The Landscape of CAR-T Cell Clinical Trials against Solid Tumors—A Comprehensive Overview


Simple Summary
Abstract
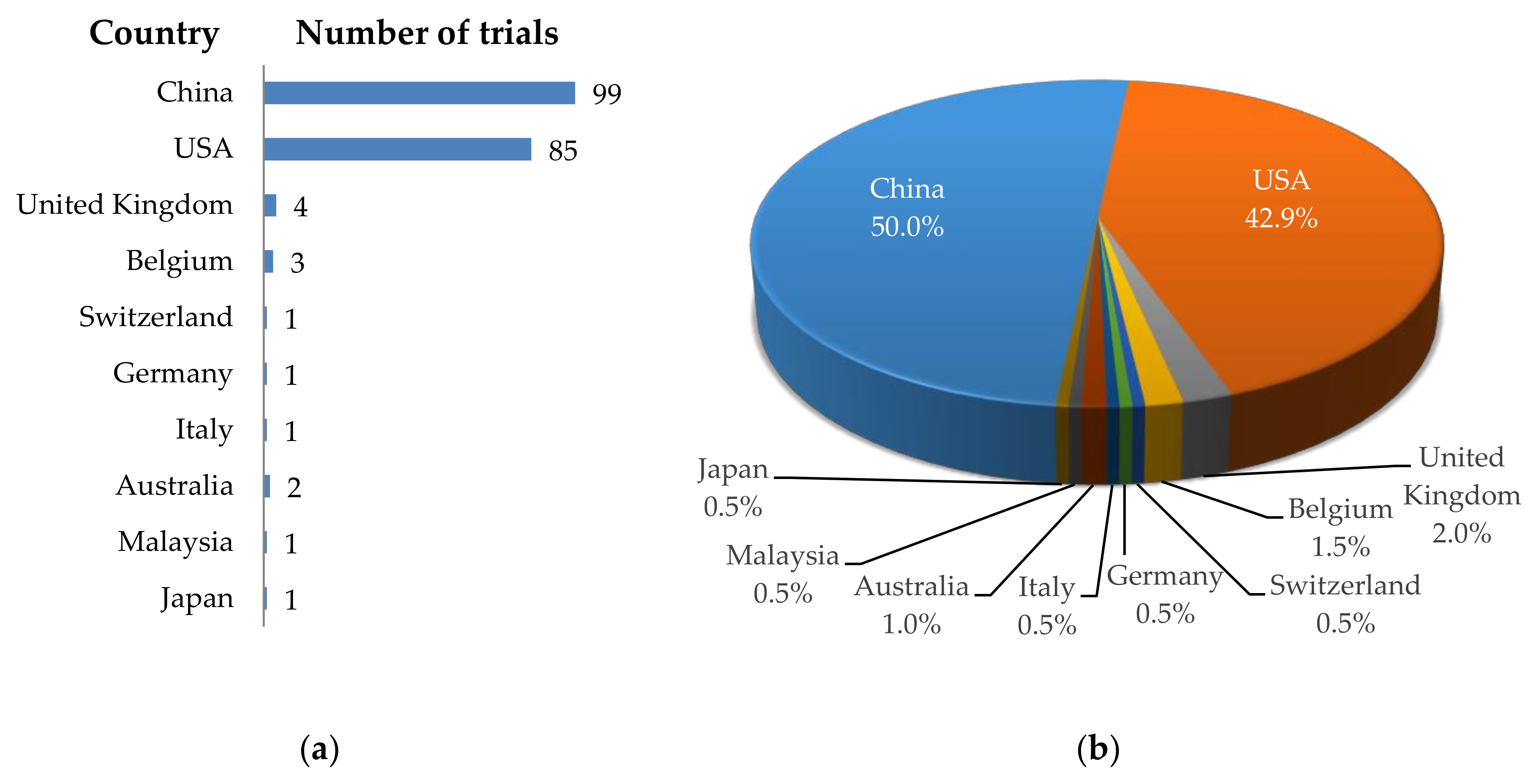
1. Introduction
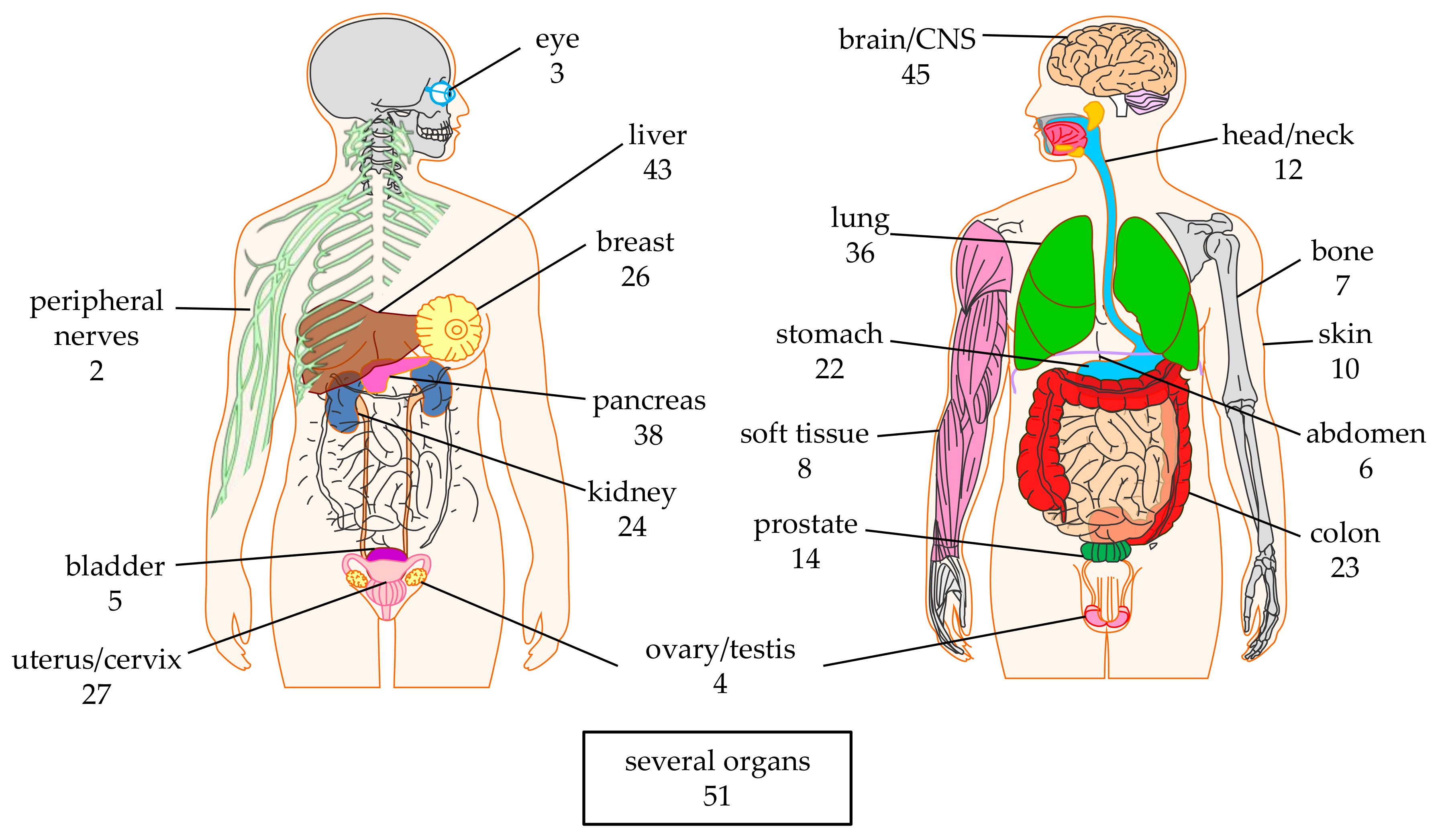
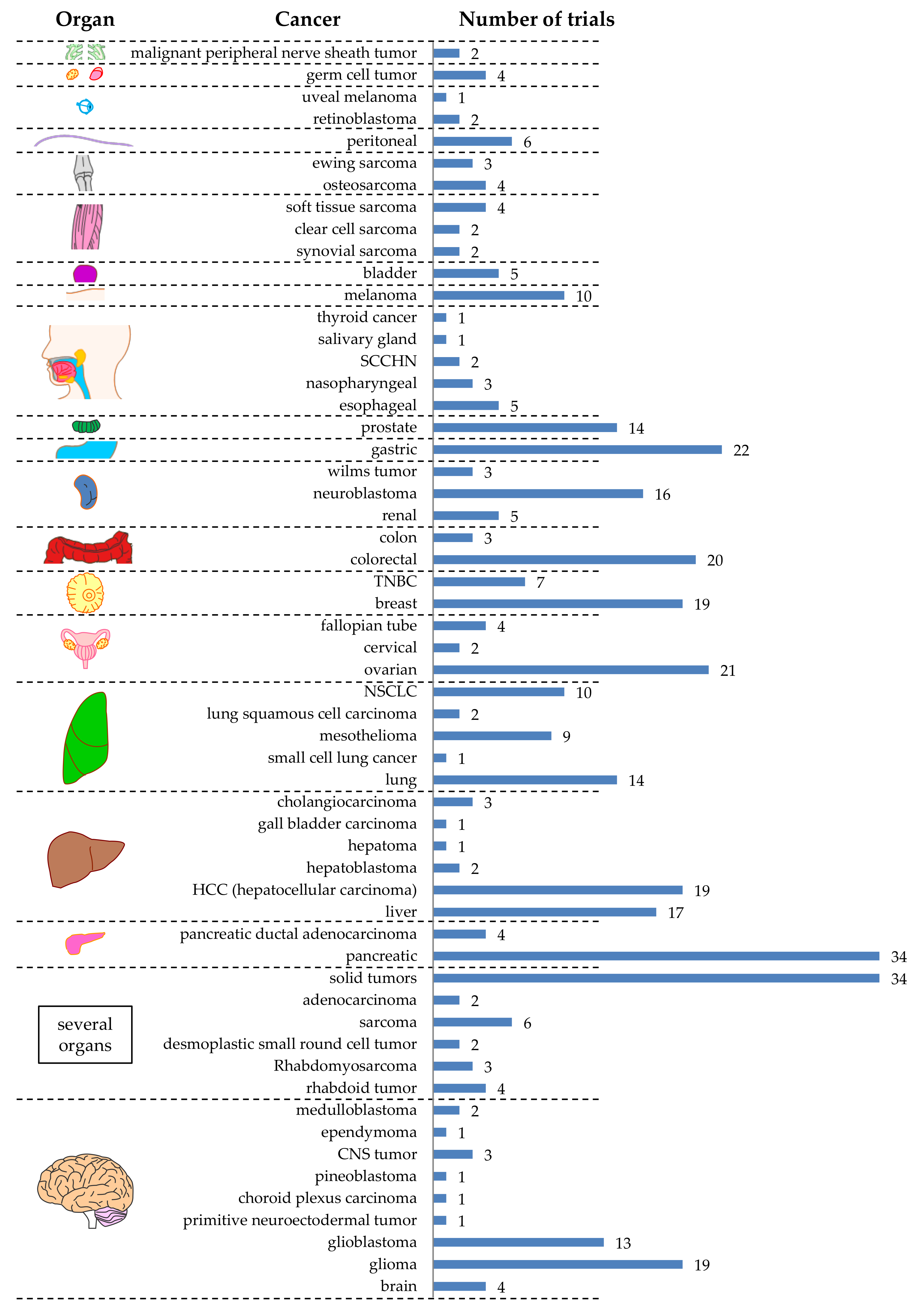
2. CAR-T Cell Clinical Trials against Solid Tumors—Organs, Tumor Entities, Antigens
2.1. Targeted Organs
2.2. Targeted Tumor Entities
2.3. Targeted Antigens
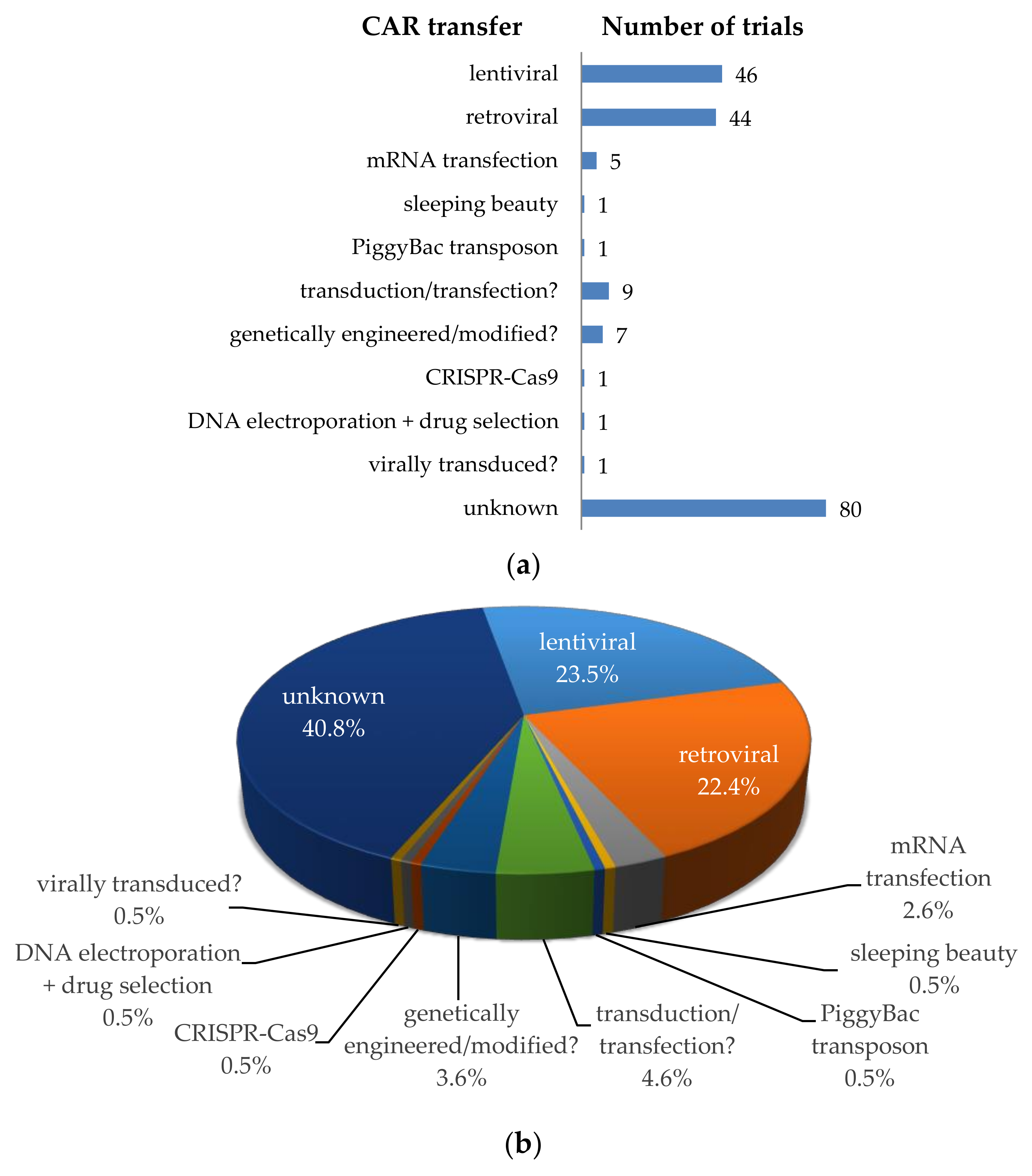
3. CAR-T Cell Clinical Trials against Solid Tumors—CAR Transfer Methods, CAR Formats, Extra Features
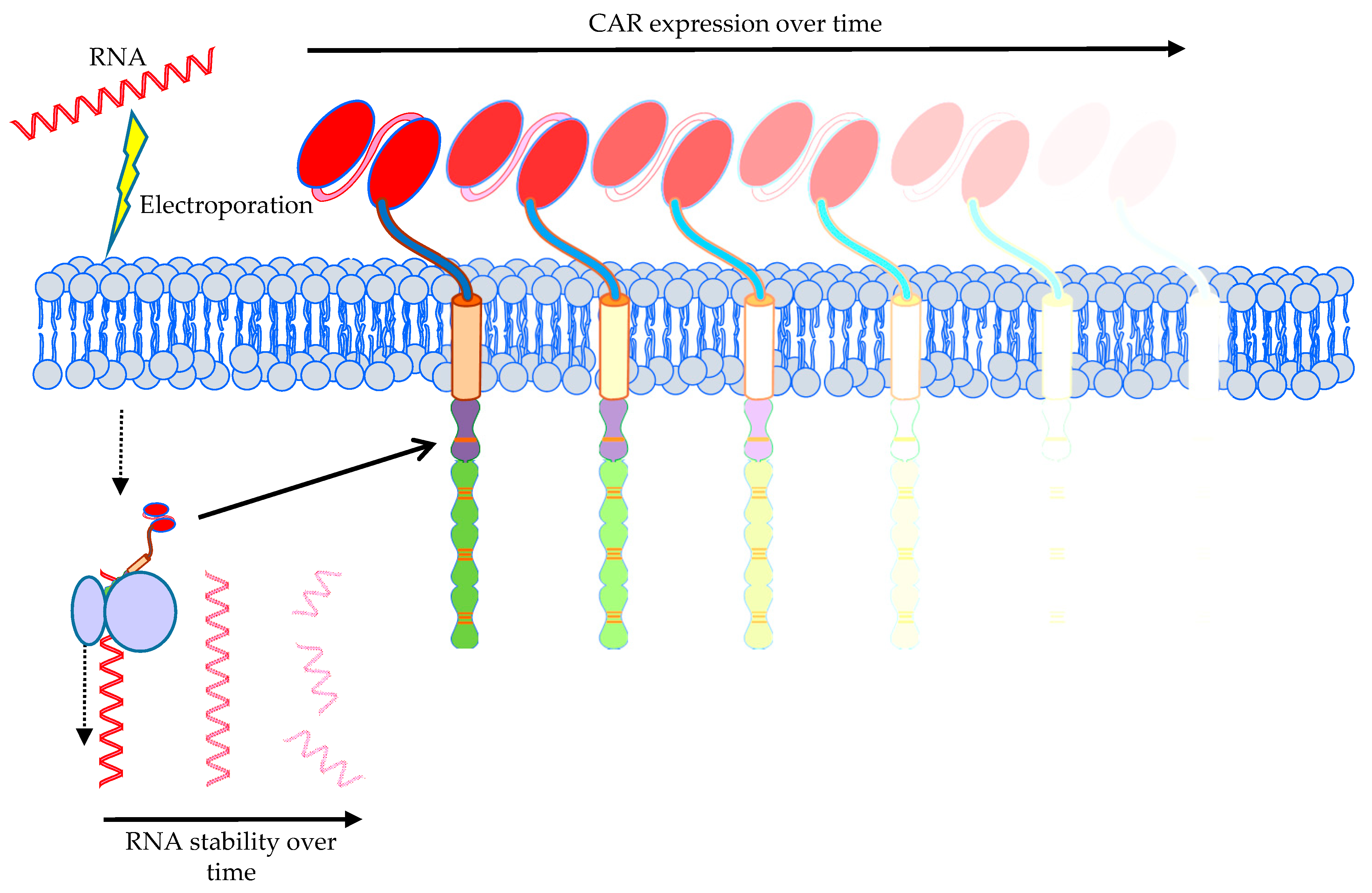
3.1. Transfer Methods to Introduce the CAR into T Cells
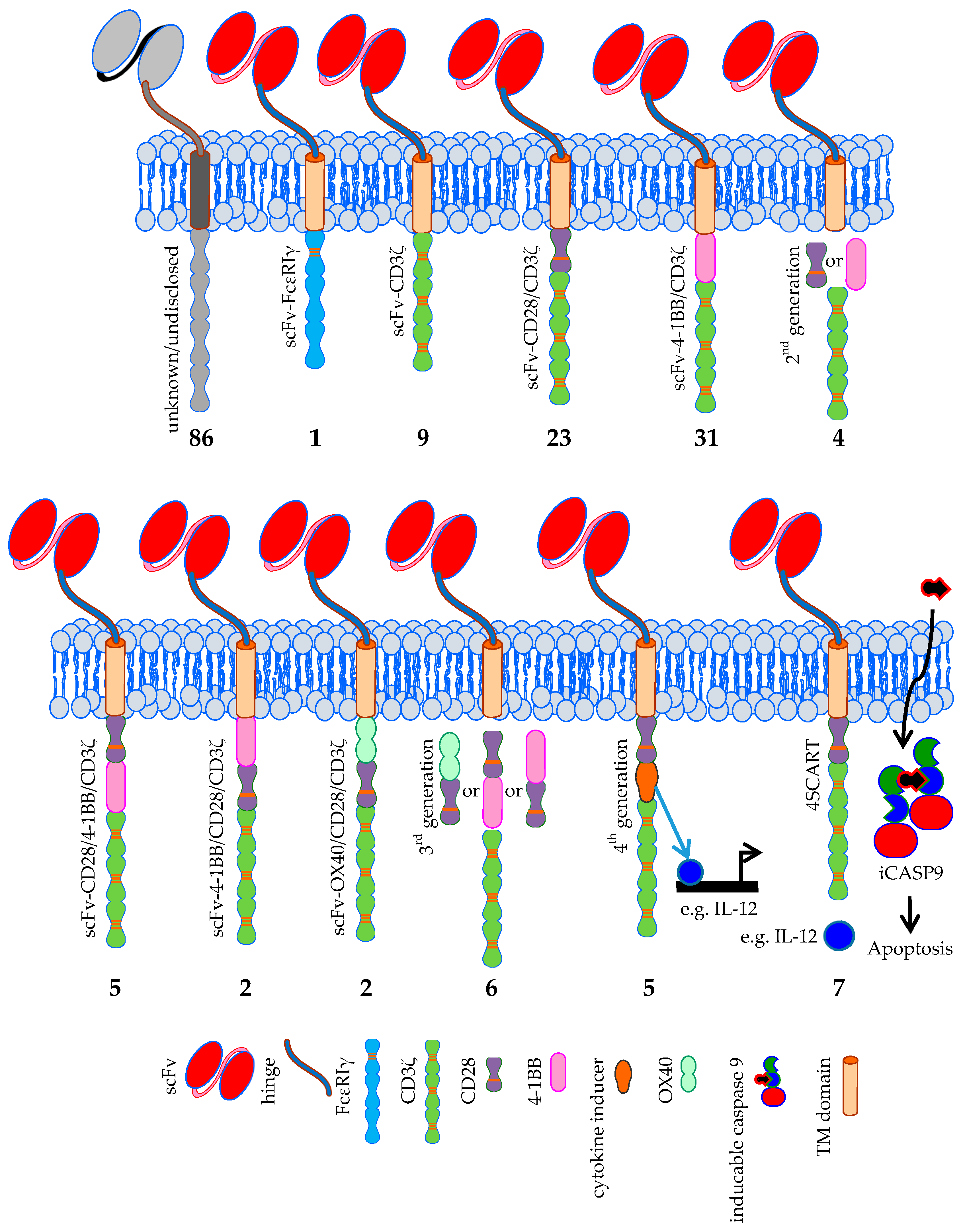
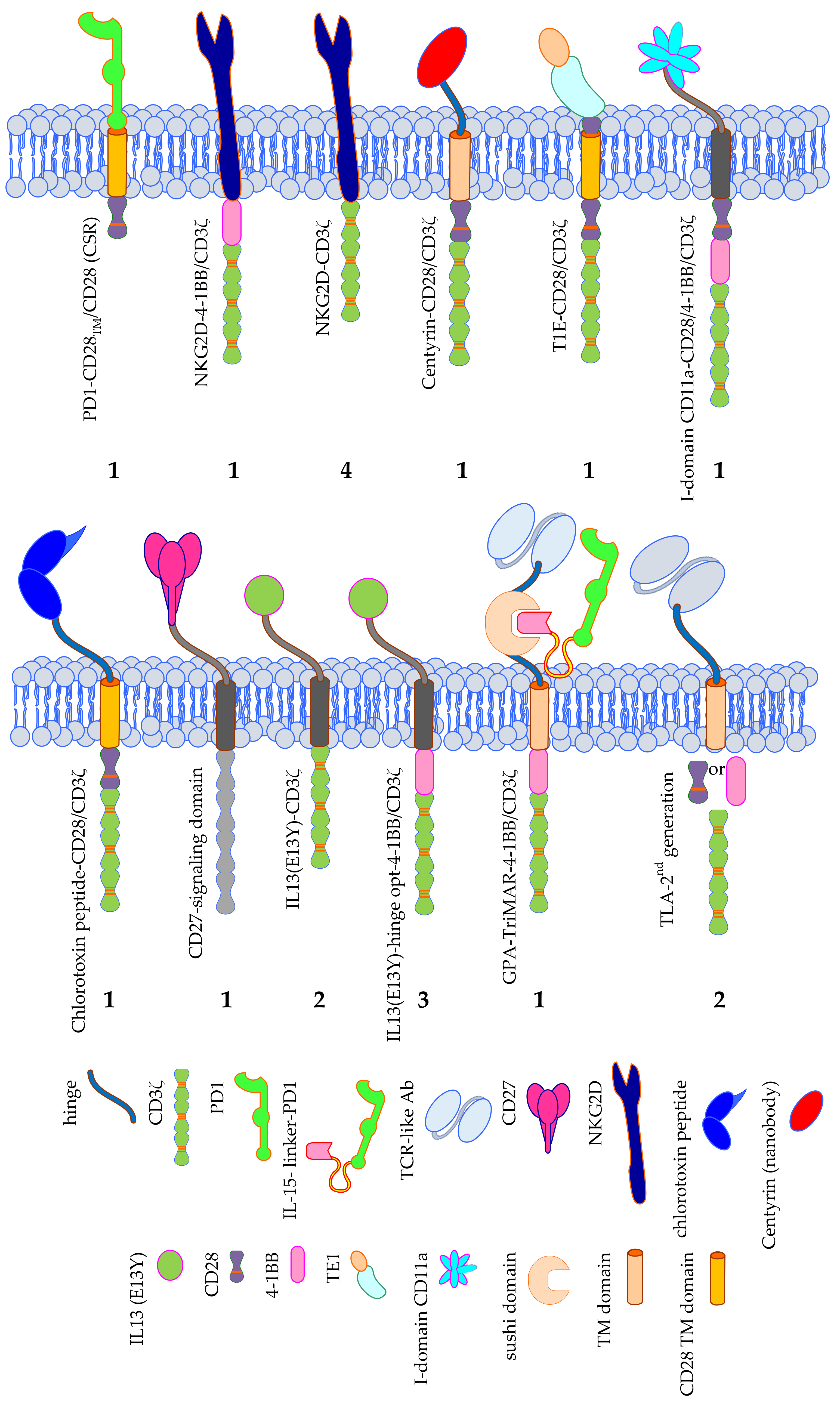
3.2. CAR Formats; the Classical and the More Exotic Models
3.2.1. CARs: The Classical Models
3.2.2. CARs: The More Exotic Models
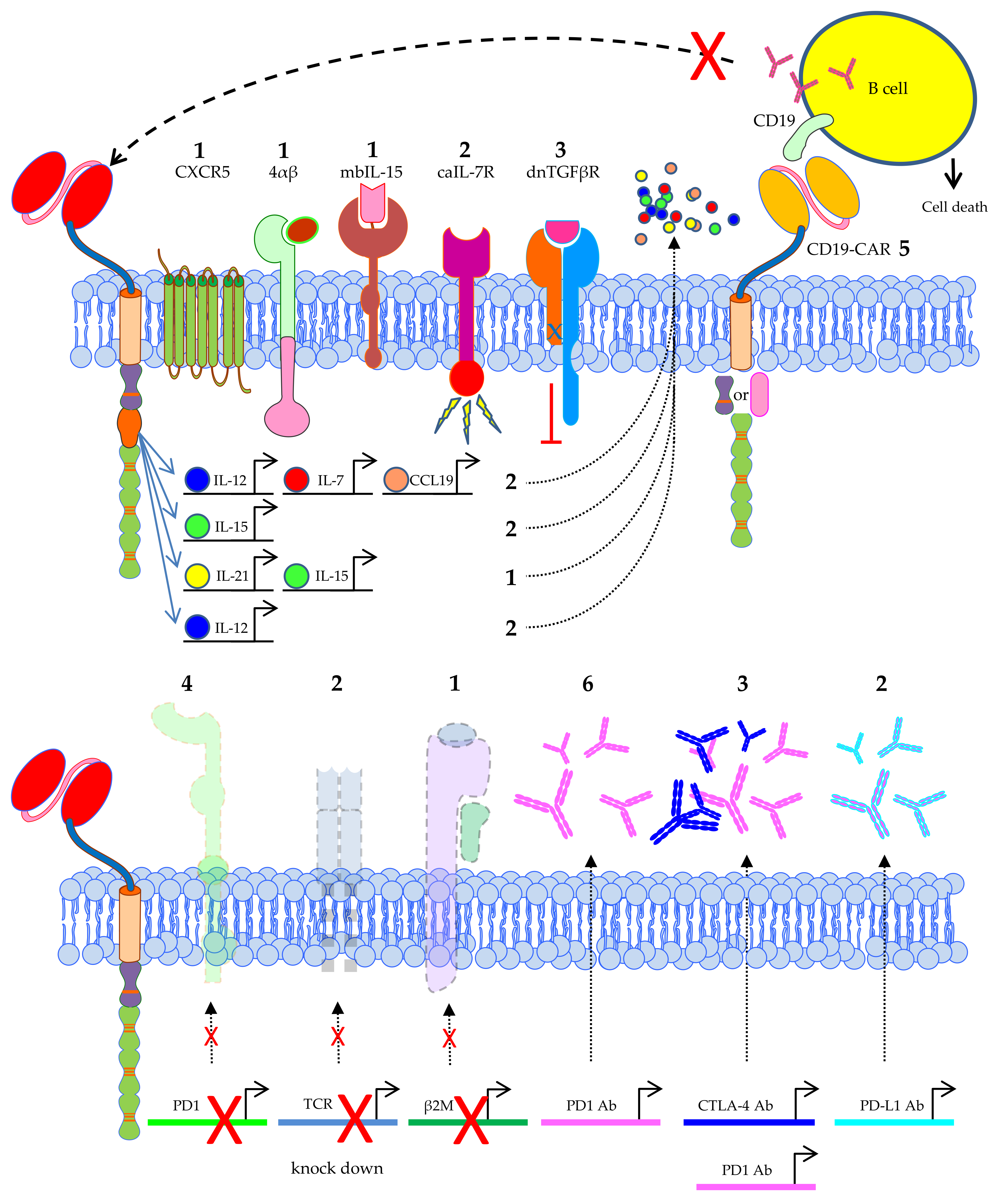
3.3. Add-Ons; T-Cell Populations Used for Transfer or Extra Features Introduced into CAR-T Cells
3.3.1. T-Cell Populations Used for CAR-T-Cell Therapy
3.3.2. Extra Features Introduced into CAR-T Cells
4. CAR-T Cell Clinical Trials against Solid Tumors—Patient Pretreatments, Injection Sites, Safety Measurements, Clinical Outcomes
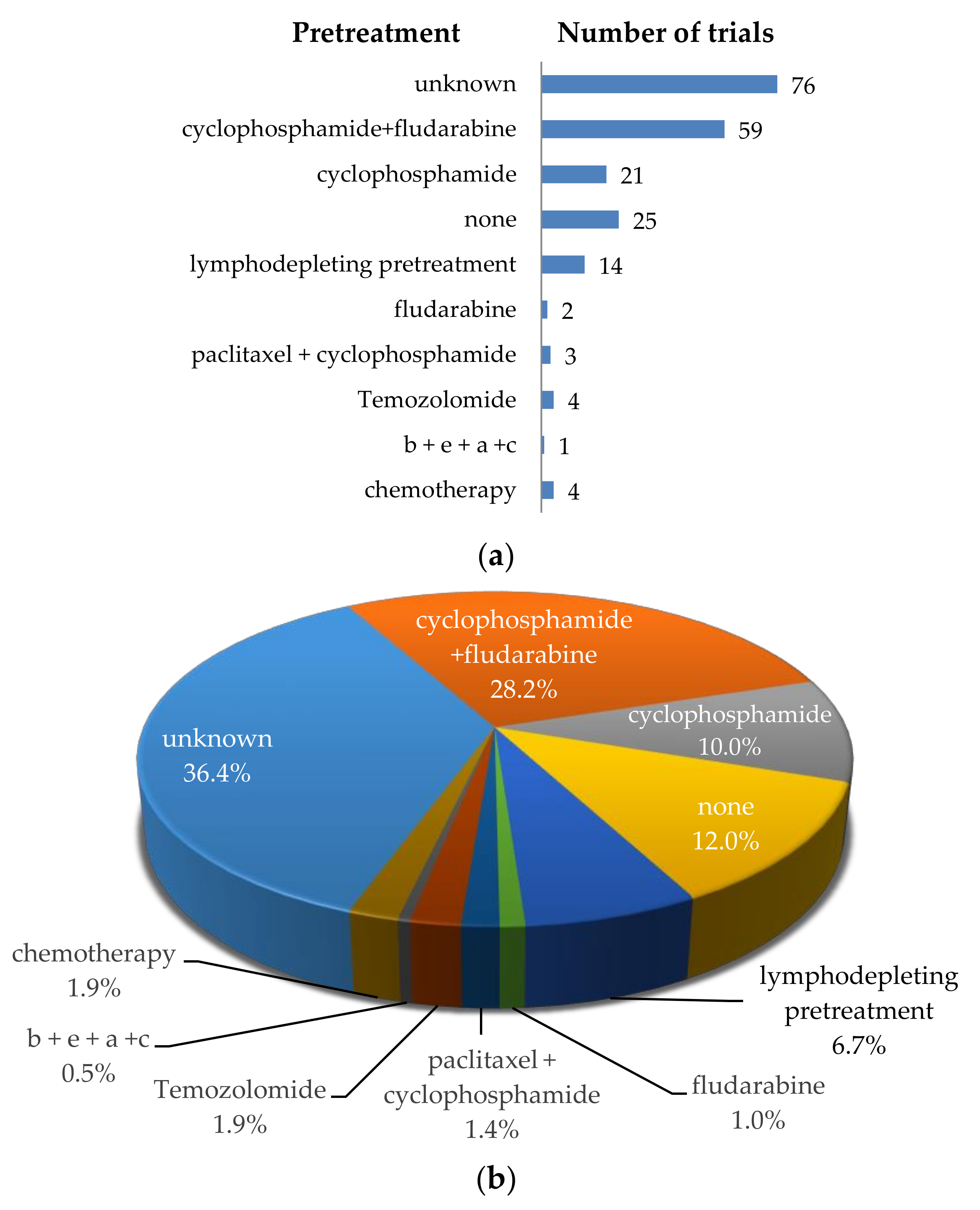
4.1. Treatments of Patients before CAR-T-Cell Transfer
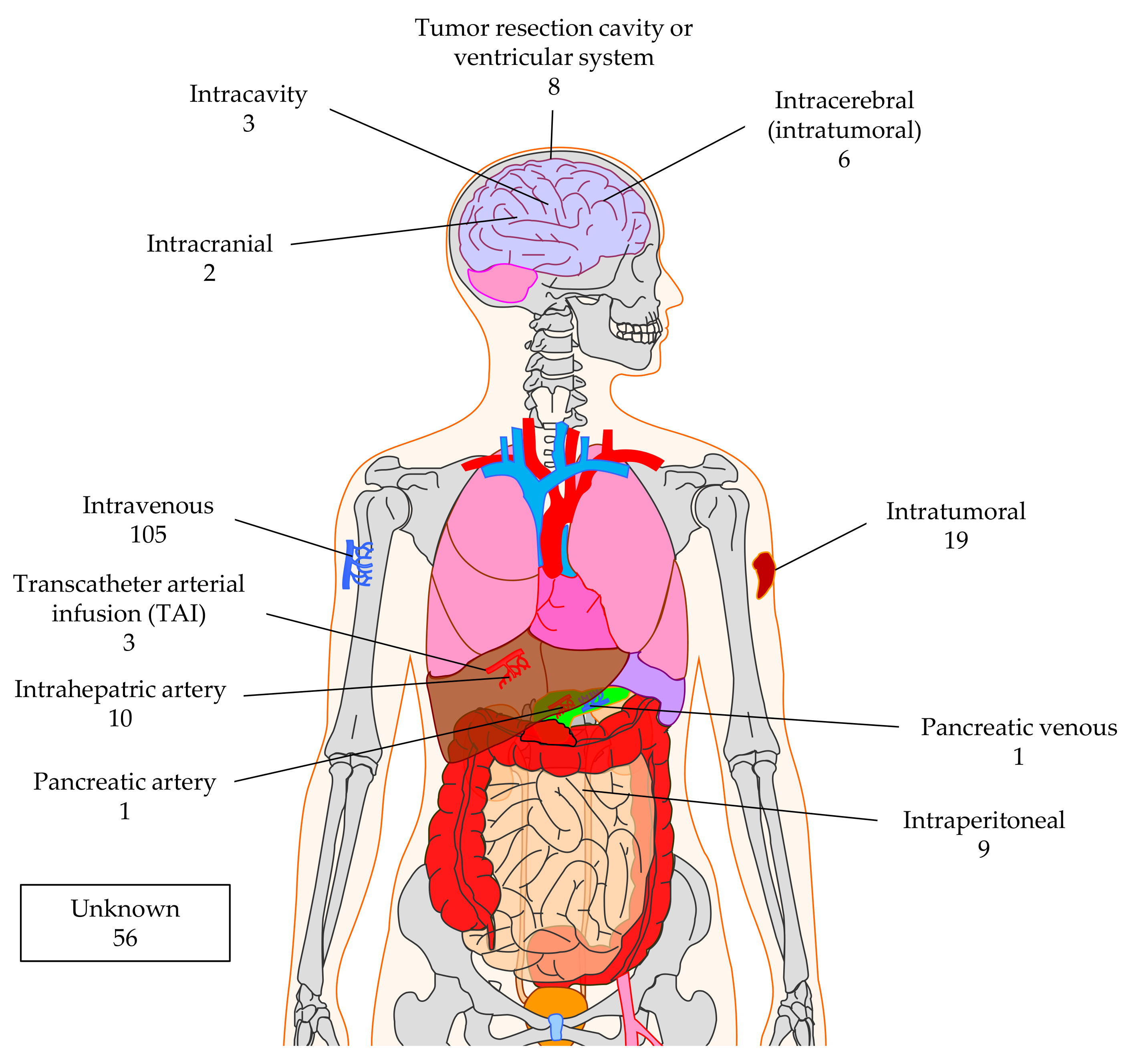
4.2. Injection Sites for CAR-T Cell Application
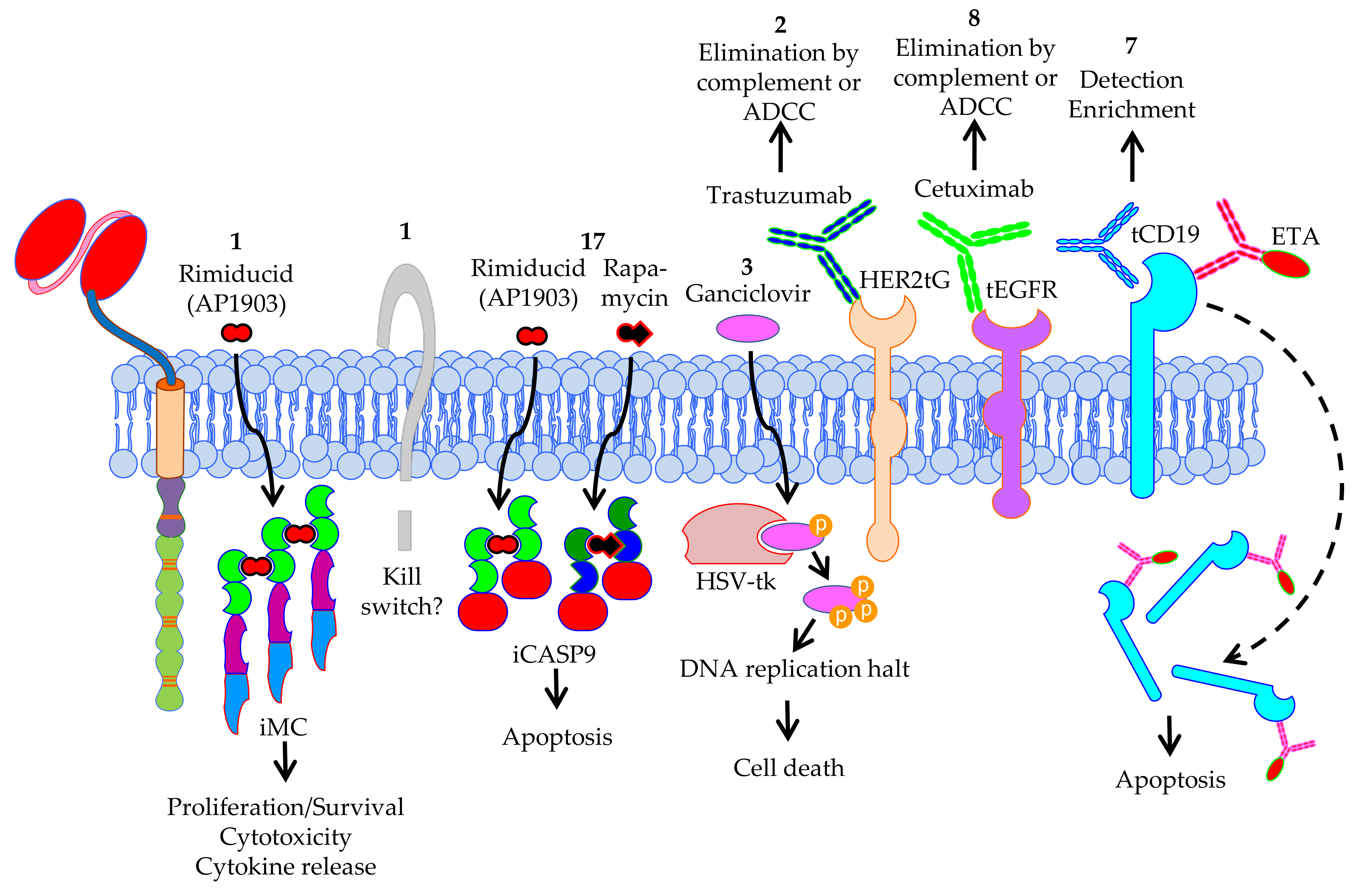
4.3. Safety Measurements to Control Negative Effects of CAR-T Cells in the Patient
4.4. Clinical Outcomes and Adverse Events of CAR-T-Cell Therapy of Solid Tumors
4.4.1. Clinical Outcomes
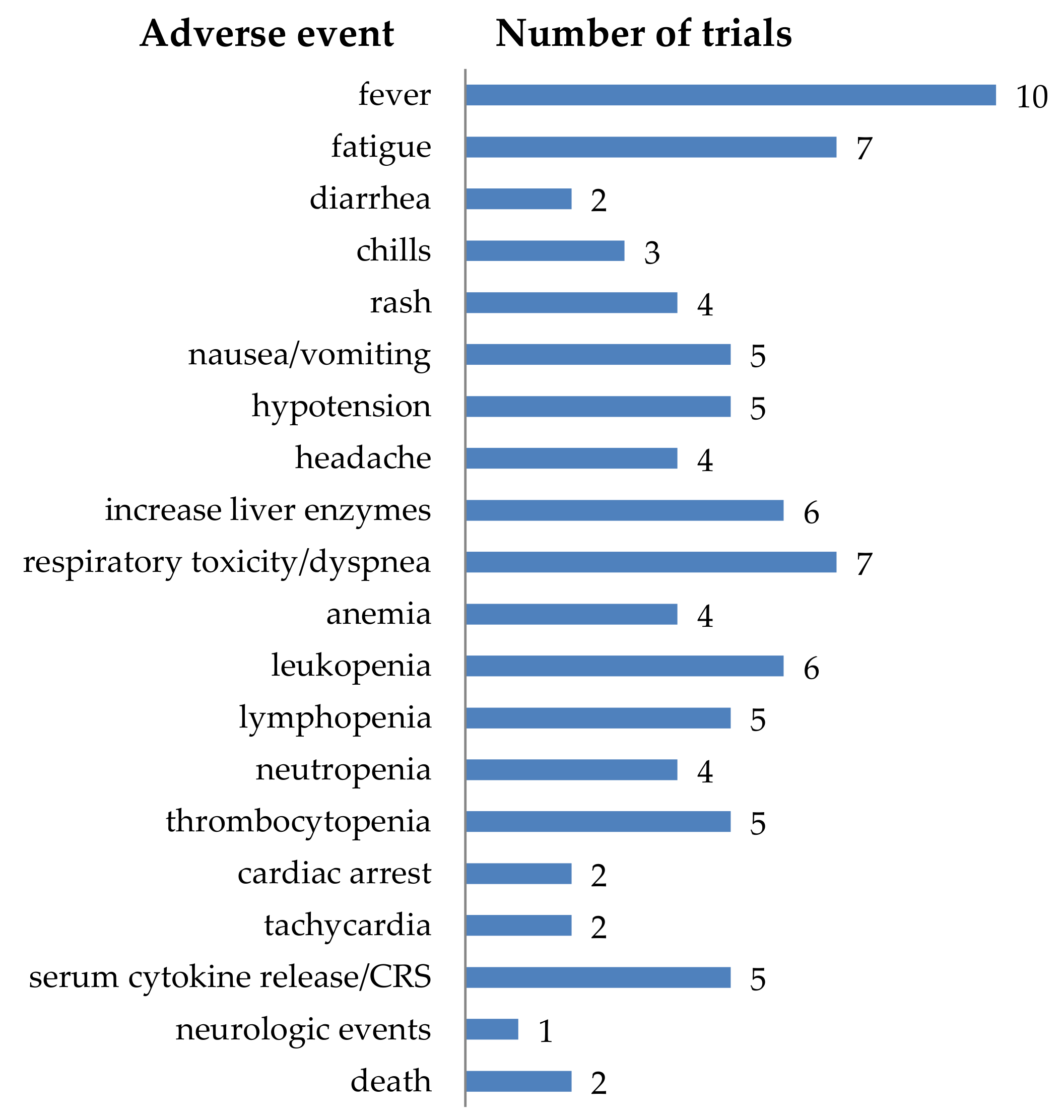
4.4.2. Adverse Events
5. Conclusions
Supplementary Materials
Funding
Acknowledgments
Conflicts of Interest
References
- Gross, G.; Gorochov, G.; Waks, T.; Eshhar, Z. Generation of effector T cells expressing chimeric T cell receptor with antibody type-specificity. Transpl. Proc. 1989, 21, 127–130. [Google Scholar]
- Gross, G.; Waks, T.; Eshhar, Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc. Natl. Acad. Sci. USA 1989, 86, 10024–10028. [Google Scholar] [CrossRef] [PubMed]
- June, C.H.; Sadelain, M. Chimeric antigen receptor therapy. N. Engl. J. Med. 2018, 379, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Neelapu, S.S.; Locke, F.L.; Go, W.Y. CAR T-Cell Therapy in large B-cell lymphoma. N. Engl. J. Med. 2018, 378, 1065. [Google Scholar] [CrossRef]
- Schuster, S.J.; Bishop, M.R.; Tam, C.S.; Waller, E.K.; Borchmann, P.; McGuirk, J.P.; Jager, U.; Jaglowski, S.; Andreadis, C.; Westin, J.R.; et al. Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. N. Engl. J. Med. 2019, 380, 45–56. [Google Scholar] [CrossRef]
- Wang, Z.; Guo, Y.; Han, W. Current status and perspectives of chimeric antigen receptor modified T cells for cancer treatment. Protein Cell 2017, 8, 896–925. [Google Scholar] [CrossRef]
- Han, S.; Latchoumanin, O.; Wu, G.; Zhou, G.; Hebbard, L.; George, J.; Qiao, L. Recent clinical trials utilizing chimeric antigen receptor T cells therapies against solid tumors. Cancer Lett. 2017, 390, 188–200. [Google Scholar] [CrossRef]
- Yeku, O.; Li, X.; Brentjens, R.J. Adoptive T-cell therapy for solid tumors. Am. Soc. Clin. Oncol. Educ. Book 2017, 37, 193–204. [Google Scholar] [CrossRef]
- Arabi, F.; Torabi-Rahvar, M.; Shariati, A.; Ahmadbeigi, N.; Naderi, M. Antigenic targets of CAR T Cell Therapy. A retrospective view on clinical trials. Exp. Cell Res. 2018, 369, 1–10. [Google Scholar] [CrossRef]
- Lamers, C.H.J.; Sleijfer, S.; Steenbergen, S.V.; Elzakker, P.V.; Krimpen, B.V.; Groot, C.; Vulto, A.; Bakker, M.D.; Oosterwijk, E.; Debets, R.; et al. Treatment of metastatic renal cell carcinoma with CAIX CAR-engineered T cells: Clinical evaluation and management of on-target toxicity. Mol. Ther. 2013, 21, 904–912. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.A.; Yang, J.C.; Kitano, M.; Dudley, M.E.; Laurencot, C.M.; Rosenberg, S.A. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther. 2010, 18, 843–851. [Google Scholar] [CrossRef] [PubMed]
- Sridhar, P.; Petrocca, F. Regional delivery of chimeric antigen receptor (CAR) T-cells for cancer therapy. Cancers 2017, 9, 92. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Luo, F.; Yang, J.; Zhao, C.; Chu, Y. New chimeric antigen receptor design for solid tumors. Front. Immunol. 2017, 8, 1934. [Google Scholar] [CrossRef]
- Heyman, B.; Yang, Y. Chimeric antigen receptor T cell therapy for solid tumors: Current status, obstacles and future strategies. Cancers 2019, 11, 191. [Google Scholar] [CrossRef]
- Titov, A.; Valiullina, A.; Zmievskaya, E.; Zaikova, E.; Petukhov, A.; Miftakhova, R.; Bulatov, E.; Rizvanov, A. Advancing CAR T-Cell therapy for solid tumors: Lessons learned from lymphoma treatment. Cancers 2020, 12, 125. [Google Scholar] [CrossRef]
- Yu, W.L.; Hua, Z.C. Chimeric Antigen Receptor T-cell (CAR T) Therapy for hematologic and solid malignancies: Efficacy and safety-a systematic review with meta-analysis. Cancers 2019, 11, 47. [Google Scholar] [CrossRef]
- Essand, M.; Loskog, A.S. Genetically engineered T cells for the treatment of cancer. J. Intern. Med. 2013, 273, 166–181. [Google Scholar] [CrossRef]
- Mirzaei, H.R.; Rodriguez, A.; Shepphird, J.; Brown, C.E.; Badie, B. chimeric antigen receptors T cell therapy in solid tumor: Challenges and clinical applications. Front. Immunol. 2017, 8, 1850. [Google Scholar] [CrossRef]
- Yu, S.; Li, A.; Liu, Q.; Li, T.; Yuan, X.; Han, X.; Wu, K. Chimeric antigen receptor T cells: A novel therapy for solid tumors. J. Hematol. Oncol. 2017, 10, 78. [Google Scholar] [CrossRef]
- Springuel, L.; Lonez, C.; Alexandre, B.; Cutsem, E.V.; Machiels, J.H.; Eynde, M.V.D.; Prenen, H.; Hendlisz, A.; Shaza, L.; Carrasco, J.; et al. Chimeric antigen receptor-T cells for targeting solid tumors: Current challenges and existing strategies. BioDrugs 2019, 33, 515–537. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Li, W.; Huang, K.; Zhang, Y.; Kupfer, G.; Zhao, Q. Chimeric antigen receptor T cell (CAR-T) immunotherapy for solid tumors: Lessons learned and strategies for moving forward. J. Hematol. Oncol. 2018, 11, 22. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Li, X.; Zhou, W.L.; Huang, Y.; Liang, X.; Jiang, L.; Yang, X.; Sun, J.; Li, Z.; Han, W.D.; et al. Genetically engineered T cells for cancer immunotherapy. Signal. Transduct Target. 2019, 4, 35. [Google Scholar] [CrossRef] [PubMed]
- Holzinger, A.; Barden, M.; Abken, H. The growing world of CAR T cell trials: A systematic review. Cancer Immunol. Immunother. 2016, 65, 1433–1450. [Google Scholar] [CrossRef]
- Shi, D.; Shi, Y.; Kaseb, A.O.; Qi, X.; Zhang, Y.; Chi, J.; Lu, Q.; Gao, H.; Jiang, H.; Wang, H.; et al. Chimeric antigen receptor-glypican-3 T-cell therapy for advanced hepatocellular carcinoma: Results of phase I Trials. Clin. Cancer Res. 2020. [Google Scholar] [CrossRef]
- Fucà, G.; Reppel, L.; Landoni, E.; Savoldo, B.; Dotti, G. Enhancing chimeric antigen receptor T-cell efficacy in solid tumors. Clin. Cancer Res. 2020, 26, 2444–2451. [Google Scholar] [CrossRef]
- Zhao, Z.; Xiao, X.; Saw, P.E.; Wu, W.; Huang, H.; Chen, J.; Nie, Y. Chimeric antigen receptor T cells in solid tumors: A war against the tumor microenvironment. Sci. China Life Sci. 2020, 63, 180–205. [Google Scholar] [CrossRef]
- Akhavan, D.; Alizadeh, D.; Wang, D.; Weist, M.R.; Shepphird, J.K.; Brown, C.E. CAR T cells for brain tumors: Lessons learned and road ahead. Immunol. Rev. 2019, 290, 60–84. [Google Scholar] [CrossRef]
- Majzner, R.G.; Mackall, C.L. Clinical lessons learned from the first leg of the CAR T cell journey. Nat. Med. 2019, 25, 1341–1355. [Google Scholar] [CrossRef]
- Holzinger, A.; Abken, H. CAR T cells targeting solid tumors: Carcinoembryonic antigen (CEA) proves to be a safe target. Cancer Immunol. Immunother. 2017, 66, 1505–1507. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424, Erratum in 2020, 70, 313, doi:10.3322/caac.21609. [Google Scholar] [CrossRef] [PubMed]
- WCRF/AICR. Worldwide Cancer Data; Global Cancer Statistics for the Most Common Cancers. Available online: https://www.wcrf.org/dietandcancer/cancer-trends/worldwide-cancer-data (accessed on 4 August 2020).
- WHO. Cancer. Available online: https://www.who.int/news-room/fact-sheets/detail/cancer (accessed on 4 August 2020).
- Feng, R.M.; Zong, Y.N.; Cao, S.M.; Xu, R.H. Current cancer situation in China: Good or bad news from the 2018 Global Cancer Statistics? Cancer Commun. 2019, 39, 22. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Lai, Y.; Li, J.; Qin, L.; Xu, Y.; Zhao, R.; Li, B.; Lin, S.; Wang, S.; Wu, Q.; et al. PSCA and MUC1 in non-small-cell lung cancer as targets of chimeric antigen receptor T cells. Oncoimmunology 2017, 6, e1284722. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.C.; Zeng, Z.; Huang, Y.N.; Deng, Y.C.; Fu, G.H. Clinical significance of TM4SF1 as a tumor suppressor gene in gastric cancer. Cancer Med. 2018, 7, 2592–2600. [Google Scholar] [CrossRef]
- Guo, Y.; Feng, K.; Liu, Y.; Wu, Z.; Dai, H.; Yang, Q.; Wang, Y.; Jia, H.; Han, W. Phase I study of chimeric antigen receptor-modified T cells in patients with EGFR-positive advanced biliary tract cancers. Clin. Cancer Res. 2018, 24, 1277–1286. [Google Scholar] [CrossRef]
- Feng, K.; Guo, Y.; Dai, H.; Wang, Y.; Li, X.; Jia, H.; Han, W. Chimeric antigen receptor-modified T cells for the immunotherapy of patients with EGFR-expressing advanced relapsed/refractory non-small cell lung cancer. Sci. China Life Sci. 2016, 59, 468–479. [Google Scholar] [CrossRef]
- Feng, K.C.; Guo, Y.L.; Liu, Y.; Dai, H.R.; Wang, Y.; Lv, H.Y.; Huang, J.H.; Yang, Q.M.; Han, W.D. Cocktail treatment with EGFR-specific and CD133-specific chimeric antigen receptor-modified T cells in a patient with advanced cholangiocarcinoma. J. Hematol. Oncol. 2017, 10, 4. [Google Scholar] [CrossRef]
- Pampusch, M.S.; Skinner, P.J. Transduction and expansion of primary T cells in nine days with maintenance of central memory phenotype. J. Vis. Exp. 2020. [Google Scholar] [CrossRef]
- Hattinger, C.M.; Patrizio, M.P.; Magagnoli, F.; Luppi, S.; Serra, M. An update on emerging drugs in osteosarcoma: Towards tailored therapies? Expert Opin. Emerg. Drugs 2019, 24, 153–171. [Google Scholar] [CrossRef]
- Hendlisz, A.P. 33rd Annual Meeting & Pre-Conference Programs of the Society for Immunotherapy of Cancer (SITC 2018): Washington, DC, USA, 7–11 November 2018. J. Immunother. Cancer 2018, 6, 114. [Google Scholar] [CrossRef]
- Lonez, C.; Verma, B.; Hendlisz, A.; Aftimos, P.; Awada, A.; Neste, E.V.D.; Catala, G.; Machiels, J.H.; Piette, F.; Brayer, J.B.; et al. Study protocol for THINK: A multinational open-label phase I study to assess the safety and clinical activity of multiple administrations of NKR-2 in patients with different metastatic tumour types. BMJ Open 2017, 7, e017075. [Google Scholar] [CrossRef] [PubMed]
- Feng, K.; Liu, Y.; Guo, Y.; Qiu, J.; Wu, Z.; Dai, H.; Yang, Q.; Wang, Y.; Han, W. Phase I study of chimeric antigen receptor modified T cells in treating HER2-positive advanced biliary tract cancers and pancreatic cancers. Protein Cell 2018, 9, 838–847. [Google Scholar] [CrossRef] [PubMed]
- Badhiwala, J.; Decker, W.K.; Berens, M.E.; Bhardwaj, R.D. Clinical trials in cellular immunotherapy for brain/CNS tumors. Expert Rev. Neurother. 2013, 13, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.; Brawley, V.; Hegde, M.; Bielamowicz, K.; Kalra, M.; Landi, D.; Robertson, C.; Gray, T.L.; Diouf, O.; Wakefield, A.; et al. HER2-Specific Chimeric Antigen Receptor-Modified Virus-Specific T Cells for Progressive Glioblastoma: A Phase 1 Dose-Escalation Trial. JAMA Oncol. 2017, 3, 1094–1101. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.; Brawley, V.; Hegde, M.; Bielamowicz, K.; Wakefield, A.; Ghazi, A.; Ashoori, A.; Diouf, O.; Gerken, C.; Landi, D.; et al. Autologous HER2 CMV bispecific CAR T cells are safe and demonstrate clinical benefit for glioblastoma in a Phase I trial. J. Immunother. Cancer 2015, 3. [Google Scholar] [CrossRef]
- Ahmed, N.; Brawley, V.S.; Hegde, M.; Robertson, C.; Ghazi, A.; Gerken, C.; Liu, E.; Dakhova, O.; Ashoori, A.; Corder, A.; et al. Human Epidermal Growth Factor Receptor 2 (HER2) -Specific Chimeric Antigen Receptor-Modified T Cells for the Immunotherapy of HER2-Positive Sarcoma. J. Clin. Oncol. 2015, 33, 1688–1696. [Google Scholar] [CrossRef]
- Navai, S.A.; Derenzo, C.; Joseph, S.; Sanber, K.; Byrd, T.; Zhang, H.; Mata, M.; Gerken, C.; Shree, A.; Mathew, P.R.; et al. Abstract LB-147: Administration of HER2-CAR T cells after lymphodepletion safely improves T cell expansion and induces clinical responses in patients with advanced sarcomas. Cancer Res. 2019, 79, LB-147. [Google Scholar] [CrossRef]
- Hegde, M.; DeRenzo, C.C.; Zhang, H.; Mata, M.; Gerken, C.; Shree, A.; Yi, Z.; Brawley, V.; Dakhova, O.; Wu, M.-F.; et al. Expansion of HER2-CAR T cells after lymphodepletion and clinical responses in patients with advanced sarcoma. J. Clin. Oncol. 2017, 35, 10508. [Google Scholar] [CrossRef]
- Katz, S.C.; Burga, R.A.; McCormack, E.; Wang, L.J.; Mooring, W.; Point, G.R.; Khare, P.D.; Thorn, M.; Ma, Q.; Stainken, B.F.; et al. Phase I Hepatic Immunotherapy for Metastases Study of Intra-Arterial Chimeric Antigen Receptor-Modified T-cell Therapy for CEA+ Liver Metastases. Clin. Cancer Res. 2015, 21, 3149–3159. [Google Scholar] [CrossRef]
- Saied, A.; Licata, L.; Burga, R.A.; Thorn, M.; McCormack, E.; Stainken, B.F.; Assanah, E.O.; Khare, P.D.; Davies, R.; Espat, N.J.; et al. Neutrophil:lymphocyte ratios and serum cytokine changes after hepatic artery chimeric antigen receptor-modified T-cell infusions for liver metastases. Cancer Gene 2014, 21, 457–462. [Google Scholar] [CrossRef]
- Appelbaum, J.S.; Pinto, N.; Orentas, R.J. Chapter 11 - Promising Chimeric Antigen Receptors for Non-B-Cell Hematological Malignancies, Pediatric Solid Tumors, and Carcinomas. In Chimeric Antigen Receptor T-Cell Therapies for Cancer; Lee, D.W., Shah, N.N., Eds.; Elsevier: Amsterdam, The Netherlands, 2020; pp. 137–163. [Google Scholar]
- Zhang, C.; Wang, Z.; Yang, Z.; Wang, M.; Li, S.; Li, Y.; Zhang, R.; Xiong, Z.; Wei, Z.; Shen, J.; et al. Phase I Escalating-Dose Trial of CAR-T Therapy Targeting CEA(+) Metastatic Colorectal Cancers. Mol. Ther. 2017, 25, 1248–1258. [Google Scholar] [CrossRef] [PubMed]
- Katz, S.C.; Hardaway, J.; Prince, E.; Guha, P.; Cunetta, M.; Moody, A.; Wang, L.J.; Armenio, V.; Espat, N.J.; Junghans, R.P. HITM-SIR: Phase Ib trial of intraarterial chimeric antigen receptor T-cell therapy and selective internal radiation therapy for CEA(+) liver metastases. Cancer Gene 2020, 27, 341–355. [Google Scholar] [CrossRef] [PubMed]
- Katz, S.C.; Prince, E.; Cunetta, M.; Guha, P.; Moody, A.; Armenio, V.; Wang, L.J.; Espat, N.J.; Junghans, R.P. Abstract CT109: HITM-SIR: Phase Ib trial of CAR-T hepatic artery infusions and selective internal radiation therapy for liver metastases. Cancer Res. 2017, 77, CT109. [Google Scholar] [CrossRef]
- Moroz, K. Anti-CEA CAR-T Demonstrates Significant Therapeutic Effect in Pancreatic Cancer Patients With Liver Metastases. Available online: https://www.interventionaloncology360.com/news/anti-cea-car-t-demonstrates-significant-therapeutic-effect-pancreatic-cancer-patients-liver (accessed on 4 August 2020).
- Thistlethwaite, F.C.; Gilham, D.E.; Guest, R.D.; Rothwell, D.G.; Pillai, M.; Burt, D.J.; Byatte, A.J.; Kirillova, N.; Valle, J.W.; Sharma, S.K.; et al. The clinical efficacy of first-generation carcinoembryonic antigen (CEACAM5)-specific CAR T cells is limited by poor persistence and transient pre-conditioning-dependent respiratory toxicity. Cancer Immunol. Immunother. 2017, 66, 1425–1436. [Google Scholar] [CrossRef]
- Purba, E.R.; Saita, E.I.; Maruyama, I.N. Activation of the EGF Receptor by Ligand Binding and Oncogenic Mutations: The “Rotation Model”. Cells 2017, 6, 13. [Google Scholar] [CrossRef]
- Huang, H.S.; Nagane, M.; Klingbeil, C.K.; Lin, H.; Nishikawa, R.; Ji, X.D.; Huang, C.M.; Gill, G.N.; Wiley, H.S.; Cavenee, W.K. The enhanced tumorigenic activity of a mutant epidermal growth factor receptor common in human cancers is mediated by threshold levels of constitutive tyrosine phosphorylation and unattenuated signaling. J. Biol. Chem. 1997, 272, 2927–2935. [Google Scholar] [CrossRef]
- Shtiegman, K.; Kochupurakkal, B.S.; Zwang, Y.; Pines, G.; Starr, A.; Vexler, A.; Citri, A.; Katz, M.; Lavi, S.; Ben-Basat, Y.; et al. Defective ubiquitinylation of EGFR mutants of lung cancer confers prolonged signaling. Oncogene 2007, 26, 6968–6978. [Google Scholar] [CrossRef]
- Mitri, Z.; Constantine, T.; O’Regan, R. The HER2 Receptor in Breast Cancer: Pathophysiology, Clinical Use, and New Advances in Therapy. Chemother. Res. Pr. 2012, 2012, 743193. [Google Scholar] [CrossRef]
- Slamon, D.J.; Godolphin, W.; Jones, L.A.; Holt, J.A.; Wong, S.G.; Keith, D.E.; Levin, W.J.; Stuart, S.G.; Udove, J.; Ullrich, A.; et al. Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science 1989, 244, 707–712. [Google Scholar] [CrossRef]
- Zhang, J.G.; Kruse, C.A.; Driggers, L.; Hoa, N.; Wisoff, J.; Allen, J.C.; Zagzag, D.; Newcomb, E.W.; Jadus, M.R. Tumor antigen precursor protein profiles of adult and pediatric brain tumors identify potential targets for immunotherapy. J. NeuroOncol. 2008, 88, 65–76. [Google Scholar] [CrossRef]
- Ogasawara, K.; Lanier, L.L. NKG2D in NK and T cell-mediated immunity. J. Clin. Immunol. 2005, 25, 534–540. [Google Scholar] [CrossRef] [PubMed]
- Obeidy, P.; Sharland, A.F. NKG2D and its ligands. Int. J. Biochem. Cell Biol. 2009, 41, 2364–2367. [Google Scholar] [CrossRef] [PubMed]
- Waldhauer, I.; Steinle, A. NK cells and cancer immunosurveillance. Oncogene 2008, 27, 5932–5943. [Google Scholar] [CrossRef] [PubMed]
- López-Soto, A.; Huergo-Zapico, L.; Acebes-Huerta, A.; Villa-Alvarez, M.; Gonzalez, S. NKG2D signaling in cancer immunosurveillance. Int. J. Cancer 2015, 136, 1741–1750. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Mandai, M.; Hamanishi, J.; Matsumura, N.; Suzuki, A.; Yagi, H.; Yamaguchi, K.; Baba, T.; Fujii, S.; Konishi, I. Clinical significance of the NKG2D ligands, MICA/B and ULBP2 in ovarian cancer: High expression of ULBP2 is an indicator of poor prognosis. Cancer Immunol. Immunother. 2009, 58, 641–652. [Google Scholar] [CrossRef]
- Liu, H.; Wang, S.; Xin, J.; Wang, J.; Yao, C.; Zhang, Z. Role of NKG2D and its ligands in cancer immunotherapy. Am. J. Cancer Res. 2019, 9, 2064–2078. [Google Scholar]
- Chapoval, A.I.; Ni, J.; Lau, J.S.; Wilcox, R.A.; Flies, D.B.; Liu, D.; Dong, H.; Sica, G.L.; Zhu, G.; Tamada, K.; et al. B7-H3: A costimulatory molecule for T cell activation and IFN-gamma production. Nat. Immunol. 2001, 2, 269–274. [Google Scholar] [CrossRef]
- Hofmeyer, K.A.; Ray, A.; Zang, X. The contrasting role of B7-H3. Proc. Natl. Acad. Sci. USA 2008, 105, 10277–10278. [Google Scholar] [CrossRef]
- Seaman, S.; Zhu, Z.; Saha, S.; Zhang, X.M.; Yang, M.Y.; Hilton, M.B.; Morris, K.; Szot, C.; Morris, H.; Swing, D.A.; et al. Eradication of tumors through simultaneous ablation of CD276/B7-H3-positive tumor cells and tumor vasculature. Cancer Cell 2017, 31, 501–515.e8. [Google Scholar] [CrossRef]
- Castriconi, R.; Dondero, A.; Augugliaro, R.; Cantoni, C.; Carnemolla, B.; Sementa, A.R.; Negri, F.; Conte, R.; Corrias, M.V.; Moretta, L.; et al. Identification of 4Ig-B7-H3 as a neuroblastoma-associated molecule that exerts a protective role from an NK cell-mediated lysis. Proc. Natl. Acad. Sci. USA 2004, 101, 12640–12645. [Google Scholar] [CrossRef]
- Inamura, K.; Yokouchi, Y.; Kobayashi, M.; Sakakibara, R.; Ninomiya, H.; Subat, S.; Nagano, H.; Nomura, K.; Okumura, S.; Shibutani, T.; et al. Tumor B7-H3 (CD276) expression and smoking history in relation to lung adenocarcinoma prognosis. Lung Cancer 2017, 103, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Loos, M.; Hedderich, D.M.; Friess, H.; Kleeff, J. B7-h3 and its role in antitumor immunity. Clin. Dev. Immunol. 2010, 2010, 683875. [Google Scholar] [CrossRef] [PubMed]
- Loos, M.; Hedderich, D.M.; Ottenhausen, M.; Giese, N.A.; Laschinger, M.; Esposito, I.; Kleeff, J.; Friess, H. Expression of the costimulatory molecule B7-H3 is associated with prolonged survival in human pancreatic cancer. BMC Cancer 2009, 9, 463. [Google Scholar] [CrossRef] [PubMed]
- Picarda, E.; Ohaegbulam, K.C.; Zang, X. Molecular pathways: Targeting B7-H3 (CD276) for human cancer immunotherapy. Clin. Cancer Res. 2016, 22, 3425–3431. [Google Scholar] [CrossRef]
- Yamato, I.; Sho, M.; Nomi, T.; Akahori, T.; Shimada, K.; Hotta, K.; Kanehiro, H.; Konishi, N.; Yagita, H.; Nakajima, Y. Clinical importance of B7-H3 expression in human pancreatic cancer. Br. J. Cancer 2009, 101, 1709–1716. [Google Scholar] [CrossRef]
- Benzon, B.; Zhao, S.G.; Haffner, M.C.; Takhar, M.; Erho, N.; Yousefi, K.; Hurley, P.; Bishop, J.L.; Tosoian, J.; Ghabili, K.; et al. Correlation of B7-H3 with androgen receptor, immune pathways and poor outcome in prostate cancer: An expression-based analysis. Prostate Cancer Prostatic Dis. 2017, 20, 28–35. [Google Scholar] [CrossRef]
- Parker, A.S.; Heckman, M.G.; Sheinin, Y.; Wu, K.J.; Hilton, T.W.; Diehl, N.N.; Pisansky, T.M.; Schild, S.E.; Kwon, E.D.; Buskirk, S.J. Evaluation of B7-H3 expression as a biomarker of biochemical recurrence after salvage radiation therapy for recurrent prostate cancer. Int. J. Radiat Oncol. Biol. Phys. 2011, 79, 1343–1349. [Google Scholar] [CrossRef]
- Qin, X.; Zhang, H.; Ye, D.; Dai, B.; Zhu, Y.; Shi, G. B7-H3 is a new cancer-specific endothelial marker in clear cell renal cell carcinoma. Onco Targets 2013, 6, 1667–1673. [Google Scholar] [CrossRef]
- Roth, T.J.; Sheinin, Y.; Lohse, C.M.; Kuntz, S.M.; Frigola, X.; Inman, B.A.; Krambeck, A.E.; McKenney, M.E.; Karnes, R.J.; Blute, M.L.; et al. B7-H3 ligand expression by prostate cancer: A novel marker of prognosis and potential target for therapy. Cancer Res. 2007, 67, 7893–7900. [Google Scholar] [CrossRef]
- Zang, X.; Sullivan, P.S.; Soslow, R.A.; Waitz, R.; Reuter, V.E.; Wilton, A.; Thaler, H.T.; Arul, M.; Slovin, S.F.; Wei, J.; et al. Tumor associated endothelial expression of B7-H3 predicts survival in ovarian carcinomas. Mod. Pathol. 2010, 23, 1104–1112. [Google Scholar] [CrossRef]
- Zang, X.; Thompson, R.H.; Al-Ahmadie, H.A.; Serio, A.M.; Reuter, V.E.; Eastham, J.A.; Scardino, P.T.; Sharma, P.; Allison, J.P. B7-H3 and B7x are highly expressed in human prostate cancer and associated with disease spread and poor outcome. Proc. Natl. Acad. Sci. USA 2007, 104, 19458–19463. [Google Scholar] [CrossRef] [PubMed]
- Dhar, P.; McAuley, J. The Role of the Cell Surface Mucin MUC1 as a Barrier to Infection and Regulator of Inflammation. Front. Cell Infect. Micro. Biol. 2019, 9, 117. [Google Scholar] [CrossRef] [PubMed]
- Wagner, C.E.; Wheeler, K.M.; Ribbeck, K. Mucins and their role in shaping the functions of mucus barriers. Annu. Rev. Cell Dev. Biol. 2018, 34, 189–215. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, R.; Gautam, S.K.; Cannon, A.; Thompson, C.; Hall, B.R.; Aithal, A.; Banerjee, K.; Jain, M.; Solheim, J.C.; Kumar, S.; et al. Cancer-associated mucins: Role in immune modulation and metastasis. Cancer Metastasis Rev. 2019, 38, 223–236. [Google Scholar] [CrossRef]
- Guo, M.; You, C.; Dou, J. Role of transmembrane glycoprotein mucin 1 (MUC1) in various types of colorectal cancer and therapies: Current research status and updates. Biomed. Pharm. 2018, 107, 1318–1325. [Google Scholar] [CrossRef]
- Taylor-Papadimitriou, J.; Burchell, J.M.; Graham, R.; Beatson, R. Latest developments in MUC1 immunotherapy. Biochem. Soc. Trans. 2018, 46, 659–668. [Google Scholar] [CrossRef]
- Hammarström, S. The carcinoembryonic antigen (CEA) family: Structures, suggested functions and expression in normal and malignant tissues. Semin. Cancer Biol. 1999, 9, 67–81. [Google Scholar] [CrossRef]
- Nap, M.; Mollgard, K.; Burtin, P.; Fleuren, G.J. Immunohistochemistry of carcino-embryonic antigen in the embryo, fetus and adult. Tumour Biol. 1988, 9, 145–153. [Google Scholar] [CrossRef]
- Han, Z.W.; Lyv, Z.W.; Cui, B.; Wang, Y.Y.; Cheng, J.T.; Zhang, Y.; Cai, W.Q.; Zhou, Y.; Ma, Z.W.; Wang, X.W.; et al. The old CEACAMs find their new role in tumor immunotherapy. Investig. New Drugs 2020. [Google Scholar] [CrossRef]
- Beauchemin, N.; Arabzadeh, A. Carcinoembryonic antigen-related cell adhesion molecules (CEACAMs) in cancer progression and metastasis. Cancer Metastasis Rev. 2013, 32, 643–671. [Google Scholar] [CrossRef]
- Yan, Z.; Deng, X.; Chen, M.; Xu, Y.; Ahram, M.; Sloane, B.F.; Friedman, E. Oncogenic c-Ki-ras but not oncogenic c-Ha-ras up-regulates CEA expression and disrupts basolateral polarity in colon epithelial cells. J. Biol. Chem. 1997, 272, 27902–27907. [Google Scholar] [CrossRef] [PubMed]
- Nolan, K.F.; Yun, C.O.; Akamatsu, Y.; Murphy, J.C.; Leung, S.O.; Beecham, E.J.; Junghans, R.P. Bypassing immunization: Optimized design of “designer T cells” against carcinoembryonic antigen (CEA)-expressing tumors, and lack of suppression by soluble CEA. Clin. Cancer Res. 1999, 5, 3928–3941. [Google Scholar] [PubMed]
- Wu, C.; Orozco, C.; Boyer, J.; Leglise, M.; Goodale, J.; Batalov, S.; Hodge, C.L.; Haase, J.; Janes, J.; Huss, J.W., 3rd; et al. BioGPS: An extensible and customizable portal for querying and organizing gene annotation resources. Genome Biol. 2009, 10, R130. [Google Scholar] [CrossRef] [PubMed]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Casucci, M.; Hawkins, R.E.; Dotti, G.; Bondanza, A. Overcoming the toxicity hurdles of genetically targeted T cells. Cancer Immunol. Immunother. 2015, 64, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Slamon, D.J.; Leyland-Jones, B.; Shak, S.; Fuchs, H.; Paton, V.; Bajamonde, A.; Fleming, T.; Eiermann, W.; Wolter, J.; Pegram, M.; et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N. Engl. J. Med. 2001, 344, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Lamers, C.H.; Willemsen, R.; Elzakker, P.v.; Steenbergen-Langeveld, S.v.; Broertjes, M.; Oosterwijk-Wakka, J.; Oosterwijk, E.; Sleijfer, S.; Debets, R.; Gratama, J.W. Immune responses to transgene and retroviral vector in patients treated with ex vivo-engineered T cells. Blood 2011, 117, 72–82. [Google Scholar] [CrossRef]
- Milone, M.C.; O’Doherty, U. Clinical use of lentiviral vectors. Leukemia 2018, 32, 1529–1541. [Google Scholar] [CrossRef]
- Vargas, J.E.; Chicaybam, L.; Stein, R.T.; Tanuri, A.; Delgado-Cañedo, A.; Bonamino, M.H. Retroviral vectors and transposons for stable gene therapy: Advances, current challenges and perspectives. J. Transl. Med. 2016, 14, 288. [Google Scholar] [CrossRef]
- Schambach, A.; Morgan, M. Retroviral vectors for cancer gene therapy. Recent Results Cancer Res. 2016, 209, 17–35. [Google Scholar] [CrossRef]
- Magnani, C.F.; Tettamanti, S.; Alberti, G.; Pisani, I.; Biondi, A.; Serafini, M.; Gaipa, G. Transposon-based CAR T cells in acute leukemias: Where are we going? Cells 2020, 9, 1337. [Google Scholar] [CrossRef] [PubMed]
- Hudecek, M.; Ivics, Z. Non-viral therapeutic cell engineering with the Sleeping Beauty transposon system. Curr. Opin. Genet. Dev. 2018, 52, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Tipanee, J.; Driessche, T.V.D.; Chuah, M.K. Transposons: Moving forward from preclinical studies to clinical trials. Hum. Gene 2017, 28, 1087–1104. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [PubMed]
- Birkholz, K.; Hombach, A.; Krug, C.; Reuter, S.; Kershaw, M.; Kampgen, E.; Schuler, G.; Abken, H.; Schaft, N.; Dorrie, J. Transfer of mRNA encoding recombinant immunoreceptors reprograms CD4+ and CD8+ T cells for use in the adoptive immunotherapy of cancer. Gene 2009, 16, 596–604. [Google Scholar] [CrossRef]
- Holzinger, A.; Abken, H. CAR T Cells: A Snapshot on the growing options to design a CAR. Hemasphere 2019, 3, e172. [Google Scholar] [CrossRef]
- Boomer, J.S.; Green, J.M. An enigmatic tail of CD28 signaling. Cold Spring Harb. Perspect. Biol. 2010, 2, a002436. [Google Scholar] [CrossRef]
- Milone, M.C.; Fish, J.D.; Carpenito, C.; Carroll, R.G.; Binder, G.K.; Teachey, D.; Samanta, M.; Lakhal, M.; Gloss, B.; Danet-Desnoyers, G.; et al. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol. Ther. 2009, 17, 1453–1464. [Google Scholar] [CrossRef]
- Kawalekar, O.U.; O’Connor, R.S.; Fraietta, J.A.; Guo, L.; McGettigan, S.E.; Posey, A.D., Jr.; Patel, P.R.; Guedan, S.; Scholler, J.; Keith, B.; et al. Distinct signaling of coreceptors regulates specific metabolism pathways and impacts memory development in CAR T cells. Immunity 2016, 44, 380–390. [Google Scholar] [CrossRef]
- Roselli, E.; Frieling, J.S.; Thorner, K.; Ramello, M.C.; Lynch, C.C.; Abate-Daga, D. CAR-T engineering: Optimizing signal transduction and effector mechanisms. BioDrugs 2019, 33, 647–659. [Google Scholar] [CrossRef]
- Hombach, A.A.; Holzinger, A.; Abken, H. The weal and woe of costimulation in the adoptive therapy of cancer with chimeric antigen receptor (CAR)-redirected T cells. Curr. Mol. Med. 2013, 13, 1079–1088. [Google Scholar] [CrossRef] [PubMed]
- Sadelain, M.; Brentjens, R.; Riviere, I. The basic principles of chimeric antigen receptor design. Cancer Discov. 2013, 3, 388–398. [Google Scholar] [CrossRef] [PubMed]
- Redeker, A.; Arens, R. Improving adoptive T cell therapy: The particular role of T Cell costimulation, cytokines, and post-transfer vaccination. Front. Immunol. 2016, 7, 345. [Google Scholar] [CrossRef] [PubMed]
- Weinkove, R.; George, P.; Dasyam, N.; McLellan, A.D. Selecting costimulatory domains for chimeric antigen receptors: Functional and clinical considerations. Clin. Transl. Immunol. 2019, 8, e1049. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; Wei, R.; Ma, Q.; Shi, L.; He, F.; Shi, Z.; Jin, T.; Xie, R.; Wei, B.; Chen, J.; et al. In vivo expansion and antitumor activity of coinfused CD28- and 4-1BB-Engineered CAR-T cells in patients with B Cell leukemia. Mol. Ther. 2018, 26, 976–985. [Google Scholar] [CrossRef]
- Zhang, T.; Wu, M.R.; Sentman, C.L. An NKp30-based chimeric antigen receptor promotes T cell effector functions and antitumor efficacy in vivo. J. Immunol. 2012, 189, 2290–2299. [Google Scholar] [CrossRef]
- Krug, C.; Birkholz, K.; Paulus, A.; Schwenkert, M.; Schmidt, P.; Hoffmann, N.; Hombach, A.; Fey, G.; Abken, H.; Schuler, G.; et al. Stability and activity of MCSP-specific chimeric antigen receptors (CARs) depend on the scFv antigen-binding domain and the protein backbone. Cancer Immunol. Immunother. 2015, 64, 1623–1635. [Google Scholar] [CrossRef]
- Frigault, M.J.; Lee, J.; Basil, M.C.; Carpenito, C.; Motohashi, S.; Scholler, J.; Kawalekar, O.U.; Guedan, S.; McGettigan, S.E.; Posey, A.D., Jr.; et al. Identification of chimeric antigen receptors that mediate constitutive or inducible proliferation of T cells. Cancer Immunol. Res. 2015, 3, 356–367. [Google Scholar] [CrossRef]
- Long, A.H.; Haso, W.M.; Shern, J.F.; Wanhainen, K.M.; Murgai, M.; Ingaramo, M.; Smith, J.P.; Walker, A.J.; Kohler, M.E.; Venkateshwara, V.R.; et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat. Med. 2015, 21, 581–590. [Google Scholar] [CrossRef]
- Park, J.H.; Riviere, I.; Gonen, M.; Wang, X.; Senechal, B.; Curran, K.J.; Sauter, C.; Wang, Y.; Santomasso, B.; Mead, E.; et al. Long-term follow-up of CD19 CAR therapy in acute lymphoblastic leukemia. N. Engl. J. Med. 2018, 378, 449–459. [Google Scholar] [CrossRef]
- Davila, M.L.; Bouhassira, D.C.; Park, J.H.; Curran, K.J.; Smith, E.L.; Pegram, H.J.; Brentjens, R. Chimeric antigen receptors for the adoptive T cell therapy of hematologic malignancies. Int. J. Hematol. 2014, 99, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Cannons, J.L.; Choi, Y.; Watts, T.H. Role of TNF receptor-associated factor 2 and p38 mitogen-activated protein kinase activation during 4-1BB-dependent immune response. J. Immunol. 2000, 165, 6193–6204. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.W.; Nam, K.O.; Park, S.J.; Kwon, B.S. 4-1BB enhances CD8+ T cell expansion by regulating cell cycle progression through changes in expression of cyclins D and E and cyclin-dependent kinase inhibitor p27kip1. Eur. J. Immunol. 2003, 33, 2133–2141. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Condomines, M.; Stegen, S.J.C.v.d.; Perna, F.; Kloss, C.C.; Gunset, G.; Plotkin, J.; Sadelain, M. Structural design of engineered costimulation determines tumor rejection kinetics and persistence of CAR T cells. Cancer Cell 2015, 28, 415–428. [Google Scholar] [CrossRef] [PubMed]
- Lim, W.A.; June, C.H. The principles of engineering immune cells to treat cancer. Cell 2017, 168, 724–740. [Google Scholar] [CrossRef]
- Fraietta, J.A.; Lacey, S.F.; Orlando, E.J.; Pruteanu-Malinici, I.; Gohil, M.; Lundh, S.; Boesteanu, A.C.; Wang, Y.; O’Connor, R.S.; Hwang, W.T.; et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat. Med. 2018, 24, 563–571. [Google Scholar] [CrossRef]
- Petrausch, U.; Schuberth, P.C.; Hagedorn, C.; Soltermann, A.; Tomaszek, S.; Stahel, R.; Weder, W.; Renner, C. Re-directed T cells for the treatment of fibroblast activation protein (FAP)-positive malignant pleural mesothelioma (FAPME-1). BMC Cancer 2012, 12, 615. [Google Scholar] [CrossRef]
- Schuberth, P.C.; Hagedorn, C.; Jensen, S.M.; Gulati, P.; Broek, M.v.d.; Mischo, A.; Soltermann, A.; Jüngel, A.; Marroquin Belaunzaran, O.; Stahel, R.; et al. Treatment of malignant pleural mesothelioma by fibroblast activation protein-specific re-directed T cells. J. Transl. Med. 2013, 11, 187. [Google Scholar] [CrossRef]
- Ceppi, F.; Rivers, J.; Annesley, C.; Pinto, N.; Park, J.R.; Lindgren, C.; Mgebroff, S.; Linn, N.; Delaney, M.; Gardner, R.A. Lymphocyte apheresis for chimeric antigen receptor T-cell manufacturing in children and young adults with leukemia and neuroblastoma. Transfusion 2018, 58, 1414–1420. [Google Scholar] [CrossRef]
- Li, W.; Guo, L.; Rathi, P.; Marinova, E.; Gao, X.; Wu, M.F.; Liu, H.; Dotti, G.; Gottschalk, S.; Metelitsa, L.S.; et al. Redirecting T cells to Glypican-3 with 4-1BB zeta chimeric antigen receptors results in Th1 polarization and potent antitumor activity. Hum. Gene 2017, 28, 437–448. [Google Scholar] [CrossRef]
- Chmielewski, M.; Hombach, A.A.; Abken, H. Of CARs and TRUCKs: Chimeric antigen receptor (CAR) T cells engineered with an inducible cytokine to modulate the tumor stroma. Immunol. Rev. 2014, 257, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Chmielewski, M.; Kopecky, C.; Hombach, A.A.; Abken, H. IL-12 release by engineered T cells expressing chimeric antigen receptors can effectively Muster an antigen-independent macrophage response on tumor cells that have shut down tumor antigen expression. Cancer Res. 2011, 71, 5697–5706. [Google Scholar] [CrossRef] [PubMed]
- Pegram, H.J.; Lee, J.C.; Hayman, E.G.; Imperato, G.H.; Tedder, T.F.; Sadelain, M.; Brentjens, R.J. Tumor-targeted T cells modified to secrete IL-12 eradicate systemic tumors without need for prior conditioning. Blood 2012, 119, 4133–4141. [Google Scholar] [CrossRef] [PubMed]
- Koneru, M.; Purdon, T.J.; Spriggs, D.; Koneru, S.; Brentjens, R.J. IL-12 secreting tumor-targeted chimeric antigen receptor T cells eradicate ovarian tumors in vivo. Oncoimmunology 2015, 4, e994446. [Google Scholar] [CrossRef]
- Koneru, M.; O’Cearbhaill, R.; Pendharkar, S.; Spriggs, D.R.; Brentjens, R.J. A phase I clinical trial of adoptive T cell therapy using IL-12 secreting MUC-16(ecto) directed chimeric antigen receptors for recurrent ovarian cancer. J. Transl. Med. 2015, 13, 102. [Google Scholar] [CrossRef]
- Xu, A.; Bhanumathy, K.K.; Wu, J.; Ye, Z.; Freywald, A.; Leary, S.C.; Li, R.; Xiang, J. IL-15 signaling promotes adoptive effector T-cell survival and memory formation in irradiation-induced lymphopenia. Cell Bio. Sci. 2016, 6, 30. [Google Scholar] [CrossRef]
- Liu, X.; Ranganathan, R.; Jiang, S.; Fang, C.; Sun, J.; Kim, S.; Newick, K.; Lo, A.; June, C.H.; Zhao, Y.; et al. A chimeric switch-receptor targeting PD1 augments the efficacy of second-generation CAR T cells in advanced solid tumors. Cancer Res. 2016, 76, 1578–1590. [Google Scholar] [CrossRef]
- Davies, D.M.; Foster, J.; Stegen, S.J.V.D.; Parente-Pereira, A.C.; Chiapero-Stanke, L.; Delinassios, G.J.; Burbridge, S.E.; Kao, V.; Liu, Z.; Bosshard-Carter, L.; et al. Flexible targeting of ErbB dimers that drive tumorigenesis by using genetically engineered T cells. Mol. Med. 2012, 18, 565–576. [Google Scholar] [CrossRef]
- Schalkwyk, M.C.v.; Papa, S.E.; Jeannon, J.P.; Guerrero Urbano, T.; Spicer, J.F.; Maher, J. Design of a phase I clinical trial to evaluate intratumoral delivery of ErbB-targeted chimeric antigen receptor T-cells in locally advanced or recurrent head and neck cancer. Hum. Gene Clin. Dev. 2013, 24, 134–142. [Google Scholar] [CrossRef]
- Papa, S.; Schalkwyk, M.v.; Maher, J. Clinical Evaluation of ErbB-Targeted CAR T-Cells, Following Intracavity Delivery in Patients with ErbB-Expressing Solid Tumors. In Gene Therapy of Solid Cancers: Methods and Protocols; Walther, W., Stein, U., Eds.; Springer: New York, NY, USA, 2015; pp. 365–382. [Google Scholar]
- Wang, D.; Starr, R.; Chang, W.C.; Aguilar, B.; Alizadeh, D.; Wright, S.L.; Yang, X.; Brito, A.; Sarkissian, A.; Ostberg, J.R.; et al. Chlorotoxin-directed CAR T cells for specific and effective targeting of glioblastoma. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef]
- Law, C.L.; Gordon, K.A.; Toki, B.E.; Yamane, A.K.; Hering, M.A.; Cerveny, C.G.; Petroziello, J.M.; Ryan, M.C.; Smith, L.; Simon, R.; et al. Lymphocyte activation antigen CD70 expressed by renal cell carcinoma is a potential therapeutic target for anti-CD70 antibody-drug conjugates. Cancer Res. 2006, 66, 2328–2337. [Google Scholar] [CrossRef] [PubMed]
- Jilaveanu, L.B.; Sznol, J.; Aziz, S.A.; Duchen, D.; Kluger, H.M.; Camp, R.L. CD70 expression patterns in renal cell carcinoma. Hum. Pathol. 2012, 43, 1394–1399. [Google Scholar] [CrossRef] [PubMed]
- Wischhusen, J.; Jung, G.; Radovanovic, I.; Beier, C.; Steinbach, J.P.; Rimner, A.; Huang, H.; Schulz, J.B.; Ohgaki, H.; Aguzzi, A.; et al. Identification of CD70-mediated apoptosis of immune effector cells as a novel immune escape pathway of human glioblastoma. Cancer Res. 2002, 62, 2592–2599. [Google Scholar] [PubMed]
- Claus, C.; Riether, C.; Schürch, C.; Matter, M.S.; Hilmenyuk, T.; Ochsenbein, A.F. CD27 signaling increases the frequency of regulatory T cells and promotes tumor growth. Cancer Res. 2012, 72, 3664–3676. [Google Scholar] [CrossRef]
- Diegmann, J.; Junker, K.; Gerstmayer, B.; Bosio, A.; Hindermann, W.; Rosenhahn, J.; von Eggeling, F. Identification of CD70 as a diagnostic biomarker for clear cell renal cell carcinoma by gene expression profiling, real-time RT-PCR and immunohistochemistry. Eur. J. Cancer 2005, 41, 1794–1801. [Google Scholar] [CrossRef]
- Brown, C.E.; Badie, B.; Barish, M.E.; Weng, L.; Ostberg, J.R.; Chang, W.C.; Naranjo, A.; Starr, R.; Wagner, J.; Wright, C.; et al. Bioactivity and safety of IL13Rα2-Redirected chimeric antigen receptor CD8+ T Cells in patients with recurrent glioblastoma. Clin. Cancer Res. 2015, 21, 4062–4072. [Google Scholar] [CrossRef]
- Yaghoubi, S.S.; Jensen, M.C.; Satyamurthy, N.; Budhiraja, S.; Paik, D.; Czernin, J.; Gambhir, S.S. Noninvasive detection of therapeutic cytolytic T cells with 18F-FHBG PET in a patient with glioma. Nat. Clin. Pr. Oncol. 2009, 6, 53–58. [Google Scholar] [CrossRef]
- Keu, K.V.; Witney, T.H.; Yaghoubi, S.; Rosenberg, J.; Kurien, A.; Magnusson, R.; Williams, J.; Habte, F.; Wagner, J.R.; Forman, S.; et al. Reporter gene imaging of targeted T cell immunotherapy in recurrent glioma. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef]
- Brown, C.E.; Alizadeh, D.; Starr, R.; Weng, L.; Wagner, J.R.; Naranjo, A.; Ostberg, J.R.; Blanchard, M.S.; Kilpatrick, J.; Simpson, J.; et al. Regression of glioblastoma after chimeric antigen receptor T-Cell Therapy. N. Engl. J. Med. 2016, 375, 2561–2569. [Google Scholar] [CrossRef]
- Zhang, G.; Wang, L.; Cui, H.; Wang, X.; Zhang, G.; Ma, J.; Han, H.; He, W.; Wang, W.; Zhao, Y.; et al. Anti-melanoma activity of T cells redirected with a TCR-like chimeric antigen receptor. Sci. Rep. 2014, 4, 3571. [Google Scholar] [CrossRef]
- Zhang, G.; Liu, R.; Zhu, X.; Wang, L.; Ma, J.; Han, H.; Wang, X.; Zhang, G.; He, W.; Wang, W.; et al. Retargeting NK-92 for anti-melanoma activity by a TCR-like single-domain antibody. Immunol. Cell Biol. 2013, 91, 615–624. [Google Scholar] [CrossRef]
- Timmune. TriCAR-T Technology. Available online: http://www.timmune.com/services/show.php?lang=en&id=142 (accessed on 4 August 2020).
- Eureka Therapeutics. Eureka Therapeutics Achieves Regression of Metastatic Liver Cancer using ET140202 T-cell Therapy. Available online: https://www.eurekatherapeutics.com/media/press-releases/090518/ (accessed on 4 August 2020).
- Tanaka, M.; Tashiro, H.; Omer, B.; Lapteva, N.; Ando, J.; Ngo, M.; Mehta, B.; Dotti, G.; Kinchington, P.R.; Leen, A.M.; et al. Vaccination Targeting native receptors to enhance the function and proliferation of chimeric antigen receptor (CAR)-modified T Cells. Clin. Cancer Res. 2017, 23, 3499–3509. [Google Scholar] [CrossRef] [PubMed]
- Louis, C.U.; Savoldo, B.; Dotti, G.; Pule, M.; Yvon, E.; Myers, G.D.; Rossig, C.; Russell, H.V.; Diouf, O.; Liu, E.; et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood 2011, 118, 6050–6056. [Google Scholar] [CrossRef]
- Pule, M.A.; Savoldo, B.; Myers, G.D.; Rossig, C.; Russell, H.V.; Dotti, G.; Huls, M.H.; Liu, E.; Gee, A.P.; Mei, Z.; et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: Persistence and antitumor activity in individuals with neuroblastoma. Nat. Med. 2008, 14, 1264–1270. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Berger, C.; Wong, C.W.; Forman, S.J.; Riddell, S.R.; Jensen, M.C. Engraftment of human central memory-derived effector CD8+ T cells in immunodeficient mice. Blood 2011, 117, 1888–1898. [Google Scholar] [CrossRef]
- Wang, X.; Naranjo, A.; Brown, C.E.; Bautista, C.; Wong, C.W.; Chang, W.C.; Aguilar, B.; Ostberg, J.R.; Riddell, S.R.; Forman, S.J.; et al. Phenotypic and functional attributes of lentivirus-modified CD19-specific human CD8+ central memory T cells manufactured at clinical scale. J. Immunother. 2012, 35, 689–701. [Google Scholar] [CrossRef] [PubMed]
- Morita, C.T.; Beckman, E.M.; Bukowski, J.F.; Tanaka, Y.; Band, H.; Bloom, B.R.; Golan, D.E.; Brenner, M.B. Direct presentation of nonpeptide prenyl pyrophosphate antigens to human gamma delta T cells. Immunity 1995, 3, 495–507. [Google Scholar] [CrossRef]
- Tanaka, Y.; Morita, C.T.; Tanaka, Y.; Nieves, E.; Brenner, M.B.; Bloom, B.R. Natural and synthetic non-peptide antigens recognized by human gamma delta T cells. Nature 1995, 375, 155–158. [Google Scholar] [CrossRef]
- Oevermann, L.; Lang, P.; Feuchtinger, T.; Schumm, M.; Teltschik, H.M.; Schlegel, P.; Handgretinger, R. Immune reconstitution and strategies for rebuilding the immune system after haploidentical stem cell transplantation. Ann. N. Y. Acad. Sci. 2012, 1266, 161–170. [Google Scholar] [CrossRef]
- Baitsch, L.; Baumgaertner, P.; Devêvre, E.; Raghav, S.K.; Legat, A.; Barba, L.; Wieckowski, S.; Bouzourene, H.; Deplancke, B.; Romero, P.; et al. Exhaustion of tumor-specific CD8⁺ T cells in metastases from melanoma patients. J. Clin. Investig. 2011, 121, 2350–2360. [Google Scholar] [CrossRef]
- Öhrmalm, L.; Smedman, C.; Wong, M.; Broliden, K.; Tolfvenstam, T.; Norbeck, O. Decreased functional T lymphocyte-mediated cytokine responses in patients with chemotherapy-induced neutropenia. J. Intern. Med. 2013, 274, 363–370. [Google Scholar] [CrossRef] [PubMed]
- Gober, H.J.; Kistowska, M.; Angman, L.; Jenö, P.; Mori, L.; De Libero, G. Human T cell receptor gammadelta cells recognize endogenous mevalonate metabolites in tumor cells. J. Exp. Med. 2003, 197, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Coleman, R.E.; Winter, M.C.; Cameron, D.; Bell, R.; Dodwell, D.; Keane, M.M.; Gil, M.; Ritchie, D.; Passos-Coelho, J.L.; Wheatley, D.; et al. The effects of adding zoledronic acid to neoadjuvant chemotherapy on tumour response: Exploratory evidence for direct anti-tumour activity in breast cancer. Br. J. Cancer 2010, 102, 1099–1105. [Google Scholar] [CrossRef] [PubMed]
- Bansal, R.; Reshef, R. Revving the CAR–Combination strategies to enhance CAR T cell effectiveness. Blood Rev. 2020. [Google Scholar] [CrossRef]
- Donnadieu, E.; Dupré., L.; Pinho, L.G.; Cotta-de-Almeida, V. Surmounting the obstacles that impede effective CAR T cell trafficking to solid tumors. J. Leukoc. Biol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Greenbaum, U.; Yalniz, F.F.; Srour, S.A.; Rezvani, K.; Singh, H.; Olson, A.; Blumenschein, G., Jr.; Hong, D.S.; Shpall, E.J.; Kebriaei, P. Chimeric Antigen Receptor Therapy: How Are We Driving in Solid Tumors? Biol. Blood Marrow Transplant. 2020. [Google Scholar] [CrossRef]
- Lesch, S.; Benmebarek, M.R.; Cadilha, B.L.; Stoiber, S.; Subklewe, M.; Endres, S.; Kobold, S. Determinants of response and resistance to CAR T cell therapy. Semin. Cancer Biol. 2019. [Google Scholar] [CrossRef]
- Schurich, A.; Magalhaes, I.; Mattsson, J. Metabolic regulation of CAR T cell function by the hypoxic microenvironment in solid tumors. Immunotherapy 2019, 11, 335–345. [Google Scholar] [CrossRef]
- Scharping, N.E.; Delgoffe, G.M. Tumor microenvironment metabolism: A new checkpoint. for anti-tumor immunity. Vaccines 2016, 4, 46. [Google Scholar] [CrossRef]
- Beatty, G.L.; Moon, E.K. Chimeric antigen receptor T cells are vulnerable to immunosuppressive mechanisms present within the tumor microenvironment. Oncoimmunology 2014, 3, e970027. [Google Scholar] [CrossRef]
- Haas, A.R.; Tanyi, J.L.; O’Hara, M.H.; Gladney, W.L.; Lacey, S.F.; Torigian, D.A.; Soulen, M.C.; Tian, L.; McGarvey, M.; Nelson, A.M.; et al. Phase I study of lentiviral-transduced chimeric antigen receptor-modified T cells recognizing mesothelin in advanced solid cancers. Mol. Ther. 2019, 27, 1919–1929. [Google Scholar] [CrossRef] [PubMed]
- Hege, K.M.; Bergsland, E.K.; Fisher, G.A.; Nemunaitis, J.J.; Warren, R.S.; McArthur, J.G.; Lin, A.A.; Schlom, J.; June, C.H.; Sherwin, S.A. Safety, tumor trafficking and immunogenicity of chimeric antigen receptor (CAR)-T cells specific for TAG-72 in colorectal cancer. J. Immunother. Cancer 2017, 5, 22. [Google Scholar] [CrossRef] [PubMed]
- Adachi, K.; Kano, Y.; Nagai, T.; Okuyama, N.; Sakoda, Y.; Tamada, K. IL-7 and CCL19 expression in CAR-T cells improves immune cell infiltration and CAR-T cell survival in the tumor. Nat. Biotechnol. 2018, 36, 346–351. [Google Scholar] [CrossRef] [PubMed]
- Lo, A.; Wang, L.S.; Scholler, J.; Monslow, J.; Avery, D.; Newick, K.; O’Brien, S.; Evans, R.A.; Bajor, D.J.; Clendenin, C.; et al. Tumor-Promoting Desmoplasia Is Disrupted by Depleting FAP-Expressing Stromal Cells. Cancer Res. 2015, 75, 2800–2810. [Google Scholar] [CrossRef]
- Wang, L.C.; Lo, A.; Scholler, J.; Sun, J.; Majumdar, R.S.; Kapoor, V.; Antzis, M.; Cotner, C.E.; Johnson, L.A.; Durham, A.C.; et al. Targeting fibroblast activation protein in tumor stroma with chimeric antigen receptor T cells can inhibit tumor growth and augment host immunity without severe toxicity. Cancer Immunol. Res. 2014, 2, 154–166. [Google Scholar] [CrossRef]
- Jiang, Z.; Jiang, X.; Chen, S.; Lai, Y.; Wei, X.; Li, B.; Lin, S.; Wang, S.; Wu, Q.; Liang, Q.; et al. Anti-GPC3-CAR T cells suppress the growth of tumor cells in patient-derived xenografts of hepatocellular carcinoma. Front. Immunol. 2016, 7, 690. [Google Scholar] [CrossRef]
- Pearson, C.; Silva, A.; Seddon, B. Exogenous amino acids are essential for interleukin-7 induced CD8 T cell growth. PLoS ONE 2012, 7, e33998. [Google Scholar] [CrossRef]
- Robbiani, D.F.; Finch, R.A.; Jäger, D.; Muller, W.A.; Sartorelli, A.C.; Randolph, G.J. The leukotriene C(4) transporter MRP1 regulates CCL19 (MIP-3beta, ELC)-dependent mobilization of dendritic cells to lymph nodes. Cell 2000, 103, 757–768. [Google Scholar] [CrossRef]
- Reif, K.; Ekland, E.H.; Ohl, L.; Nakano, H.; Lipp, M.; Förster, R.; Cyster, J.G. Balanced responsiveness to chemoattractants from adjacent zones determines B-cell position. Nature 2002, 416, 94–99. [Google Scholar] [CrossRef]
- Bromley, S.K.; Thomas, S.Y.; Luster, A.D. Chemokine receptor CCR7 guides T cell exit from peripheral tissues and entry into afferent lymphatics. Nat. Immunol. 2005, 6, 895–901. [Google Scholar] [CrossRef]
- Shum, T.; Omer, B.; Tashiro, H.; Kruse, R.L.; Wagner, D.L.; Parikh, K.; Yi, Z.; Sauer, T.; Liu, D.; Parihar, R.; et al. Constitutive signaling from an engineered IL7 receptor promotes durable tumor elimination by tumor-redirected T cells. Cancer Discov. 2017, 7, 1238–1247. [Google Scholar] [CrossRef] [PubMed]
- Hurton, L.V.; Singh, H.; Najjar, A.M.; Switzer, K.C.; Mi, T.; Maiti, S.; Olivares, S.; Rabinovich, B.; Huls, H.; Forget, M.A.; et al. Tethered IL-15 augments antitumor activity and promotes a stem-cell memory subset in tumor-specific T cells. Proc. Natl. Acad. Sci. USA 2016, 113, E7788–E7797. [Google Scholar] [CrossRef]
- Wilkie, S.; Burbridge, S.E.; Chiapero-Stanke, L.; Pereira, A.C.; Cleary, S.; Stegen, S.J.v.d.; Spicer, J.F.; Davies, D.M.; Maher, J. Selective expansion of chimeric antigen receptor-targeted T-cells with potent effector function using interleukin-4. J. Biol. Chem. 2010, 285, 25538–25544. [Google Scholar] [CrossRef] [PubMed]
- Siegel, P.M.; Massagué, J. Cytostatic and apoptotic actions of TGF-beta in homeostasis and cancer. Nat. Rev. Cancer 2003, 3, 807–821. [Google Scholar] [CrossRef] [PubMed]
- Travis, M.A.; Sheppard, D. TGF-β activation and function in immunity. Annu. Rev. Immunol. 2014, 32, 51–82. [Google Scholar] [CrossRef]
- Wieser, R.; Attisano, L.; Wrana, J.L.; Massagué, J. Signaling activity of transforming growth factor beta type II receptors lacking specific domains in the cytoplasmic region. Mol. Cell Biol. 1993, 13, 7239–7247. [Google Scholar] [CrossRef]
- Gorelik, L.; Flavell, R.A. Immune-mediated eradication of tumors through the blockade of transforming growth factor-beta signaling in T cells. Nat. Med. 2001, 7, 1118–1122. [Google Scholar] [CrossRef]
- Kloss, C.C.; Lee, J.; Zhang, A.; Chen, F.; Melenhorst, J.J.; Lacey, S.F.; Maus, M.V.; Fraietta, J.A.; Zhao, Y.; June, C.H. Dominant-negative TGF-β receptor enhances PSMA-targeted human CAR T cell proliferation and augments prostate cancer eradication. Mol. Ther. 2018, 26, 1855–1866. [Google Scholar] [CrossRef]
- Goff, S.L.; Morgan, R.A.; Yang, J.C.; Sherry, R.M.; Robbins, P.F.; Restifo, N.P.; Feldman, S.A.; Lu, Y.C.; Lu, L.; Zheng, Z.; et al. Pilot trial of adoptive transfer of chimeric antigen receptor-transduced T cells targeting EGFRvIII in patients with glioblastoma. J. Immunother. 2019, 42, 126–135. [Google Scholar] [CrossRef]
- Adusumilli, P.S.; Zauderer, M.G.; Rusch, V.W.; O’Cearbhaill, R.; Zhu, A.; Ngai, D.; McGee, E.; Chintala, N.; Messinger, J.; Cheema, W.; et al. Regional delivery of mesothelin-targeted CAR T cells for pleural cancers: Safety and preliminary efficacy in combination with anti-PD-1 agent. J. Clin. Oncol. 2019, 37, 2511. [Google Scholar] [CrossRef]
- Schietinger, A.; Philip, M.; Krisnawan, V.E.; Chiu, E.Y.; Delrow, J.J.; Basom, R.S.; Lauer, P.; Brockstedt, D.G.; Knoblaugh, S.E.; Hämmerling, G.J.; et al. Tumor-specific T cell dysfunction is a dynamic antigen-driven differentiation program initiated early during tumorigenesis. Immunity 2016, 45, 389–401. [Google Scholar] [CrossRef] [PubMed]
- Heczey, A.; Louis, C.U.; Savoldo, B.; Dakhova, O.; Durett, A.; Grilley, B.; Liu, H.; Wu, M.F.; Mei, Z.; Gee, A.; et al. CAR T Cells administered in combination with lymphodepletion and PD-1 inhibition to patients with neuroblastoma. Mol. Ther. 2017, 25, 2214–2224. [Google Scholar] [CrossRef] [PubMed]
- Brentjens, R.J.; Rivière, I.; Park, J.H.; Davila, M.L.; Wang, X.; Stefanski, J.; Taylor, C.; Yeh, R.; Bartido, S.; Borquez-Ojeda, O.; et al. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood 2011, 118, 4817–4828. [Google Scholar] [CrossRef]
- Gattinoni, L.; Finkelstein, S.E.; Klebanoff, C.A.; Antony, P.A.; Palmer, D.C.; Spiess, P.J.; Hwang, L.N.; Yu, Z.; Wrzesinski, C.; Heimann, D.M.; et al. Removal of homeostatic cytokine sinks by lymphodepletion enhances the efficacy of adoptively transferred tumor-specific CD8+ T cells. J. Exp. Med. 2005, 202, 907–912. [Google Scholar] [CrossRef]
- Dudley, M.E.; Wunderlich, J.R.; Yang, J.C.; Sherry, R.M.; Topalian, S.L.; Restifo, N.P.; Royal, R.E.; Kammula, U.; White, D.E.; Mavroukakis, S.A.; et al. Adoptive cell transfer therapy following non-myeloablative but lymphodepleting chemotherapy for the treatment of patients with refractory metastatic melanoma. J. Clin. Oncol. 2005, 23, 2346–2357. [Google Scholar] [CrossRef]
- Suryadevara, C.M.; Desai, R.; Abel, M.L.; Riccione, K.A.; Batich, K.A.; Shen, S.H.; Chongsathidkiet, P.; Gedeon, P.C.; Elsamadicy, A.A.; Snyder, D.J.; et al. Temozolomide lymphodepletion enhances CAR abundance and correlates with antitumor efficacy against established glioblastoma. Oncoimmunology 2018, 7, e1434464. [Google Scholar] [CrossRef] [PubMed]
- Stavrou, M.; Philip, B.; Traynor-White, C.; Davis, C.G.; Onuoha, S.; Cordoba, S.; Thomas, S.; Pule, M. A Rapamycin-Activated Caspase 9-Based Suicide Gene. Mol. Ther. 2018, 26, 1266–1276. [Google Scholar] [CrossRef]
- Di Stasi, A.; Tey, S.K.; Dotti, G.; Fujita, Y.; Kennedy-Nasser, A.; Martinez, C.; Straathof, K.; Liu, E.; Durett, A.G.; Grilley, B.; et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N. Engl. J. Med. 2011, 365, 1673–1683. [Google Scholar] [CrossRef]
- Foster, A.E.; Mahendravada, A.; Shinners, N.P.; Chang, W.C.; Crisostomo, J.; Lu, A.; Khalil, M.; Morschl, E.; Shaw, J.L.; Saha, S.; et al. Regulated expansion and survival of chimeric antigen receptor-modified T cells using small molecule-dependent inducible MyD88/CD40. Mol. Ther. 2017, 25, 2176–2188. [Google Scholar] [CrossRef]
- Becerra, C.R.; Manji, G.A.; Kim, D.W.; Gardner, O.; Malankar, A.; Shaw, J.; Blass, D.; Yi, X.; Foster, A.E.; Woodard, P. Ligand-inducible, prostate stem cell antigen (PSCA)-directed GoCAR-T cells in advanced solid tumors: Preliminary results with cyclophosphamide (Cy) ± fludarabine (Flu) lymphodepletion (LD). J. Clin. Oncol. 2019, 37, 2536. [Google Scholar] [CrossRef]
- Moolten, F.L. Tumor chemosensitivity conferred by inserted herpes thymidine kinase genes: Paradigm for a prospective cancer control strategy. Cancer Res. 1986, 46, 5276–5281. [Google Scholar] [PubMed]
- Beltinger, C.; Fulda, S.; Kammertoens, T.; Meyer, E.; Uckert, W.; Debatin, K.M. Herpes simplex virus thymidine kinase/ganciclovir-induced apoptosis involves ligand-independent death receptor aggregation and activation of caspases. Proc. Natl. Acad. Sci. USA 1999, 96, 8699–8704. [Google Scholar] [CrossRef] [PubMed]
- Traversari, C.; Marktel, S.; Magnani, Z.; Mangia, P.; Russo, V.; Ciceri, F.; Bonini, C.; Bordignon, C. The potential immunogenicity of the TK suicide gene does not prevent full clinical benefit associated with the use of TK-transduced donor lymphocytes in HSCT for hematologic malignancies. Blood 2007, 109, 4708–4715. [Google Scholar] [CrossRef] [PubMed]
- Park, J.R.; Digiusto, D.L.; Slovak, M.; Wright, C.; Naranjo, A.; Wagner, J.; Meechoovet, H.B.; Bautista, C.; Chang, W.C.; Ostberg, J.R.; et al. Adoptive transfer of chimeric antigen receptor re-directed cytolytic T lymphocyte clones in patients with neuroblastoma. Mol. Ther. 2007, 15, 825–833. [Google Scholar] [CrossRef] [PubMed]
- Summers, C.; Grier, A.; Gardner, R.; Delaney, C.; Jensen, M.C. Multiplexed Engineering of CD19CAR T Cells for Post-Transplant Consolidative Immunotherapy. Blood 2016, 128, 1159. [Google Scholar] [CrossRef]
- Wang, X.; Chang, W.-C.; Wong, C.W.; Colcher, D.; Sherman, M.; Ostberg, J.R.; Forman, S.J.; Riddell, S.R.; Jensen, M.C. A transgene-encoded cell surface polypeptide for selection, in vivo tracking, and ablation of engineered cells. Blood 2011, 118, 1255–1263. [Google Scholar] [CrossRef]
- Budde, L.E.; Berger, C.; Lin, Y.; Wang, J.; Lin, X.; Frayo, S.E.; Brouns, S.A.; Spencer, D.M.; Till, B.G.; Jensen, M.C.; et al. Combining a CD20 chimeric antigen receptor and an inducible caspase 9 suicide switch to improve the efficacy and safety of T cell adoptive immunotherapy for lymphoma. PLoS ONE 2013, 8, e82742. [Google Scholar] [CrossRef]
- Budde, L.E.; Mardiros, A.; Chang, W.-C.; Wang, X.; Berger, C.; Brown, C.; Riddell, S.R.; Press, O.W.; Forman, S.J. Truncated cell-surface CD19 as a conditional suicide switch for adoptive T Cell immunotherapy. Blood 2013, 122, 1660. [Google Scholar] [CrossRef]
- Harrer, D.C.; Simon, B.; Fujii, S.I.; Shimizu, K.; Uslu, U.; Schuler, G.; Gerer, K.F.; Hoyer, S.; Dorrie, J.; Schaft, N. RNA-transfection of gamma/delta T cells with a chimeric antigen receptor or an alpha/beta T-cell receptor: A safer alternative to genetically engineered alpha/beta T cells for the immunotherapy of melanoma. BMC Cancer 2017, 17, 551. [Google Scholar] [CrossRef]
- Dorrie, J.; Babalija, L.; Hoyer, S.; Gerer, K.F.; Schuler, G.; Heinzerling, L.; Schaft, N. BRAF and MEK Inhibitors influence the function of reprogrammed t cells: Consequences for adoptive T-Cell therapy. Int. J. Mol. Sci. 2018, 19, 289. [Google Scholar] [CrossRef]
- Harrer, D.C.; Schuler, G.; Dorrie, J.; Schaft, N. CSPG4-Specific CAR T cells for high-risk childhood B cell precursor Leukemia. Int. J. Mol. Sci. 2019, 20, 2764. [Google Scholar] [CrossRef] [PubMed]
- Uslu, U.; Schuler, G.; Dorrie, J.; Schaft, N. Combining a chimeric antigen receptor and a conventional T-cell receptor to generate T cells expressing two additional receptors (TETARs) for a multi-hit immunotherapy of melanoma. Exp. Derm. 2016, 25, 872–879. [Google Scholar] [CrossRef] [PubMed]
- Wiesinger, M.; Marz, J.; Kummer, M.; Schuler, G.; Dorrie, J.; Schuler-Thurner, B.; Schaft, N. Clinical-Scale production of CAR-T cells for the treatment of melanoma patients by mRNA transfection of a CSPG4-specific CAR under full GMP compliance. Cancers 2019, 11, 1198. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Ma, Y.; Zhu, J.; Chen, Y.; Sun, Y.; Yao, Y.; Yang, Z.; Xie, J. A Review on electroporation-based intracellular delivery. Molecules 2018, 23, 3044. [Google Scholar] [CrossRef]
- Tchou, J.; Zhao, Y.; Levine, B.L.; Zhang, P.J.; Davis, M.M.; Melenhorst, J.J.; Kulikovskaya, I.; Brennan, A.L.; Liu, X.; Lacey, S.F.; et al. Safety and Efficacy of intratumoral injections of chimeric antigen receptor (CAR) T cells in metastatic breast cancer. Cancer Immunol. Res. 2017, 5, 1152–1161. [Google Scholar] [CrossRef]
- Beatty, G.L.; Haas, A.R.; Maus, M.V.; Torigian, D.A.; Soulen, M.C.; Plesa, G.; Chew, A.; Zhao, Y.; Levine, B.L.; Albelda, S.M.; et al. Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce anti-tumor activity in solid malignancies. Cancer Immunol. Res. 2014, 2, 112–120. [Google Scholar] [CrossRef]
- Beatty, G.L.; O’Hara, M.H.; Lacey, S.F.; Torigian, D.A.; Nazimuddin, F.; Chen, F.; Kulikovskaya, I.M.; Soulen, M.C.; McGarvey, M.; Nelson, A.M.; et al. Activity of mesothelin-specific chimeric antigen receptor t cells against pancreatic carcinoma metastases in a phase 1 trial. Gastroenterology 2018, 155, 29–32. [Google Scholar] [CrossRef]
- Maus, M.V.; Haas, A.R.; Beatty, G.L.; Albelda, S.M.; Levine, B.L.; Liu, X.; Zhao, Y.; Kalos, M.; June, C.H. T cells expressing chimeric antigen receptors can cause anaphylaxis in humans. Cancer Immunol. Res. 2013, 1, 26–31. [Google Scholar] [CrossRef]
- Svoboda, J.; Rheingold, S.R.; Gill, S.I.; Grupp, S.A.; Lacey, S.F.; Kulikovskaya, I.; Suhoski, M.M.; Melenhorst, J.J.; Loudon, B.; Mato, A.R.; et al. Nonviral RNA chimeric antigen receptor-modified T cells in patients with Hodgkin lymphoma. Blood 2018, 132, 1022–1026. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, M.; Wu, Z.; Tong, C.; Dai, H.; Guo, Y.; Liu, Y.; Huang, J.; Lv, H.; Luo, C.; et al. CD133-directed CAR T cells for advanced metastasis malignancies: A phase I trial. Oncoimmunology 2018, 7, e1440169. [Google Scholar] [CrossRef]
- Zhan, X.; Wang, B.; Li, Z.; Li, J.; Wang, H.; Chen, L.; Jiang, H.; Wu, M.; Xiao, J.; Peng, X.; et al. Phase I trial of Claudin 18.2-specific chimeric antigen receptor T cells for advanced gastric and pancreatic adenocarcinoma. J. Clin. Oncol. 2019, 37, 2509. [Google Scholar] [CrossRef]
- Morgan, R.A.; Johnson, L.A.; Davis, J.L.; Zheng, Z.; Woolard, K.D.; Reap, E.A.; Feldman, S.A.; Chinnasamy, N.; Kuan, C.T.; Song, H.; et al. Recognition of glioma stem cells by genetically modified T cells targeting EGFRvIII and development of adoptive cell therapy for glioma. Hum. Gene 2012, 23, 1043–1053. [Google Scholar] [CrossRef] [PubMed]
- Maus, M.V. Designing CAR T cells for glioblastoma. Oncoimmunology 2015, 4, e1048956. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.J.D.; Martinez-Lage, M.; Brem, S.; Maloney, E.; Shen, A.; et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Boesteanu, A.C.; Binder, Z.A.; Xu, C.; Reid, R.A.; Rodriguez, J.L.; Cook, D.R.; Thokala, R.; Blouch, K.; McGettigan-Croce, B.; et al. CheckpoInt. Blockade Reverses Anergy in IL-13Rα2 Humanized scFv-Based CAR T Cells to Treat Murine and Canine Gliomas. Mol. Oncolytics 2018, 11, 20–38. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.A.; Scholler, J.; Ohkuri, T.; Kosaka, A.; Patel, P.R.; McGettigan, S.E.; Nace, A.K.; Dentchev, T.; Thekkat, P.; Loew, A.; et al. Rational development and characterization of humanized anti-EGFR variant III chimeric antigen receptor T cells for glioblastoma. Sci. Transl. Med. 2015, 7, 275ra222. [Google Scholar] [CrossRef]
- Kershaw, M.H.; Westwood, J.A.; Parker, L.L.; Wang, G.; Eshhar, Z.; Mavroukakis, S.A.; White, D.E.; Wunderlich, J.R.; Canevari, S.; Rogers-Freezer, L.; et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin. Cancer Res. 2006, 12, 6106–6115. [Google Scholar] [CrossRef]
- Lee, D.W.; Barrett, D.M.; Mackall, C.; Orentas, R.; Grupp, S.A. The future is now: Chimeric antigen receptors as new targeted therapies for childhood cancer. Clin. Cancer Res. 2012, 18, 2780–2790, Correction in 2017, 23, 611, doi:10.1158/1078-0432.Ccr-16-2684. [Google Scholar] [CrossRef]
- Gargett, T.; Yu, W.; Dotti, G.; Yvon, E.S.; Christo, S.N.; Hayball, J.D.; Lewis, I.D.; Brenner, M.K.; Brown, M.P. GD2-specific CAR T Cells Undergo Potent Activation and Deletion Following Antigen Encounter but can be Protected From Activation-induced Cell Death by PD-1 Blockade. Mol. Ther. 2016, 24, 1135–1149. [Google Scholar] [CrossRef]
- Stroncek, D.F.; Lee, D.W.; Ren, J.; Sabatino, M.; Highfill, S.; Khuu, H.; Shah, N.N.; Kaplan, R.N.; Fry, T.J.; Mackall, C.L. Elutriated lymphocytes for manufacturing chimeric antigen receptor T cells. J. Transl. Med. 2017, 15, 59. [Google Scholar] [CrossRef]
- Straathof, K.; Flutter, B.; Wallace, R.; Thomas, S.; Cheung, G.; Collura, A.; Gileadi, T.; Barton, J.; Wright, G.; Inglott, S.; et al. Abstract CT145: A cancer research UK phase I trial of anti-GD2 chimeric antigen receptor (CAR) transduced T-cells (1RG-CART) in patients with relapsed or refractory neuroblastoma. Cancer Res. 2018, 78, CT145. [Google Scholar] [CrossRef]
- Zhai, B.; Shi, D.; Gao, H.; Qi, X.; Jiang, H.; Zhang, Y.; Chi, J.; Ruan, H.; Wang, H.; Ru, Q.C.; et al. A phase I study of anti-GPC3 chimeric antigen receptor modified T cells (GPC3 CAR-T) in Chinese patients with refractory or relapsed GPC3+ hepatocellular carcinoma (r/r GPC3+ HCC). J. Clin. Oncol. 2017, 35, 3049. [Google Scholar] [CrossRef]
- Tanyi, J.L.; Stashwick, C.; Plesa, G.; Morgan, M.A.; Porter, D.; Maus, M.V.; June, C.H. Possible compartmental cytokine release syndrome in a patient with recurrent ovarian cancer after treatment with mesothelin-targeted CAR-T Cells. J. Immunother. 2017, 40, 104–107. [Google Scholar] [CrossRef] [PubMed]
- Tanyi, J.L.; Haas, A.R.; Beatty, G.L.; Stashwick, C.J.; O’Hara, M.H.; Morgan, M.A.; Porter, D.L.; Melenhorst, J.J.; Plesa, G.; Lacey, S.F.; et al. Anti-mesothelin chimeric antigen receptor T cells in patients with epithelial ovarian cancer. J. Clin. Oncol. 2016, 34, 5511. [Google Scholar] [CrossRef]
- Prasad, S.; Adusumilli, M.G.Z.; Valerie, W.; Rusch, R.E.; Amy, Z.; Daniel, A.; Ngai, E.; Navin, K.; Chintala, J.C.; Elizabeth, F.; et al. CT036—A Phase I Clinical Trial of Malignant Pleural Disease Treated with Regionally Delivered Autologous Mesothelin-Targeted Car T Cells: Safety and Efficacy. Available online: https://www.abstractsonline.com/pp8/#!/6812/presentation/9837 (accessed on 4 August 2020).
- You, F.; Jiang, L.; Zhang, B.; Lu, Q.; Zhou, Q.; Liao, X.; Wu, H.; Du, K.; Zhu, Y.; Meng, H.; et al. Phase 1 clinical trial demonstrated that MUC1 positive metastatic seminal vesicle cancer can be effectively eradicated by modified Anti-MUC1 chimeric antigen receptor transduced T cells. Sci. China Life Sci. 2016, 59, 386–397. [Google Scholar] [CrossRef]
- Junghans, R.P.; Ma, Q.; Rathore, R.; Gomes, E.M.; Bais, A.J.; Lo, A.S.; Abedi, M.; Davies, R.A.; Cabral, H.J.; Al-Homsi, A.S.; et al. Phase I trial of anti-PSMA designer CAR-T cells in prostate cancer: Possible role for interacting interleukin 2-T cell pharmacodynamics as a determinant of clinical response. Prostate 2016, 76, 1257–1270. [Google Scholar] [CrossRef]
- Ma, Q.; Safar, M.; Holmes, E.; Wang, Y.; Boynton, A.L.; Junghans, R.P. Anti-prostate specific membrane antigen designer T cells for prostate cancer therapy. Prostate 2004, 61, 12–25. [Google Scholar] [CrossRef]
- Specht, J.M.; Lee, S.; Turtle, C.J.; Berger, C.; Baladrishnan, A.; Srivastava, S.; Voillet, V.; Veatch, J.; Gooley, T.; Mullane, E.; et al. Abstract CT131: A phase I study of adoptive immunotherapy for advanced ROR1+ malignancies with defined subsets of autologous T cells expressing a ROR1-specific chimeric antigen receptor (ROR1-CAR). Cancer Res. 2018, 78, CT131. [Google Scholar] [CrossRef]
- Specht, J.M.L.S.; Turtle, C.; Berger, C.; Balakrishnan, A.; Srivastava, S.; Viollet, V.; Veatch, J.; Gooley, T.; Mullane, E.; Chaney, C.N.; et al. A Phase I Study of Adoptive Immunotherapy for Ror1+ Advanced Triple Negative Breast Cancer (Tnbc) with Defined Subsets of Autologous T Cells Expressing A Ror1-Specific Chimeric Antigen Receptor (ROR1-CAR). Available online: https://www.abstracts2view.com/sabcs/view.php?nu=SABCS18L_1227 (accessed on 4 August 2020).
- Shimabukuro-Vornhagen, A.; Godel, P.; Subklewe, M.; Stemmler, H.J.; Schlosser, H.A.; Schlaak, M.; Kochanek, M.; Boll, B.; Bergwelt-Baildon, M.S.v. Cytokine release syndrome. J. Immunother. Cancer 2018, 6, 56. [Google Scholar] [CrossRef]
- Perales, M.A.; Kebriaei, P.; Kean, L.S.; Sadelain, M. Building a safer and faster CAR: Seatbelts, airbags and crispr. Biol. Blood Marrow Transplant. 2017. [Google Scholar] [CrossRef]
- Stoiber, S.; Cadilha, B.L.; Benmebarek, M.R.; Lesch, S.; Endres, S.; Kobold, S. Limitations in the design of chimeric antigen receptors for cancer therapy. Cells 2019, 8, 472. [Google Scholar] [CrossRef]
- Sievers, N.M.; Dörrie, J.; Schaft, N. CARs: Beyond T Cells and T Cell-derived signaling domains. Int. J. Mol. Sci. 2020, 21, 3525. [Google Scholar] [CrossRef] [PubMed]
- Harrer, D.C.; Dorrie, J.; Schaft, N. Chimeric antigen receptors in different cell types: New vehicles join the race. Hum. Gene 2018, 29, 547–558. [Google Scholar] [CrossRef] [PubMed]
- Montagner, I.M.; Penna, A.; Fracasso, G.; Carpanese, D.; Dalla Pieta, A.; Barbieri, V.; Zuccolotto, G.; Rosato, A. Anti-PSMA CAR-engineered NK-92 Cells: An Off-the-shelf cell therapy for prostate cancer. Cells 2020, 9, 1382. [Google Scholar] [CrossRef] [PubMed]
- Cutsem, E.V.; Machiels, J.; Eynde, M.V.D.; Prenen, H.; Hendlisz, A.; Shaza, L.; Carrasco, J.; Canon, J.; Sotiropoulou, P.; Breman, E.; et al. SO-009-Phase 1 studies assessing the safety and clinical activity of autologous and allogeneic NKG2D-based CAR-T therapy in metastatic colorectal cancer. Ann. Oncol. 2019, 30, iv124–iv125. [Google Scholar] [CrossRef]
- Santoro, S.P.; Kim, S.; Motz, G.T.; Alatzoglou, D.; Li, C.; Irving, M.; Powell, D.J., Jr.; Coukos, G. T cells bearing a chimeric antigen receptor against prostate-specific membrane antigen mediate vascular disruption and result in tumor regression. Cancer Immunol. Res. 2015, 3, 68–84. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antigen |  | Several Organs |  |  |  |  |  |  |  |  |  |  |  |  |  |  |  |  |  |  | Number of Trials |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AFP peptide/A2 | √ | 2 | |||||||||||||||||||
| B7-H3 | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | 6 | ||||||||||
| CD20 | √ | 1 | |||||||||||||||||||
| CD44v6 | √ | √ | √ | 2 | |||||||||||||||||
| CD70 | √ | √ | √ | √ | √ | 2 | |||||||||||||||
| CD133 | √ | √ | √ | √ | √ | √ | 3 | ||||||||||||||
| CD147 (EMMPRIN) | √ | √ | 2 | ||||||||||||||||||
| CD171 (L1CAM) | √ | 2 | |||||||||||||||||||
| CEA | √ | √ | √ | √ | √ | √ | √ | √ | √ | 16 | |||||||||||
| claudin 18.2 (CLD18) | √ | √ | √ | 6 | |||||||||||||||||
| c-Met/hepatocyte growth factor receptor | √ | √ | 4 | ||||||||||||||||||
| DLL3 (delta-like protein 3) | √ | 1 | |||||||||||||||||||
| EGFR | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | 8 | ||||||
| EGFR family member | √ | 1 | |||||||||||||||||||
| EGFRvIII | √ | 11 | |||||||||||||||||||
| EGFR806 | √ | 1 | |||||||||||||||||||
| EpCAM | √ | √ | √ | √ | √ | √ | √ | √ | 6 | ||||||||||||
| EphA2 | √ | 2 | |||||||||||||||||||
| ErbB2 dimers | √ | 1 | |||||||||||||||||||
| FAP | √ | 1 | |||||||||||||||||||
| FBP (folate binding protein) | √ | 3 | |||||||||||||||||||
| GD2 | √ | √ | √ | √ | √ | √ | 24 | ||||||||||||||
| gp100 (209-217/HLA-A2) | √ | 1 | |||||||||||||||||||
| GPC3 (glypican-3) | √ | √ | √ | √ | √ | √ | 18 | ||||||||||||||
| HER2 | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | 17 | |||||||||
| ICAM1 | √ | 1 | |||||||||||||||||||
| IL13Rα2 | √ | √ | 6 | ||||||||||||||||||
| Lewis Y | √ | 2 | |||||||||||||||||||
| ligands of chlorotoxin | √ | 1 | |||||||||||||||||||
| LMP1 (EBV) | √ | 1 | |||||||||||||||||||
| mesothelin | √ | √ | √ | √ | √ | √ | 32 | ||||||||||||||
| MG7 | √ | 1 | |||||||||||||||||||
| MUC1 | √ | √ | √ | √ | √ | √ | √ | √ | √ | 11 | |||||||||||
| Muc1 (cleaved form) | √ | 1 | |||||||||||||||||||
| MUC16ecto | √ | √ | √ | 1 | |||||||||||||||||
| TnMuc1 | √ | √ | √ | √ | √ | 1 | |||||||||||||||
| Nectin4/FAP | √ | √ | √ | √ | √ | √ | 1 | ||||||||||||||
| NKG2D-ligands (MIC-A,-B, ULBP-1,-2,-3,-4,-5,-6) | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | 6 | |||||||||
| PD-L1 | √ | √ | 5 | ||||||||||||||||||
| PSCA | √ | √ | √ | √ | 4 | ||||||||||||||||
| PSMA | √ | √ | √ | √ | 11 | ||||||||||||||||
| ROR1 | √ | √ | √ | 1 | |||||||||||||||||
| ROR2 | √ | √ | √ | √ | √ | 2 | |||||||||||||||
| VEGFR2 | √ | √ | 1 | ||||||||||||||||||
| Several Ags | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | 16 | ||||||
| undisclosed antigen | √ | √ | 2 |
| Organ | Cancer Type | Targeted Antigens |
|---|---|---|
| brain/CNS | brain | CD133, HER2, PSMA |
| glioma | B7-H3, CD147, EGFR, EGFRvIII, EphA2, GD2, HER2, IL13Rα2, MUC1, CD133 | |
| glioblastoma | B7-H3, ligands of chlorotoxin, EGFRvIII, HER2, IL13Rα2, NKG2D-Ligands, PD-L1 | |
| primitive neuroectodermal tumor | B7-H3 | |
| choroid plexus carcinoma | B7-H3 | |
| pineoblastoma | B7-H3 | |
| CNS tumor | B7-H3, EGFR806, HER2 | |
| ependymoma | B7-H3 | |
| medulloblastoma | B7-H3, NKG2D-Ligands | |
| several organs | rhabdoid tumor | B7-H3, EGFR, GPC3 |
| Rhabdomyosarcoma | B7-H3, EGFR, GPC3 | |
| desmoplastic small round cell tumor | B7-H3, EGFR | |
| sarcoma | GD2, HER2, NKG2D-Ligands, CD133, MUC1, CD117 | |
| adenocarcinoma | CEA | |
| solid tumors | B7-H3, CEA, claudin 18.2, EGFR, EGFR family member, GD2, GPC3, HER2, Lewis Y, mesothelin, MUC1, MUC16ecto, TnMuc1, Nectin4, ROR2 | |
| pancreas | pancreatic | CD70, CD133, CEA, claudin 18.2, EGFR, EpCAM, HER2, mesothelin, MUC1, Nectin4, NKG2D-Ligands, PSCA, ROR2, EGFRvIII |
| pancreatic ductal adenocarcinoma | claudin 18.2, mesothelin, TnMuc1 | |
| liver | liver | CD133, CEA, EGFR, EpCAM, GPC3, MG7, NKG2D-Ligands |
| HCC (hepatocellular carcinoma) | AFP/HLA-A2, CD147, GPC3, MUC1, NKG2D-Ligands, c-MET, PD-L1 | |
| hepatoblastoma | B7-H3, EGFR | |
| hepatoma | several | |
| gall bladder carcinoma | EGFR | |
| cholangiocarcinoma | EGFR, HER2, MUC1 | |
| lung | lung | CEA, EGFR, HER2, mesothelin, Lewis Y, PSCA, MUC1, PD-L1, CD80/86, MAGE-A1, MAGE-A4, GD2 |
| small cell lung cancer | DLL3 | |
| mesothelioma | FAP, mesothelin | |
| lung squamous cell carcinoma | GPC3 | |
| NSCLC | EGFR, mesothelin, MUC1, TnMuc1, Nectin4, PD-L1, ROR1, CD80/86 | |
| uterus/cervix | ovarian | CD70, CD133, CEA, EGFR, FBP, HER2, mesothelin, TnMuc1, Nectin4, NKG2D-Ligands |
| cervical | mesothelin, GD2, PSMA, MUC1, mesothelin | |
| fallopian tube | mesothelin, TnMuc1 | |
| breast | breast | CD44v6, CD70, CD133, CEA, c-MET, EpCAM, HER2, mesothelin, Muc1 (cleaved from), Nectin4, GD2 |
| TNBC | c-MET, mesothelin, MUC1, TnMuc1, NKG2D-Ligands, ROR1 | |
| colon | colorectal | CD133, CEA, EGFR, HER2, MUC1, NKG2D-Ligands |
| colon | EpCAM, HER2, NKG2D-Ligands | |
| kidney | renal | CD70, EGFR, VEGFR2, ROR2, AXL |
| neuroblastoma | B7-H3, CD171, EGFR, GD2, PSMA | |
| wilms tumor | B7-H3, EGFR, GPC3 | |
| stomach | gastric | CD44v6, CEA, claudin 18.2, EGFR, EpCAM, HER2, MUC1, NKG2D-Ligands, PSCA, ROR2 |
| prostate | prostate | CD44v6, EpCAM, NKG2D-Ligands, PSCA, PSMA |
| head/neck | esophageal | EpCAM, HER2, MUC1 |
| nasopharyngeal | EpCAM, LMP1, NKG2D-Ligands | |
| SCCHN | ErbB dimers, HER2 | |
| salivary gland | HER2 | |
| thyroid cancer | ICAM1 | |
| skin | melanoma | B7-H3, CD20, CD70, c-MET, GD2, gp100/HLA-A2, IL13Rα2, VEGFR2 |
| bladder | bladder | HER2, Nectin4, NKG2D-Ligands, ROR2, PSMA, FBP |
| soft tissue | synovial sarcoma | B7-H3, EGFR |
| clear cell sarcoma | B7-H3, EGFR | |
| soft tissue sarcoma | B7-H3, EGFR, GPC3, ROR2 | |
| bone | osteosarcoma | B7-H3, EGFR, GD2 |
| ewing sarcoma | B7-H3, EGFR | |
| abdomen | peritoneal | CEA, EpCAM, mesothelin |
| eye | retinoblastoma | B7-H3, EGFR |
| uveal melanoma | GD2 | |
| ovary/testis | germ cell tumor | B7-H3, EGFR, GPC3 |
| peripheral nerves | malignant peripheral nerve sheath tumor | B7-H3, EGFR |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schaft, N. The Landscape of CAR-T Cell Clinical Trials against Solid Tumors—A Comprehensive Overview. Cancers 2020, 12, 2567. https://doi.org/10.3390/cancers12092567
Schaft N. The Landscape of CAR-T Cell Clinical Trials against Solid Tumors—A Comprehensive Overview. Cancers. 2020; 12(9):2567. https://doi.org/10.3390/cancers12092567
Chicago/Turabian StyleSchaft, Niels. 2020. "The Landscape of CAR-T Cell Clinical Trials against Solid Tumors—A Comprehensive Overview" Cancers 12, no. 9: 2567. https://doi.org/10.3390/cancers12092567
APA StyleSchaft, N. (2020). The Landscape of CAR-T Cell Clinical Trials against Solid Tumors—A Comprehensive Overview. Cancers, 12(9), 2567. https://doi.org/10.3390/cancers12092567