
EmbedDTI: Enhancing the Molecular Representations via Sequence Embedding and Graph Convolutional Network for the Prediction of Drug-Target Interaction


Abstract
:1. Introduction
2. Materials and Methods
2.1. Metrics of Binding Affinity
2.2. Datasets
2.3. Corpus for Pretraining Protein Embeddings
3. Methods
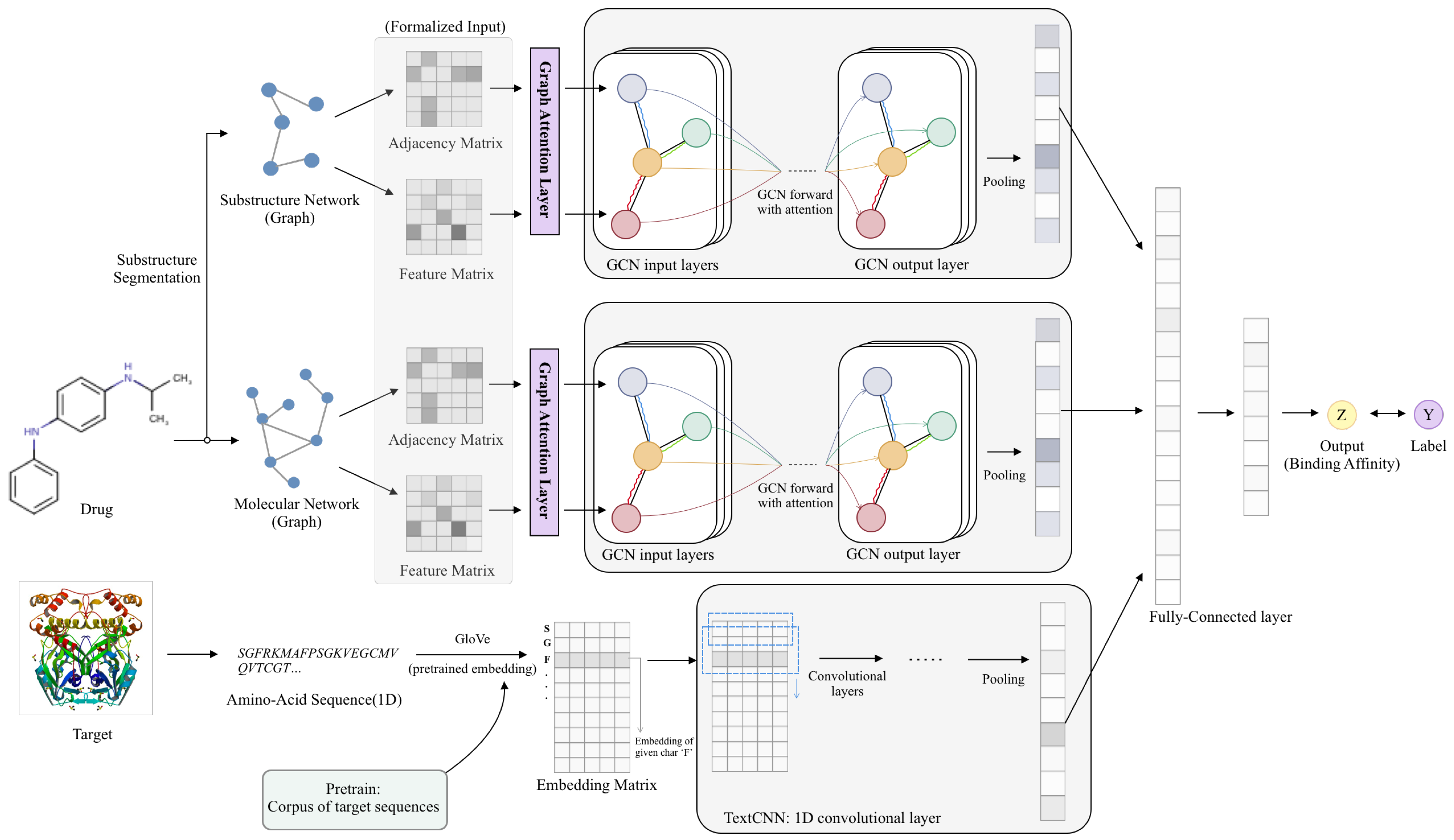
3.1. Model Overview
3.2. Initial Feature Extraction
3.2.1. Input Features of Target Proteins
3.2.2. Input Features of Drugs
Atom-Level Representation
Substructure-Level Representation
| Algorithm 1 Segmentation of substructures for molecule |
Input: SMILES strings of compounds Output: Vocabulary of substructures C
|
3.3. Feature Learning Using Deep Neural Networks
3.3.1. Target Feature Learning via CNN
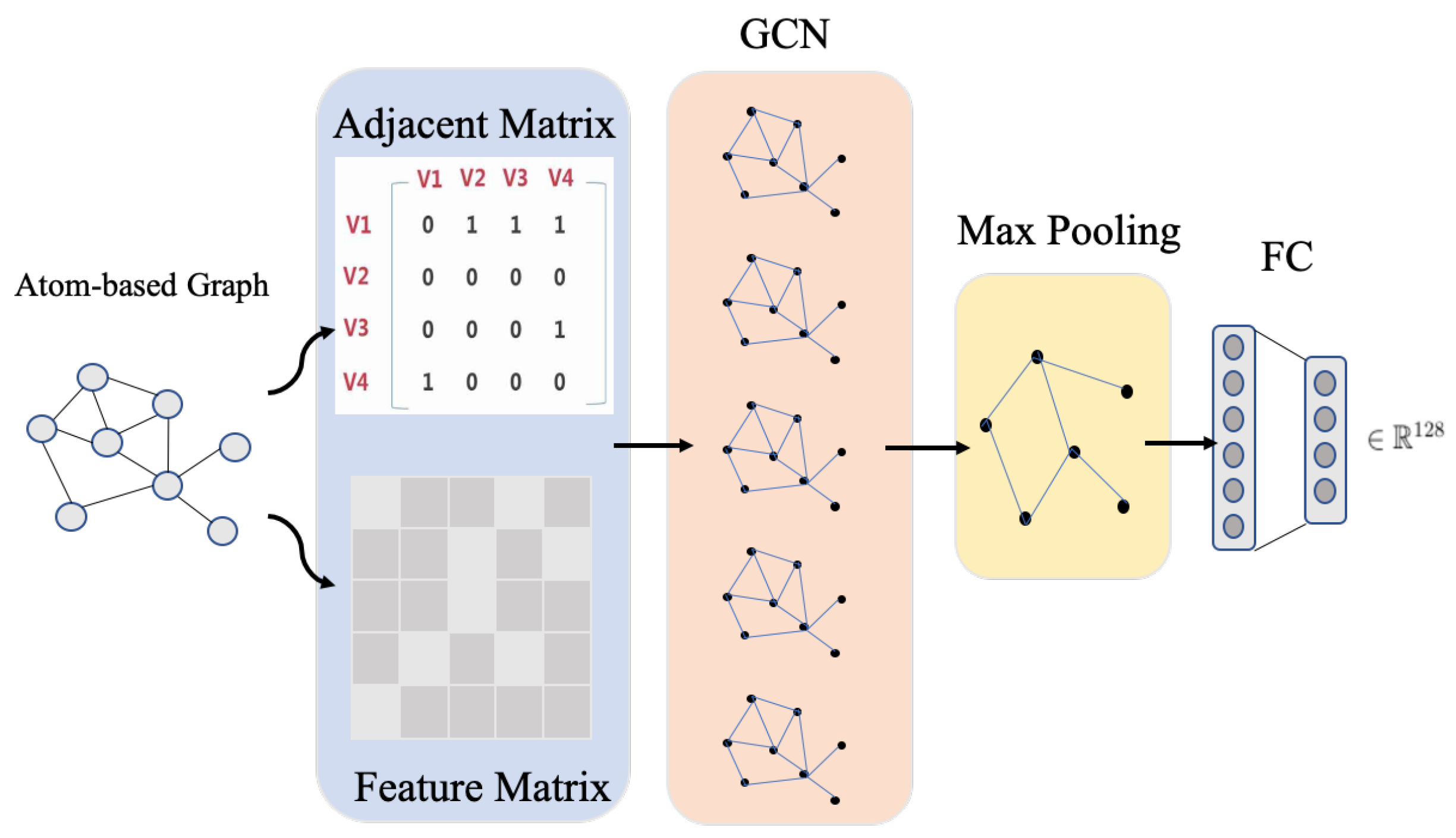
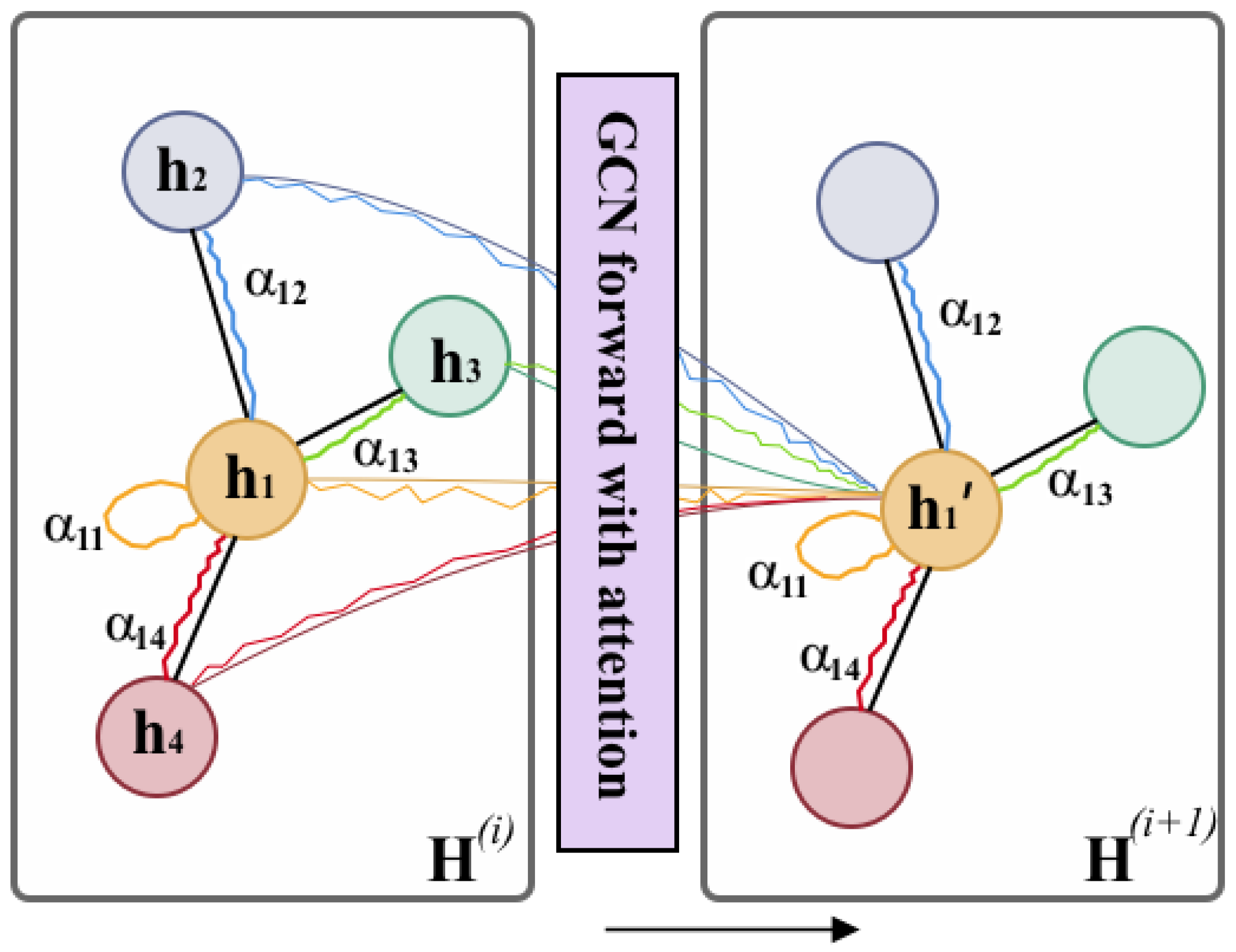
3.3.2. Drug Feature Learning via GCN
3.4. Prediction Model
4. Results
4.1. Experimental Settings
4.2. Evaluation Metrics
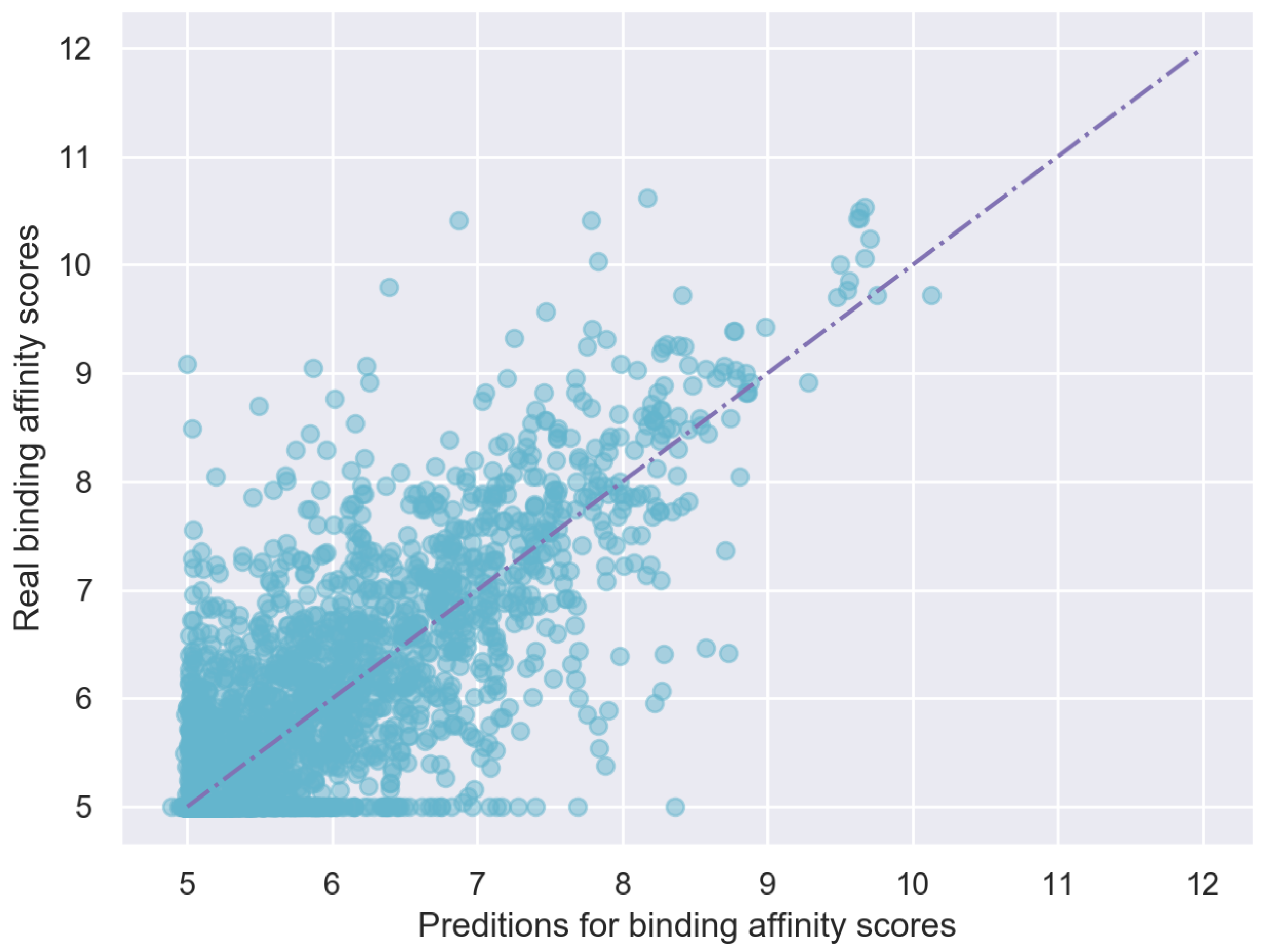
4.3. Results on Davis Dataset
- KronRLS [14]. It adopts Smith-Waterman algorithm to compute similarity between proteins and the PubChem structure clustering server to compute similarity between drug compounds. Then it uses a kernel-based method to calculate Kronecker products and integrates multiple heterogeneous information sources within a least squares regression (RLS) framework.
- SimBoost algorithms [15]. Its representation of proteins and drug compounds is the same as that of KronRLS. It constructs features for drugs, targets, and drug-target pairs, and extracts the feature vectors of drug-target pairs through feature engineering to train a gradient boosting machine to predict binding affinity.
- DeepDTA [23]. It encodes the original one-dimensional protein sequences and SMILES sequences. The encoded vector is passed through two independent CNN blocks to obtain the corresponding representation vector, and after concatenating, the predicted binding affinity is output through the fully connected layer.
- WideDTA [26]. It adds protein domains and motifs, and maximum common substructure words based on DeepDTA, a total of four parts of the original information training model.
- GraphDTA [25]. It uses TextCNN to perform feature learning on one-dimensional protein sequences. For the SMILES sequence, it uses four models of GCN, GAT, GIN, and GAT_GCN to obtain the representation vector of SMILES sequence.
- EmbedDTI_noPre: no pretraining for protein sequences.
- EmbedDTI_noSub: no substructure graph representation for drug compounds.
- EmbedDTI_noAttn: no attention module in the GCN.
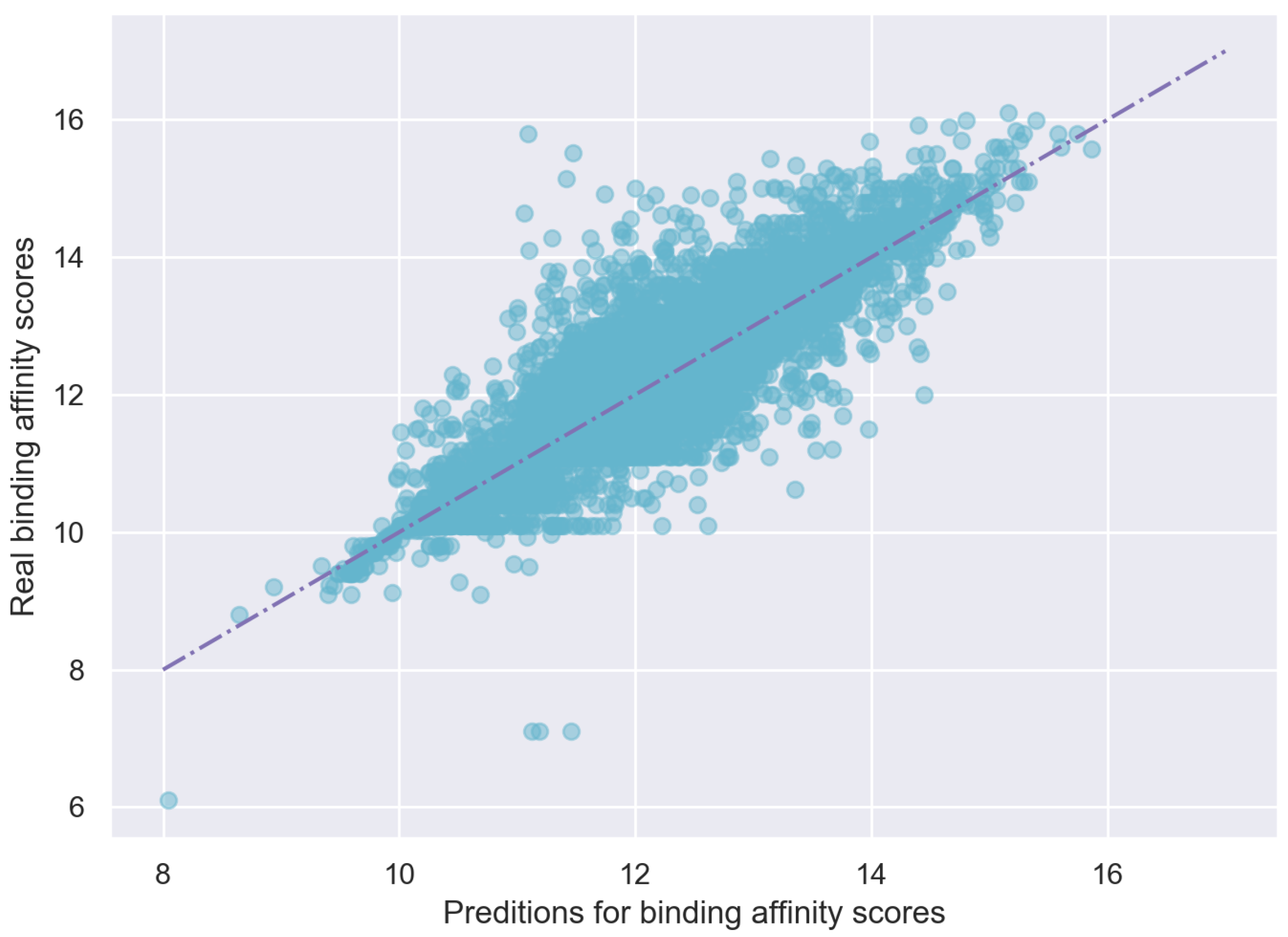
4.4. Results on KIBA Dataset
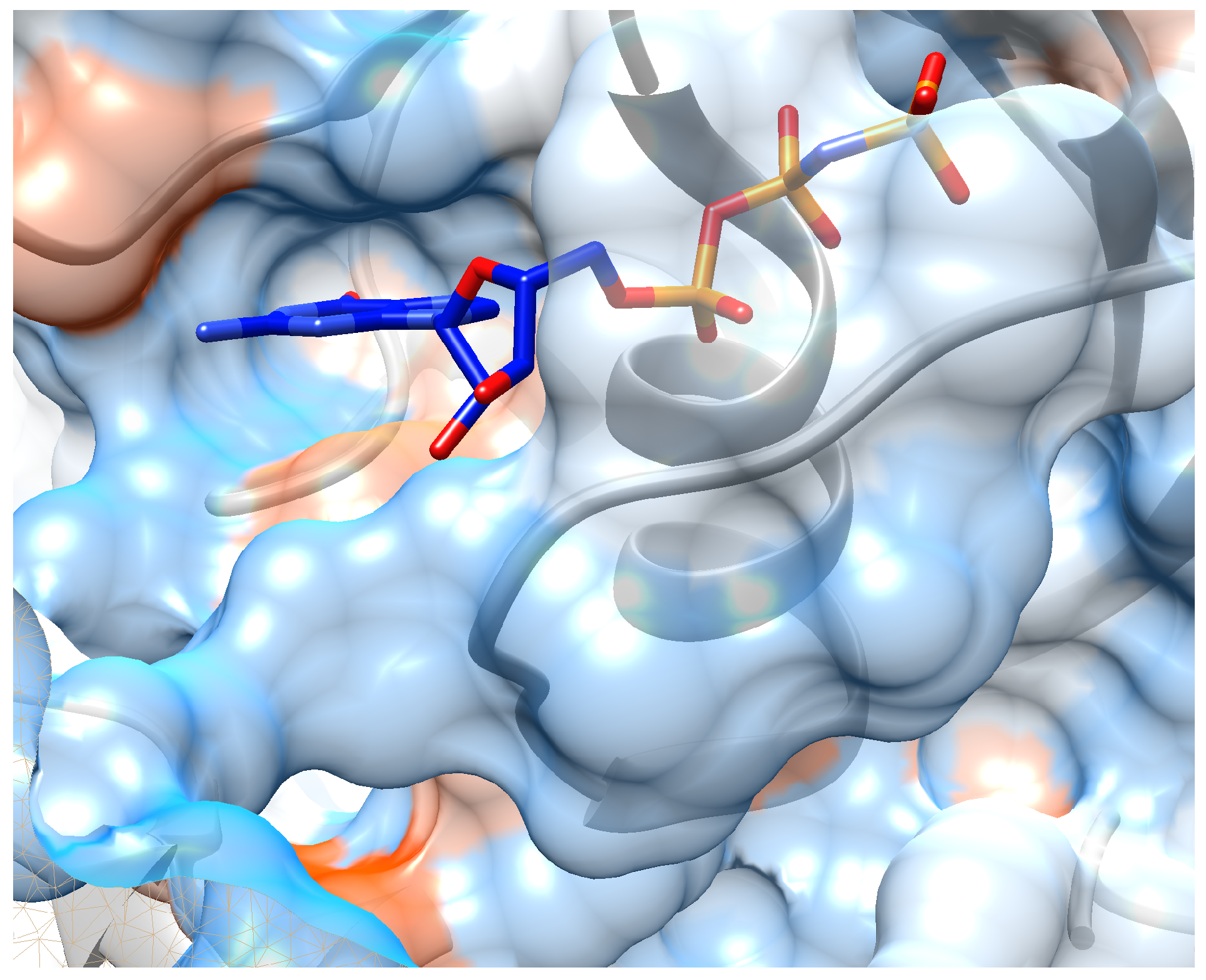
5. Case Study: Inhibitor Design for K-Ras Target
5.1. Molecular Evaluation Metrics
5.2. Implementation Details and Results
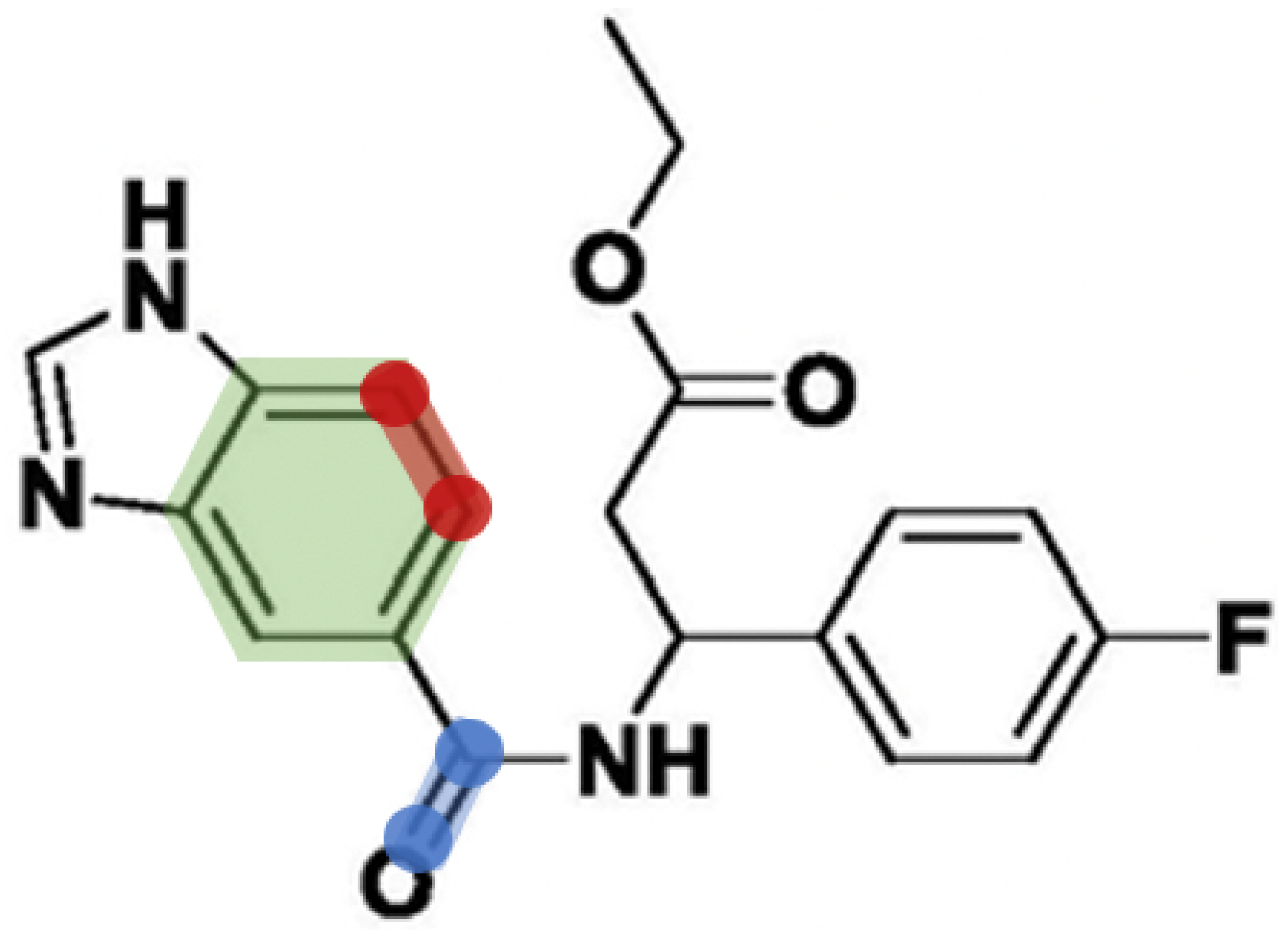
6. Investigation on the Model Attention
7. Discussion
- To exploit abundant structural information from drugs, we model each drug molecule as both a graph of atoms and a graph of substructures (groups of nodes). And we propose algorithms for segmenting out the substructures and extracting their features. The experimental results show that the two-level graph representation contributes to the performance improved significantly.
- To fully use protein sequence information, we pre-train amino acid sequences via a large database using word embedding methods from natural language processing. The pre-trained embeddings are dense continuous vectors, which can represent the latent semantic correlation between amino acids. Moreover, a deep CNN is further employed to learn high-level abstract features of proteins. The enhanced protein representation also improves model performance.
- To interpret the learning ability of EmbedDTI, we add an attention mechanism to the GCN for learning atom-based graphs and substructure-based graphs. Different attention weights are assigned to the nodes in the molecule graph to evaluate their contributions. It can recognize important nodes as well as their interactions in the graphs, which provide useful hints in drug discovery.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Politis, S.N.; Colombo, P.; Colombo, G.; Rekkas, D.M. Design of experiments (DoE) in pharmaceutical development. Drug Dev. Ind. Pharm. 2017, 43, 889–901. [Google Scholar] [CrossRef] [PubMed]
- Kapetanovic, I. Computer-aided drug discovery and development (CADDD): In silico-chemico-biological approach. Chem.-Biol. Interact. 2008, 171, 165–176. [Google Scholar] [CrossRef] [Green Version]
- Heifetz, A.; Southey, M.; Morao, I.; Townsend-Nicholson, A. Computational Methods Used in Hit-to-Lead and Lead Optimization Stages of Structure-Based Drug Discovery. Methods Mol. Biol. 2018, 1705, 375–394. [Google Scholar] [PubMed]
- Gaulton, A.; Bellis, L.J.; Bento, A.P.; Chambers, J.; Davies, M.; Hersey, A.; Light, Y.; McGlinchey, S.; Michalovich, D.; Al-Lazikani, B.; et al. ChEMBL: A large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012, 40, D1100–D1107. [Google Scholar] [CrossRef] [Green Version]
- Wishart, D.S.; Knox, C.; Guo, A.C.; Cheng, D.; Shrivastava, S.; Tzur, D.; Gautam, B.; Hassanali, M. DrugBank: A knowledgebase for drugs, drug actions and drug targets. Nucleic Acids Res. 2008, 36, D901–D906. [Google Scholar] [CrossRef] [PubMed]
- Günther, S.; Kuhn, M.; Dunkel, M.; Campillos, M.; Senger, C.; Petsalaki, E.; Ahmed, J.; Urdiales, E.G.; Gewiess, A.; Jensen, L.J.; et al. SuperTarget and Matador: Resources for exploring drug-target relationships. Nucleic Acids Res. 2007, 36, D919–D922. [Google Scholar] [CrossRef] [PubMed]
- Ragoza, M.; Hochuli, J.; Idrobo, E.; Sunseri, J.; Koes, D.R. Protein–Ligand Scoring with Convolutional Neural Networks. J. Chem. Inf. Model 2017, 57, 942–957. [Google Scholar] [CrossRef] [Green Version]
- Gowthaman, R.; Miller, S.A.; Rogers, S.; Khowsathit, J.; Lan, L.; Bai, N.; Johnson, D.K.; Liu, C.; Xu, L.; Anbanandam, A.A. DARC: Mapping Surface Topography by Ray-Casting for Effective Virtual Screening at Protein Interaction Sites. J. Med. Chem. 2015, 59, 4152–4170. [Google Scholar] [CrossRef] [Green Version]
- Verdonk, M.L.; Cole, J.C.; Hartshorn, M.J.; Murray, C.W.; Taylor, R.D. Improved protein–ligand docking using GOLD. Proteins-Struct. Funct. Bioinform. 2010, 52, 609–623. [Google Scholar] [CrossRef]
- Paul, D.S.; Gautham, N. MOLS 2.0: Software package for peptide modeling and protein–ligand docking. J. Mol. Model. 2016, 22, 239. [Google Scholar] [CrossRef] [Green Version]
- Ballesteros, J.A.; Palczewski, K. G protein-coupled receptor drug discovery: Implications from the crystal structure of rhodopsin. Curr. Opin. Drug Discov. Dev. 2001, 4, 561–574. [Google Scholar]
- Yamanishi, Y.; Araki, M.; Gutteridge, A.; Honda, W.; Kanehisa, M. Prediction of drug–target interaction networks from the integration of chemical and genomic spaces. Bioinformatics 2008, 24, i232–i240. [Google Scholar] [CrossRef] [PubMed]
- Bleakley, K.; Yamanishi, Y. Supervised Prediction of Drug–Target Interactions Using Bipartite Local Models; Oxford University Press: Oxford, UK, 2009. [Google Scholar]
- Pahikkala, T.; Airola, A.; Pietilä, S.; Shakyawar, S.; Szwajda, A.; Tang, J.; Aittokallio, T. Toward more realistic drug–target interaction predictions. Briefings Bioinform. 2015, 16, 325–337. [Google Scholar] [CrossRef] [PubMed]
- He, T.; Heidemeyer, M.; Ban, F.; Cherkasov, A.; Ester, M. SimBoost: A read-across approach for predicting drug–target binding affinities using gradient boosting machines. J. Cheminform. 2017, 9, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Cobanoglu, M.C.; Liu, C.; Hu, F.; Oltvai, Z.N.; Bahar, I. Predicting drug-target interactions using probabilistic matrix factorization. J. Chem. Inf. Model. 2013, 53, 3399–3409. [Google Scholar] [CrossRef]
- Ezzat, A.; Zhao, P.; Wu, M.; Li, X.; Kwoh, C.K. Drug-Target Interaction Prediction with Graph Regularized Matrix Factorization. IEEE/ACM Trans. Comput. Biol. Bioinform. (TCBB) 2017, 14, 646–656. [Google Scholar] [CrossRef]
- Zheng, X.; Ding, H.; Mamitsuka, H.; Zhu, S. Collaborative matrix factorization with multiple similarities for predicting drug-target interactions. In Proceedings of the 19th ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, Chicago, IL, USA, 11–14 August 2013; pp. 1025–1033. [Google Scholar]
- Luo, Y.; Zhao, X.; Zhou, J.; Yang, J.; Zhang, Y.; Kuang, W.; Peng, J.; Chen, L.; Zeng, J. A network integration approach for drug-target interaction prediction and computational drug repositioning from heterogeneous information. Nat. Commun. 2017, 8, 573. [Google Scholar] [CrossRef] [Green Version]
- Cheng, F.; Zhou, Y.; Li, J.; Li, W.; Liu, G.; Tang, Y. Prediction of chemical–protein interactions: Multitarget-QSAR versus computational chemogenomic methods. Mol. Biosyst. 2012, 8, 2373–2384. [Google Scholar] [CrossRef]
- Wang, F.; Liu, D.; Wang, H.; Luo, C.; Zheng, M.; Liu, H.; Zhu, W.; Luo, X.; Zhang, J.; Jiang, H. Computational screening for active compounds targeting protein sequences: Methodology and experimental validation. J. Chem. Inf. Model. 2011, 51, 2821–2828. [Google Scholar] [CrossRef]
- He, Z.; Zhang, J.; Shi, X.H.; Hu, L.L.; Kong, X.; Cai, Y.D.; Chou, K.C. Predicting drug-target interaction networks based on functional groups and biological features. PLoS ONE 2010, 5, e9603. [Google Scholar] [CrossRef]
- Öztürk, H.; Özgür, A.; Ozkirimli, E. DeepDTA: Deep drug–target binding affinity prediction. Bioinformatics 2018, 34, i821–i829. [Google Scholar] [CrossRef] [Green Version]
- Lee, I.; Keum, J.; Nam, H. DeepConv-DTI: Prediction of drug-target interactions via deep learning with convolution on protein sequences. PLoS Comput. Biol. 2019, 15, e1007129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, T.; Le, H.; Venkatesh, S. GraphDTA: Prediction of drug–target binding affinity using graph convolutional networks. BioRxiv 2019, 684662. [Google Scholar] [CrossRef] [Green Version]
- Öztürk, H.; Ozkirimli, E.; Özgür, A. WideDTA: Prediction of drug-target binding affinity. arXiv 2019, arXiv:1902.04166. [Google Scholar]
- Feng, Q.; Dueva, E.; Cherkasov, A.; Ester, M. Padme: A deep learning-based framework for drug-target interaction prediction. arXiv 2018, arXiv:1807.09741. [Google Scholar]
- Shin, B.; Park, S.; Kang, K.; Ho, J.C. Self-attention based molecule representation for predicting drug-target interaction. arXiv 2019, arXiv:1908.06760. [Google Scholar]
- Davis, M.I.; Hunt, J.P.; Herrgard, S.; Ciceri, P.; Wodicka, L.M.; Pallares, G.; Hocker, M.; Treiber, D.K.; Zarrinkar, P.P. Comprehensive analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2011, 29, 1046–1051. [Google Scholar] [CrossRef]
- Tang, J.; Szwajda, A.; Shakyawar, S.; Xu, T.; Hintsanen, P.; Wennerberg, K.; Aittokallio, T. Making sense of large-scale kinase inhibitor bioactivity data sets: A comparative and integrative analysis. J. Chem. Inf. Model. 2014, 54, 735–743. [Google Scholar] [CrossRef] [PubMed]
- Consortium, T.U. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2020, 49, D480–D489. [Google Scholar] [CrossRef] [PubMed]
- Pennington, J.; Socher, R.; Manning, C.D. GloVe: Global Vectors for Word Representation. In Proceedings of the Empirical Methods in Natural Language Processing (EMNLP), Doha, Qatar, 25–29 October 2014; pp. 1532–1543. [Google Scholar]
- Weininger, D. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J. Chem. Inf. Comput. Sci. 1988, 28, 31–36. [Google Scholar] [CrossRef]
- Landrum, G. RDKit: Open-Source Cheminformatics; 2006. Available online: http://www.rdkit.org/ (accessed on 16 October 2021).
- Jin, W.; Barzilay, R.; Jaakkola, T. Junction tree variational autoencoder for molecular graph generation. In Proceedings of the 35th International Conference on Machine Learning (PMLR), Stockholm, Sweden, 10–15 July 2018; Volume 80, pp. 2323–2332. [Google Scholar]
- Kipf, T.N.; Welling, M. Semi-supervised classification with graph convolutional networks. arXiv 2016, arXiv:1609.02907. [Google Scholar]
- Bickerton, G.R.; Paolini, G.V.; Besnard, J.; Muresan, S.; Hopkins, A.L. Quantifying the chemical beauty of drugs. Nat. Chem. 2012, 4, 90–98. [Google Scholar] [CrossRef] [Green Version]
- Baber, J.; Feher, M. Predicting synthetic accessibility: Application in drug discovery and development. Mini Rev. Med. Chem. 2004, 4, 681–692. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Shi, C.; Zhou, H.; Yang, Y.; Zhang, W.; Yu, Y.; Li, L. Mars: Markov molecular sampling for multi-objective drug discovery. arXiv 2021, arXiv:2103.10432. [Google Scholar]
- Kamel, M.; Zaghary, W.; Al-Wabli, R.; Anwar, M. Synthetic approaches and potential bioactivity of different functionalized quinazoline and quinazolinone scaffolds. Egypt. Pharm. J. 2016, 15, 34–98. [Google Scholar]
- Gatadi, S.; Lakshmi, T.V.; Nanduri, S. 4(3H)-Quinazolinone derivatives: Promising antibacterial drug leads. Eur. J. Med. Chem. 2019, 170, 157–172. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dataset | # of Compounds | # of Proteins | # of DT Interactions |
|---|---|---|---|
| Davis | 68 | 442 | 30,056 |
| KIBA | 2111 | 229 | 118,254 |
| Parameters | Value |
|---|---|
| Batch size | 512 |
| Learning rate | 0.0005 |
| # epoch | 1500 |
| Dropout | 0.2 |
| Optimizer | Adam |
| # filters of the 3 layers in CNN | 1000, 256, 32 |
| Filter sizes of the 3 layers in CNN | 8, 8, 3 |
| Input Dim. of the 3 layers in GCN | N, N, |
| Output Dim. of the 3 layers in GCN | N, , |
| # hidden units in final FC layers | 1024, 512 |
| Max length of protein sequences | 1000 |
| Models | Protein Rep. | Drug Pep. | MSE | CI |
|---|---|---|---|---|
| Baseline Models | ||||
| KronRLS | Smith-Waterman | Pubchem-Sim | 0.379 | 0.871 |
| SimBoost | Smith-Waterman | Pubchem-Sim | 0.282 | 0.872 |
| DeepDTA | 1D | 1D | 0.261 | 0.878 |
| WideDTA | 1D + PDM | 1D + LMCS | 0.262 | 0.886 |
| GraphDTA_GCN | 1D | Graph | 0.254 | 0.880 |
| Our Proposed Models | ||||
| EmbedDTI_noPre | 1D | Graph + Graph | 0.236 | 0.892 |
| EmbedDTI_noSub | 1D | Graph | 0.235 | 0.896 |
| EmbedDTI_noAttn | 1D | Graph + Graph | 0.233 | 0.898 |
| EmbedDTI | 1D | Graph + Graph | 0.230 | 0.900 |
| Models | Protein Rep. | Drug Rep. | MSE | CI |
|---|---|---|---|---|
| Baseline Models | ||||
| KronRLS | Smith-Waterman | Pubchem-Sim | 0.411 | 0.782 |
| SimBoost | Smith-Waterman | Pubchem-Sim | 0.222 | 0.836 |
| DeepDTA | 1D | 1D | 0.194 | 0.863 |
| WideDTA | 1D + PDM | 1D + LMCS | 0.179 | 0.875 |
| GraphDTA_GCN | 1D | Graph | 0.139 | 0.889 |
| Our Proposed Models | ||||
| EmbedDTI_noPre | 1D | Graph + Graph | 0.134 | 0.896 |
| EmbedDTI_noSub | 1D | Graph | 0.134 | 0.893 |
| EmbedDTI_noAttn | 1D | Graph + Graph | 0.131 | 0.901 |
| EmbedDTI | 1D | Graph + Graph | 0.133 | 0.897 |
| Rank Index | Canonical SMILES |
|---|---|
| 1 | Oc1ccc(-c2cncc(C(c3nc4c(C5NC6CCC5C6)cccc4[nH]3)c3cccc4ocnc34)c2)cc1 |
| 2 | NC1CCCN(c2ccccc2S(=O)(=O)c2cc(C=Cc3ccccc3)cc(Cc3ccc4c(c3)OCO4)c2)C1 |
| 3 | [O-]C1CNCCC1C1COc2ccc(CN3CCOCC3c3cnc4ccc(F)c(C(F)(F)F)c4c3)cc2O1 |
| 4 | C=Cc1ccc(-c2cc(NC(=O)[O-])nc(-c3ccc(C4CC(=O)N(F)C4c4ccc(F)cc4)cc3)n2)cc1F |
| 5 | CC(=O)N1CCC(c2cccc(NNc3cc(Cl)cc(C4OCCC(C(=O)N5CCCCCC5)C4F)c3)c2)CC1 |
| 6 | Oc1cnc(C2COC(c3ccc(Cl)c4c3OCC(c3cc(F)c(F)c5c3OCO5)O4)C(F)C2O)c(F)c1 |
| 7 | O=C(C1CCc2cc(Nc3cc([O-])c(F)c(C4CN(c5ccc(F)cc5)CCO4)c3)cc(F)c21)N1CCNCC1 |
| 8 | Fc1cc(Cc2ccc(-c3nc4ccc(F)c(F)c4s3)cc2)ccc1Nc1ccccc1-c1ccccc1 |
| 9 | [O-]c1ccc(Nc2ccc(Cc3nc(-c4cccnc4)no3)c(Cc3cc(F)cc(-c4nnc([O-])o4)c3)c2)cc1 |
| 10 | OC1C=C(c2cccc(C(F)(F)F)c2)CC(C2CCNC(C3CCOC(c4ccccn4)C3)C2)C1 |
| Rank Index | Prediction Score | QED Score | SA Score | Docking Score (by SMINA) |
|---|---|---|---|---|
| 1 | ||||
| 2 | ||||
| 3 | ||||
| 4 | ||||
| 5 | ||||
| 6 | ||||
| 7 | ||||
| 8 | ||||
| 9 | ||||
| 10 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jin, Y.; Lu, J.; Shi, R.; Yang, Y. EmbedDTI: Enhancing the Molecular Representations via Sequence Embedding and Graph Convolutional Network for the Prediction of Drug-Target Interaction. Biomolecules 2021, 11, 1783. https://doi.org/10.3390/biom11121783
Jin Y, Lu J, Shi R, Yang Y. EmbedDTI: Enhancing the Molecular Representations via Sequence Embedding and Graph Convolutional Network for the Prediction of Drug-Target Interaction. Biomolecules. 2021; 11(12):1783. https://doi.org/10.3390/biom11121783
Chicago/Turabian StyleJin, Yuan, Jiarui Lu, Runhan Shi, and Yang Yang. 2021. "EmbedDTI: Enhancing the Molecular Representations via Sequence Embedding and Graph Convolutional Network for the Prediction of Drug-Target Interaction" Biomolecules 11, no. 12: 1783. https://doi.org/10.3390/biom11121783
APA StyleJin, Y., Lu, J., Shi, R., & Yang, Y. (2021). EmbedDTI: Enhancing the Molecular Representations via Sequence Embedding and Graph Convolutional Network for the Prediction of Drug-Target Interaction. Biomolecules, 11(12), 1783. https://doi.org/10.3390/biom11121783