
MassGenie: A Transformer-Based Deep Learning Method for Identifying Small Molecules from Their Mass Spectra


Abstract
:1. Introduction
2. Materials and Methods
2.1. Small Molecule Datasets Used
2.2. In Silico Fragmentation Method
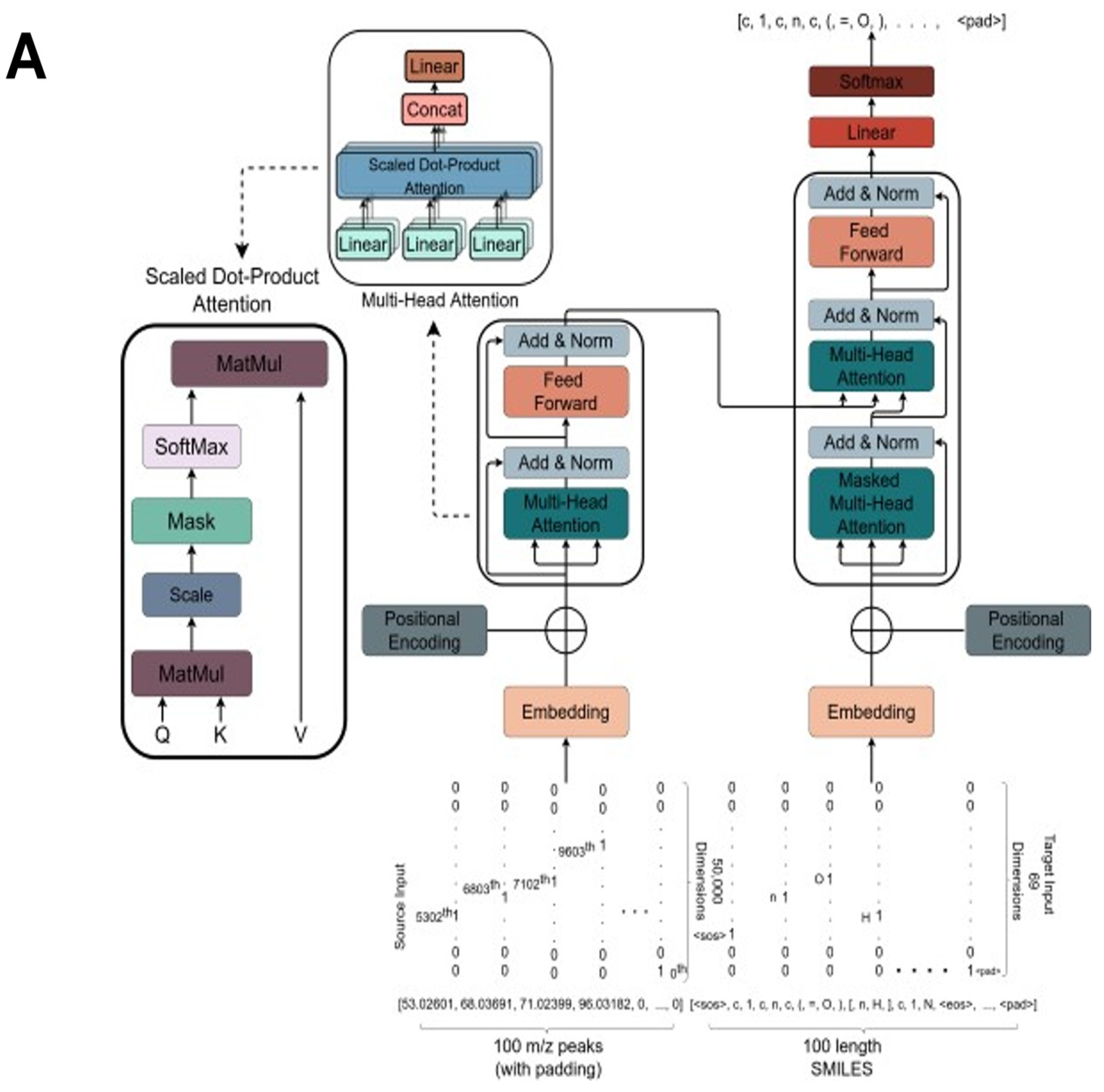
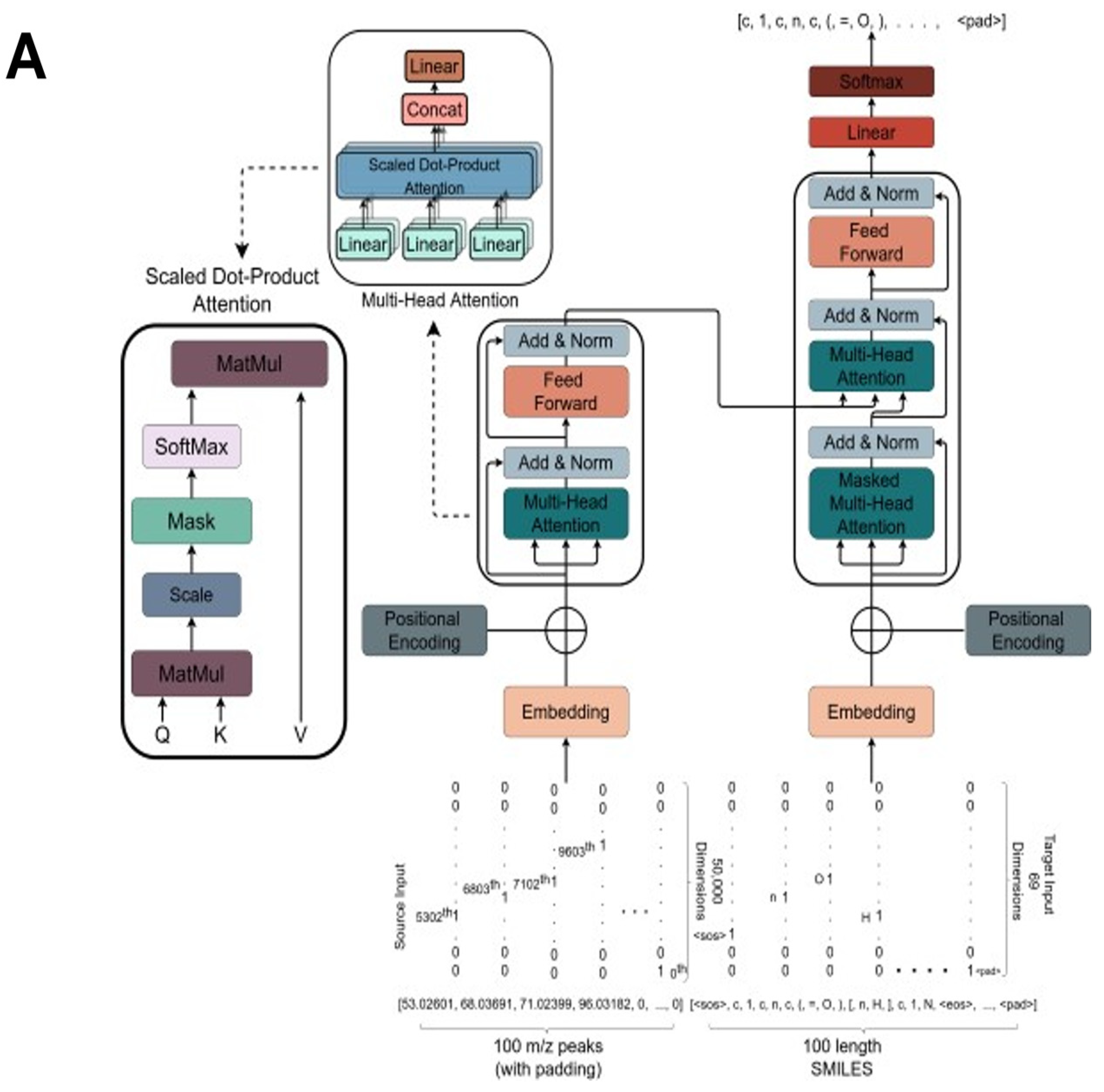
2.3. The Transformer Model
2.4. Training Settings
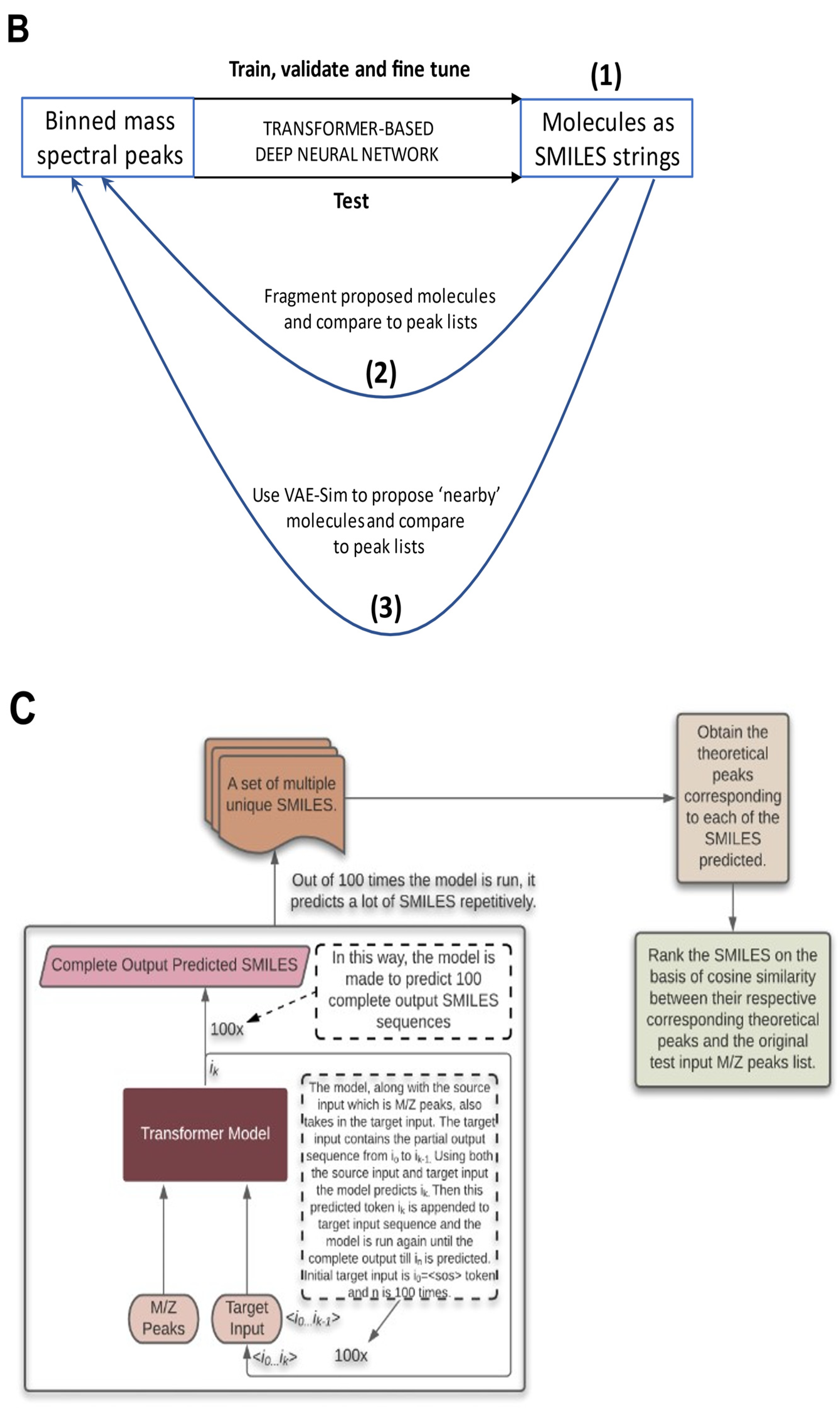
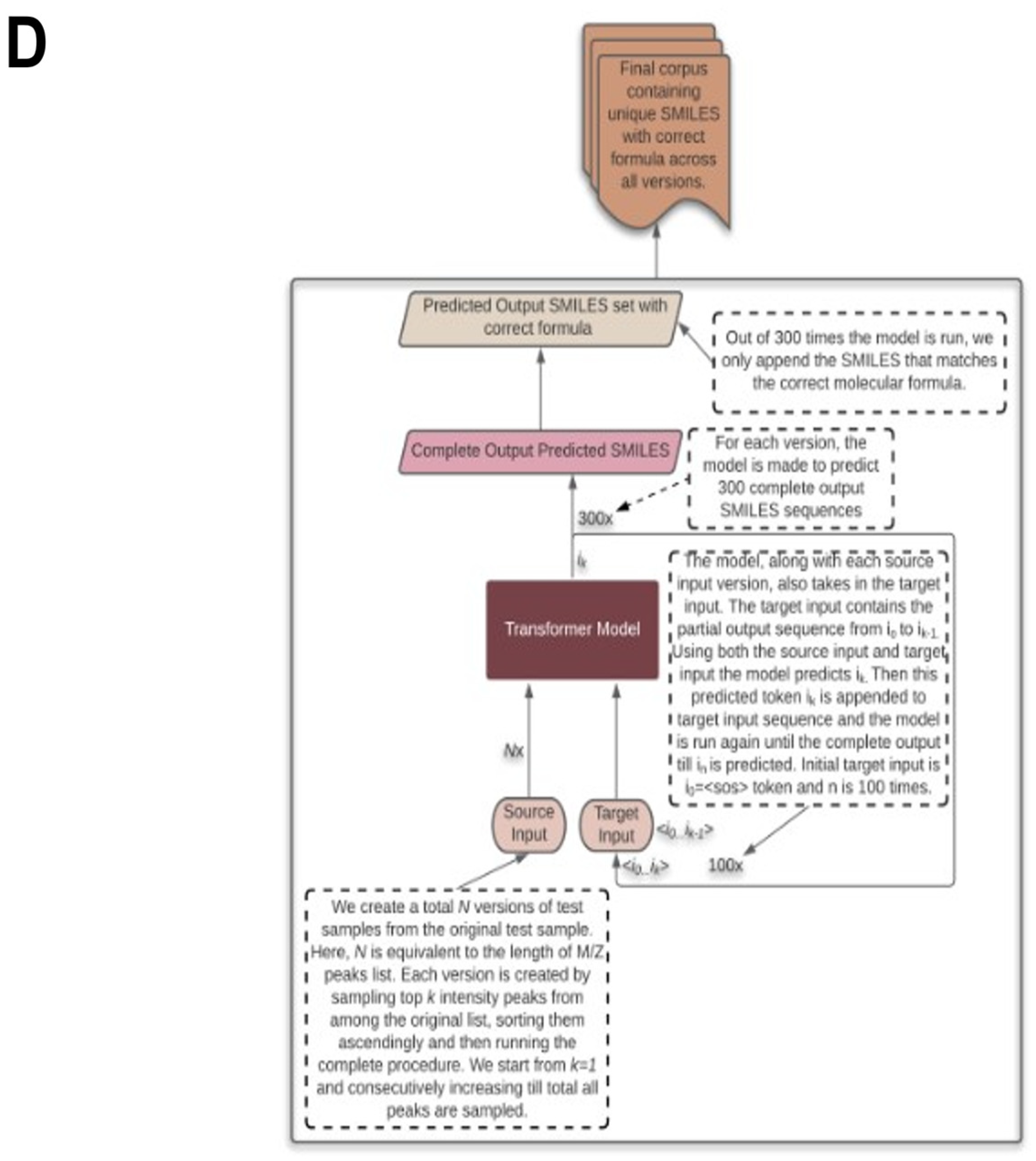
2.5. Testing the Results
2.6. Testing with CASMI Data
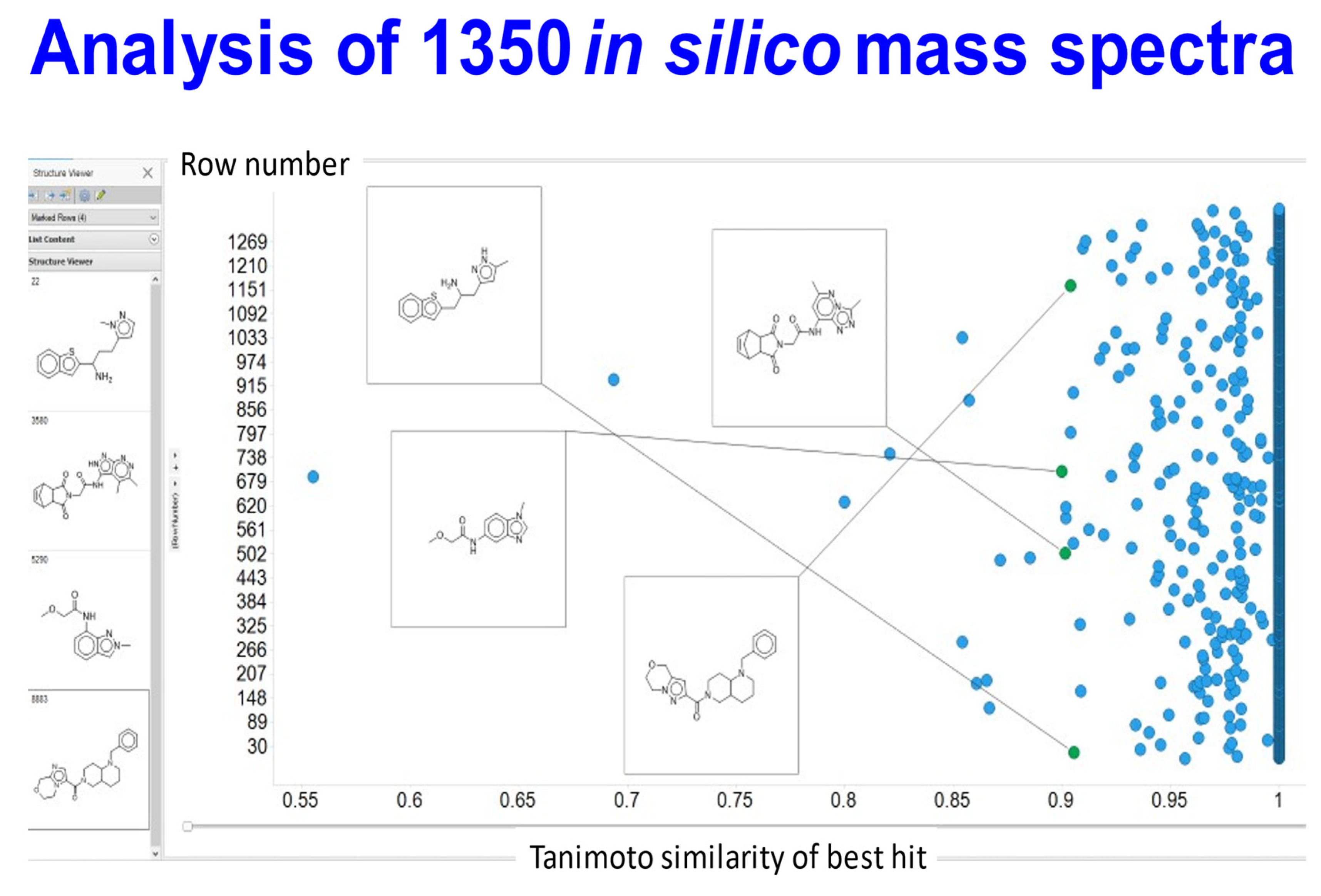
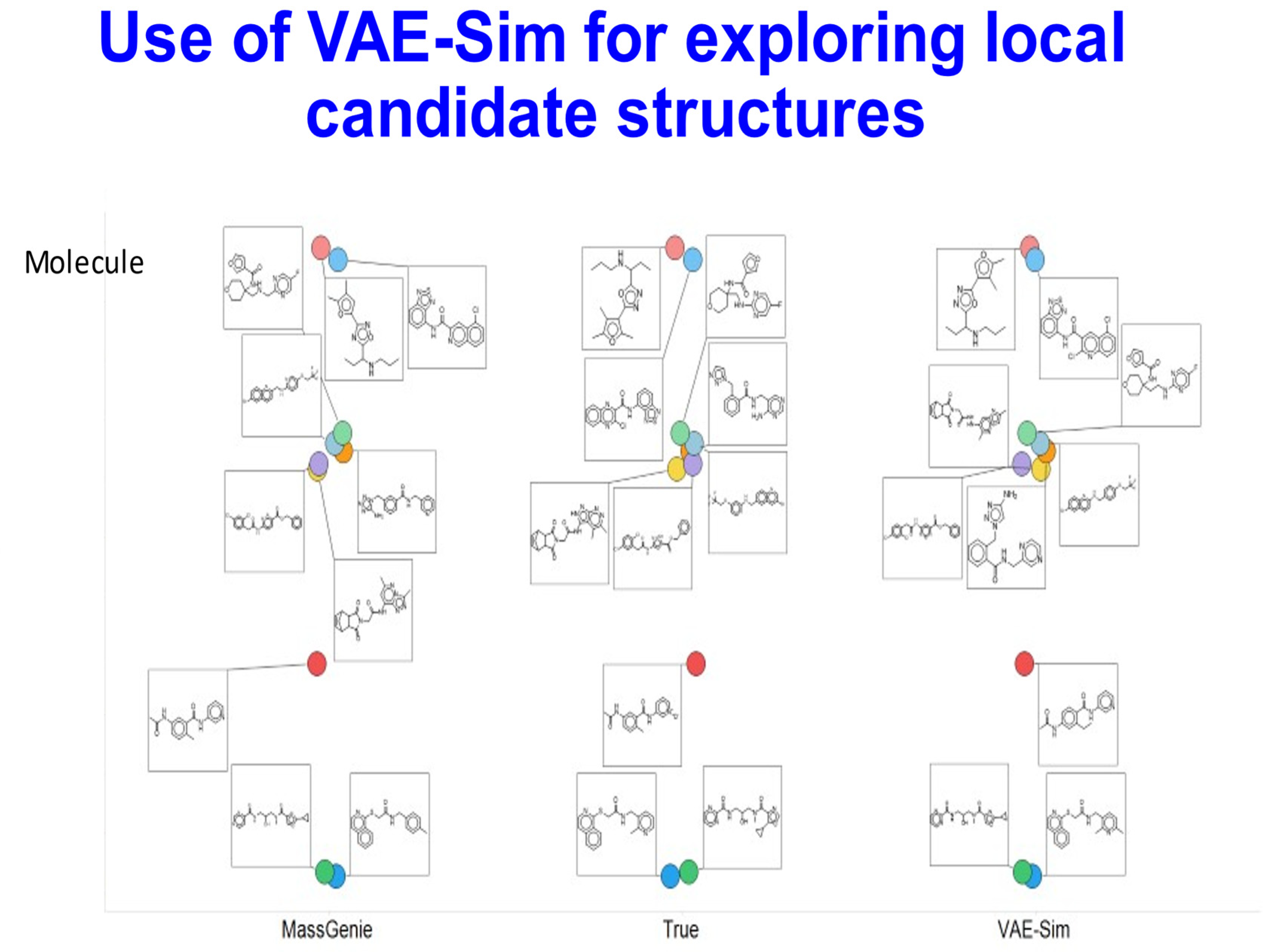
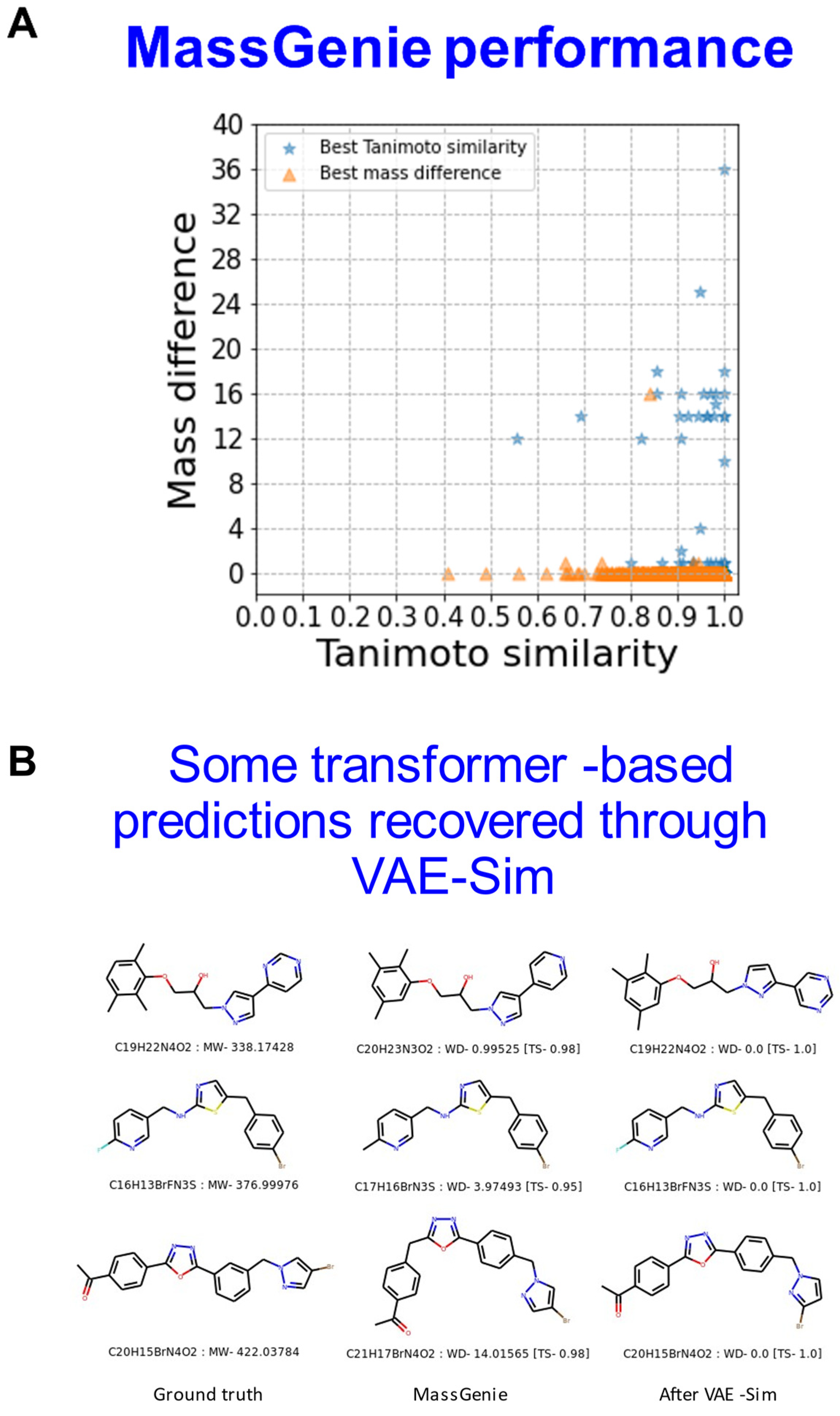
3. Results
| [60.02059, 66.02125, 70.00248, 81.03214, 81.04474, 96.05563, 109.99178, 112.04313, 116.018036, 125.002686, 127.05403, 127.91178, 131.02895, 131.04152, 134.03624, 135.01643, 137.9867, 146.05243, 149.04715, 150.02734, 150.03992, 152.9976, 153.98161, 165.05083, 168.99251, 201.92743, 205.04015, 216.93832, 229.0846, 229.92235, 233.03505, 236.89629, 239.06648, 244.93324, 245.91727, 249.02995, 251.90718, 255.03694, 259.01187, 260.9282, 264.8912, 267.0614, 274.03534, 279.08142, 279.9021, 283.03186, 283.05634, 287.00677, 295.05185, 296.9758, 299.02676, 302.03024, 303.00168, 316.92114, 324.97076, 331.94464, 340.96567, 340.9657, 344.9161, 346.97266, 350.94757, 359.9396, 360.91104, 362.94308, 365.97107, 366.918, 374.9676, 378.9425, 381.9415, 385.9164, 386.98758, 390.93802, 390.9625, 390.96252, 393.966, 394.91293, 394.93744, 400.9399, 402.958, 406.93292, 406.93295, 409.93643, 410.90787, 413.91135, 428.93484, 444.92978, 459.95325] |
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Griffin, J.L. The Cinderella story of metabolic profiling: Does metabolomics get to go to the functional genomics ball? Philos. Trans. R. Soc. Lond. B Biol. Sci. 2006, 361, 147–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliver, S.G.; Winson, M.K.; Kell, D.B.; Baganz, F. Systematic functional analysis of the yeast genome. Trends Biotechnol. 1998, 16, 373–378. [Google Scholar] [CrossRef]
- Dunn, W.B.; Broadhurst, D.; Begley, P.; Zelena, E.; Francis-McIntyre, S.; Anderson, N.; Brown, N.; Knowles, J.; Halsall, A.; Haselden, J.N.; et al. The Husermet consortium, Procedures for large-scale metabolic profiling of serum and plasma using gas chromatography and liquid chromatography coupled to mass spectrometry. Nat. Protoc. 2011, 6, 1060–1083. [Google Scholar] [CrossRef]
- Dunn, W.B.; Erban, A.; Weber, R.J.M.; Creek, D.J.; Brown, M.; Breitling, R.; Hankemeier, T.; Goodacre, R.; Neumann, S.; Kopka, J.; et al. Mass Appeal: Metabolite identification in mass spectrometry-focused untargeted metabolomics. Metabolites 2013, 9, S44–S66. [Google Scholar] [CrossRef] [Green Version]
- Arús-Pous, J.; Awale, M.; Probst, D.; Reymond, J.L. Exploring Chemical Space with Machine Learning. Chimia 2019, 73, 1018–1023. [Google Scholar] [CrossRef]
- Bohacek, R.S.; McMartin, C.; Guida, W.C. The art and practice of structure-based drug design: A molecular modeling perspective. Med. Res. Rev. 1996, 16, 3–50. [Google Scholar] [CrossRef]
- Polishchuk, P.G.; Madzhidov, T.I.; Varnek, A. Estimation of the size of drug-like chemical space based on GDB-17 data. J. Comput. Aided Mol. Des. 2013, 27, 675–679. [Google Scholar] [CrossRef]
- Sterling, T.; Irwin, J.J. ZINC 15—Ligand Discovery for Everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef]
- Pitt, W.R.; Parry, D.M.; Perry, B.G.; Groom, C.R. Heteroaromatic Rings of the Future. J. Med. Chem. 2009, 52, 2952–2963. [Google Scholar] [CrossRef] [PubMed]
- Hastie, T.; Tibshirani, R.; Friedman, J. The Elements of Statistical Learning: Data Mining, Inference and Prediction, 2nd ed.; Springer: Berlin, Germany, 2009. [Google Scholar]
- Nash, W.J.; Dunn, W.B. From mass to metabolite in human untargeted metabolomics: Recent advances in annotation of metabolites applying liquid chromatography-mass spectrometry data. Trends Anal. Chem. 2019, 120, 115324. [Google Scholar] [CrossRef]
- Sindelar, M.; Patti, G.J. Chemical Discovery in the Era of Metabolomics. J. Am. Chem. Soc. 2020, 142, 9097–9105. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Wang, R.; Xiong, X.; Yin, Y.; Cai, Y.; Ma, Z.; Liu, N.; Zhu, Z.J. Metabolic reaction network-based recursive metabolite annotation for untargeted metabolomics. Nat. Commun. 2019, 10, 1516. [Google Scholar] [CrossRef] [Green Version]
- Misra, B.B.; van der Hooft, J.J.J. Updates in metabolomics tools and resources: 2014–2015. Electrophoresis 2016, 37, 86–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Misra, B.B. New software tools, databases, and resources in metabolomics: Updates from 2020. J. Metab. 2021, 17, 49. [Google Scholar] [CrossRef]
- Dunn, W.B.; Lin, W.; Broadhurst, D.; Begley, P.; Brown, M.; Zelena, E.; Vaughan, A.A.; Halsall, A.; Harding, N.; Knowles, J.D.; et al. Molecular phenotyping of a UK population: Defining the human serum metabolome. J. Metab. 2015, 11, 9–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganna, A.; Fall, T.; Salihovic, S.; Lee, W.; Broeckling, C.D.; Kumar, J.; Hägg, S.; Stenemo, M.; Magnusson, P.K.E.; Prenni, J.E.; et al. Large-scale non-targeted metabolomic profiling in three human population-based studies. J. Metab. 2016, 12, 4. [Google Scholar] [CrossRef]
- Wright Muelas, M.; Roberts, I.; Mughal, F.; O’Hagan, S.; Day, P.J.; Kell, D.B. An untargeted metabolomics strategy to measure differences in metabolite uptake and excretion by mammalian cell lines. J. Metab. 2020, 16, 107. [Google Scholar] [CrossRef]
- Borges, R.M.; Colby, S.M.; Das, S.; Edison, A.S.; Fiehn, O.; Kind, T.; Lee, J.; Merrill, A.T.; Merz, K.M., Jr.; Metz, T.O.; et al. Quantum Chemistry Calculations for Metabolomics. Chem. Rev. 2021, 121, 5633–5670. [Google Scholar] [CrossRef]
- Peisl, B.Y.L.; Schymanski, E.L.; Wilmes, P. Dark matter in host-microbiome metabolomics: Tackling the unknowns—A review. Anal. Chim. Acta 2018, 1037, 13–27. [Google Scholar] [CrossRef] [PubMed]
- De Vijlder, T.; Valkenborg, D.; Lemiere, F.; Romijn, E.P.; Laukens, K.; Cuyckens, F. A tutorial in small molecule identification via electrospray ionization-mass spectrometry: The practical art of structural elucidation. Mass Spectrom. Rev. 2018, 37, 607–629. [Google Scholar] [CrossRef]
- Dührkop, K.; Fleischauer, M.; Ludwig, M.; Aksenov, A.A.; Melnik, A.V.; Meusel, M.; Dorrestein, P.C.; Rousu, J.; Böcker, S. SIRIUS 4: A rapid tool for turning tandem mass spectra into metabolite structure information. Nat. Methods 2019, 16, 299–302. [Google Scholar] [CrossRef] [Green Version]
- Kind, T.; Tsugawa, H.; Cajka, T.; Ma, Y.; Lai, Z.; Mehta, S.S.; Wohlgemuth, G.; Barupal, D.K.; Showalter, M.R.; Arita, M.; et al. Identification of small molecules using accurate mass MS/MS search. Mass Spectrom. Rev. 2018, 37, 513–532. [Google Scholar] [CrossRef]
- Vinaixa, M.; Schymanski, E.L.; Neumann, S.; Navarro, M.; Salek, R.M.; Yanes, O. Mass spectral databases for LC/MS- and GC/MS-based metabolomics: State of the field and future prospects. Trends Anal. Chem. 2016, 78, 23–35. [Google Scholar] [CrossRef] [Green Version]
- Neumann, S.; Bocker, S. Computational mass spectrometry for metabolomics: Identification of metabolites and small molecules. Anal. Bioanal. Chem. 2010, 398, 2779–2788. [Google Scholar] [CrossRef] [Green Version]
- Blaženović, I.; Kind, T.; Ji, J.; Fiehn, O. Software Tools and Approaches for Compound Identification of LC-MS/MS Data in Metabolomics. Metabolites 2018, 8, 31. [Google Scholar] [CrossRef] [Green Version]
- Creek, D.J.; Dunn, W.B.; Fiehn, O.; Griffin, J.L.; Hall, R.D.; Lei, Z.; Mistrik, R.; Neumann, S.; Schymanski, E.L.; Sumner, L.W.; et al. Metabolite identification: Are you sure? And how do your peers gauge your confidence? Metabolites 2014, 10, 350–353. [Google Scholar] [CrossRef]
- Peters, K.; Bradbury, J.; Bergmann, S.; Capuccini, M.; Cascante, M.; de Atauri, P.; Ebbels, T.M.D.; Foguet, C.; Glen, R.; Gonzalez-Beltran, A.; et al. PhenoMeNal: Processing and analysis of metabolomics data in the cloud. Gigascience 2019, 8, giy149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bingol, K.; Bruschweiler-Li, L.; Li, D.; Zhang, B.; Xie, M.; Brüschweiler, R. Emerging new strategies for successful metabolite identification in metabolomics. Bioanalysis 2016, 8, 557–573. [Google Scholar] [CrossRef] [Green Version]
- Blaženović, I.; Kind, T.; Torbašinović, H.; Obrenović, S.; Mehta, S.S.; Tsugawa, H.; Wermuth, T.; Schauer, N.; Jahn, M.; Biedendieck, R.; et al. Comprehensive comparison of in silico MS/MS fragmentation tools of the CASMI contest: Database boosting is needed to achieve 93% accuracy. J. Cheminform. 2017, 9, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Djoumbou-Feunang, Y.; Pon, A.; Karu, N.; Zheng, J.; Li, C.; Arndt, D.; Gautam, M.; Allen, F.; Wishart, D.S. CFM-ID 3.0: Significantly Improved ESI-MS/MS Prediction and Compound Identification. Metabolites 2019, 9, 72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Djoumbou-Feunang, Y.; Fiamoncini, J.; Gil-de-la-Fuente, A.; Greiner, R.; Manach, C.; Wishart, D.S. BioTransformer: A comprehensive computational tool for small molecule metabolism prediction and metabolite identification. J. Cheminform. 2019, 11, 2. [Google Scholar] [CrossRef] [Green Version]
- Alexandrov, T. Spatial Metabolomics and Imaging Mass Spectrometry in the Age of Artificial Intelligence. Annu. Rev. Biomed. Data Sci. 2020, 3, 61–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ludwig, M.; Nothias, L.-F.; Dührkop, K.; Koester, I.; Fleischauer, M.; Hoffmann, M.A.; Petras, D.; Vargas, F.; Morsy, M.; Aluwihare, L.; et al. Database-independent molecular formula annotation using Gibbs sampling through ZODIAC. Nat. Mach. Intell. 2020, 2, 629–641. [Google Scholar] [CrossRef]
- McEachran, A.D.; Chao, A.; Al-Ghoul, H.; Lowe, C.; Grulke, C.; Sobus, J.R.; Williams, A.J. Revisiting Five Years of CASMI Contests with EPA Identification Tools. Metabolites 2020, 10, 260. [Google Scholar] [CrossRef]
- Bowen, B.P.; Northen, T.R. Dealing with the unknown: Metabolomics and metabolite atlases. J. Am. Soc. Mass Spectrom. 2010, 21, 1471–1476. [Google Scholar] [CrossRef] [Green Version]
- Bhatia, A.; Sarma, S.J.; Lei, Z.; Sumner, L.W. UHPLC-QTOF-MS/MS-SPE-NMR: A Solution to the Metabolomics Grand Challenge of Higher-Throughput, Confident Metabolite Identifications. Methods Mol. Biol. 2019, 2037, 113–133. [Google Scholar] [PubMed]
- Liu, Y.; De Vijlder, T.; Bittremieux, W.; Laukens, K.; Heyndrickx, W. Current and future deep learning algorithms for tandem mass spectrometry (MS/MS)-based small molecule structure elucidation. Rapid Commun. Mass Spectrom. 2021, e9120. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, A.; Vázquez-Baeza, Y.; Gauglitz, J.M.; Wang, M.; Dührkop, K.; Nothias-Esposito, M.; Acharya, D.D.; Ernst, M.; van der Hooft, J.J.J.; Zhu, Q.; et al. Chemically informed analyses of metabolomics mass spectrometry data with Qemistree. Nat. Chem. Biol. 2021, 17, 146–151. [Google Scholar] [CrossRef]
- Stravs, M.A.; Dührkop, K.; Böcker, S.; Zamboni, N. MSNovelist: De novo structure generation from mass spectra. bioRxiv 2021, 450875. [Google Scholar]
- Buchanan, B.G.; Feigenbaum, E.A. DENDRAL and META-DENDRAL: Their application dimensions. Artif. Intell. 1978, 11, 5–24. [Google Scholar] [CrossRef]
- Feigenbaum, E.A.; Buchanan, B.G. DENDRAL and META-DENDRAL: Roots of knowledge systems and expert system applications. Artif. Intell. 1993, 59, 223–240. [Google Scholar] [CrossRef]
- Lindsay, R.K.; Buchanan, B.G.; Feigenbaum, E.A.; Lederberg, J. DENDRAL—A Case study of the first expert system for scientific hypothesis formation. Artif. Intell. 1993, 61, 209–261. [Google Scholar] [CrossRef] [Green Version]
- Kell, D.B.; Oliver, S.G. Here is the evidence, now what is the hypothesis? The complementary roles of inductive and hypothesis-driven science in the post-genomic era. Bioessays 2004, 26, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Ruddigkeit, L.; van Deursen, R.; Blum, L.C.; Reymond, J.L. Enumeration of 166 billion organic small molecules in the chemical universe database GDB-17. J. Chem. Inf. Model. 2012, 52, 2864–2875. [Google Scholar] [CrossRef]
- Gómez-Bombarelli, R.; Wei, J.N.; Duvenaud, D.; Hernández-Lobato, J.M.; Sánchez-Lengeling, B.; Sheberla, D.; Aguilera-Iparraguirre, J.; Hirzel, T.D.; Adams, R.P.; Aspuru-Guzik, A. Automatic Chemical Design Using a Data-Driven Continuous Representation of Molecules. ACS Cent. Sci. 2018, 4, 268–276. [Google Scholar] [CrossRef]
- LeCun, Y.; Bengio, Y.; Hinton, G. Deep learning. Nature 2015, 521, 436–444. [Google Scholar] [CrossRef]
- Schmidhuber, J. Deep learning in neural networks: An overview. Neural Netw. 2015, 61, 85–117. [Google Scholar] [CrossRef] [Green Version]
- Arús-Pous, J.; Blaschke, T.; Ulander, S.; Reymond, J.L.; Chen, H.; Engkvist, O. Exploring the GDB-13 chemical space using deep generative models. J. Cheminform. 2019, 11, 20. [Google Scholar] [CrossRef]
- David, L.; Arús-Pous, J.; Karlsson, J.; Engkvist, O.; Bjerrum, E.J.; Kogej, T.; Kriegl, J.M.; Beck, B.; Chen, H. Applications of Deep-Learning in Exploiting Large-Scale and Heterogeneous Compound Data in Industrial Pharmaceutical Research. Front. Pharm. 2019, 10, 1303. [Google Scholar] [CrossRef] [Green Version]
- Grisoni, F.; Schneider, G. De novo Molecular Design with Generative Long Short-term Memory. Chimia 2019, 73, 1006–1011. [Google Scholar] [CrossRef]
- Schneider, G. Generative models for artificially-intelligent molecular design. Mol. Inform. 2018, 37, 188031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Lengeling, B.; Aspuru-Guzik, A. Inverse molecular design using machine learning: Generative models for matter engineering. Science 2018, 361, 360–365. [Google Scholar] [CrossRef] [Green Version]
- Segler, M.H.S.; Kogej, T.; Tyrchan, C.; Waller, M.P. Generating Focused Molecule Libraries for Drug Discovery with Recurrent Neural Networks. ACS Cent. Sci. 2018, 4, 120–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elton, D.C.; Boukouvalas, Z.; Fuge, M.D.; Chung, P.W. Deep learning for molecular design: A review of the state of the art. Mol. Syst. Des. Eng. 2019, 4, 828–849. [Google Scholar] [CrossRef] [Green Version]
- Kell, D.B.; Samanta, S.; Swainston, N. Deep learning and generative methods in cheminformatics and chemical biology: Navigating small molecule space intelligently. J. Biochem. 2020, 477, 4559–4580. [Google Scholar] [CrossRef]
- Jiménez-Luna, J.; Grisoni, F.; Weskamp, N.; Schneider, G. Artificial intelligence in drug discovery: Recent advances and future perspectives. Expert Opin. Drug Discov. 2021, 16, 949–959. [Google Scholar] [CrossRef]
- Skinnider, M.; Wang, F.; Pasin, D.; Greiner, R.; Foster, L.; Dalsgaard, P.; Wishart, D.S. A Deep Generative Model Enables Automated Structure Elucidation of Novel Psychoactive Substances. ChemRxiv 2021, 1–23. [Google Scholar] [CrossRef]
- Bepler, T.; Berger, B. Learning the protein language: Evolution, structure, and function. Cell Syst. 2021, 12, 654–666.e3. [Google Scholar] [CrossRef] [PubMed]
- Samanta, S.; O’Hagan, S.; Swainston, N.; Roberts, T.J.; Kell, D.B. VAE-Sim: A novel molecular similarity measure based on a variational autoencoder. Molecules 2020, 25, 3446. [Google Scholar] [CrossRef]
- Grimme, S. Towards first principles calculation of electron impact mass spectra of molecules. Angew. Chem. Int. Ed. Engl. 2013, 52, 6306–6312. [Google Scholar] [CrossRef]
- Scheubert, K.; Hufsky, F.; Böcker, S. Computational mass spectrometry for small molecules. J. Cheminform. 2013, 5, 12. [Google Scholar] [CrossRef] [Green Version]
- Ridder, L.; van der Hooft, J.J.J.; Verhoeven, S. Automatic Compound Annotation from Mass Spectrometry Data Using MAGMa. J. Mass Spectrom. 2014, 3, S0033. [Google Scholar] [CrossRef] [Green Version]
- Ruttkies, C.; Schymanski, E.L.; Wolf, S.; Hollender, J.; Neumann, S. MetFrag relaunched: Incorporating strategies beyond in silico fragmentation. J. Cheminform 2016, 8, 3. [Google Scholar] [CrossRef] [Green Version]
- Ruttkies, C.; Neumann, S.; Posch, S. Improving MetFrag with statistical learning of fragment annotations. BMC Bioinform. 2019, 20, 376. [Google Scholar] [CrossRef]
- da Silva, R.R.; Wang, M.; Nothias, L.F.; van der Hooft, J.J.J.; Caraballo-Rodríguez, A.M.; Fox, E.; Balunas, M.J.; Klassen, J.L.; Lopes, N.P.; Dorrestein, P.C. Propagating annotations of molecular networks using in silico fragmentation. PLoS Comput. Biol. 2018, 14, e1006089. [Google Scholar] [CrossRef] [PubMed]
- Wandy, J.; Davies, V.; van der Hooft, J.J.J.; Weidt, S.; Daly, R.; Rogers, S. In Silico Optimization of Mass Spectrometry Fragmentation Strategies in Metabolomics. Metabolites 2019, 9, 219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ernst, M.; Kang, K.B.; Caraballo-Rodriguez, A.M.; Nothias, L.F.; Wandy, J.; Chen, C.; Wang, M.; Rogers, S.; Medema, M.H.; Dorrestein, P.C.; et al. MolNetEnhancer: Enhanced Molecular Networks by Integrating Metabolome Mining and Annotation Tools. Metabolites 2019, 9, 144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, F.; Pon, A.; Wilson, M.; Greiner, R.; Wishart, D. CFM-ID: A web server for annotation, spectrum prediction and metabolite identification from tandem mass spectra. Nucleic Acids Res. 2014, 42, W94–W99. [Google Scholar] [CrossRef] [Green Version]
- Schüler, J.A.; Neumann, S.; Müller-Hannemann, M.; Brandt, W. ChemFrag: Chemically meaningful annotation of fragment ion mass spectra. J. Mass Spectrom. 2018, 53, 1104–1115. [Google Scholar] [CrossRef]
- Hoffmann, M.A.; Nothias, L.F.; Ludwig, M.; Fleischauer, M.; Gentry, E.C.; Witting, M.; Dorrestein, P.C.; Dührkop, K.; Böcker, S. High-confidence structural annotation of metabolites absent from spectral libraries. Nat. Biotechnol. 2021. [Google Scholar] [CrossRef]
- Feunang, Y.D.; Eisner, R.; Knox, C.; Chepelev, L.; Hastings, J.; Owen, G.; Fahy, E.; Steinbeck, C.; Subramanian, S.; Bolton, E.; et al. ClassyFire: Automated chemical classification with a comprehensive, computable taxonomy. J. Cheminform. 2016, 8, 61. [Google Scholar]
- Hassanpour, N.; Alden, N.; Menon, R.; Jayaraman, A.; Lee, K.; Hassoun, S. Biological Filtering and Substrate Promiscuity Prediction for Annotating Untargeted Metabolomics. Metabolites 2020, 10, 160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krizhevsky, A.; Sutskever, I.; Hinton, G.E. ImageNet Classification with Deep Convolutional Neural Networks. Adv. Neural Inf. Process. Syst. 2012, 25, 1090–1098. [Google Scholar] [CrossRef]
- Brown, T.B.; Mann, B.; Ryder, N.; Subbiah, M.; Kaplan, J.; Dhariwal, P.; Neelakantan, A.; Shyam, P.; Sastry, G.; Askell, A.; et al. Language Models are Few-Shot Learners. arXiv 2020, arXiv:2005.14165. [Google Scholar]
- Shardlow, M.; Ju, M.; Li, M.; O’Reilly, C.; Iavarone, E.; McNaught, J.; Ananiadou, S. A Text Mining Pipeline Using Active and Deep Learning Aimed at Curating Information in Computational Neuroscience. Neuroinformatics 2019, 17, 391–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaswani, A.; Shazeer, N.; Parmar, N.; Uszkoreit, J.; Jones, L.; Gomez, A.N.; Kaiser, L.; Polosukhin, I. Attention Is All You Need. arXiv 2017, arXiv:1706.03762. [Google Scholar]
- Devlin, J.; Chang, M.-W.; Lee, K.; Toutanova, K. BERT: Pre-training of Deep Bidirectional Transformers for Language Understanding. arXiv 2018, arXiv:1810.04805. [Google Scholar]
- Hutson, M. The language machines. Nature 2021, 591, 22–25. [Google Scholar] [CrossRef]
- Singh, S.; Mahmood, A. The NLP Cookbook: Modern Recipes for Transformer based Deep Learning Architectures. arXiv 2021, arXiv:2104.10640. [Google Scholar] [CrossRef]
- Topal, M.O.; Bas, A.; van Heerden, I. Exploring Transformers in Natural Language Generation: GPT, BERT, and XLNet. arXiv 2021, arXiv:2102.08036. [Google Scholar]
- Kaiser, L.; Gomez, A.N.; Shazeer, N.; Vaswani, A.; Parmar, N.; Jones, L.; Uszkoreit, J. One Model To Learn Them All. arXiv 2017, arXiv:1706.05137. [Google Scholar]
- Lu, K.; Grover, A.; Abbeel, P.; Mordatch, I. Pretrained Transformers as Universal Computation Engines. arXiv 2021, arXiv:2103.05247. [Google Scholar]
- Shrivastava, A.D.; Swainston, N.; Samanta, S.; Roberts, I.; Wright Muelas, M.; Kell, D.B. MassGenie: A transformer-based deep learning method for identifying small molecules from their mass spectra. bioRxiv 2021. [Google Scholar] [CrossRef]
- O’Hagan, S.; Swainston, N.; Handl, J.; Kell, D.B. A ‘rule of 0.5′ for the metabolite-likeness of approved pharmaceutical drugs. Metabolites 2015, 11, 323–339. [Google Scholar] [CrossRef]
- O’Hagan, S.; Kell, D.B. Consensus rank orderings of molecular fingerprints illustrate the ‘most genuine’ similarities between marketed drugs and small endogenous human metabolites, but highlight exogenous natural products as the most important ‘natural’ drug transporter substrates. ADMET DMPK 2017, 5, 85–125. [Google Scholar]
- Roberts, I.; Wright Muelas, M.; Taylor, J.M.; Davison, A.S.; Xu, Y.; Grixti, J.M.; Gotts, N.; Sorokin, A.; Goodacre, R.; Kell, D.B. Untargeted metabolomics of COVID-19 patient serum reveals potential prognostic markers of both severity and outcome. medRxiv 2020. [Google Scholar] [CrossRef]
- Willighagen, E.L.; Mayfield, J.W.; Alvarsson, J.; Berg, A.; Carlsson, L.; Jeliazkova, N.; Kuhn, S.; Pluskal, T.; Rojas-Cherto, M.; Spjuth, O.; et al. The Chemistry Development Kit (CDK) v2.0: Atom typing, depiction, molecular formulas, and substructure searching. J. Cheminform. 2017, 9, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goyal, P.; Dollár, P.; Girshick, R.; Noordhuis, P.; Wesolowski, L.; Kyrola, A.; Tulloch, A.; Jia, Y.; He, K. Accurate, Large Minibatch SGD: Training ImageNet in 1 Hour. arXiv 2017, arXiv:1706.02677. [Google Scholar]
- Sumner, L.W.; Amberg, A.; Barrett, D.; Beale, M.H.; Beger, R.; Daykin, C.A.; Fan, T.W.; Fiehn, O.; Goodacre, R.; Griffin, J.L.; et al. Proposed minimum reporting standards for chemical analysis Chemical Analysis Working Group (CAWG) Metabolomics Standards Initiative (MSI). Metabolites 2007, 3, 211–221. [Google Scholar]
- Bender, A.; Glen, R.C. Molecular similarity: A key technique in molecular informatics. Org. Biomol. Chem. 2004, 2, 3204–3218. [Google Scholar] [CrossRef] [PubMed]
- Maggiora, G.; Vogt, M.; Stumpfe, D.; Bajorath, J. Molecular Similarity in Medicinal Chemistry. J. Med. Chem. 2014, 57, 3186–3204. [Google Scholar] [CrossRef] [PubMed]
- Todeschini, R.; Consonni, V.; Xiang, H.; Holliday, J.; Buscema, M.; Willett, P. Similarity Coefficients for Binary Chemoinformatics Data: Overview and Extended Comparison Using Simulated and Real Data Sets. J. Chem. Inf. Model. 2012, 52, 2884–2901. [Google Scholar] [CrossRef] [PubMed]
- Jeffryes, J.G.; Colastani, R.L.; Elbadawi-Sidhu, M.; Kind, T.; Niehaus, T.D.; Broadbelt, L.J.; Hanson, A.D.; Fiehn, O.; Tyo, K.E.; Henry, C.S. MINEs: Open access databases of computationally predicted enzyme promiscuity products for untargeted metabolomics. J. Cheminform. 2015, 7, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, H.; Zhou, J. Privacy Leakage of SIFT Features via Deep Generative Model based Image Reconstruction. arXiv 2020, arXiv:2009.01030. [Google Scholar] [CrossRef]
- Schymanski, E.L.; Neumann, S. The Critical Assessment of Small Molecule Identification (CASMI): Challenges and Solutions. Metabolites 2013, 3, 517–538. [Google Scholar] [CrossRef] [PubMed]
- Mendez, K.M.; Broadhurst, D.I.; Reinke, S.N. The application of artificial neural networks in metabolomics: A historical perspective. Metabolites 2019, 15, 142. [Google Scholar] [CrossRef] [PubMed]
- Kind, T.; Fiehn, O. Seven Golden Rules for heuristic filtering of molecular formulas obtained by accurate mass spectrometry. BMC Bioinform. 2007, 8, 105. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Sengupta, S.; Lakshminarayanan, V. Explainable deep learning models in medical image analysis. arXiv 2020, arXiv:2005.13799. [Google Scholar]
- Trieu, H.L.; Tran, T.T.; Duong, K.N.A.; Nguyen, A.; Miwa, M.; Ananiadou, S. DeepEventMine: End-to-end neural nested event extraction from biomedical texts. Bioinformatics 2020, 36, 4910–4917. [Google Scholar] [CrossRef]
- Ertl, P. Cheminformatics analysis of organic substituents: Identification of the most common substituents, calculation of substituent properties, and automatic identification of drug-like bioisosteric groups. J. Chem Inf. Comput. Sci. 2003, 43, 374–380. [Google Scholar] [CrossRef]
- Ananiadou, S.; Kell, D.B.; Tsujii, J.-I. Text Mining and its potential applications in Systems Biology. Trends Biotechnol. 2006, 24, 571–579. [Google Scholar] [CrossRef]
- Ju, M.; Nguyen, N.T.H.; Miwa, M.; Ananiadou, S. An ensemble of neural models for nested adverse drug events and medication extraction with subwords. J. Am. Med. Inform. Assoc. 2019, 27, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Babai, L. Monte-Carlo Algorithms in Graph Isomorphism Testing; D.M.S. No. 79–10; University De Montréal: Montréal, QC, Canada, 1979. [Google Scholar]
- Luby, M.; Sinclair, A.; Zuckerman, D. Optimal speedup of Las Vegas algorithms. Inf. Proc. Lett. 1993, 47, 173–180. [Google Scholar] [CrossRef]
- Sze, S.H.; Pevzner, P.A. Las Vegas algorithms for gene recognition: Suboptimal and error-tolerant spliced alignment. J. Comput. Biol. 1997, 4, 297–309. [Google Scholar] [CrossRef]
- Zhai, X.; Kolesnikov, A.; Houlsby, N.; Beyer, L. Scaling Vision Transformers. arXiv 2021, arXiv:2106.04560. [Google Scholar]
- Sun, C.; Shrivastava, A.; Singh, S.; Gupta, A. Revisiting Unreasonable Effectiveness of Data in Deep Learning Era. arXiv 2017, arXiv:1707.02968. [Google Scholar]
- Kaplan, J.; McCandlish, S.; Henighan, T.; Brown, T.B.; Chess, B.; Child, R.; Gray, S.; Radford, A.; Wu, J.; Amodei, D. Scaling Laws for Neural Language Models. arXiv 2020, arXiv:2001.08361. [Google Scholar]
- Henighan, T.; Kaplan, J.; Katz, M.; Chen, M.; Hesse, C.; Jackson, J.; Jun, H.; Brown, T.B.; Dhariwal, P.; Gray, S.; et al. Scaling Laws for Autoregressive Generative Modeling. arXiv 2020, arXiv:2010.14701. [Google Scholar]
- Sharma, U.; Kaplan, J. A Neural Scaling Law from the Dimension of the Data Manifold. arXiv 2021, arXiv:2004.10802. [Google Scholar]
- Domingos, P. Every Model Learned by Gradient Descent Is Approximately a Kernel Machine. arXiv 2020, arXiv:2012.00152. [Google Scholar]
- Fedus, W.; Zoph, B.; Shazeer, N. Switch Transformers: Scaling to Trillion Parameter Models with Simple and Efficient Sparsity. arXiv 2021, arXiv:2101.03961. [Google Scholar]
- Dwivedi, V.P.; Joshi, C.K.; Laurent, T.; Bengio, Y.; Bresson, X. Benchmarking Graph Neural Networks. arXiv 2020, arXiv:2003.00982. [Google Scholar]
- Khemchandani, Y.; O’Hagan, S.; Samanta, S.; Swainston, N.; Roberts, T.J.; Bollegala, D.; Kell, D.B. DeepGraphMolGen, a multiobjective, computational strategy for generating molecules with desirable properties: A graph convolution and reinforcement learning approach. J. Cheminform. 2020, 12, 53. [Google Scholar] [CrossRef]
- Lim, J.; Hwang, S.Y.; Moon, S.; Kim, S.; Kim, W.Y. Scaffold-based molecular design with a graph generative model. Chem. Sci. 2020, 11, 1153–1164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, C.; Xu, M.; Zhu, Z.; Zhang, W.; Zhang, M.; Tang, J. GraphAF: A Flow-based Autoregressive Model for Molecular Graph Generation. arXiv 2020, arXiv:2001.09382. [Google Scholar]
- Wu, Z.; Pan, S.; Chen, F.; Long, G.; Zhang, C.; Yu, P.S. A Comprehensive Survey on Graph Neural Networks. IEEE Trans. Neural Netw. Learn. Syst. 2020, 32, 4–24. [Google Scholar] [CrossRef] [Green Version]
- David, L.; Thakkar, A.; Mercado, R.; Engkvist, O. Molecular representations in AI-driven drug discovery: A review and practical guide. J. Cheminform. 2020, 12, 56. [Google Scholar] [CrossRef]
- Elsken, T.; Metzen, J.H.; Hutter, F. Neural Architecture Search: A Survey. arXiv 2018, arXiv:1808.05377. [Google Scholar]
- Pham, H.; Guan, M.Y.; Zoph, B.; Le, Q.V.; Dean, J. Efficient Neural Architecture Search via Parameter Sharing. arXiv 2018, arXiv:1802.03268. [Google Scholar]
- Chithrananda, S.; Grand, G.; Ramsundar, B. ChemBERTa: Large-Scale Self-Supervised Pretraining for Molecular Property Prediction. arXiv 2020, arXiv:2010.09885. [Google Scholar]
- Tay, Y.; Dehghani, M.; Bahri, D.; Metzler, D. Efficient Transformers: A Survey. arXiv 2020, arXiv:2009.06732v2. [Google Scholar]
- Lin, T.; Wang, Y.; Liu, X.; Qiu, X. A Survey of Transformers. arXiv 2021, arXiv:2106.04554. [Google Scholar]
- Irie, K.; Schlag, I.; Csordás, R.; Schmidhuber, J. Going Beyond Linear Transformers with Recurrent Fast Weight Programmers. arXiv 2021, arXiv:2106.06295. [Google Scholar]
- Cahyawijaya, S. Greenformers: Improving Computation and Memory Efficiency in Transformer Models via Low-Rank Approximation. arXiv 2021, arXiv:2108.10808. [Google Scholar]
- Katharopoulos, A.; Vyas, A.; Pappas, N.; Fleuret, F. Transformers are RNNs: Fast Autoregressive Transformers with Linear Attention. arXiv 2020, arXiv:2006.16236. [Google Scholar]
- Khan, S.; Naseer, M.; Hayat, M.; Zamir, S.W.; Khan, F.S.; Shah, M. Transformers in Vision: A Survey. arXiv 2021, arXiv:2101.01169. [Google Scholar]
- Zhu, C.; Ping, W.; Xiao, C.; Shoeybi, M.; Goldstein, T.; Anandkumar, A.; Catanzaro, B. Long-Short Transformer: Efficient Transformers for Language and Vision. arXiv 2021, arXiv:2107.02192. [Google Scholar]
- Wang, S.; Li, B.Z.; Khabsa, M.; Fang, H.; Ma, H. Linformer: Self-Attention with Linear Complexity. arXiv 2020, arXiv:2006.04768. [Google Scholar]
- Kitaev, N.; Kaiser, Ł.; Levskaya, A. Reformer: The Efficient Transformer. arXiv 2020, arXiv:2001.04451. [Google Scholar]
- Shleifer, S.; Weston, J.; Ott, M. NormFormer: Improved Transformer Pretraining with Extra Normalization. arXiv 2021, arXiv:2110.09456. [Google Scholar]
- Tlusty, T.; Libchaber, A.; Eckmann, J.-P. Physical model of the sequence-to-function map of proteins. bioRxiv 2016, 069039. [Google Scholar] [CrossRef]


{kind=link}
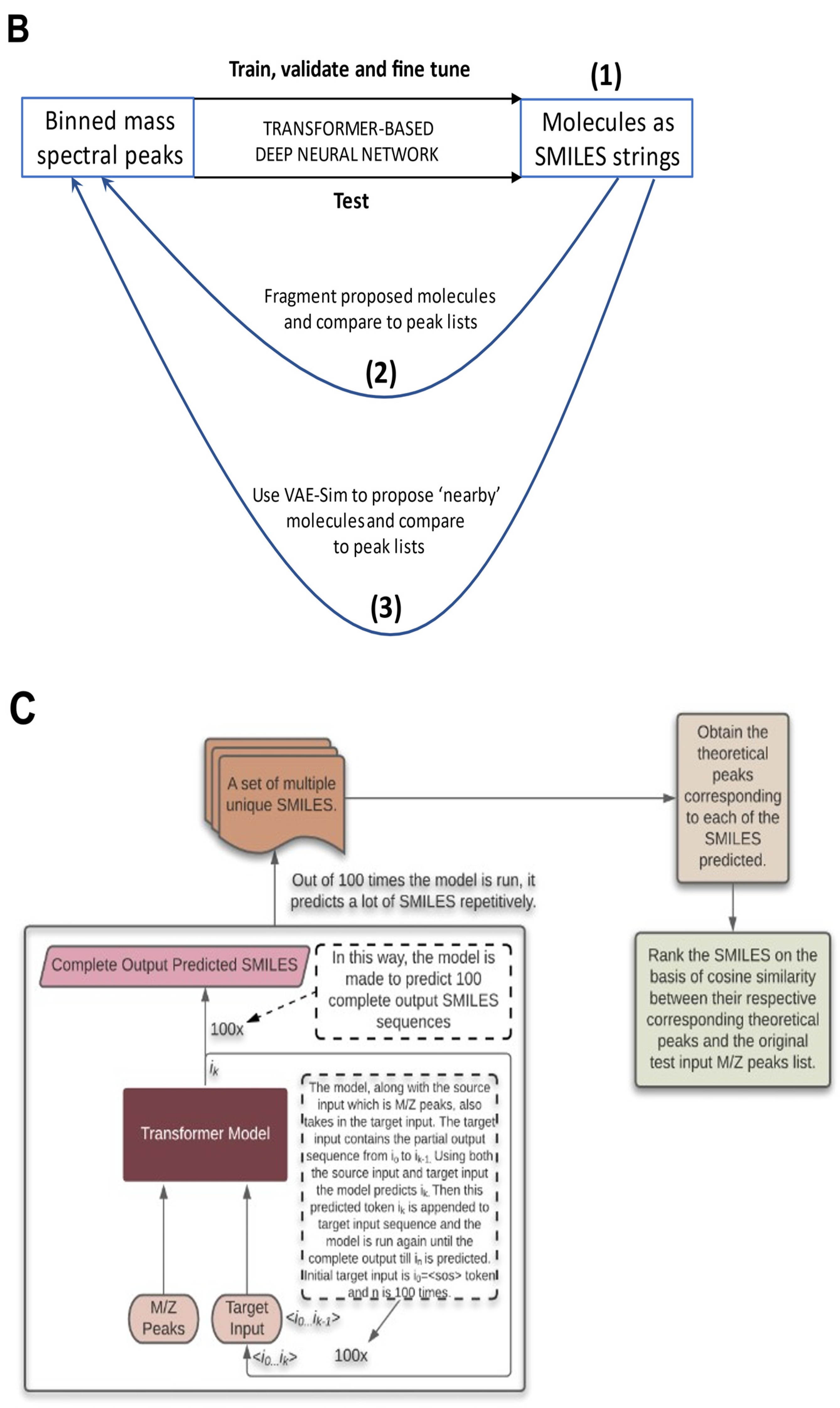{kind=link}
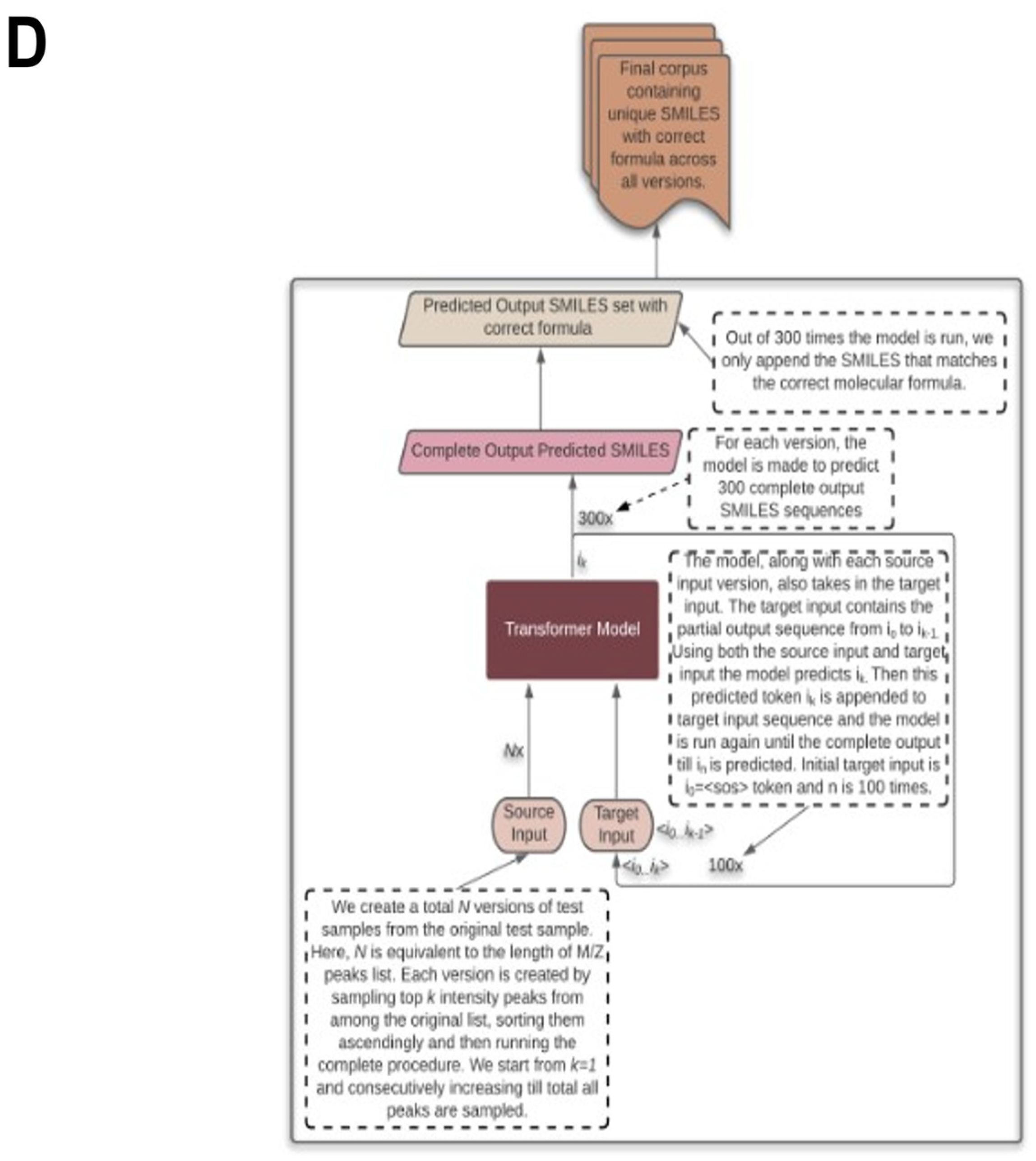{kind=link}
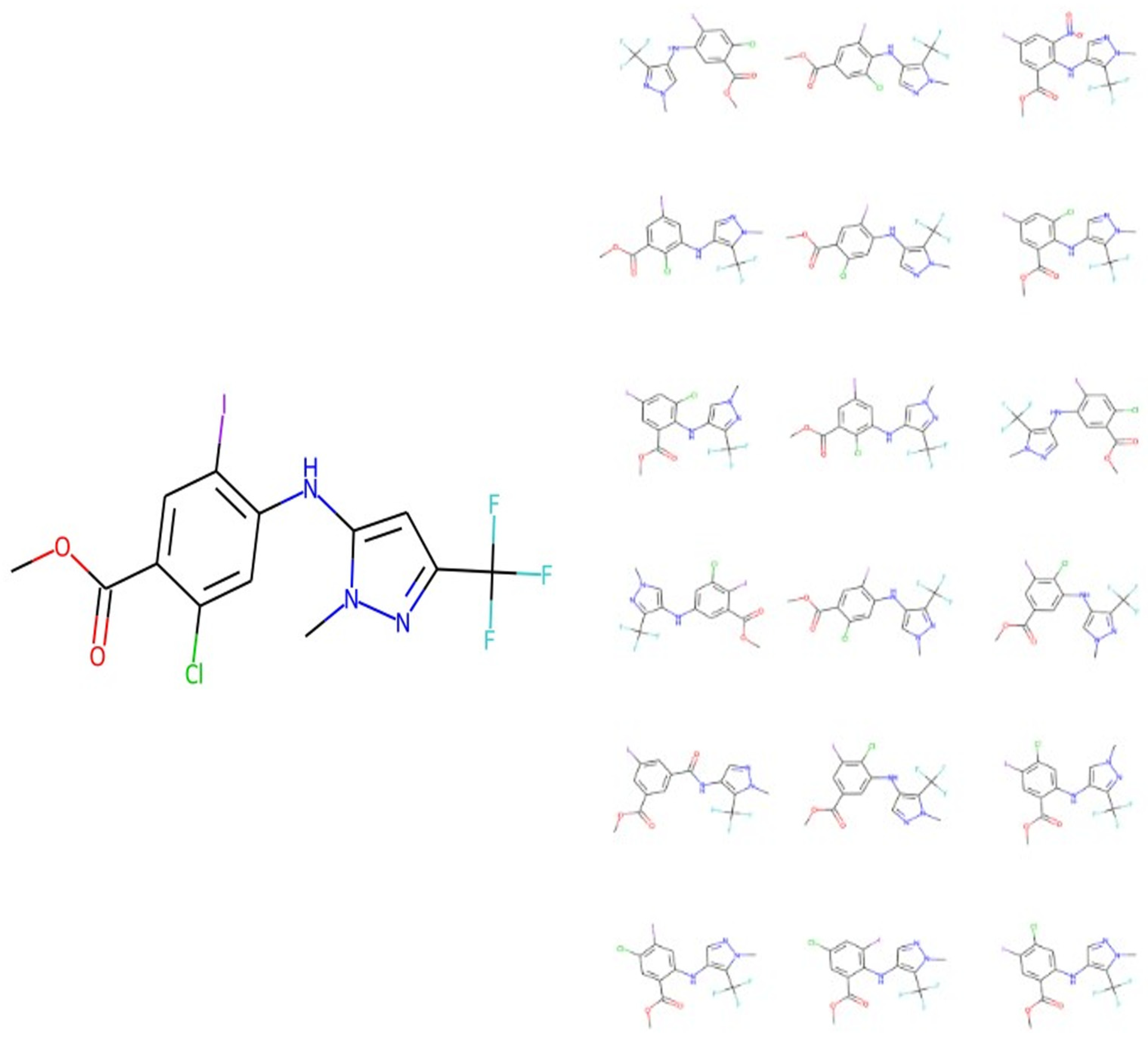{kind=link}
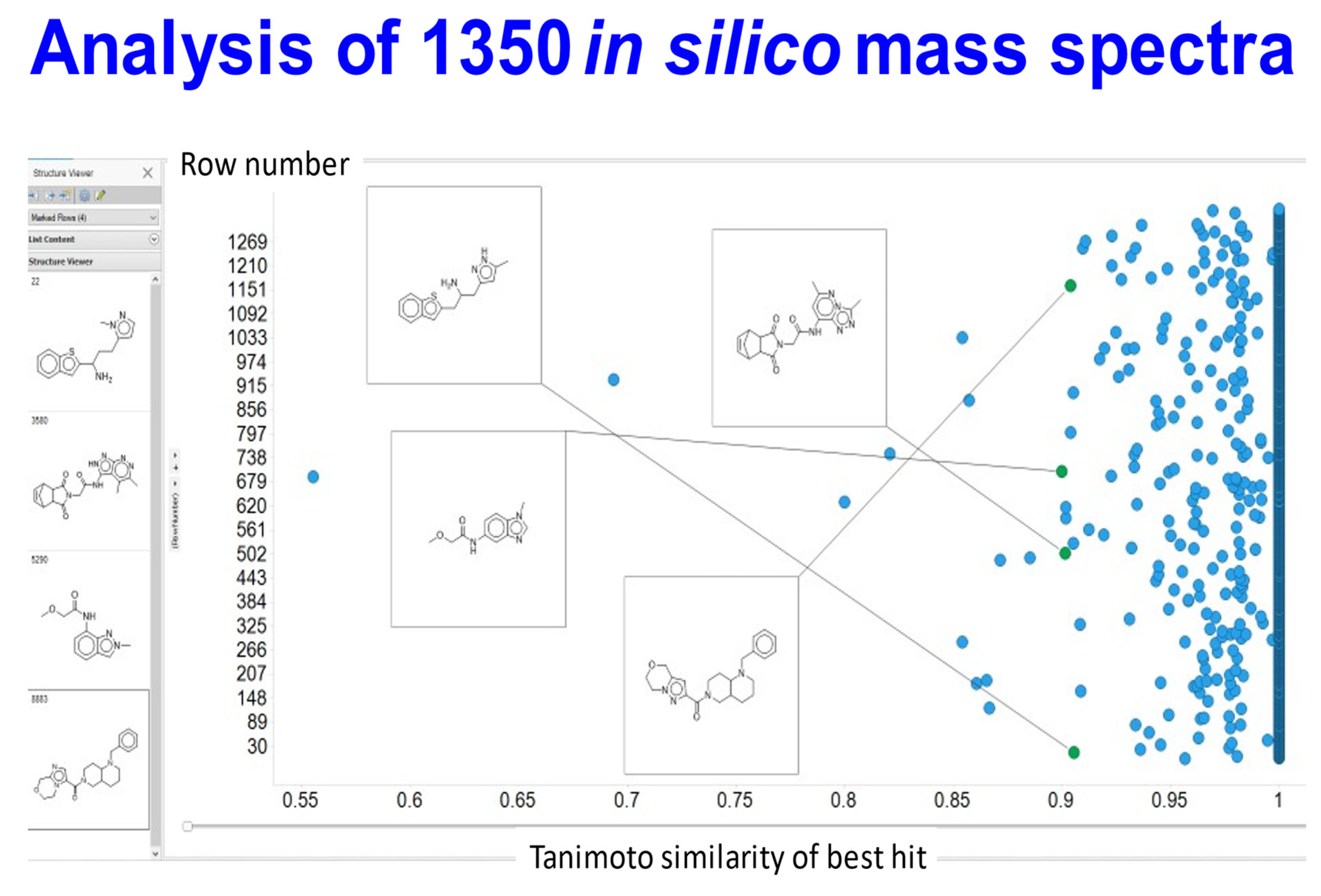{kind=link}
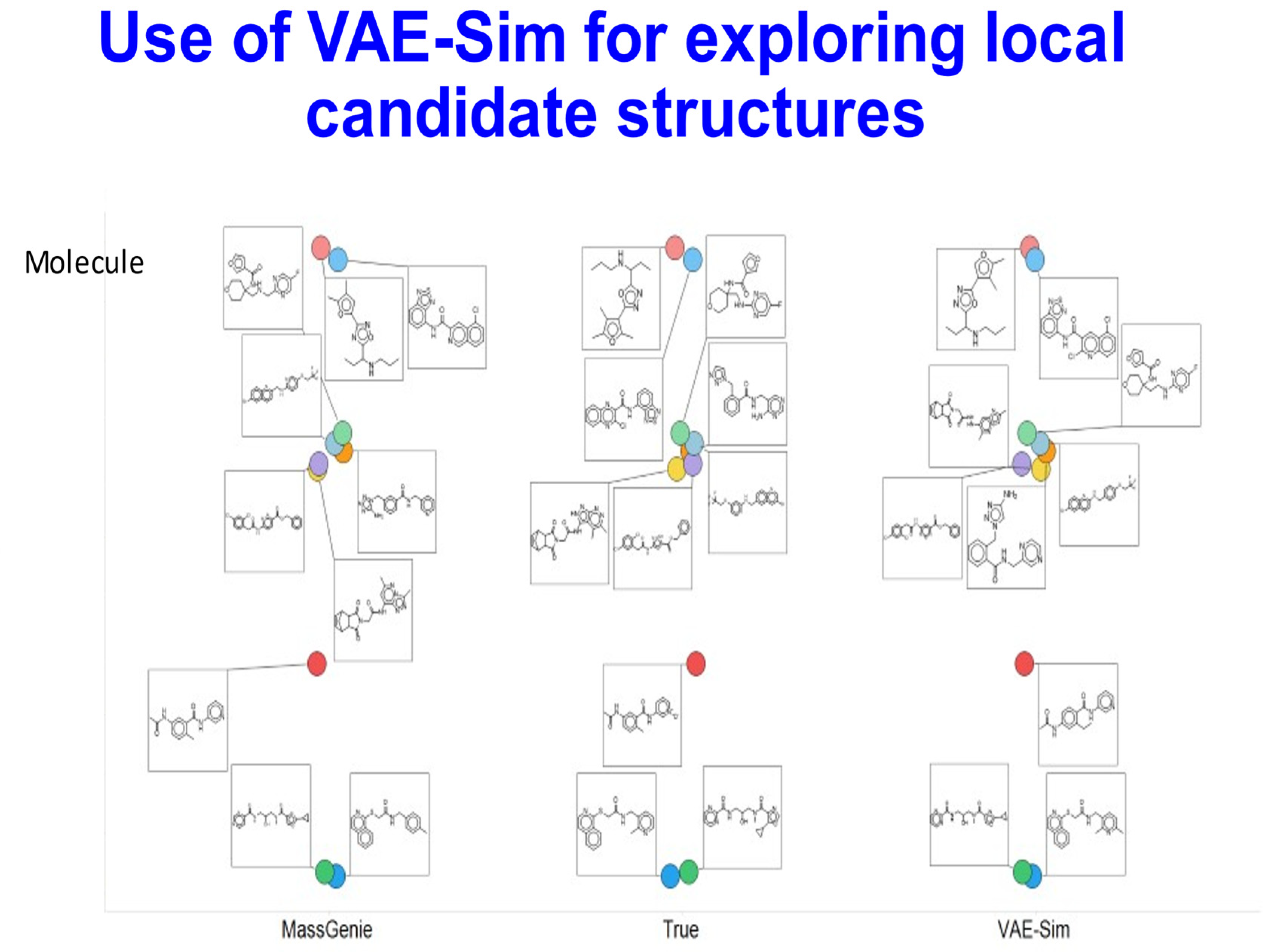{kind=link}
{kind=link}
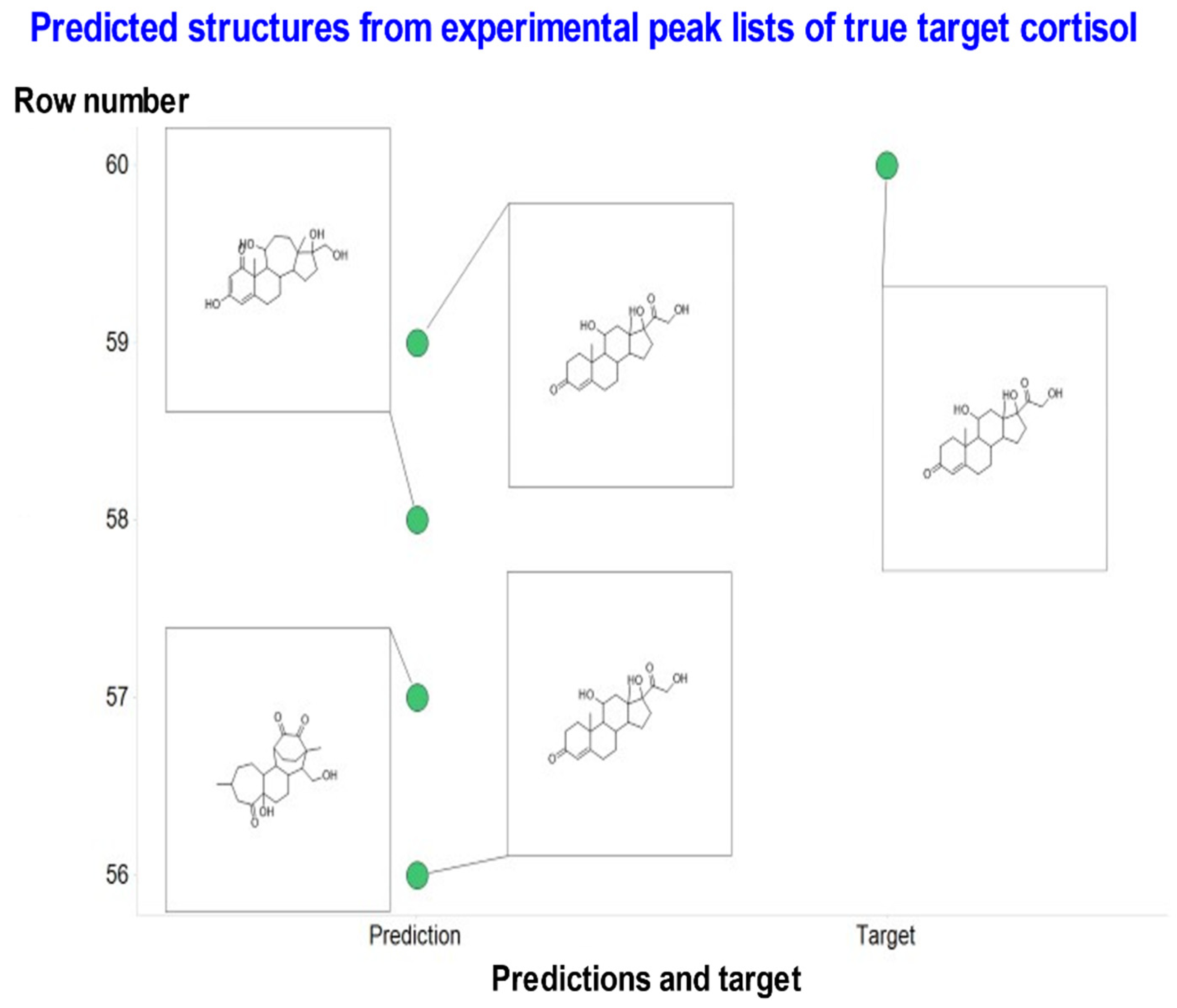{kind=link}
| Data Source | Recon 2 | Natural Products | Marketed Drugs | FLUOROPHORES | ZINC15-subset1 | ZINC15-subset2 | Total |
|---|---|---|---|---|---|---|---|
| Original smiles count | 1112 | 158,809 | 1381 | 143 | 249,455 | 6,202,415 | 6,613,315 |
| Canonical smiles count | 1056 | 148,971 | 1357 | 115 | 247,804 | 4,425,891 | 4,825,194 (4,774,258-unique) |
| Compound | Formula | Monoisotopic Mass | M + H | Matrix | Number of Peaks/Fragments | SMILES | Result | Comment |
|---|---|---|---|---|---|---|---|---|
| (Predicted by MS Software and Correct by Standard) | Canonical No Isotopic or Chiral Markings | |||||||
| Lauryl-l-carnitine | C19H37NO4 | 343.2723 | 344.2801 | STD in water | 11 | CCCCCCCCCCCC(=O)OC(C[N+](C)(C)C)CC(=O)[O-] | Part of options | Part of the options (3 out of 28) in ChemSpider |
| Stearoyl-l-carnitine | C25H49NO4 | 427.3662 | 428.374 | STD in water | 7 | CCCCCCCCCCCCCCCCCC(=O)OC(C[N+](C)(C)C)CC(=O)[O-] | Part of options | Part of the options (2 out of 2) in ChemSpider |
| Nicotinamide | C6H6N2O | 122.048012 | 123.055 | Serum | 8 | OC(=N)c1cccnc1 | Matched (single option) | Correct (based on Pubchem=—ChemSpider has an incorrect structure) |
| Glutamine | C5H10N2O3 | 146.069138 | 147.076 | Serum | 6 | NC(=O)CCC(C(=O)O)N | Part of options | Part of the options (3 out of 15) |
| 3-Nitro-L-Tyrosine | C9H10N2O5 | 226.058975 | 277.066 | STD in water | 13 | OC(=O)C(Cc1ccc(c(c1)[N+](=O)[O-])O)N | No prediction | No predictions |
| Asparagine | C4H8N2O3 | 132.053497 | 133.06 | STD in water | 5 | NC(=O)CC(C(=O)O)N | Part of options | proposed ureidopropionic acid; no asparagine |
| Glycylglycine | C4H8N2O3 | 132.053497 | 133.06 | STD in water | 3 | NCC(=NCC(=O)O)O | Part of options | Offers asparagine, ureido propionic acid and glycylglycine between (10) |
| Lactose | C12H22O11 | 342.1162115 | 343.1232 | STD in water | 9 | OCC1OC(O)C(C(C1OC1OC(CO)C(C(C1O)O)O)O)O | 41/54 | Identified it is a sugar |
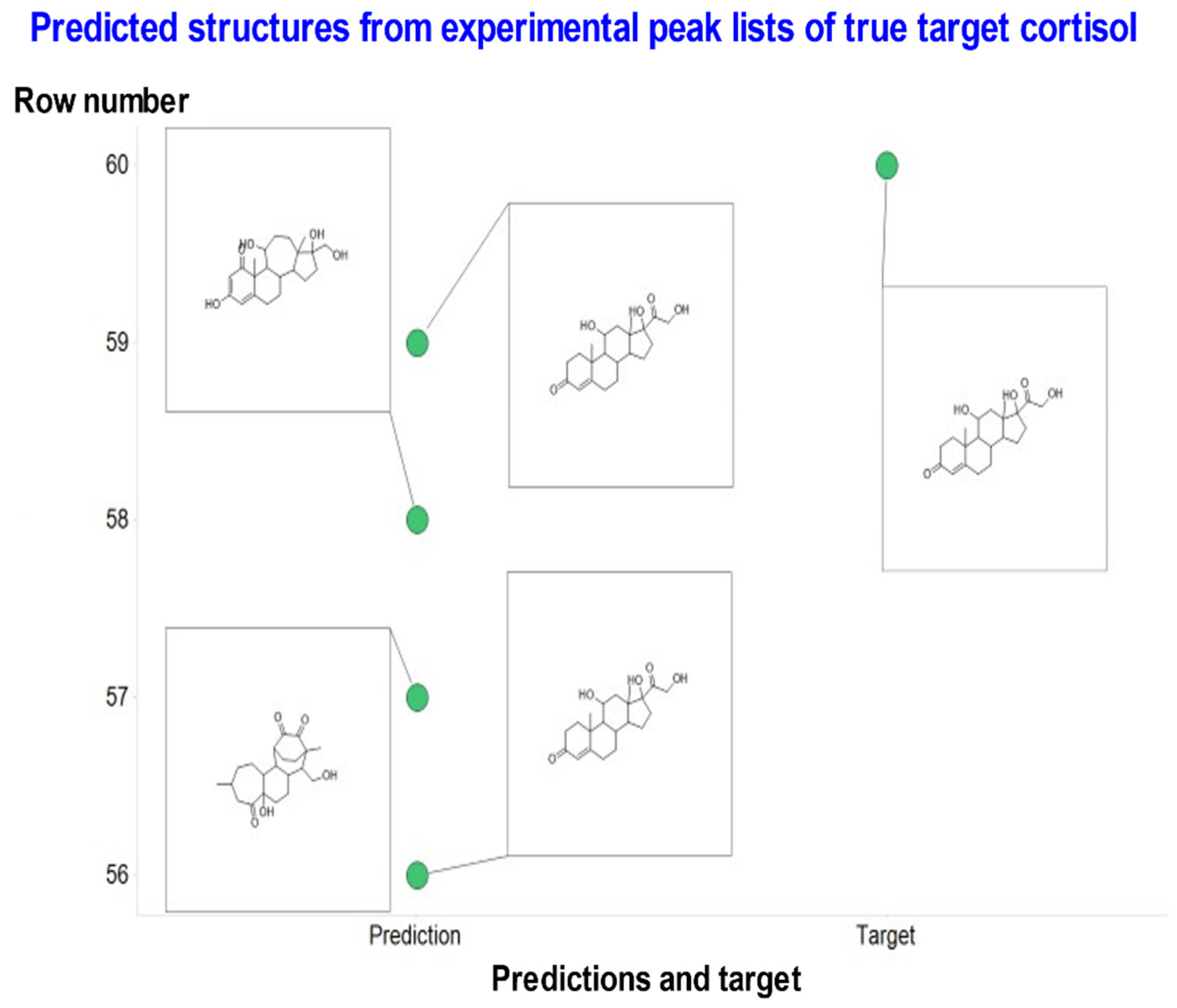
| Cortisol | C21H30O5 | 362.2093241 | 363.2161 | STD in water | 14 | OCC(=O)C1(O)CCC2C1(C)CC(O)C1C2CCC2=CC(=O)CCC12C | 2 out of 4 | Most common is correct |
| Corticosterone | C21H30O4 | 346.2144094 | 347.2216 | STD in water | 10 | OCC(=O)C1CCC2C1(C)CC(O)C1C2CCC2=CC(=O)CCC12C | 3 out of 8 | Most common is correct |
| Cortisone | C21H28O5 | 360.193674 | 361.2011 | STD in water | 16 | OCC(=O)C1(O)CCC2C1(C)CC(=O)C1C2CCC2=CC(=O)CCC12C | 2 out of 3 | Most common is correct |
| Protirelina | C16H22N6O4 | 362.170254 | 363.1767 | STD in water | 10 | O=C1CCC(N1)C(=O)NC(C(=O)N1CCCC1C(=O)N)Cc1[nH]cnc1 | No prediction with the correct formula | |
| Cortisol-21-acetate | C23H32O6 | 404.21989 | 405.2262 | STD in water | 12 | CC(=O)OCC(=O)C1(O)CCC2C1(C)CC(O)C1C2CCC2=CC(=O)CCC12C | ||
| Rosmarinate | C18H16O8 | 360.0845175 | 361.092 | STD in water | 11 | O=C(OC(C(=O)O)Cc1ccc(c(c1)O)O)C=Cc1ccc(c(c1)O)O | ||
| Deoxycorticosterone acetate | C23H32O4 | 372.23006 | 373.2369 | STD in water | 15 | CC(=O)OCC(=O)C1CCC2C1(C)CCC1C2CCC2=CC(=O)CCC12C | ||
| Cytosine | C4H5N3O | 111.04326 | 112.0505 | Serum | 5 | Nc1ccnc(=O)[nH]1 | Part of options | 1 out of 17 options Non-blind test |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shrivastava, A.D.; Swainston, N.; Samanta, S.; Roberts, I.; Wright Muelas, M.; Kell, D.B. MassGenie: A Transformer-Based Deep Learning Method for Identifying Small Molecules from Their Mass Spectra. Biomolecules 2021, 11, 1793. https://doi.org/10.3390/biom11121793
Shrivastava AD, Swainston N, Samanta S, Roberts I, Wright Muelas M, Kell DB. MassGenie: A Transformer-Based Deep Learning Method for Identifying Small Molecules from Their Mass Spectra. Biomolecules. 2021; 11(12):1793. https://doi.org/10.3390/biom11121793
Chicago/Turabian StyleShrivastava, Aditya Divyakant, Neil Swainston, Soumitra Samanta, Ivayla Roberts, Marina Wright Muelas, and Douglas B. Kell. 2021. "MassGenie: A Transformer-Based Deep Learning Method for Identifying Small Molecules from Their Mass Spectra" Biomolecules 11, no. 12: 1793. https://doi.org/10.3390/biom11121793
APA StyleShrivastava, A. D., Swainston, N., Samanta, S., Roberts, I., Wright Muelas, M., & Kell, D. B. (2021). MassGenie: A Transformer-Based Deep Learning Method for Identifying Small Molecules from Their Mass Spectra. Biomolecules, 11(12), 1793. https://doi.org/10.3390/biom11121793