar] [CrossRef][Green Version]
- Mato, J.M.; Corrales, F.J.; Lu, S.C.; Avila, M.A. S-Adenosylmethionine: A control switch that regulates liver function. FASEB J. 2002, 16, 15–26. [Google Scholar] [CrossRef]
- Li, N.; Zhang, H.H.; Wang, S.H.; Zhu, W.M.; Ren, J.A.; Li, J.S. S-adenosylmethionine in treatment of cholestasis after total parenteral nutrition: Laboratory investigation and clinical application. Hepatobiliary Pancreat Dis. Int. 2002, 1, 96–100. [Google Scholar]
- Silveri, M.M.; Parow, A.M.; Villafuerte, R.A.; Damico, K.E.; Goren, J.; Stoll, A.L.; Cohen, B.M.; Renshaw, P.F. S-adenosyl-L-methionine: Effects on brain bioenergetic status and transverse relaxation time in healthy subjects. Biol. Psychiatry 2003, 54, 833–839. [Google Scholar] [CrossRef]
- Soeken, K.L.; Lee, W.L.; Bausell, R.B.; Agelli, M.; Berman, B.M. Safety and efficacy of S-adenosylmethionine (SAMe) for osteoarthritis. J. Fam. Pract. 2002, 51, 425–430. [Google Scholar] [PubMed]
- Pino, M.S.; Chung, D.C. The chromosomal instability pathway in colon cancer. Gastroenterology 2010, 138, 2059–2072. [Google Scholar] [CrossRef] [PubMed]
- Menter, D.G.; Davis, J.S.; Broom, B.M.; Overman, M.J.; Morris, J.; Kopetz, S. Back to the Colorectal Cancer Consensus Molecular Subtype Future. Curr. Gastroenterol. Rep. 2019, 21, 5. [Google Scholar] [CrossRef] [PubMed]
- Iyer, D.R.; Rhind, N. The Intra-S Checkpoint Responses to DNA Damage. Genes 2017, 8, 74. [Google Scholar] [CrossRef] [PubMed]
- Principe, D.R.; Doll, J.A.; Bauer, J.; Jung, B.; Munshi, H.G.; Bartholin, L.; Pasche, B.; Lee, C.; Grippo, P.J. TGF-β: Duality of function between tumor prevention and carcinogenesis. J. Natl. Cancer I 2014, 106. [Google Scholar] [CrossRef]
- Du, L.; Li, J.; Lei, L.; He, H.; Chen, E.; Dong, J.; Yang, J. High Vimentin Expression Predicts a Poor Prognosis and Progression in Colorectal Cancer: A Study with Meta-Analysis and TCGA Database. Biomed. Res. Int. 2018, 2018, 6387810. [Google Scholar] [CrossRef]
- Liu, Q.Z.; Gao, X.H.; Chang, W.J.; Gong, H.F.; Fu, C.G.; Zhang, W.; Cao, G.W. Expression of ITGB1 predicts prognosis in colorectal cancer: A large prospective study based on tissue microarray. Int. J. Clin. Exp. Pathol. 2015, 8, 12802–12810. [Google Scholar]
- Vu, T.; Datta, P.K. Regulation of EMT in Colorectal Cancer: A Culprit in Metastasis. Cancers 2017, 9, 171. [Google Scholar] [CrossRef]
- Cai, X.; Liu, C.; Zhang, T.N.; Zhu, Y.W.; Dong, X.; Xue, P. Down-regulation of FN1 inhibits colorectal carcinogenesis by suppressing proliferation, migration, and invasion. J. Cell Biochem. 2018, 119, 4717–4728. [Google Scholar] [CrossRef]
- Valcz, G.; Galamb, O.; Krenács, T.; Spisák, S.; Kalmár, A.; Patai, Á.V.; Wichmann, B.; Dede, K.; Tulassay, Z.; Molnár, B. Exosomes in colorectal carcinoma formation: ALIX under the magnifying glass. Mod. Pathol. 2016, 29, 928–938. [Google Scholar] [CrossRef]
- Zhuo, C.; Wu, X.; Li, J.; Hu, D.; Jian, J.; Chen, C.; Zheng, X.; Yang, C. Chemokine (C-X-C motif) ligand 1 is associated with tumor progression and poor prognosis in patients with colorectal cancer. Biosci. Rep. 2018, 38, BSR20180580. [Google Scholar] [CrossRef]
- de Aberasturi, A.L.; Calvo, A. TMPRSS4: An emerging potential therapeutic target in cancer. Br. J. Cancer 2015, 112, 4–8. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Sun, Z.; Szyf, M. S-adenosyl-methionine (SAM) alters the transcriptome and methylome and specifically blocks growth and invasiveness of liver cancer cells. Oncotarget 2017, 8, 111866–111881. [Google Scholar] [CrossRef] [PubMed]
- Isakovic, L.; Saavedra, O.M.; Llewellyn, D.B.; Claridge, S.; Zhan, L.; Bernstein, N.; Vaisburg, A.; Elowe, N.; Petschner, A.J.; Rahil, J.; et al. Constrained (l-)-S-adenosyl-l-homocysteine (SAH) analogues as DNA methyltransferase inhibitors. Bioorg. Med. Chem. Lett. 2009, 19, 2742–2746. [Google Scholar] [CrossRef]
- Mahmood, N.; Cheishvili, D.; Arakelian, A.; Tanvir, I.; Khan, H.A.; Pépin, A.S.; Szyf, M.; Rabbani, S.A. Methyl donor S-adenosylmethionine (SAM) supplementation attenuates breast cancer growth, invasion, and metastasis in vivo; therapeutic and chemopreventive applications. Oncotarget 2017, 9, 5169–5183. [Google Scholar] [CrossRef] [PubMed]
- Sheaffer, K.L.; Elliott, E.N.; Kaestner, K.H. DNA Hypomethylation Contributes to Genomic Instability and Intestinal Cancer Initiation. Cancer Prev. Res. 2016, 9, 534–546. [Google Scholar] [CrossRef]
- Detich, N.; Hamm, S.; Just, G.; Knox, J.D.; Szyf, M. The methyl donor S-Adenosylmethionine inhibits active demethylation of DNA: A candidate novel mechanism for the pharmacological effects of S-Adenosylmethionine. J. Biol. Chem. 2003, 278, 20812–20820. [Google Scholar] [CrossRef]
- Kuo, L.J.; Yang, L.X. Gamma-H2AX—A novel biomarker for DNA double-strand breaks. In Vivo 2008, 22, 305–309. [Google Scholar]
- Samanta, S.; Dey, P. Micronucleus and its applications. Diagn Cytopathol. 2012, 40, 84–90. [Google Scholar] [CrossRef]
- Ramírez, T.; García-Montalvo, V.; Wise, C.; Cea-Olivares, R.; Poirier, L.A.; Herrera, L.A. S-adenosyl-L-methionine is able to reverse micronucleus formation induced by sodium arsenite and other cytoskeleton disrupting agents in cultured human cells. Mutat Res. 2003, 528, 61–74. [Google Scholar] [CrossRef]
- Ramírez, T.; Stopper, H.; Hock, R.; Herrera, L.A. Prevention of aneuploidy by S-adenosyl-methionine in human cells treated with sodium arsenite. Mutat Res. 2007, 617, 16–22. [Google Scholar] [CrossRef]
- Araldi, R.P.; de Melo, T.C.; Mendes, T.B.; de Sá Júnior, P.L.; Nozima, B.H.; Ito, E.T.; de Carvalho, R.F.; de Souza, E.B.; de Cassia Stocco, R. Using the comet and micronucleus assays for genotoxicity studies: A review. Biomed. Pharm. 2015, 72, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Luzhna, L.; Kathiria, P.; Kovalchuk, O. Micronuclei in genotoxicity assessment: From genetics to epigenetics and beyond. Front. Genet. 2013, 4, 131. [Google Scholar] [CrossRef] [PubMed]
- Turgeon, M.O.; Perry, N.J.S.; Poulogiannis, G. DNA Damage, Repair, and Cancer Metabolism. Front. Oncol. 2018, 8, 15. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Egervari, G.; Wang, Y.; Berger, S.L.; Lu, Z. Regulation of chromatin and gene expression by metabolic enzymes and metabolites. Nat. Rev. Mol. Cell Biol. 2018, 19, 563–578. [Google Scholar] [CrossRef]
- Terradas, M.; Martín, M.; Tusell, L.; Genescà, A. Genetic activities in micronuclei: Is the DNA entrapped in micronuclei lost for the cell? Mutat Res. 2010, 705, 60–67. [Google Scholar] [CrossRef]
- Benitez, A.; Liu, W.; Palovcak, A.; Wang, G.; Moon, J.; An, K.; Kim, A.; Zheng, K.; Zhang, Y.; Bai, F.; et al. FANCA Promotes DNA Double-Strand Break Repair by Catalyzing Single-Strand Annealing and Strand Exchange. Mol. Cell 2018, 71, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Hendriks, Y.M.; Jagmohan-Changur, S.; van der Klift, H.M.; Morreau, H.; van Puijenbroek, M.; Tops, C.; van Os, T.; Wagner, A.; Ausems, M.G.F.M.; Gomez, E.; et al. Heterozygous mutations in PMS2 cause hereditary nonpolyposis colorectal carcinoma (Lynch syndrome). Gastroenterology 2006, 130, 312–322. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.D.; Liu, N. Fau-Jasin, M.; Jasin, M. Mammalian XRCC2 promotes the repair of DNA double-strand breaks by homologous recombination. Nature 1999, 401, 397–399. [Google Scholar] [CrossRef] [PubMed]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zsigrai, S.; Kalmár, A.; Nagy, Z.B.; Barták, B.K.; Valcz, G.; Szigeti, K.A.; Galamb, O.; Dankó, T.; Sebestyén, A.; Barna, G.; et al. S-Adenosylmethionine Treatment of Colorectal Cancer Cell Lines Alters DNA Methylation, DNA Repair and Tumor Progression-Related Gene Expression. Cells 2020, 9, 1864. https://doi.org/10.3390/cells9081864
Zsigrai S, Kalmár A, Nagy ZB, Barták BK, Valcz G, Szigeti KA, Galamb O, Dankó T, Sebestyén A, Barna G, et al. S-Adenosylmethionine Treatment of Colorectal Cancer Cell Lines Alters DNA Methylation, DNA Repair and Tumor Progression-Related Gene Expression. Cells. 2020; 9(8):1864. https://doi.org/10.3390/cells9081864
Chicago/Turabian StyleZsigrai, Sára, Alexandra Kalmár, Zsófia B. Nagy, Barbara K. Barták, Gábor Valcz, Krisztina A. Szigeti, Orsolya Galamb, Titanilla Dankó, Anna Sebestyén, Gábor Barna, and et al. 2020. "S-Adenosylmethionine Treatment of Colorectal Cancer Cell Lines Alters DNA Methylation, DNA Repair and Tumor Progression-Related Gene Expression" Cells 9, no. 8: 1864. https://doi.org/10.3390/cells9081864
APA StyleZsigrai, S., Kalmár, A., Nagy, Z. B., Barták, B. K., Valcz, G., Szigeti, K. A., Galamb, O., Dankó, T., Sebestyén, A., Barna, G., Szabó, V., Pipek, O., Medgyes-Horváth, A., Csabai, I., Tulassay, Z., Igaz, P., Takács, I., & Molnár, B. (2020). S-Adenosylmethionine Treatment of Colorectal Cancer Cell Lines Alters DNA Methylation, DNA Repair and Tumor Progression-Related Gene Expression. Cells, 9(8), 1864. https://doi.org/10.3390/cells9081864