Allogenic Faecal Microbiota Transfer Induces Immune-Related Gene Sets in the Colon Mucosa of Patients with Irritable Bowel Syndrome

 , , , ,
, , , ,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Design
2.2. RNA Isolation and Microarray Processing
2.3. Biological Interpretation of Array Data
2.4. Correlations between Pathway Activity and Mucosal Microbiome Data
2.5. Protein Isolation
2.6. Cytokine Analysis
2.7. Data Availability
3. Results
3.1. Subjects
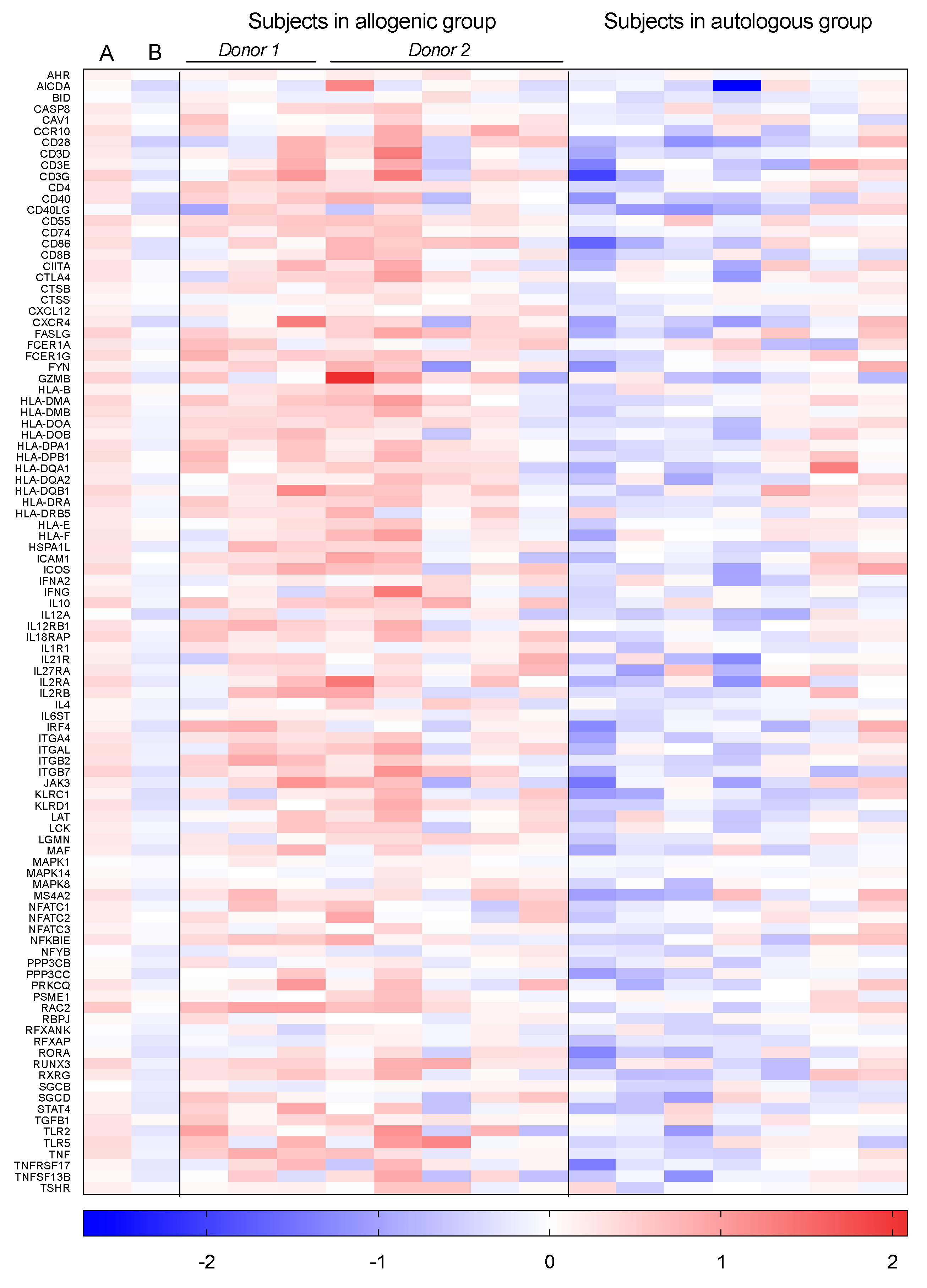
3.2. Individually Differentially Expressed Genes
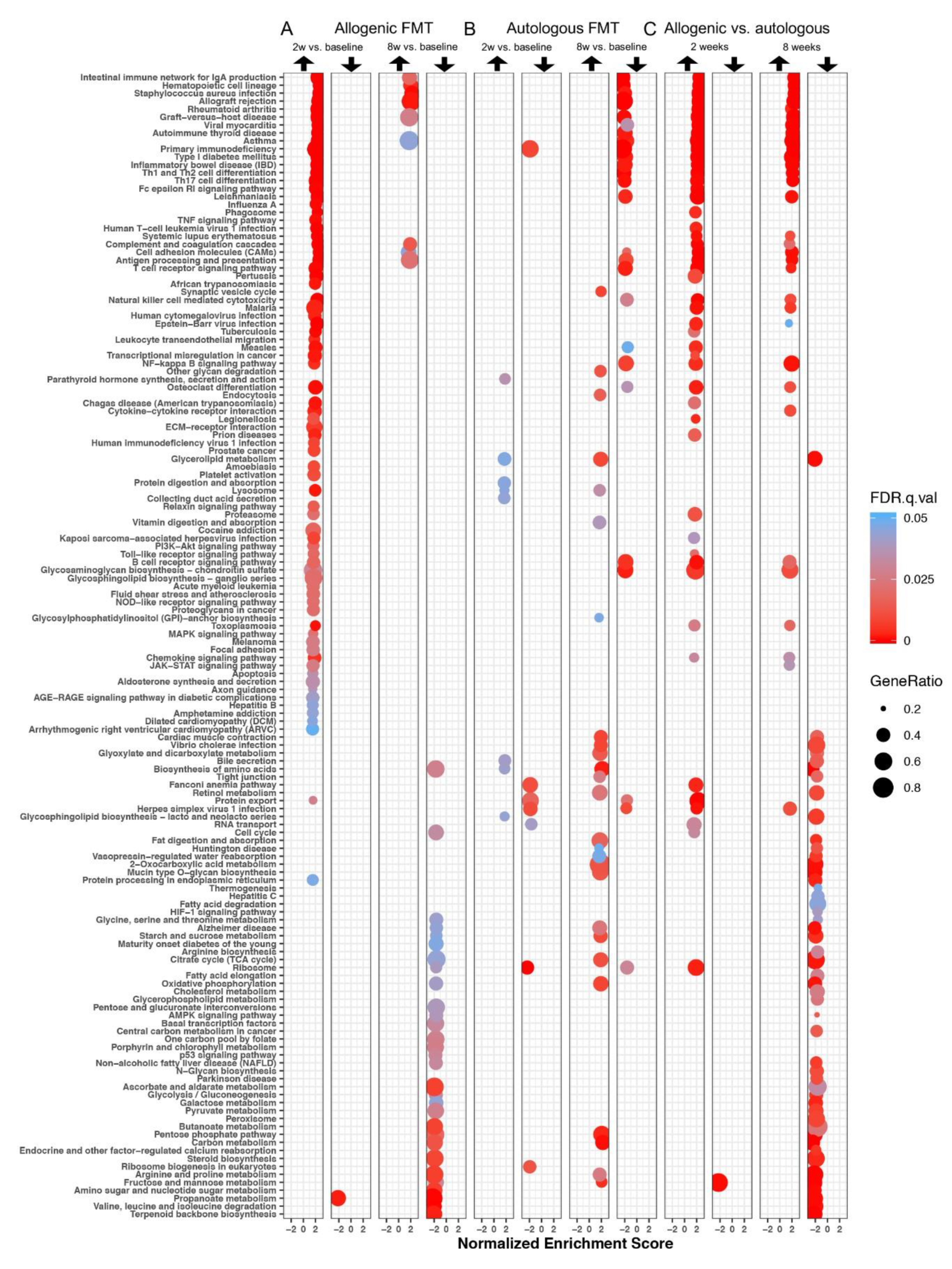
3.3. Gene Set Enrichment Analysis
3.3.1. Differentially Expressed Gene Sets after Faecal Microbiota Transfer
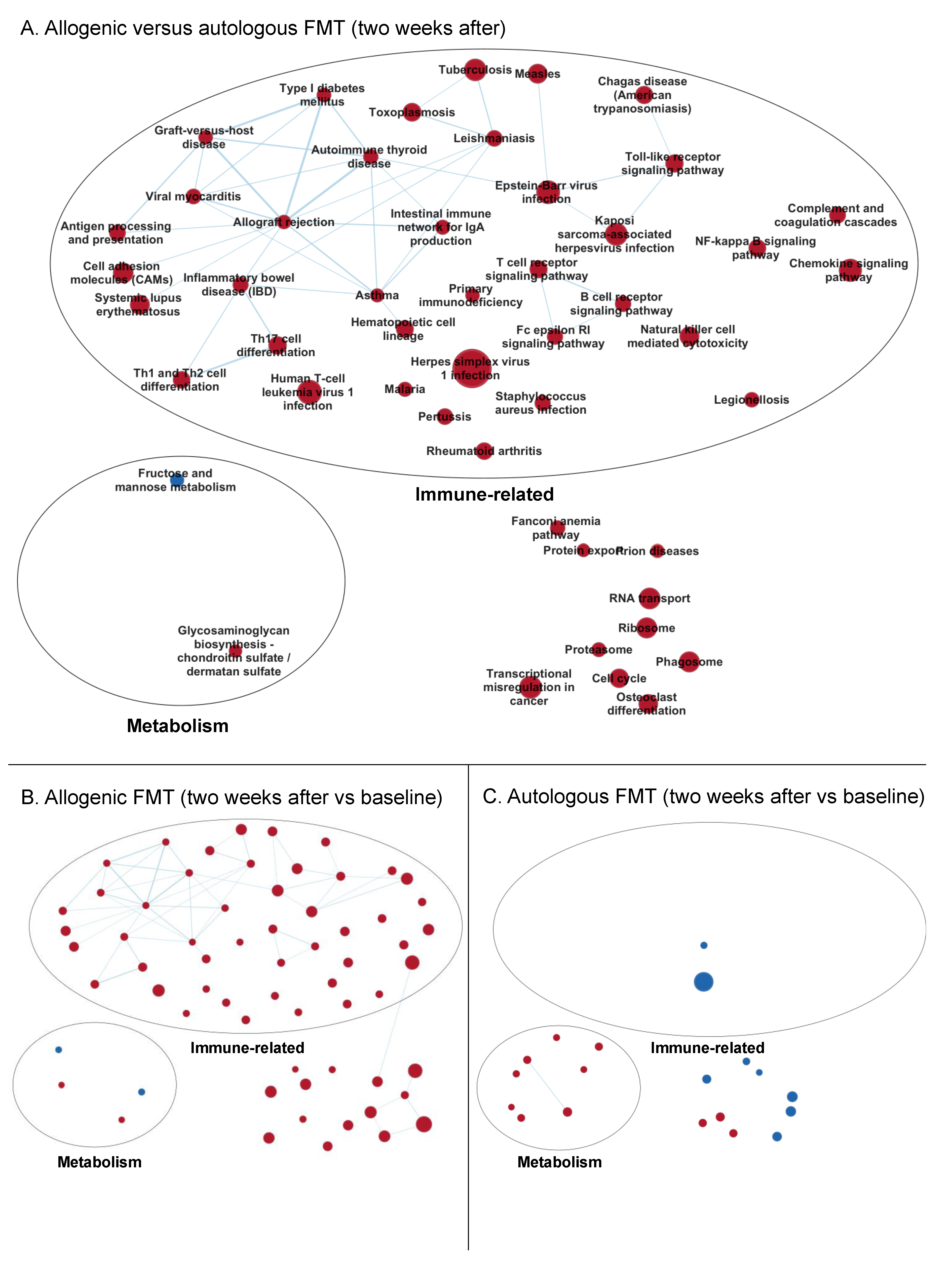
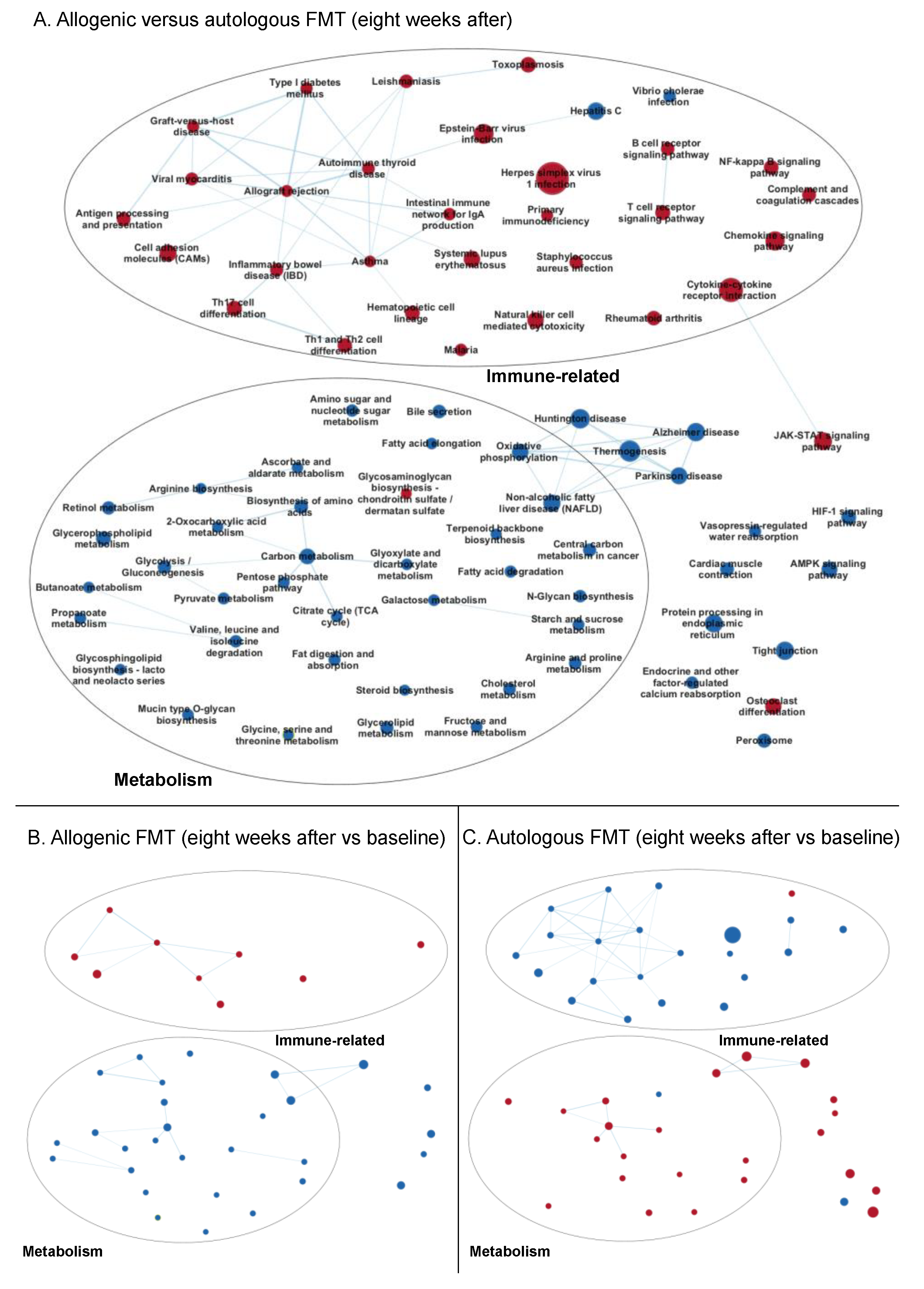
3.3.2. Enrichment Maps
Two Weeks after Faecal Microbiota Transfer
Eight Weeks after Faecal Microbiota Transfer
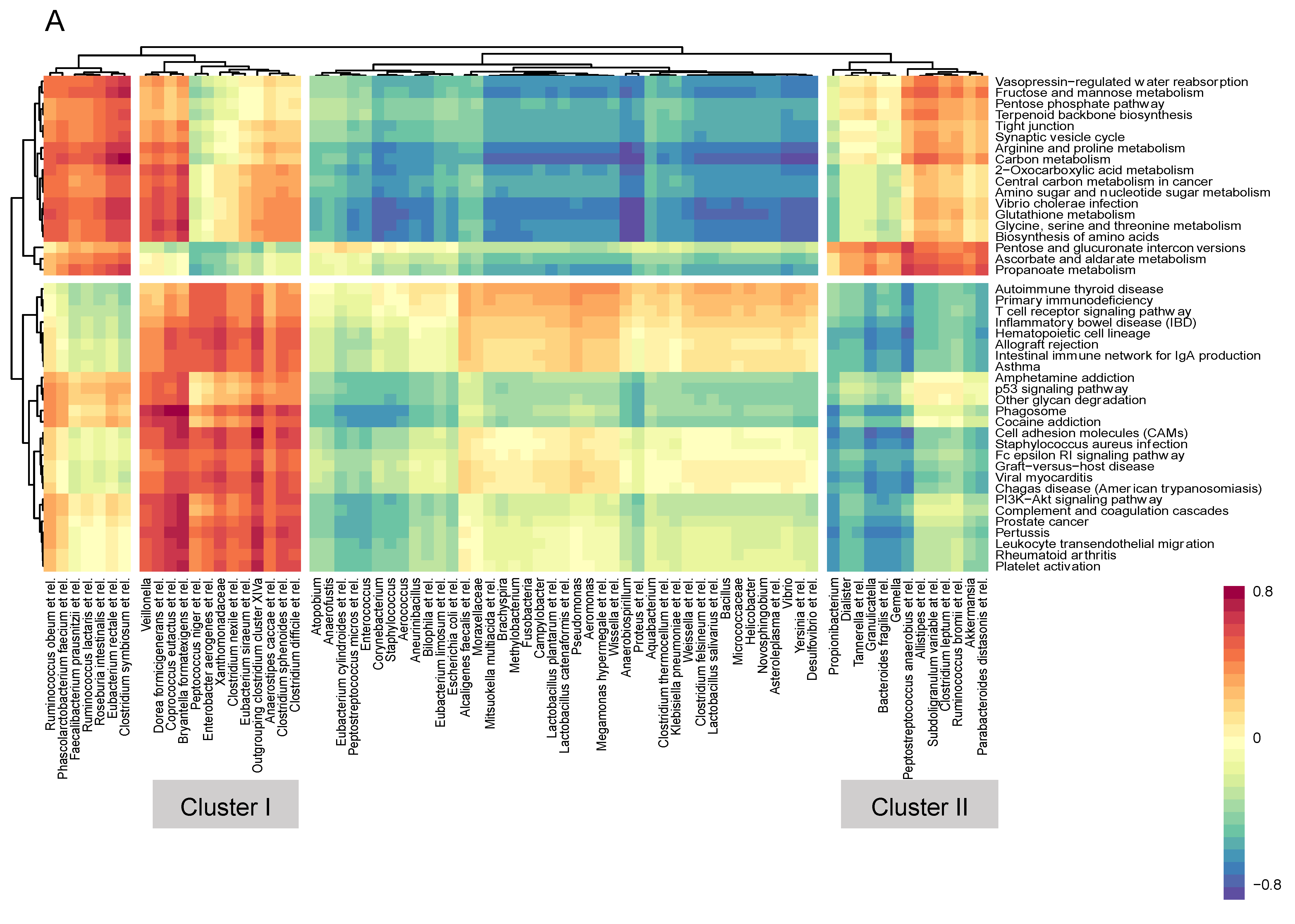
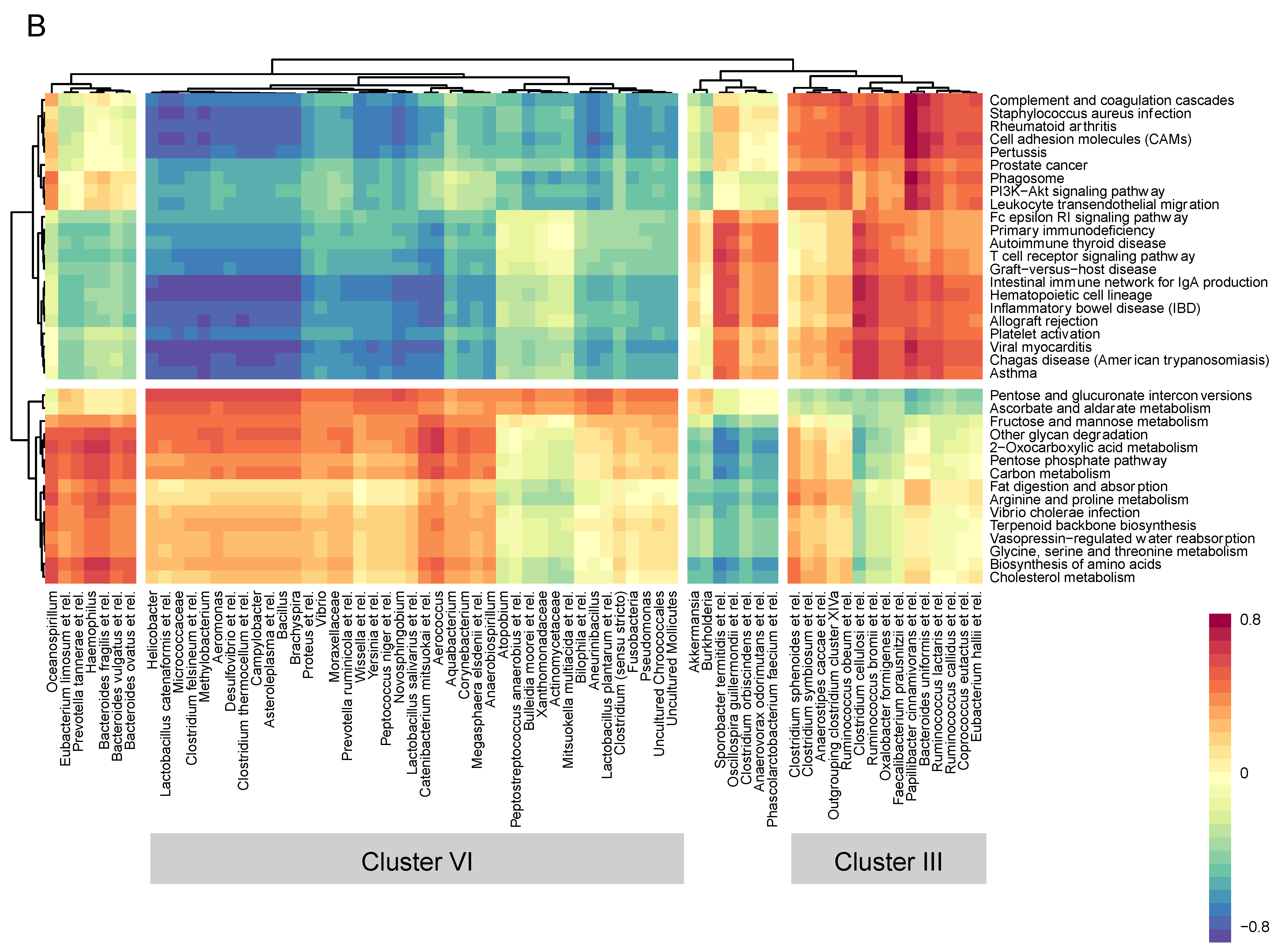
3.4. Microbiome–Gene Set Correlations
3.5. Cytokines
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Quraishi, M.N.; Widlak, M.; Bhala, N.; Moore, D.; Price, M.; Sharma, N.; Iqbal, T.H. Systematic review with meta-analysis: The efficacy of faecal microbiota transplantation for the treatment of recurrent and refractory Clostridium difficile infection. Aliment. Pharmacol. Ther. 2017, 46, 479–493. [Google Scholar] [CrossRef] [PubMed]
- Kootte, R.S.; Levin, E.; Salojarvi, J.; Smits, L.P.; Hartstra, A.V.; Udayappan, S.D.; Hermes, G.; Bouter, K.E.; Koopen, A.M.; Holst, J.J.; et al. Improvement of Insulin Sensitivity after Lean Donor Feces in Metabolic Syndrome Is Driven by Baseline Intestinal Microbiota Composition. Cell Metab. 2017, 26, 611–619. [Google Scholar] [CrossRef] [PubMed]
- Moayyedi, P.; Surette, M.G.; Kim, P.T.; Libertucci, J.; Wolfe, M.; Onischi, C.; Armstrong, D.; Marshall, J.K.; Kassam, Z.; Reinisch, W.; et al. Fecal Microbiota Transplantation Induces Remission in Patients With Active Ulcerative Colitis in a Randomized Controlled Trial. Gastroenterology 2015, 149, 102–109. [Google Scholar] [CrossRef] [PubMed]
- Paramsothy, S.; Kamm, M.A.; Kaakoush, N.O.; Walsh, A.J.; van den Bogaerde, J.; Samuel, D.; Leong, R.W.L.; Connor, S.; Ng, W.; Paramsothy, R.; et al. Multidonor intensive faecal microbiota transplantation for active ulcerative colitis: a randomised placebo-controlled trial. Lancet 2017, 389, 1218–1228. [Google Scholar] [CrossRef]
- Rossen, N.G.; Fuentes, S.; van der Spek, M.J.; Tijssen, J.G.; Hartman, J.H.; Duflou, A.; Lowenberg, M.; van den Brink, G.R.; Mathus-Vliegen, E.M.; de Vos, W.M.; et al. Findings From a Randomized Controlled Trial of Fecal Transplantation for Patients With Ulcerative Colitis. Gastroenterology 2015, 149, 110–118. [Google Scholar] [CrossRef]
- Vrieze, A.; Van Nood, E.; Holleman, F.; Salojarvi, J.; Kootte, R.S.; Bartelsman, J.F.; Dallinga-Thie, G.M.; Ackermans, M.T.; Serlie, M.J.; Oozeer, R.; et al. Transfer of intestinal microbiota from lean donors increases insulin sensitivity in individuals with metabolic syndrome. Gastroenterology 2012, 143, 913–916. [Google Scholar] [CrossRef]
- Lovell, R.M.; Ford, A.C. Global prevalence of and risk factors for irritable bowel syndrome: A meta-analysis. Clin. Gastroenterol. Hepatol. 2012, 10, 712–721. [Google Scholar] [CrossRef]
- Sperber, A.D.; Dumitrascu, D.; Fukudo, S.; Gerson, C.; Ghoshal, U.C.; Gwee, K.A.; Hungin, A.P.S.; Kang, J.Y.; Minhu, C.; Schmulson, M.; et al. The global prevalence of IBS in adults remains elusive due to the heterogeneity of studies: A Rome Foundation working team literature review. Gut 2017, 66, 1075–1082. [Google Scholar] [CrossRef]
- Johnsen, P.H.; Hilpusch, F.; Cavanagh, J.P.; Leikanger, I.S.; Kolstad, C.; Valle, P.C.; Goll, R. Faecal microbiota transplantation versus placebo for moderate-to-severe irritable bowel syndrome: a double-blind, randomised, placebo-controlled, parallel-group, single-centre trial. Lancet Gastroenterol. Hepatol. 2018, 3, 17–24. [Google Scholar] [CrossRef]
- Halkjaer, S.I.; Christensen, A.H.; Lo, B.Z.S.; Browne, P.D.; Gunther, S.; Hansen, L.H.; Petersen, A.M. Faecal microbiota transplantation alters gut microbiota in patients with irritable bowel syndrome: Results from a randomised, double-blind placebo-controlled study. Gut 2018. [Google Scholar] [CrossRef]
- Holster, S.; Lindqvist, C.M.; Repsilber, D.; Salonen, A.; de Vos, W.M.; König, J.; Brummer, R.J. The Effect of Allogenic Versus Autologous Fecal Microbiota Transfer on Symptoms, Visceral Perception and Fecal and Mucosal Microbiota in Irritable Bowel Syndrome: A Randomized Controlled Study. Clin. Transl. Gastroenterol. 2019, 10, e00034. [Google Scholar] [CrossRef] [PubMed]
- Suez, J.; Zmora, N.; Zilberman-Schapira, G.; Mor, U.; Dori-Bachash, M.; Bashiardes, S.; Zur, M.; Regev-Lehavi, D.; Ben-Zeev Brik, R.; Federici, S.; et al. Post-Antibiotic Gut Mucosal Microbiome Reconstitution Is Impaired by Probiotics and Improved by Autologous FMT. Cell 2018, 174, 1406–1423. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.; Kools, H.; de Groot, P.J.; Gavai, A.K.; Basnet, R.K.; Cheng, F.; Wu, J.; Wang, X.; Lommen, A.; Hooiveld, G.J.; et al. MADMAX-Management and analysis database for multiple ~omics experiments. J. Integr. Bioinform. 2011, 8, 160. [Google Scholar] [CrossRef] [PubMed]
- Irizarry, R.A.; Hobbs, B.; Collin, F.; Beazer-Barclay, Y.D.; Antonellis, K.J.; Scherf, U.; Speed, T.P. Exploration, normalization, and summaries of high-density oligonucleotide array probe level data. Biostatistics 2003, 4, 249–264. [Google Scholar] [CrossRef]
- Dai, M.; Wang, P.; Boyd, A.D.; Kostov, G.; Athey, B.; Jones, E.G.; Bunney, W.E.; Myers, R.M.; Speed, T.P.; Akil, H.; et al. Evolving gene/transcript definitions significantly alter the interpretation of GeneChip data. Nucleic Acids Res. 2005, 33, e175. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Sartor, M.A.; Tomlinson, C.R.; Wesselkamper, S.C.; Sivaganesan, S.; Leikauf, G.D.; Medvedovic, M. Intensity-based hierarchical Bayes method improves testing for differentially expressed genes in microarray experiments. BMC Bioinform. 2006, 7, 538. [Google Scholar] [CrossRef]
- Liu, R.; Holik, A.Z.; Su, S.; Jansz, N.; Chen, K.; Leong, H.S.; Blewitt, M.E.; Asselin-Labat, M.L.; Smyth, G.K.; Ritchie, M.E. Why weight? Modelling sample and observational level variability improves power in RNA-seq analyses. Nucleic Acids Res. 2015, 43, e97. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Diyagama, D.; Neilson, J.; van Laar, R.; Dobrovic, A.; Holloway, A.; Smyth, G.K. Empirical array quality weights in the analysis of microarray data. BMC Bioinform. 2006, 7, 261. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate-a Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Storey, J.D.; Tibshirani, R. Statistical significance for genomewide studies. Proc. Natl. Acad. Sci. USA 2003, 100, 9440–9445. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Abatangelo, L.; Maglietta, R.; Distaso, A.; D’Addabbo, A.; Creanza, T.M.; Mukherjee, S.; Ancona, N. Comparative study of gene set enrichment methods. BMC Bioinform. 2009, 10, 275. [Google Scholar] [CrossRef] [PubMed]
- Allison, D.B.; Cui, X.; Page, G.P.; Sabripour, M. Microarray data analysis: From disarray to consolidation and consensus. Nat. Rev. Genet. 2006, 7, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Merico, D.; Isserlin, R.; Stueker, O.; Emili, A.; Bader, G.D. Enrichment map: A network-based method for gene-set enrichment visualization and interpretation. PLoS ONE 2010, 5, e13984. [Google Scholar] [CrossRef]
- Rohart, F.; Gautier, B.; Singh, A.; Le Cao, K.A. mixOmics: An R package for ‘omics feature selection and multiple data integration. PLoS Comput. Biol. 2017, 13, e1005752. [Google Scholar] [CrossRef]
- Hanzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef]
- Salonen, A.; Nikkila, J.; Jalanka-Tuovinen, J.; Immonen, O.; Rajilic-Stojanovic, M.; Kekkonen, R.A.; Palva, A.; de Vos, W.M. Comparative analysis of fecal DNA extraction methods with phylogenetic microarray: effective recovery of bacterial and archaeal DNA using mechanical cell lysis. J. Microbiol. Methods 2010, 81, 127–134. [Google Scholar] [CrossRef]
- Rajilic-Stojanovic, M.; Heilig, H.G.; Molenaar, D.; Kajander, K.; Surakka, A.; Smidt, H.; de Vos, W.M. Development and application of the human intestinal tract chip, a phylogenetic microarray: Analysis of universally conserved phylotypes in the abundant microbiota of young and elderly adults. Environ. Microbiol. 2009, 11, 1736–1751. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, I.; Cao, K.A.; Davis, M.J.; Dejean, S. Visualising associations between paired ‘omics’ data sets. BioData Min. 2012, 5, 19. [Google Scholar] [CrossRef] [PubMed]
- Kwak, J.Y.; Lamouse-Smith, E.S.N. Can probiotics enhance vaccine-specific immunity in children and adults? Benef. Microbes 2017, 8, 657–669. [Google Scholar] [CrossRef] [PubMed]
- Richards, A.L.; Burns, M.B.; Alazizi, A.; Barreiro, L.B.; Pique-Regi, R.; Blekhman, R.; Luca, F. Genetic and transcriptional analysis of human host response to healthy gut microbiota. mSystems 2016, 1. [Google Scholar] [CrossRef]
- Kellermayer, R.; Nagy-Szakal, D.; Harris, R.A.; Luna, R.A.; Pitashny, M.; Schady, D.; Mir, S.A.; Lopez, M.E.; Gilger, M.A.; Belmont, J.; et al. Serial fecal microbiota transplantation alters mucosal gene expression in pediatric ulcerative colitis. Am. J. Gastroenterol. 2015, 110, 604–606. [Google Scholar] [CrossRef]
- Van Baarlen, P.; Troost, F.J.; van Hemert, S.; van der Meer, C.; de Vos, W.M.; de Groot, P.J.; Hooiveld, G.J.; Brummer, R.J.; Kleerebezem, M. Differential NF-kappaB pathways induction by Lactobacillus plantarum in the duodenum of healthy humans correlating with immune tolerance. Proc. Natl. Acad. Sci. USA 2009, 106, 2371–2376. [Google Scholar] [CrossRef]
- Van Baarlen, P.; Troost, F.; van der Meer, C.; Hooiveld, G.; Boekschoten, M.; Brummer, R.J.; Kleerebezem, M. Human mucosal in vivo transcriptome responses to three lactobacilli indicate how probiotics may modulate human cellular pathways. Proc. Natl. Acad. Sci. USA 2011, 108 (Suppl. 1), 4562–4569. [Google Scholar] [CrossRef]
- De Vos, P.; Mujagic, Z.; de Haan, B.J.; Siezen, R.J.; Bron, P.A.; Meijerink, M.; Wells, J.M.; Masclee, A.A.M.; Boekschoten, M.V.; Faas, M.M.; et al. Lactobacillus plantarum Strains Can Enhance Human Mucosal and Systemic Immunity and Prevent Non-steroidal Anti-inflammatory Drug Induced Reduction in T Regulatory Cells. Front. Immunol. 2017, 8, 1000. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Allogenic (n = 8) | Autologous (n = 7) | p-Value | |
|---|---|---|---|
| Age, median (IQR) | 34 (27–42) | 38 (32–45) | 0.42 |
| Sex, m/f | 5/3 | 3/4 | 0.62 |
| BMI (kg/m2), median (IQR) | 20.9 (20.2–25.1) | 23.8 (20.5–24.7) | 0.94 |
| Classification, IBS-D/IBS-C/IBS-M | 5/1/2 | 4/2/1 | 1.0/0.57/1.0 |
| Post-infectious IBS | 4 | 3 | 1.0 |
| Disease duration: unknown/1–5 y/5 y | 0/4/4 | 1/3/3 | 0.47/1.0/1.0 |
| Concomitant medication | 7 | 5 | 0.57 |
| Gut-related medication | 3 | 3 | 1.0 |
| Laxatives | 1 | 2 * | 1.0 |
| Anti-diarrhoeal | 1 | 1 | 1.0 |
| Anti-spasmodic | 1 | 1 | 1.0 |
| Antidepressants | 5 | 2 | 0.31 |
| SSRIs | 5 | 0 | 0.03 |
| NaSSAs | 0 | 1 | 0.47 |
| SSNRIs | 0 | 1# | 0.47 |
| TCAs | 0 | 1# | 0.47 |
| Allogenic FMT | Autologous FMT | ||
|---|---|---|---|
| Cluster I (+) | Cluster II (−) | Cluster III (+) | Cluster IV (−) |
| Peptococcus niger et rel. | Propionibacterium | Anaerovorax odorimutans et rel. | Aneurinibacillus |
| Enterobacter aerogenes et rel. | Bacteroides fragilis et rel. | Phascolarctobacterium faecium et rel. | Bilophila et rel. |
| Xanthomonadaceae | Gemella | Clostridium orbiscindens et rel. | Lactobacillus plantarum et rel. |
| Clostridium nexile et rel. | Granulicatella | Sporobacter termitidis et rel. | Clostridium (sensu stricto) |
| Outgrouping clostridium cluster XIVa | Dialister | Oscillospira guillermondii et rel. | Uncultured Chroococcales |
| Eubacterium siraeum et rel. | Tannerella et rel. | Oxalobacter formigenes et rel. | Fusobacteria |
| Clostridium sphenoides et rel. | Peptostreptococcus anaerobius et rel. | Faecalibacterium prausnitzii et rel. | Pseudomonas |
| Clostridium difficile et rel. | Akkermansia | Ruminococcus bromii et rel. | Uncultured Mollicutes |
| Anaerostipes caccae et rel. | Parabacteroides distasonis et rel. | Clostridium cellulosi et rel. | Novosphingobium |
| Coprococcus eutactus et rel. | Clostridium leptum et rel. | Ruminococcus lactaris et rel. | Wissella et rel. |
| Bryantella formatexigens et rel. | Ruminococcus bromii et rel. | Ruminococcus callidus et rel. | Yersinia et rel. |
| Veillonella | Allistipes et rel. | Coprococcus eutactus et rel. | Peptococcus niger et rel. |
| Dorea formicigenerans et rel. | Subdoligranulum variable at rel. | Eubacterium hallii et rel. | Proteus et rel. |
| Papillibacter cinnamivorans et rel. | Vibrio | ||
| Bacteroides uniformis et rel. | Moraxellaceae | ||
| Clostridium sphenoides et rel. | Prevotella ruminicola et rel. | ||
| Outgrouping clostridium cluster XIVa | Lactobacillus catenaformis et rel. | ||
| Ruminococcus obeum et rel. | Micrococcaceae | ||
| Clostridium symbiosum et rel. | Helicobacter | ||
| Anaerostipes caccae et rel. | Clostridium felsineum et rel. | ||
| Methylobacterium | |||
| Lactobacillus salivarius et rel. | |||
| Aeromonas | |||
| Desulfovibrio et rel. | |||
| Clostridium thermocellum et rel. | |||
| Campylobacter | |||
| Asteroleplasma et rel. | |||
| Bacillus | |||
| Brachyspira | |||
| Actinomycetaceae | |||
| Aerococcus | |||
| Anaerobiospirillum | |||
| Aquabacterium | |||
| Atopobium | |||
| Bulleidia moorei et rel. | |||
| Catenibacterium mitsuokai et rel. | |||
| Corynebacterium | |||
| Megasphaera elsdenii et rel. | |||
| Mitsuokella multiacida et rel. | |||
| Peptostreptococcus anaerobius et rel. | |||
| Xanthomonadaceae | |||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Holster, S.; Hooiveld, G.J.; Repsilber, D.; de Vos, W.M.; Brummer, R.J.; König, J. Allogenic Faecal Microbiota Transfer Induces Immune-Related Gene Sets in the Colon Mucosa of Patients with Irritable Bowel Syndrome. Biomolecules 2019, 9, 586. https://doi.org/10.3390/biom9100586
Holster S, Hooiveld GJ, Repsilber D, de Vos WM, Brummer RJ, König J. Allogenic Faecal Microbiota Transfer Induces Immune-Related Gene Sets in the Colon Mucosa of Patients with Irritable Bowel Syndrome. Biomolecules. 2019; 9(10):586. https://doi.org/10.3390/biom9100586
Chicago/Turabian StyleHolster, Savanne, Guido J. Hooiveld, Dirk Repsilber, Willem M. de Vos, Robert J. Brummer, and Julia König. 2019. "Allogenic Faecal Microbiota Transfer Induces Immune-Related Gene Sets in the Colon Mucosa of Patients with Irritable Bowel Syndrome" Biomolecules 9, no. 10: 586. https://doi.org/10.3390/biom9100586
APA StyleHolster, S., Hooiveld, G. J., Repsilber, D., de Vos, W. M., Brummer, R. J., & König, J. (2019). Allogenic Faecal Microbiota Transfer Induces Immune-Related Gene Sets in the Colon Mucosa of Patients with Irritable Bowel Syndrome. Biomolecules, 9(10), 586. https://doi.org/10.3390/biom9100586