Reelin Functions, Mechanisms of Action and Signaling Pathways During Brain Development and Maturation


{kind=link}
{kind=link}
Abstract
1. Introduction
2. Neuronal Migration Phenotype of the Reeler Mutant Mouse
2.1. Cell Mispositioning in the Neocortex
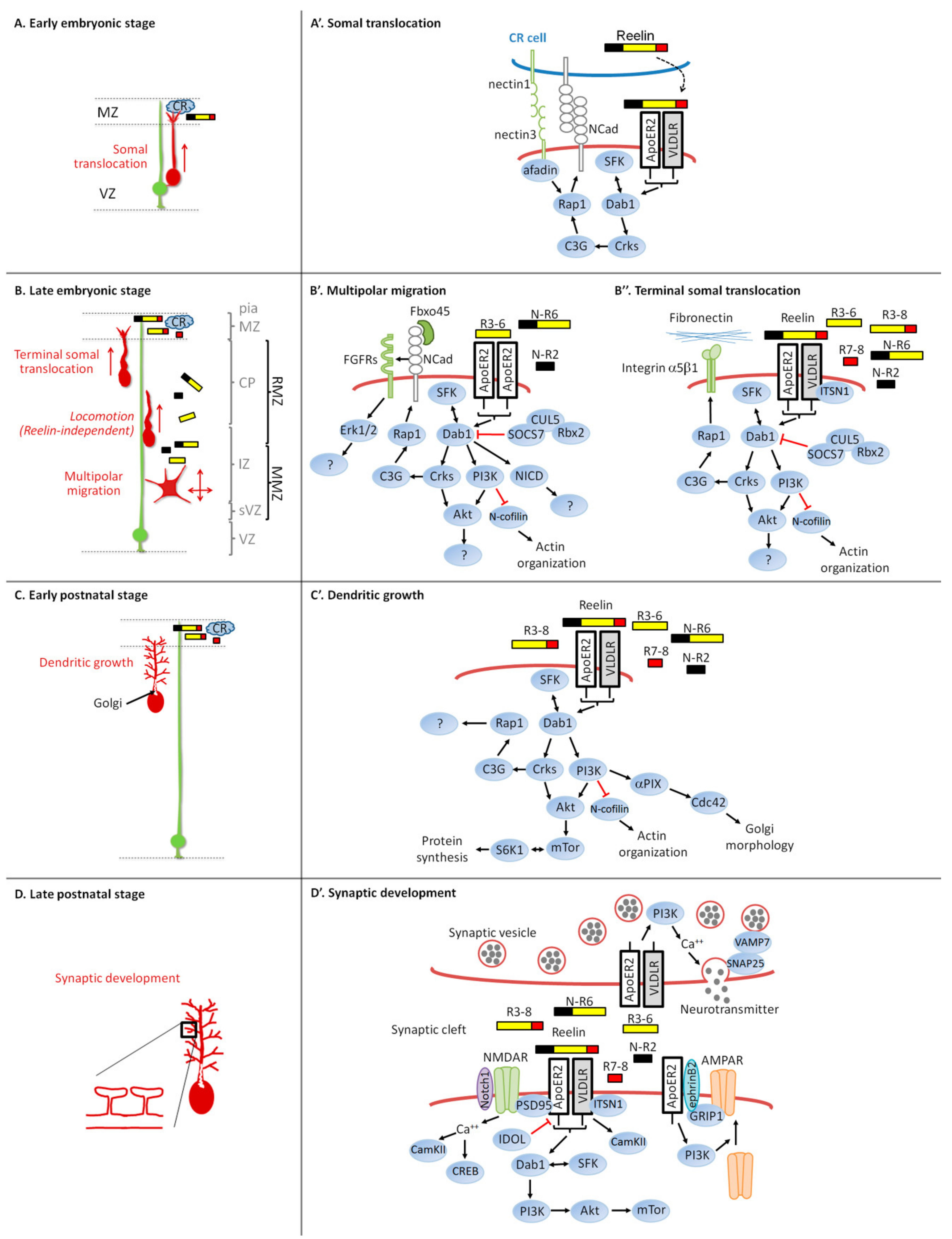
2.2. Mechanism of Action of Reelin During Neocortical Neuron Migration
2.3. Cell Mispositioning and Mechanism of Action of Reelin in the Hippocampus
2.4. Cell Mispositioning and Mechanism of Action of Reelin in the Cerebellum
2.5. Cell Mispositioning and Mechanism of Action of Reelin in the Olfactory Bulb
3. Reelin Signaling and Neuronal Migration
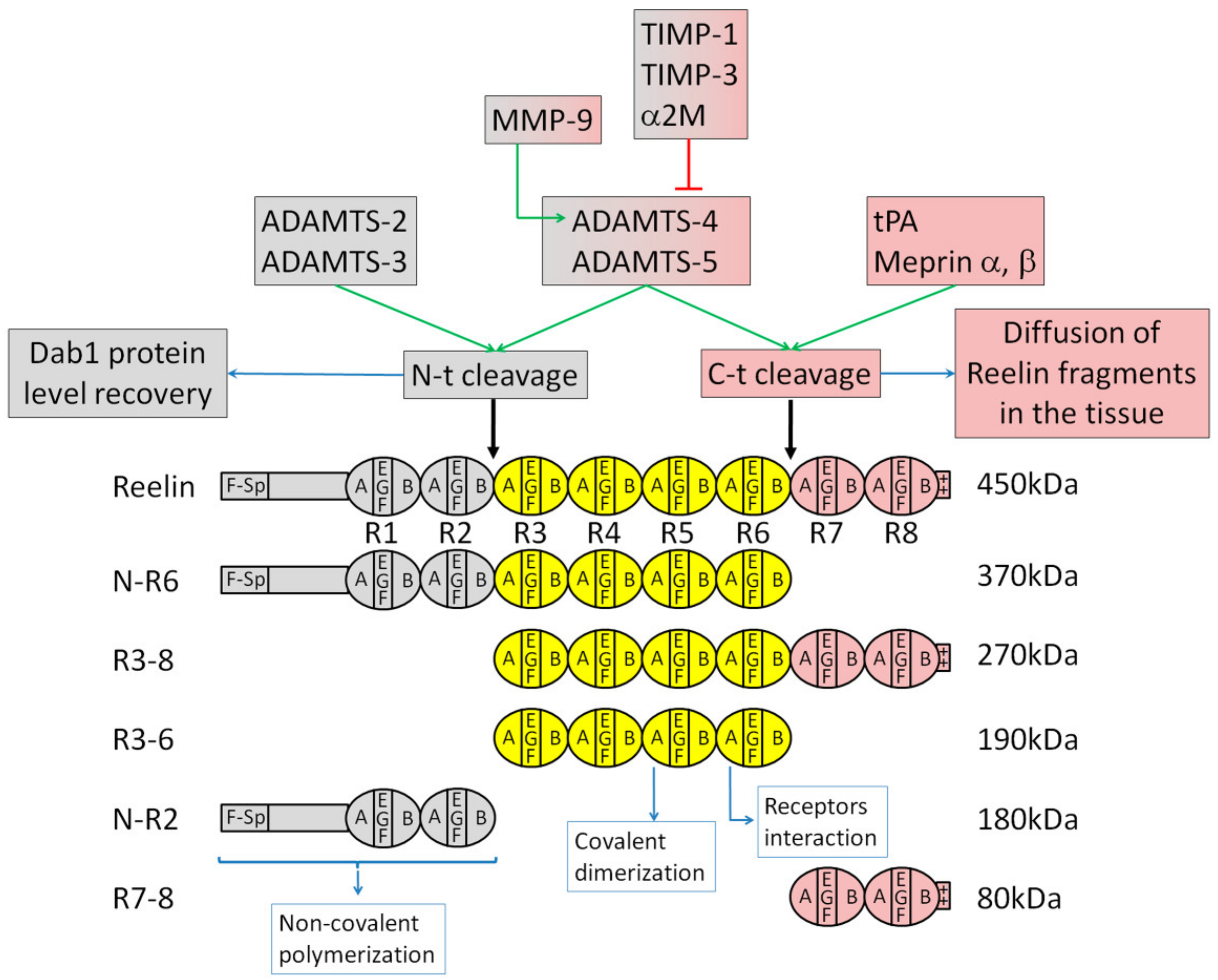
3.1. Reelin Structure and Proteolytic Processing
3.2. Intracellular Signaling Pathway and Neuronal Migration
4. Dendritic Growth Defect in the Reeler Mouse
5. Reelin Signaling and Dendritic Growth
6. Synaptic Plasticity Defect in the Reeler Mouse
7. Reelin Signaling and Synaptic Plasticity
8. Reelin and Human Neurological Diseases
9. Concluding Remarks
Funding
Acknowledgments
Conflicts of Interest
References
- Falconer, D.S. Two new mutants, ‘trembler’ and ‘reeler’, with neurological actions in the house mouse (Mus musculus L.). J. Genet. 1951, 50, 192–205. [Google Scholar] [CrossRef] [PubMed]
- Caviness, V.S.; So, D.K.; Sidman, R.L. The Hybrid Reeler Mouse. J. Hered. 1972, 63, 241–246. [Google Scholar] [CrossRef]
- Hamburgh, M. Analysis of the postnatal developmental effects of “reeler,” a neurological mutation in mice. A study in developmental genetics. Dev. Biol. 1963, 8, 165–185. [Google Scholar] [CrossRef]
- De Rouvroit, C.L.; Goffinet, A.M. The Reeler Mouse as a Model of Brain Development. In Factors Influencing Mammalian Kidney Development: Implications for Health in Adult Life; Springer Science and Business Media: Berlin/Heidelberg, Germany, 1998; Volume 150, pp. 1–106. [Google Scholar] [CrossRef]
- Rice, D.S.; Nusinowitz, S.; Azimi, A.M.; Martínez, A.; Soriano, E.; Curran, T. The Reelin Pathway Modulates the Structure and Function of Retinal Synaptic Circuitry. Neuron 2001, 31, 929–941. [Google Scholar] [CrossRef]
- Yip, J.W.; Yip, Y.P.L.; Nakajima, K.; Capriotti, C. Reelin controls position of autonomic neurons in the spinal cord. Proc. Natl. Acad. Sci. USA 2000, 97, 8612–8616. [Google Scholar] [CrossRef] [PubMed]
- Phelps, P.E.; Rich, R.; Dupuy-Davies, S.; Ríos, Y.; Wong, T. Evidence for a Cell-Specific Action of Reelin in the Spinal Cord. Dev. Biol. 2002, 244, 180–198. [Google Scholar] [CrossRef]
- Boyle, M.P.; Bernard, A.; Thompson, C.L.; Ng, L.; Boe, A.; Mortrud, M.; Hawrylycz, M.J.; Jones, A.R.; Hevner, R.F.; Lein, E.S. Cell-type-specific consequences of reelin deficiency in the mouse neocortex, hippocampus, and amygdala. J. Comp. Neurol. 2011, 519, 2061–2089. [Google Scholar] [CrossRef]
- Khialeeva, E.; Carpenter, E.M. Nonneuronal roles for the reelin signaling pathway. Dev. Dyn. 2016, 246, 217–226. [Google Scholar] [CrossRef]
- Takahashi, T.; Nowakowski, R.S.; Caviness, V. The cell cycle of the pseudostratified ventricular epithelium of the embryonic murine cerebral wall. J. Neurosci. 1995, 15, 6046–6057. [Google Scholar] [CrossRef]
- Zecevic, N.; Chen, Y.; Filipovic, R. Contributions of cortical subventricular zone to the development of the human cerebral cortex. J. Comp. Neurol. 2005, 491, 109–122. [Google Scholar] [CrossRef]
- Wood, J.G.; Martin, S.; Price, D.J. Evidence that the earliest generated cells of the murine cerebral cortex form a transient population in the subplate and marginal zone. Dev. Brain Res. 1992, 66, 137–140. [Google Scholar] [CrossRef]
- Meyer, G.; Schaaps, J.P.; Moreau, L.; Goffinet, A.M. Embryonic and Early Fetal Development of the Human Neocortex. J. Neurosci. 2000, 20, 1858–1868. [Google Scholar] [CrossRef] [PubMed]
- Marín-Padilla, M. Structural organization of the human cerebral cortex prior to the appearance of the cortical plate. Brain Struct. Funct. 1983, 168, 21–40. [Google Scholar] [CrossRef] [PubMed]
- Pedraza, M.; Hoerder-Suabedissen, A.; Albert-Maestro, M.A.; Molnar, Z.; De Carlos, J.A. Extracortical origin of some murine subplate cell populations. Proc. Natl. Acad. Sci. USA 2014, 111, 8613–8618. [Google Scholar] [CrossRef]
- Garcia-Moreno, F.; López-Mascaraque, L.; De Carlos, J.A. Origins and migratory routes of murine Cajal-Retzius cells. J. Comp. Neurol. 2006, 500, 419–432. [Google Scholar] [CrossRef] [PubMed]
- Morante, J.; Carleton, A.; Ortino, B.; Kremer, E.J.; Fairén, A.; Lledo, P.-M. Subpallial origin of a population of projecting pioneer neurons during corticogenesis. Proc. Natl. Acad. Sci. USA 2003, 100, 12468–12473. [Google Scholar] [CrossRef] [PubMed]
- Soriano, E.; Del Rio, J.A. The Cells of Cajal-Retzius: Still a Mystery One Century After. Neuron 2005, 46, 389–394. [Google Scholar] [CrossRef] [PubMed]
- Mukhtar, T.; Taylor, V. Untangling Cortical Complexity During Development. J. Exp. Neurosci. 2018, 12. [Google Scholar] [CrossRef] [PubMed]
- Kumamoto, T.; Hanashima, C. Neuronal subtype specification in establishing mammalian neocortical circuits. Neurosci. Res. 2014, 86, 37–49. [Google Scholar] [CrossRef]
- Caviness, V.S., Jr. Neocortical histogenesis in normal and reeler mice: A developmental study based upon [3H]thymidine autoradiography. Dev. Brain Res. 1982, 4, 293–302. [Google Scholar] [CrossRef]
- Sheppard, A.; Pearlman, A. Abnormal reorganization of preplate neurons and their associated extracellular matrix: An early manifestation of altered neocortical development in the reeler mutant mouse. J. Comp. Neurol. 1997, 378, 173–179. [Google Scholar] [CrossRef]
- Miyata, T.; Kawaguchi, A.; Okano, H.; Ogawa, M. Asymmetric Inheritance of Radial Glial Fibers by Cortical Neurons. Neuron 2001, 31, 727–741. [Google Scholar] [CrossRef]
- Kosodo, Y.; Toida, K.; Dubreuil, V.; Alexandre, P.; Schenk, J.; Kiyokage, E.; Attardo, A.; Mora-Bermúdez, F.; Arii, T.; Clarke, J.D.W.; et al. Cytokinesis of neuroepithelial cells can divide their basal process before anaphase. EMBO J. 2008, 27, 3151–3163. [Google Scholar] [CrossRef] [PubMed]
- Nadarajah, B.; Brunstrom, J.E.; Grutzendler, J.; Wong, R.O.L.; Pearlman, A.L. Two modes of radial migration in early development of the cerebral cortex. Nat. Neurosci. 2001, 4, 143–150. [Google Scholar] [CrossRef]
- Itoh, Y.; Moriyama, Y.; Hasegawa, T.; A Endo, T.; Toyoda, T.; Gotoh, Y. Scratch regulates neuronal migration onset via an epithelial-mesenchymal transition–like mechanism. Nat. Neurosci. 2013, 16, 416–425. [Google Scholar] [CrossRef]
- Rousso, D.L.; Pearson, C.A.; Gaber, Z.B.; Miquelajauregui, A.; Li, S.; Portera-Cailliau, C.; Morrisey, E.E.; Novitch, B.G. Foxp-Mediated Suppression of N-Cadherin Regulates Neuroepithelial Character and Progenitor Maintenance in the CNS. Neuron 2012, 74, 314–330. [Google Scholar] [CrossRef]
- Nichols, A.J.; Olson, E.C. Reelin Promotes Neuronal Orientation and Dendritogenesis during Preplate Splitting. Cereb. Cortex 2010, 20, 2213–2223. [Google Scholar] [CrossRef]
- DeFelipe, J. The Evolution of the Brain, the Human Nature of Cortical Circuits, and Intellectual Creativity. Front. Neuroanat. 2011, 5, 29. [Google Scholar] [CrossRef]
- Tabata, H.; Nakajima, K. Multipolar Migration: The Third Mode of Radial Neuronal Migration in the Developing Cerebral Cortex. J. Neurosci. 2003, 23, 9996–10001. [Google Scholar] [CrossRef]
- Noctor, S.C.; Martínez-Cerdeño, V.; Ivic, L.; Kriegstein, A.R. Cortical neurons arise in symmetric and asymmetric division zones and migrate through specific phases. Nat. Neurosci. 2004, 7, 136–144. [Google Scholar] [CrossRef]
- Jossin, Y. Molecular mechanisms of cell polarity in a range of model systems and in migrating neurons. Mol. Cell Neurosci. 2020, 106, 103503. [Google Scholar] [CrossRef] [PubMed]
- Elias, L.A.; Wang, R.D.; Kriegstein, A.R. Gap junction adhesion is necessary for radial migration in the neocortex. Nat. 2007, 448, 901–907. [Google Scholar] [CrossRef] [PubMed]
- Caviness, V.S.; Rakic, P. Mechanisms of Cortical Development: A View From Mutations in Mice. Annu. Rev. Neurosci. 1978, 1, 297–326. [Google Scholar] [CrossRef] [PubMed]
- Olson, E.C.; Park, P.J. Smooth, rough and upside-down neocortical development. Curr. Opin. Genet. Dev. 2002, 12, 320–327. [Google Scholar] [CrossRef]
- Jossin, Y. Neuronal Migration and the Role of Reelin During Early Development of the Cerebral Cortex. Mol. Neurobiol. 2004, 30, 225–252. [Google Scholar] [CrossRef]
- Jaglin, X.H.; Chelly, J. Tubulin-related cortical dysgeneses: Microtubule dysfunction underlying neuronal migration defects. Trends Genet. 2009, 25, 555–566. [Google Scholar] [CrossRef]
- Caviness, V.S.; Sidman, R.L. Time of origin of corresponding cell classes in the cerebral cortex of normal and reeler mutant mice: An autoradiographic analysis. J. Comp. Neurol. 1973, 148, 141–151. [Google Scholar] [CrossRef]
- Dekimoto, H.; Terashima, T.; Katsuyama, Y. Dispersion of the neurons expressing layer specific markers in the reeler brain. Dev. Growth Differ. 2010, 52, 181–193. [Google Scholar] [CrossRef]
- D’Arcangelo, G.; Miao, G.G.; Chen, S.-C.; Scares, H.D.; Morgan, J.I.; Curran, T. A protein related to extracellular matrix proteins deleted in the mouse mutant reeler. Nature 1995, 374, 719–723. [Google Scholar] [CrossRef]
- Gilmore, E.C.; Herrup, K. Cortical development: Receiving Reelin. Curr. Biol. 2000, 10, R162–R166. [Google Scholar] [CrossRef]
- Pinto-Lord, M.; Evrard, P.; Caviness, V.S., Jr. Obstructed neuronal migration along radial glial fibers in the neocortex of the reeler mouse: A golgi-EM analysis. Dev. Brain Res. 1982, 4, 379–393. [Google Scholar] [CrossRef]
- Dulabon, L.; Olson, E.C.; Taglienti, M.G.; Eisenhuth, S.; McGrath, B.; A Walsh, C.; A Kreidberg, J.; Anton, E. Reelin Binds α3β1 Integrin and Inhibits Neuronal Migration. Neuron 2000, 27, 33–44. [Google Scholar] [CrossRef]
- Sanada, K.; Gupta, A.; Tsai, L.-H. Disabled-1-Regulated Adhesion of Migrating Neurons to Radial Glial Fiber Contributes to Neuronal Positioning during Early Corticogenesis. Neuron 2004, 42, 197–211. [Google Scholar] [CrossRef]
- Trommsdorff, M.; Gotthardt, M.; Hiesberger, T.; Shelton, J.; Stockinger, W.; Nimpf, J.; E Hammer, R.; Richardson, J.A.; Herz, J. Reeler/Disabled-like disruption of neuronal migration in knockout mice lacking the VLDL receptor and ApoE receptor 2. Cell 1999, 97, 689–701. [Google Scholar] [CrossRef]
- Cooper, J.A. A mechanism for inside-out lamination in the neocortex. Trends Neurosci. 2008, 31, 113–119. [Google Scholar] [CrossRef]
- Olson, E.C.; Kim, S.; Walsh, C.A. Impaired Neuronal Positioning and Dendritogenesis in the Neocortex after Cell-Autonomous Dab1 Suppression. J. Neurosci. 2006, 26, 1767–1775. [Google Scholar] [CrossRef]
- Sekine, K.; Honda, T.; Kawauchi, T.; Kubo, K.; Nakajima, K. The Outermost Region of the Developing Cortical Plate Is Crucial for Both the Switch of the Radial Migration Mode and the Dab1-Dependent “Inside-Out” Lamination in the Neocortex. J. Neurosci. 2011, 31, 9426–9439. [Google Scholar] [CrossRef]
- Borrell, V.; Kaspar, B.K.; Gage, F.H.; Callaway, E.M. In vivo Evidence for Radial Migration of Neurons by Long-Distance Somal Translocation in the Developing Ferret Visual Cortex. Cereb. Cortex 2005, 16, 1571–1583. [Google Scholar] [CrossRef]
- Magdaleno, S.; Keshvara, L.; Curran, T. Rescue of ataxia and preplate splitting by ectopic expression of Reelin in reeler mice. Neuron 2002, 33, 573–586. [Google Scholar] [CrossRef]
- Jossin, Y.; Ignatova, N.; Hiesberger, T.; Herz, J.; De Rouvroit, C.L.; Goffinet, A.M. The Central Fragment of Reelin, Generated by Proteolytic Processing In Vivo, Is Critical to Its Function during Cortical Plate Development. J. Neurosci. 2004, 24, 514–521. [Google Scholar] [CrossRef]
- Jossin, Y.; Gui, L.; Goffinet, A.M. Processing of Reelin by Embryonic Neurons Is Important for Function in Tissue But Not in Dissociated Cultured Neurons. J. Neurosci. 2007, 27, 4243–4252. [Google Scholar] [CrossRef] [PubMed]
- Uchida, T.; Baba, A.; Martínez, F.J.P.; Hibi, T.; Miyata, T.; Luque, J.M.; Nakajima, K.; Hattori, M. Downregulation of Functional Reelin Receptors in Projection Neurons Implies That Primary Reelin Action Occurs atEarly/Premigratory Stages. J. Neurosci. 2009, 29, 10653–10662. [Google Scholar] [CrossRef] [PubMed]
- Meyer, G.; De Rouvroit, C.L.; Goffinet, A.M.; Wahle, P. Disabled-1 mRNA and protein expression in developing human cortex. Eur. J. Neurosci. 2003, 17, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Hirota, Y.; Kubo, K.; Katayama, K.-I.; Honda, T.; Fujino, T.; Yamamoto, T.T.; Nakajima, K. Reelin receptors ApoER2 and VLDLR are expressed in distinct spatiotemporal patterns in developing mouse cerebral cortex. J. Comp. Neurol. 2014, 523, 463–478. [Google Scholar] [CrossRef] [PubMed]
- Jossin, Y.; Cooper, J.A. Reelin, Rap1 and N-cadherin orient the migration of multipolar neurons in the developing neocortex. Nat. Neurosci. 2011, 14, 697–703. [Google Scholar] [CrossRef] [PubMed]
- Kon, E.; Calvo-Jimenez, E.; Cossard, A.; Na, Y.; A Cooper, J.; Jossin, Y. N-cadherin-regulated FGFR ubiquitination and degradation control mammalian neocortical projection neuron migration. eLife 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Jossin, Y. Polarization of migrating cortical neurons by Rap1 and N-cadherin. Small GTPases 2011, 2, 322–328. [Google Scholar] [CrossRef] [PubMed]
- Kon, E.; Cossard, A.; Jossin, Y. Neuronal Polarity in the Embryonic Mammalian Cerebral Cortex. Front. Cell. Neurosci. 2017, 11, 163. [Google Scholar] [CrossRef] [PubMed]
- Yano, M.; Hayakawa-Yano, Y.; Mele, A.; Darnell, R.B. Nova2 Regulates Neuronal Migration through an RNA Switch in Disabled-1 Signaling. Neuron 2010, 66, 848–858. [Google Scholar] [CrossRef]
- Kitazawa, A.; Kubo, K.; Hayashi, K.; Matsunaga, Y.; Ishii, K.; Nakajima, K. Hippocampal Pyramidal Neurons Switch from a Multipolar Migration Mode to a Novel “Climbing” Migration Mode during Development. J. Neurosci. 2014, 34, 1115–1126. [Google Scholar] [CrossRef]
- Bayer, S.A. Development of the hippocampal region in the rat I. Neurogenesis examined with3H-thymidine autoradiography. J. Comp. Neurol. 1980, 190, 87–114. [Google Scholar] [CrossRef] [PubMed]
- Stanfield, B.B.; Cowan, W.M. The development of the hippocampus and dentate gyrus in normal and reeler mice. J. Comp. Neurol. 1979, 185, 423–459. [Google Scholar] [CrossRef] [PubMed]
- Khalaf-Nazzal, R.; Francis, F. Hippocampal development—Old and new findings. Neurosci. 2013, 248, 225–242. [Google Scholar] [CrossRef] [PubMed]
- Rickmann, M.; Amaral, D.G.; Cowan, W.M. Organization of radial glial cells during the development of the rat dentate gyrus. J. Comp. Neurol. 1987, 264, 449–479. [Google Scholar] [CrossRef] [PubMed]
- Berg, D.; Bond, A.M.; Ming, G.-L.; Song, H. Radial glial cells in the adult dentate gyrus: What are they and where do they come from? F1000Research 2018, 7, 277. [Google Scholar] [CrossRef]
- Nelson, B.R.; Hodge, R.D.; Daza, R.A.; Tripathi, P.P.; Arnold, S.J.; Millen, K.J.; Hevner, R. Intermediate progenitors support migration of neural stem cells into dentate gyrus outer neurogenic niches. eLife 2020, 9, 9. [Google Scholar] [CrossRef]
- Förster, E.; Jossin, Y.; Zhao, S.; Chai, X.; Frotscher, M.; Goffinet, A.M. Recent progress in understanding the role of Reelin in radial neuronal migration, with specific emphasis on the dentate gyrus. Eur. J. Neurosci. 2006, 23, 901–909. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Chai, X.; Förster, E.; Frotscher, M. Reelin is a positional signal for the lamination of dentate granule cells. Development 2004, 131, 5117–5125. [Google Scholar] [CrossRef]
- Wang, S.; Brunne, B.; Zhao, S.; Chai, X.; Li, J.; Lau, J.; Failla, A.V.; Zobiak, B.; Sibbe, M.; Westbrook, G.L.; et al. Trajectory Analysis Unveils Reelin’s Role in the Directed Migration of Granule Cells in the Dentate Gyrus. J. Neurosci. 2017, 38, 137–148. [Google Scholar] [CrossRef]
- Hong, S.E.; Shugart, Y.Y.; Huang, D.T.; Al Shahwan, S.; Grant, P.E.; Hourihane, J.O.; Martin, N.D.; Walsh, C.A.; A Shahwan, S. Autosomal recessive lissencephaly with cerebellar hypoplasia is associated with human RELN mutations. Nat. Genet. 2000, 26, 93–96. [Google Scholar] [CrossRef]
- Chang, B.S.; Düzcan, F.; Kim, S.; Cinbis, M.; Aggarwal, A.; Apse, K.A.; Özdel, O.; Atmaca, M.; Zencir, S.; Bagci, H.; et al. The role ofRELN in lissencephaly and neuropsychiatric disease. Am. J. Med Genet. Part B Neuropsychiatr. Genet. 2006, 144, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Nishibe, M.; Katsuyama, Y.; Yamashita, T. Developmental abnormality contributes to cortex-dependent motor impairments and higher intracortical current requirement in the reeler homozygous mutants. Brain Struct. Funct. 2018, 223, 2575–2587. [Google Scholar] [CrossRef] [PubMed]
- Miale, I.L.; Sidman, R.L. An autoradiographic analysis of histogenesis in the mouse cerebellum. Exp. Neurol. 1961, 4, 277–296. [Google Scholar] [CrossRef]
- Rakic, P.; Sidman, R.L. Histogenesis of cortical layers in human cerebellum, particularly the lamina dissecans. J. Comp. Neurol. 1970, 139, 473–500. [Google Scholar] [CrossRef] [PubMed]
- Goffinet, A.M. The embryonic development of the cerebellum in normal and reeler mutant mice. Brain Struct. Funct. 1983, 168, 73–86. [Google Scholar] [CrossRef] [PubMed]
- Yuasa, S.; Kawamura, K.; Kuwano, R.; Ono, K. Neuron-glia interrelations during migration of Purkinje cells in the mouse embryonic cerebellum. Int. J. Dev. Neurosci. 1996, 14, 429–438. [Google Scholar] [CrossRef]
- Miyata, T.; Ono, Y.; Okamoto, M.; Masaoka, M.; Sakakibara, A.; Kawaguchi, A.; Hashimoto, M.; Ogawa, M. Migration, early axonogenesis, and Reelin-dependent layer-forming behavior of early/posterior-born Purkinje cells in the developing mouse lateral cerebellum. Neural Dev. 2010, 5, 23. [Google Scholar] [CrossRef]
- Ruiz i Altaba, A.; Palma, V.; Dahmane, N. Hedgehog-Gli signalling and the growth of the brain. Nat. Rev. Neurosci. 2002, 3, 24–33. [Google Scholar] [CrossRef]
- Kiessling, M.C.; Büttner, A.; Butti, C.; Müller-Starck, J.; Milz, S.; Hof, P.R.; Frank, H.-G.; Schmitz, C. Cerebellar granule cells are generated postnatally in humans. Brain Struct. Funct. 2013, 219, 1271–1286. [Google Scholar] [CrossRef]
- Schiffmann, S.N.; Bernier, B.; Goffinet, A.M. Reelin mRNA expression during mouse brain development. Eur. J. Neurosci. 1997, 9, 1055–1071. [Google Scholar] [CrossRef]
- Miyata, T.; Nakajima, K.; Aruga, J.; Takahashi, S.; Ikenaka, K.; Mikoshiba, K.; Ogawa, M. Distribution of a reeler gene-related antigen in the developing cerebellum: An immunohistochemical study with an allogeneic antibody CR-50 on normal and reeler mice. J. Comp. Neurol. 1996, 372, 215–228. [Google Scholar] [CrossRef]
- Yuasa, S.; Kitoh, J.; Oda, S.-I.; Kawamura, K. Obstructed migration of Purkinje cells in the developing cerebellum of the reeler mutant mouse. Brain Struct. Funct. 1993, 188, 317–329. [Google Scholar] [CrossRef] [PubMed]
- Schilling, K. Moving into shape: Cell migration during the development and histogenesis of the cerebellum. Histochem. Cell Biol. 2018, 150, 13–36. [Google Scholar] [CrossRef] [PubMed]
- Hevner, R.F. Reelin and the Cerebellum. In Reelin Glycoprotein; Springer Science and Business Media LLC.: Berlin/Heidelberg, Germany, 2008; pp. 141–158. [Google Scholar]
- Wyss, J.; Stanfield, B.; Cowan, W. Structural abnormalities in the olfactory bulb of the Reeler mouse. Brain Res. 1980, 188, 566–571. [Google Scholar] [CrossRef]
- Sun, W.; Kim, H.; Moon, Y. Control of neuronal migration through rostral migratory stream in mice. Anat. Cell Biol. 2010, 43, 269–279. [Google Scholar] [CrossRef] [PubMed]
- Lois, C.; A-Verdugo, J.-M.G.; Alvarez-Buylla, A. Chain Migration of Neuronal Precursors. Science 1996, 271, 978–981. [Google Scholar] [CrossRef] [PubMed]
- Kornack, D.R.; Rakic, P. The generation, migration, and differentiation of olfactory neurons in the adult primate brain. Proc. Natl. Acad. Sci. USA 2001, 98, 4752–4757. [Google Scholar] [CrossRef] [PubMed]
- Perez-Garcia, C.G.; Tissir, F.; Goffinet, A.M.; Meyer, G. Reelin receptors in developing laminated brain structures of mouse and human. Eur. J. Neurosci. 2004, 20, 2827–2832. [Google Scholar] [CrossRef]
- Hack, I.; Bancila, M.; Loulier, K.; Carroll, P.; Cremer, H. Reelin is a detachment signal in tangential chain-migration during postnatal neurogenesis. Nat. Neurosci. 2002, 5, 939–945. [Google Scholar] [CrossRef]
- Hellwig, S.; Hack, I.; Zucker, B.; Brunne, B.; Junghans, D. Reelin Together with ApoER2 Regulates Interneuron Migration in the Olfactory Bulb. PLoS ONE 2012, 7, e50646. [Google Scholar] [CrossRef][Green Version]
- Andrade, N.; Komnenovic, V.; Blake, S.M.; Jossin, Y.; Howell, B.; Goffinet, A.; Schneider, W.J.; Nimpf, J. ApoER2/VLDL receptor and Dab1 in the rostral migratory stream function in postnatal neuronal migration independently of Reelin. Proc. Natl. Acad. Sci. USA 2007, 104, 8508–8513. [Google Scholar] [CrossRef] [PubMed]
- Blake, S.M.; Strasser, V.; Andrade, N.; Duit, S.; Hofbauer, R.; Schneider, W.J.; Nimpf, J. Thrombospondin-1 binds to ApoER2 and VLDL receptor and functions in postnatal neuronal migration. EMBO J. 2008, 27, 3069–3080. [Google Scholar] [CrossRef] [PubMed]
- Hirotsune, S.; Takahara, T.; Sasaki, N.; Hirose, K.; Yoshiki, A.; Ohashi, T.; Kusakabe, M.; Murakami, Y.; Muramatsu, M.; Watanabe, S.; et al. The reeler gene encodes a protein with an EGF–like motif expressed by pioneer neurons. Nat. Genet. 1995, 10, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Bar, I.; De Rouvroit, C.; Royaux, I.; Krizman, D.; Dernoncourt, C.; Ruelle, D.; Beckers, M.; Goffinet, A. A YAC contig containing the reeler locus with preliminary characterization of candidate gene fragments. Genomics 1995, 26, 543–549. [Google Scholar] [CrossRef]
- De Bergeyck, V.; Nakajima, K.; De Rouvroit, C.L.; Naerhuyzen, B.; Goffinet, A.M.; Miyata, T.; Ogawa, M.; Mikoshiba, K. A truncated Reelin protein is produced but not secreted in the ’Orleans’ reeler mutation (Reln[rl-Orl]). Mol. Brain Res. 1997, 50, 85–90. [Google Scholar] [CrossRef]
- DeSilva, U.; D’Arcangelo, G.; Braden, V.V.; Chen, J.; Miao, G.G.; Curran, T.; Green, E.D. The human reelin gene: Isolation, sequencing, and mapping on chromosome 7. Genome Res. 1997, 7, 157–164. [Google Scholar] [CrossRef]
- De Rouvroit, C.L.; De Bergeyck, V.; Cortvrindt, C.; Bar, I.; Eeckhout, Y.; Goffinet, A.M. Reelin, the Extracellular Matrix Protein Deficient in Reeler Mutant Mice, Is Processed by a Metalloproteinase. Exp. Neurol. 1999, 156, 214–217. [Google Scholar] [CrossRef]
- Krstic, D.; Rodríguez, M.; Knuesel, I. Regulated Proteolytic Processing of Reelin through Interplay of Tissue Plasminogen Activator (tPA), ADAMTS-4, ADAMTS-5, and Their Modulators. PLoS ONE 2012, 7, e47793. [Google Scholar] [CrossRef]
- Trotter, J.H.; Lussier, A.L.; Psilos, K.E.; Mahoney, H.L.; Sponaugle, A.E.; Hoe, H.-S.; Rebeck, G.W.; Weeber, E.J. Extracellular proteolysis of reelin by tissue plasminogen activator following synaptic potentiation. Neuroscience 2014, 274, 299–307. [Google Scholar] [CrossRef]
- Koie, M.; Okumura, K.; Hisanaga, A.; Kamei, T.; Sasaki, K.; Deng, M.; Baba, A.; Kohno, T.; Hattori, M. Cleavage within Reelin Repeat 3 Regulates the Duration and Range of the Signaling Activity of Reelin Protein. J. Biol. Chem. 2014, 289, 12922–12930. [Google Scholar] [CrossRef]
- Sato, Y.; Kobayashi, D.; Kohno, T.; Kidani, Y.; Prox, J.; Becker-Pauly, C.; Hattori, M. Determination of cleavage site of Reelin between its sixth and seventh repeat and contribution of meprin metalloproteases to the cleavage. J. Biochem. 2015, 159, mvv102. [Google Scholar] [CrossRef][Green Version]
- Hisanaga, A.; Morishita, S.; Suzuki, K.; Sasaki, K.; Koie, M.; Kohno, T.; Hattori, M. A disintegrin and metalloproteinase with thrombospondin motifs 4 (ADAMTS-4) cleaves Reelin in an isoform-dependent manner. FEBS Lett. 2012, 586, 3349–3353. [Google Scholar] [CrossRef] [PubMed]
- Ogino, H.; Hisanaga, A.; Kohno, T.; Kondo, Y.; Okumura, K.; Kamei, T.; Sato, T.; Asahara, H.; Tsuiji, H.; Fukata, M.; et al. Secreted Metalloproteinase ADAMTS-3 Inactivates Reelin. J. Neurosci. 2017, 37, 3181–3191. [Google Scholar] [CrossRef] [PubMed]
- Yamakage, Y.; Kato, M.; Hongo, A.; Ogino, H.; Ishii, K.; Ishizuka, T.; Kamei, T.; Tsuiji, H.; Miyamoto, T.; Oishi, H.; et al. A disintegrin and metalloproteinase with thrombospondin motifs 2 cleaves and inactivates Reelin in the postnatal cerebral cortex and hippocampus, but not in the cerebellum. Mol. Cell. Neurosci. 2019, 100, 103401. [Google Scholar] [CrossRef] [PubMed]
- Kubo, K.; Mikoshiba, K.; Nakajima, K. Secreted Reelin molecules form homodimers. Neurosci. Res. 2002, 43, 381–388. [Google Scholar] [CrossRef]
- Kohno, S.; Kohno, T.; Nakano, Y.; Suzuki, K.; Ishii, M.; Tagami, H.; Baba, A.; Hattori, M. Mechanism and significance of specific proteolytic cleavage of Reelin. Biochem. Biophys. Res. Commun. 2009, 380, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Okugawa, E.; Ogino, H.; Shigenobu, T.; Yamakage, Y.; Tsuiji, H.; Oishi, H.; Kohno, T.; Hattori, M. Physiological significance of proteolytic processing of Reelin revealed by cleavage-resistant Reelin knock-in mice. Sci. Rep. 2020, 10, 1–13. [Google Scholar] [CrossRef]
- Duit, S.; Mayer, H.; Blake, S.M.; Schneider, W.J.; Nimpf, J. Differential Functions of ApoER2 and Very Low Density Lipoprotein Receptor in Reelin Signaling Depend on Differential Sorting of the Receptors*. J. Biol. Chem. 2009, 285, 4896–4908. [Google Scholar] [CrossRef]
- Dlugosz, P.; Tresky, R.; Nimpf, J. Differential Action of Reelin on Oligomerization of ApoER2 and VLDL Receptor in HEK293 Cells Assessed by Time-Resolved Anisotropy and Fluorescence Lifetime Imaging Microscopy. Front. Mol. Neurosci. 2019, 12, 53. [Google Scholar] [CrossRef]
- Tinnes, S.; Schäfer, M.K.E.; Flubacher, A.; Münzner, G.; Frotscher, M.; Haas, C. Epileptiform activity interferes with proteolytic processing of Reelin required for dentate granule cell positioning. FASEB J. 2010, 25, 1002–1013. [Google Scholar] [CrossRef]
- Duveau, V.; Madhusudan, A.; Caleo, M.; Knuesel, I.; Fritschy, J.-M. Impaired reelin processing and secretion by Cajal-Retzius cells contributes to granule cell dispersion in a mouse model of temporal lobe epilepsy. Hippocampus 2010, 21, 935–944. [Google Scholar] [CrossRef] [PubMed]
- Tinnes, S.; Ringwald, J.; Haas, C.A. TIMP-1 inhibits the proteolytic processing of Reelin in experimental epilepsy. FASEB J. 2013, 27, 2542–2552. [Google Scholar] [CrossRef] [PubMed]
- Haas, C.; Dudeck, O.; Kirsch, M.; Huszka, C.; Kann, G.; Pollak, S.; Zentner, J.; Frotscher, M. Role for Reelin in the Development of Granule Cell Dispersion in Temporal Lobe Epilepsy. J. Neurosci. 2002, 22, 5797–5802. [Google Scholar] [CrossRef]
- Orcinha, C.; Münzner, G.; Gerlach, J.; Kilias, A.; Follo, M.; Egert, U.; Haas, C. Seizure-Induced Motility of Differentiated Dentate Granule Cells Is Prevented by the Central Reelin Fragment. Front. Cell. Neurosci. 2016, 10, 7779. [Google Scholar] [CrossRef] [PubMed]
- Sáez-Valero, J.; Costell, M.; Sjögren, M.; Andreasen, N.; Blennow, K.; Luque, J.M. Altered levels of cerebrospinal fluid reelin in frontotemporal dementia and Alzheimer’s disease. J. Neurosci. Res. 2003, 72, 132–136. [Google Scholar] [CrossRef] [PubMed]
- Botella-López, A.; Cuchillo-Ibañez, I.; Cotrufo, T.; Mok, S.S.; Li, Q.-X.; Barquero, M.-S.; Dierssen, M.; Soriano, E.; Sáez-Valero, J. β-amyloid controls altered Reelin expression and processing in Alzheimer’s disease. Neurobiol. Dis. 2010, 37, 682–691. [Google Scholar] [CrossRef]
- Fatemi, S.H.; Kroll, J.L.; Stary, J.M. Altered levels of Reelin and its isoforms in schizophrenia and mood disorders. NeuroReport 2001, 12, 3209–3215. [Google Scholar] [CrossRef]
- Chin, J.; Massaro, C.M.; Palop, J.J.; Thwin, M.T.; Yu, G.Q.; Bien-Ly, N.; Bender, A.; Mucke, L. Reelin depletion in the entorhinal cortex of human amyloid precursor protein transgenic mice and humans with Alzheimer’s disease. J. Neurosci. 2007, 27, 2727–2733. [Google Scholar] [CrossRef]
- Kohno, T.; Honda, T.; Kubo, K.; Nakano, Y.; Tsuchiya, A.; Murakami, T.; Banno, H.; Nakajima, K.; Hattori, M. Importance of Reelin C-Terminal Region in the Development and Maintenance of the Postnatal Cerebral Cortex and Its Regulation by Specific Proteolysis. J. Neurosci. 2015, 35, 4776–4787. [Google Scholar] [CrossRef]
- Nakamura, K.; Beppu, M.; Sakai, K.; Yagyu, H.; Matsumaru, S.; Kohno, T.; Hattori, M. The C-terminal region of Reelin is necessary for proper positioning of a subset of Purkinje cells in the postnatal cerebellum. Neuroscience 2016, 336, 20–29. [Google Scholar] [CrossRef]
- D’Arcangelo, G.; Homayouni, R.; Keshvara, L.; Rice, D.S.; Sheldon, M.; Curran, T. Reelin Is a Ligand for Lipoprotein Receptors. Neuron 1999, 24, 471–479. [Google Scholar] [CrossRef]
- Hiesberger, T.; Trommsdorff, M.; Howell, B.W.; Goffinet, A.; Mumby, M.C.; A Cooper, J.; Herz, J. Direct Binding of Reelin to VLDL Receptor and ApoE Receptor 2 Induces Tyrosine Phosphorylation of Disabled-1 and Modulates Tau Phosphorylation. Neuron 1999, 24, 481–489. [Google Scholar] [CrossRef]
- Dlugosz, P.; Nimpf, J. The Reelin Receptors Apolipoprotein E receptor 2 (ApoER2) and VLDL Receptor. Int. J. Mol. Sci. 2018, 19, 3090. [Google Scholar] [CrossRef] [PubMed]
- Andersen, O.M.; Benhayon, D.; Curran, T.; Willnow, T.E. Differential Binding of Ligands to the Apolipoprotein E Receptor 2. Biochemistry 2003, 42, 9355–9364. [Google Scholar] [CrossRef] [PubMed]
- Yasui, N.; Kitago, Y.; Beppu, A.; Kohno, T.; Morishita, S.; Gomi, H.; Nagae, M.; Hattori, M.; Takagi, J. Functional Importance of Covalent Homodimer of Reelin Protein Linked via Its Central Region*. J. Biol. Chem. 2011, 286, 35247–35256. [Google Scholar] [CrossRef] [PubMed]
- Benhayon, D.; Magdaleno, S.; Curran, T. Binding of purified Reelin to ApoER2 and VLDLR mediates tyrosine phosphorylation of Disabled-1. Mol. Brain Res. 2003, 112, 33–45. [Google Scholar] [CrossRef]
- Yasui, N.; Nogi, T.; Takagi, J. Structural Basis for Specific Recognition of Reelin by Its Receptors. Structure 2010, 18, 320–331. [Google Scholar] [CrossRef]
- Strasser, V.; Fasching, D.; Hauser, C.; Mayer, H.; Bock, H.H.; Hiesberger, T.; Herz, J.; Weeber, E.J.; Sweatt, J.D.; Pramatarova, A.; et al. Receptor Clustering Is Involved in Reelin Signaling. Mol. Cell. Biol. 2004, 24, 1378–1386. [Google Scholar] [CrossRef]
- Hirota, Y.; Kubo, K.; Fujino, T.; Yamamoto, T.T.; Nakajima, K. ApoER2 Controls Not Only Neuronal Migration in the Intermediate Zone But Also Termination of Migration in the Developing Cerebral Cortex. Cereb. Cortex 2016, 28, 223–235. [Google Scholar] [CrossRef]
- Hack, I.; Hellwig, S.; Junghans, D.; Brunne, B.; Bock, H.H.; Zhao, S.; Frotscher, M. Divergent roles of ApoER2 and Vldlr in the migration of cortical neurons. Development 2007, 134, 3883–3891. [Google Scholar] [CrossRef]
- Gotthardt, M.; Trommsdorff, M.; Nevitt, M.F.; Shelton, J.; Richardson, J.A.; Stockinger, W.; Nimpf, J.; Herz, J. Interactions of the Low Density Lipoprotein Receptor Gene Family with Cytosolic Adaptor and Scaffold Proteins Suggest Diverse Biological Functions in Cellular Communication and Signal Transduction. J. Biol. Chem. 2000, 275, 25616–25624. [Google Scholar] [CrossRef] [PubMed]
- Minami, S.S.; Sung, Y.M.; Dumanis, S.B.; Chi, S.H.; Burns, M.P.; Ann, E.-J.; Suzuki, T.; Turner, R.S.; Park, H.-S.; Pak, D.T.S.; et al. The cytoplasmic adaptor protein X11α and extracellular matrix protein Reelin regulate ApoE receptor 2 trafficking and cell movement. FASEB J. 2009, 24, 58–69. [Google Scholar] [CrossRef] [PubMed]
- Beffert, U.; Weeber, E.J.; Durudas, A.; Qiu, S.; Masiulis, I.; Sweatt, J.D.; Li, W.-P.; Adelmann, G.; Frotscher, M.; Hammer, R.E.; et al. Modulation of Synaptic Plasticity and Memory by Reelin Involves Differential Splicing of the Lipoprotein Receptor Apoer2. Neuron 2005, 47, 567–579. [Google Scholar] [CrossRef] [PubMed]
- Howell, B.W.; Herrick, T.M.; A Cooper, J. Reelin-induced tryosine phosphorylation of Disabled 1 during neuronal positioning. Genome Res. 1999, 13, 643–648. [Google Scholar] [CrossRef]
- Howell, B.W.; Gertler, F.B.; Cooper, J.A. Mouse disabled (mDab1): A Src binding protein implicated in neuronal development. EMBO J. 1997, 16, 121–132. [Google Scholar] [CrossRef]
- Howell, B.W.; Lanier, L.M.; Frank, R.; Gertler, F.B.; A Cooper, J. The Disabled 1 Phosphotyrosine-Binding Domain Binds to the Internalization Signals of Transmembrane Glycoproteins and to Phospholipids. Mol. Cell. Biol. 1999, 19, 5179–5188. [Google Scholar] [CrossRef]
- Howell, B.W.; Hawkes, R.; Soriano, P.; Cooper, J.A. Neuronal position in the developing brain is regulated by mouse disabled-1. Nature 1997, 389, 733–737. [Google Scholar] [CrossRef]
- Sheldon, M.; Rice, D.S.; D’Arcangelo, G.; Yoneshima, H.; Nakajima, K.; Mikoshiba, K.; Howell, B.W.; Cooper, J.A.; Goldowitz, D.; Curran, T.; et al. Scrambler and yotari disrupt the disabled gene and produce a reeler -like phenotype in mice. Nature 1997, 389, 730–733. [Google Scholar] [CrossRef]
- Sweet, H.O.; Bronson, R.T.; Johnson, K.R.; Cook, S.A.; Davisson, M.T. Scrambler, a new neurological mutation of the mouse with abnormalities of neuronal migration. Mamm. Genome 1996, 7, 798–802. [Google Scholar] [CrossRef]
- Howell, B.W.; Herrick, T.M.; Hildebrand, J.D.; Zhang, Y.; A Cooper, J. Dab1 tyrosine phosphorylation sites relay positional signals during mouse brain development. Curr. Biol. 2000, 10, 877–885. [Google Scholar] [CrossRef]
- Arnaud, L.; Ballif, B.A.; Förster, E.; A Cooper, J. Fyn Tyrosine Kinase Is a Critical Regulator of Disabled-1 during Brain Development. Curr. Biol. 2003, 13, 9–17. [Google Scholar] [CrossRef]
- Wang, L.; A Cooper, J. Optogenetic control of the Dab1 signaling pathway. Sci. Rep. 2017, 7, 43760. [Google Scholar] [CrossRef] [PubMed]
- Kuo, G.; Arnaud, L.; Kronstad-O’Brien, P.; A Cooper, J. Absence of Fyn and Src Causes a Reeler-Like Phenotype. J. Neurosci. 2005, 25, 8578–8586. [Google Scholar] [CrossRef] [PubMed]
- Jossin, Y.; Ogawa, M.; Metin, C.; Tissir, F.; Goffinet, A.M. Inhibition of Src Family Kinases and Non-Classical Protein Kinases C Induce a Reeler-Like Malformation of Cortical Plate Development. J. Neurosci. 2003, 23, 9953–9959. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; A Cooper, J. Dual Functions of Dab1 during Brain Development. Mol. Cell. Biol. 2008, 29, 324–332. [Google Scholar] [CrossRef]
- Zhang, B.; Wang, W.; Zhang, Z.; Hu, Y.; Meng, F.; Wang, F.; Lou, H.; Zhu, L.; Godbout, R.; Duan, S.; et al. Alternative Splicing of Disabled-1 Controls Multipolar-to-Bipolar Transition of Migrating Neurons in the Neocortex. Cereb. Cortex 2017, 28, 3457–3467. [Google Scholar] [CrossRef]
- Bock, H.H.; Jossin, Y.; May, P.; Bergner, O.; Herz, J. Apolipoprotein E Receptors Are Required for Reelin-induced Proteasomal Degradation of the Neuronal Adaptor Protein Disabled-1. J. Biol. Chem. 2004, 279, 33471–33479. [Google Scholar] [CrossRef]
- Arnaud, L.; Ballif, B.A.; A Cooper, J. Regulation of Protein Tyrosine Kinase Signaling by Substrate Degradation during Brain Development. Mol. Cell. Biol. 2003, 23, 9293–9302. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Allen, N.S.; Simó, S.; A Cooper, J. Cullin 5 regulates Dab1 protein levels and neuron positioning during cortical development. Genome Res. 2007, 21, 2717–2730. [Google Scholar] [CrossRef]
- Simó, S.; A Cooper, J. Rbx2 regulates neuronal migration through different cullin 5-RING ligase adaptors. Dev. Cell 2013, 27, 399–411. [Google Scholar] [CrossRef]
- Lawrenson, I.D.; Krebs, D.L.; Linossi, E.M.; Zhang, J.-G.; McLennan, T.J.; Collin, C.; McRae, H.M.; Kolesnik, T.B.; Koh, K.; Britto, J.M.; et al. Cortical Layer Inversion and Deregulation of Reelin Signaling in the Absence of SOCS6 and SOCS7. Cereb. Cortex 2015, 27, 576–588. [Google Scholar] [CrossRef] [PubMed]
- Simo, S.; Jossin, Y.; A Cooper, J. Cullin 5 regulates cortical layering by modulating the speed and duration of Dab1-dependent neuronal migration. J. Neurosci. 2010, 30, 5668–5676. [Google Scholar] [CrossRef] [PubMed]
- Morimura, T.; Hattori, M.; Ogawa, M.; Mikoshiba, K. Disabled1 Regulates the Intracellular Trafficking of Reelin Receptors. J. Biol. Chem. 2005, 280, 16901–16908. [Google Scholar] [CrossRef] [PubMed]
- A Ballif, B.; Arnaud, L.; Arthur, W.T.; Guris, D.; Imamoto, A.; A Cooper, J. Activation of a Dab1/CrkL/C3G/Rap1 Pathway in Reelin-Stimulated Neurons. Curr. Biol. 2004, 14, 606–610. [Google Scholar] [CrossRef]
- Park, T.; Curran, T. Crk and CrkL play essential overlapping roles downstream of Dab1 in the Reelin pathway. J. Neurosci. 2008, 28, 13551–13562. [Google Scholar] [CrossRef]
- Simo, S.; Pujadas, L.; Segura, M.F.; La Torre, A.; Del Rio, J.A.; Urena, J.M.; Comella, J.X.; Soriano, E. Reelin induces the detachment of postnatal subventricular zone cells and the expression of the Egr-1 through Erk1/2 activation. Cereb. Cortex 2007, 17, 294–303. [Google Scholar] [CrossRef][Green Version]
- Telese, F.; Ma, Q.; Perez, P.M.; Notani, D.; Oh, S.; Li, W.; Comoletti, D.; Ohgi, K.A.; Taylor, H.; Rosenfeld, M.G. LRP8-Reelin-Regulated Neuronal Enhancer Signature Underlying Learning and Memory Formation. Neuron 2015, 86, 696–710. [Google Scholar] [CrossRef]
- Lee, G.H.; Chhangawala, Z.; Von Daake, S.; Savas, J.N.; Yates, J.R.; Comoletti, D.; D’Arcangelo, G. Reelin Induces Erk1/2 Signaling in Cortical Neurons Through a Non-canonical Pathway. J. Biol. Chem. 2014, 289, 20307–20317. [Google Scholar] [CrossRef]
- Ballif, B.A.; Arnaud, L.; A Cooper, J. Tyrosine phosphorylation of Disabled-1 is essential for Reelin-stimulated activation of Akt and Src family kinases. Mol. Brain Res. 2003, 117, 152–159. [Google Scholar] [CrossRef]
- Ohkubo, N.; Vitek, M.P.; Morishima, A.; Suzuki, Y.; Miki, T.; Maeda, N.; Mitsuda, N. Reelin signals survival through Src-family kinases that inactivate BAD activity. J. Neurochem. 2007, 103, 820–830. [Google Scholar] [CrossRef]
- Cho, S.-K.; Choi, J.-M.; Kim, J.-M.; Cho, J.Y.; Kim, S.-S.; Hong, S.; Suh-Kim, H.; Lee, Y.-D. AKT-independent Reelin signaling requires interactions of heterotrimeric Go and Src. Biochem. Biophys. Res. Commun. 2015, 467, 1063–1069. [Google Scholar] [CrossRef] [PubMed]
- Na, Y.; Jiménez, E.C.; Kon, E.; Cao, H.; Jossin, Y.; A Cooper, J. Fbxo45 binds SPRY motifs in the extracellular domain of N-cadherin and regulates neuron migration during brain development. Mol. Cell. Biol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Sekine, K.; Kawauchi, T.; Kubo, K.; Honda, T.; Herz, J.; Hattori, M.; Kinashi, T.; Nakajima, K. Reelin controls neuronal positioning by promoting cell-matrix adhesion via inside-out activation of integrin α5β1. Neuron 2012, 76, 353–369. [Google Scholar] [CrossRef] [PubMed]
- Belvindrah, R.; Graus-Porta, D.; Goebbels, S.; Nave, K.A.; Muller, U. Beta1 integrins in radial glia but not in migrating neurons are essential for the formation of cell layers in the cerebral cortex. J. Neurosci. 2007, 27, 13854–13865. [Google Scholar] [CrossRef] [PubMed]
- Franco, S.J.; Martinez-Garay, I.; Gil-Sanz, C.; Harkins-Perry, S.R.; Müller, U. Reelin Regulates Cadherin Function via Dab1/Rap1 to Control Neuronal Migration and Lamination in the Neocortex. Neuron 2011, 69, 482–497. [Google Scholar] [CrossRef] [PubMed]
- Gil-Sanz, C.; Franco, S.J.; Martinez-Garay, I.; Espinosa, A.; Harkins-Perry, S.; Müller, U. Cajal-Retzius cells instruct neuronal migration by coincidence signaling between secreted and contact-dependent guidance cues. Neuron 2013, 79, 461–477. [Google Scholar] [CrossRef]
- Bock, H.H.; Jossin, Y.; Liu, P.; Förster, E.; May, P.; Goffinet, A.M.; Herz, J. Phosphatidylinositol 3-Kinase Interacts with the Adaptor Protein Dab1 in Response to Reelin Signaling and Is Required for Normal Cortical Lamination. J. Biol. Chem. 2003, 278, 38772–38779. [Google Scholar] [CrossRef]
- Jossin, Y.; Goffinet, A.M. Reelin Signals through Phosphatidylinositol 3-Kinase and Akt To Control Cortical Development and through mTor To Regulate Dendritic Growth. Mol. Cell. Biol. 2007, 27, 7113–7124. [Google Scholar] [CrossRef]
- Beffert, U.; Morfini, G.; Bock, H.H.; Reyna, H.; Brady, S.T.; Herz, J. Reelin-mediated Signaling Locally Regulates Protein Kinase B/Akt and Glycogen Synthase Kinase 3β. J. Biol. Chem. 2002, 277, 49958–49964. [Google Scholar] [CrossRef]
- Morgan-Smith, M.; Wu, Y.; Zhu, X.; Pringle, J.; Snider, W.D. GSK-3 signaling in developing cortical neurons is essential for radial migration and dendritic orientation. eLife 2014, 3, e02663. [Google Scholar] [CrossRef]
- Gärtner, A.; Huang, X.; Hall, A. Neuronal polarity is regulated by glycogen synthase kinase-3 (GSK-3 ) independently of Akt/PKB serine phosphorylation. J. Cell Sci. 2006, 119, 3927–3934. [Google Scholar] [CrossRef] [PubMed]
- Chai, X.; Förster, E.; Zhao, S.; Bock, H.H.; Frotscher, M. Reelin Stabilizes the Actin Cytoskeleton of Neuronal Processes by Inducing n-Cofilin Phosphorylation at Serine3. J. Neurosci. 2009, 29, 288–299. [Google Scholar] [CrossRef]
- Chai, X.; Zhao, S.; Fan, L.; Zhang, W.; Lu, X.; Shao, H.; Wang, S.; Song, L.; Failla, A.V.; Zobiak, B.; et al. Reelin and cofilin cooperate during the migration of cortical neurons: A quantitative morphological analysis. Development 2016, 143, 1029–1040. [Google Scholar] [CrossRef] [PubMed]
- Jakob, B.; Kochlamazashvili, G.; Jäpel, M.; Gauhar, A.; Bock, H.H.; Maritzen, T.; Haucke, V. Intersectin 1 is a component of the Reelin pathway to regulate neuronal migration and synaptic plasticity in the hippocampus. Proc. Natl. Acad. Sci. USA 2017, 114, 5533–5538. [Google Scholar] [CrossRef]
- Hashimoto-Torii, K.; Torii, M.; Sarkisian, M.R.; Bartley, C.; Shen, J.; Radtke, F.; Gridley, T.; Sestan, N.; Rakic, P. Interaction between Reelin and Notch Signaling Regulates Neuronal Migration in the Cerebral Cortex. Neuron 2008, 60, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Sibbe, M.; Förster, E.; Basak, O.; Taylor, V.; Frotscher, M. Reelin and Notch1 Cooperate in the Development of the Dentate Gyrus. J. Neurosci. 2009, 29, 8578–8585. [Google Scholar] [CrossRef] [PubMed]
- Pramatarova, A.; Ochalski, P.G.; Chen, K.; Gropman, A.; Myers, S.; Min, K.-T.; Howell, B.W. Nckβ Interacts with Tyrosine-Phosphorylated Disabled 1 and Redistributes in Reelin-Stimulated Neurons. Mol. Cell. Biol. 2003, 23, 7210–7221. [Google Scholar] [CrossRef] [PubMed]
- Suetsugu, S.; Tezuka, T.; Morimura, T.; Hattori, M.; Mikoshiba, K.; Yamamoto, T.; Takenawa, T. Regulation of actin cytoskeleton by mDab1 through N-WASP and ubiquitination of mDab1. Biochem. J. 2004, 384, 1–8. [Google Scholar] [CrossRef]
- Homayouni, R.; Magdaleno, S.; Keshvara, L.; Rice, D.S.; Curran, T. Interaction of Disabled-1 and the GTPase activating protein Dab2IP in mouse brain. Mol. Brain Res. 2003, 115, 121–129. [Google Scholar] [CrossRef]
- Assadi, A.H.; Zhang, G.; Beffert, U.; McNeil, R.S.; Renfro, A.L.; Niu, S.; Quattrocchi, C.C.; A Antalffy, B.; Sheldon, M.; Armstrong, D.D.; et al. Interaction of reelin signaling and Lis1 in brain development. Nat. Genet. 2003, 35, 270–276. [Google Scholar] [CrossRef]
- Lord, M.C.P.; Caviness, V.S. Determinants of cell shape and orientation: A comparative Golgi analysis of cell-axon interrelationships in the developing neocortex of normal and reeler mice. J. Comp. Neurol. 1979, 187, 49–69. [Google Scholar] [CrossRef] [PubMed]
- Goffinet, A.; Lyon, G. Early histogenesis in the mouse cerebral cortex: A Golgi study. Neurosci. Lett. 1979, 14, 61–66. [Google Scholar] [CrossRef]
- Niu, S.; Renfro, A.; Quattrocchi, C.C.; Sheldon, M.; D’Arcangelo, G. Reelin promotes hippocampal dendrite development through the VLDLR/ApoER2-Dab1 pathway. Neuron 2004, 41, 71–84. [Google Scholar] [CrossRef]
- Chai, X.; Fan, L.; Shao, H.; Lu, X.; Zhang, W.; Li, J.; Wang, J.; Chen, S.; Frotscher, M.; Zhao, S. Reelin Induces Branching of Neurons and Radial Glial Cells during Corticogenesis. Cereb. Cortex 2014, 25, 3640–3653. [Google Scholar] [CrossRef] [PubMed]
- Matsuki, T.; Pramatarova, A.; Howell, B.W. Reduction of Crk and CrkL expression blocks reelin-induced dendritogenesis. J. Cell Sci. 2008, 121, 1869–1875. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, C.; Kron, M.M.; Masachs, N.; Zhang, H.; Lagace, D.C.; Martínez, A.; Reillo, I.; Duan, X.; Bosch, C.; Pujadas, L.; et al. Cell-autonomous inactivation of the reelin pathway impairs adult neurogenesis in the hippocampus. J. Neurosci. 2012, 32, 12051–12065. [Google Scholar] [CrossRef] [PubMed]
- O’Dell, R.S.; Cameron, D.A.; Zipfel, W.R.; Olson, E.C. Reelin Prevents Apical Neurite Retraction during Terminal Translocation and Dendrite Initiation. J. Neurosci. 2015, 35, 10659–10674. [Google Scholar] [CrossRef]
- O’Dell, R.S.; Ustine, C.J.M.; A Cameron, D.; Lawless, S.M.; Williams, R.M.; Zipfel, W.R.; Olson, E.C. Layer 6 cortical neurons require Reelin-Dab1 signaling for cellular orientation, Golgi deployment, and directed neurite growth into the marginal zone. Neural Dev. 2012, 7, 25. [Google Scholar] [CrossRef]
- Kupferman, J.V.; Basu, J.; Russo, M.J.; Guevarra, J.; Cheung, S.K.; A Siegelbaum, S. Reelin signaling specifies the molecular identity of the pyramidal neuron distal dendritic compartment. Cell 2014, 158, 1335–1347. [Google Scholar] [CrossRef][Green Version]
- Leemhuis, J.; Bouché, E.; Frotscher, M.; Henle, F.; Hein, L.; Herz, J.; Meyer, D.K.; Pichler, M.; Roth, G.; Schwan, C.; et al. Reelin signals through apolipoprotein E receptor 2 and Cdc42 to increase growth cone motility and filopodia formation. J. Neurosci. 2010, 30, 14759–14772. [Google Scholar] [CrossRef]
- Horton, A.C.; Rácz, B.; Monson, E.E.; Lin, A.L.; Weinberg, R.; Ehlers, M.D. Polarized Secretory Trafficking Directs Cargo for Asymmetric Dendrite Growth and Morphogenesis. Neuron 2005, 48, 757–771. [Google Scholar] [CrossRef] [PubMed]
- Matsuki, T.; Matthews, R.T.; Cooper, J.A.; Van Der Brug, M.P.; Cookson, M.R.; Hardy, J.A.; Olson, E.C.; Howell, B.W. Reelin and Stk25 Have Opposing Roles in Neuronal Polarization and Dendritic Golgi Deployment. Cell 2010, 143, 826–836. [Google Scholar] [CrossRef] [PubMed]
- Meseke, M.; Rosenberger, G.; Förster, E. Reelin and the Cdc42/Rac1 guanine nucleotide exchange factor αPIX/Arhgef6 promote dendritic Golgi translocation in hippocampal neurons. Eur. J. Neurosci. 2013, 37, 1404–1412. [Google Scholar] [CrossRef] [PubMed]
- Beffert, U.; Dillon, G.M.; Sullivan, J.M.; Stuart, C.E.; Gilbert, J.P.; Kambouris, J.A.; Ho, A. Microtubule plus-end tracking protein CLASP2 regulates neuronal polarity and synaptic function. J. Neurosci. 2012, 32, 13906–13916. [Google Scholar] [CrossRef]
- Dillon, G.M.; Tyler, W.A.; Omuro, K.C.; Kambouris, J.; Tyminski, C.; Henry, S.; Haydar, T.F.; Beffert, U.; Ho, A. CLASP2 Links Reelin To The Cytoskeleton During Neocortical Development. Neuron 2017, 93, 1344–1358.e5. [Google Scholar] [CrossRef]
- Del Rio, J.A.; Heimrich, B.; Super, H.; Borrell, V.; Frotscher, M.; Soriano, E. Differential Survival of Cajal–Retzius Cells in Organotypic Cultures of Hippocampus and Neocortex. J. Neurosci. 1996, 16, 6896–6907. [Google Scholar] [CrossRef]
- Anstötz, M.; Cosgrove, K.E.; Hack, I.; Mugnaini, E.; Maccaferri, G.; Lübke, J.H.R. Morphology, input-output relations and synaptic connectivity of Cajal-Retzius cells in layer 1 of the developing neocortex of CXCR4-EGFP mice. Brain Struct. Funct. 2013, 219, 2119–2139. [Google Scholar] [CrossRef]
- Pesold, C.; Impagnatiello, F.; Pisu, M.G.; Uzunov, D.P.; Costa, E.; Guidotti, A.; Caruncho, H.J. Reelin is preferentially expressed in neurons synthesizing -aminobutyric acid in cortex and hippocampus of adult rats. Proc. Natl. Acad. Sci. USA 1998, 95, 3221–3226. [Google Scholar] [CrossRef]
- Alcántara, S.; Ruiz, M.; D’Arcangelo, G.; Ezan, F.; De Lecea, L.; Curran, T.; Sotelo, C.; Soriano, E. Regional and Cellular Patterns of reelin mRNA Expression in the Forebrain of the Developing and Adult Mouse. J. Neurosci. 1998, 18, 7779–7799. [Google Scholar] [CrossRef]
- Niu, S.; Yabut, O.; D’Arcangelo, G. The Reelin signaling pathway promotes dendritic spine development in hippocampal neurons. J. Neurosci. 2008, 28, 10339–10348. [Google Scholar] [CrossRef]
- Iafrati, J.; Orejarena, M.J.; Lassalle, O.; Bouamrane, L.; Chavis, P. Reelin, an extracellular matrix protein linked to early onset psychiatric diseases, drives postnatal development of the prefrontal cortex via GluN2B-NMDARs and the mTOR pathway. Mol. Psychiatry 2013, 19, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.S.; Pesold, C.; Rodriguez, M.A.; Carboni, G.; Auta, J.; Lacor, P.; Larson, J.; Condie, B.G.; Guidotti, A.; Costa, E. Down-regulation of dendritic spine and glutamic acid decarboxylase 67 expressions in the reelin haploinsufficient heterozygous reeler mouse. Proc. Natl. Acad. Sci. USA 2001, 98, 3477–3482. [Google Scholar] [CrossRef] [PubMed]
- Bosch, C.; Masachs, N.; Exposito-Alonso, D.; Martínez, A.; Teixeira, C.M.; Fernaud, I.; Pujadas, L.; Ulloa, F.; Comella, J.X.; DeFelipe, J.; et al. Reelin Regulates the Maturation of Dendritic Spines, Synaptogenesis and Glial Ensheathment of Newborn Granule Cells. Cereb. Cortex 2016, 26, 4282–4298. [Google Scholar] [CrossRef] [PubMed]
- Dumanis, S.B.; Cha, H.-J.; Song, J.M.; Trotter, J.H.; Spitzer, M.H.; Lee, J.-Y.; Weeber, E.J.; Turner, R.S.; Pak, D.T.S.; Rebeck, G.W.; et al. ApoE Receptor 2 Regulates Synapse and Dendritic Spine Formation. PLoS ONE 2011, 6, e17203. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Marosi, M.; Choi, J.; Achiro, J.; Kim, S.; Li, S.; Otis, K.; Martin, K.C.; Portera-Cailliau, C.; Tontonoz, P. The E3 ubiquitin ligase IDOL regulates synaptic ApoER2 levels and is important for plasticity and learning. eLife 2017, 6. [Google Scholar] [CrossRef]
- Trotter, J.; Lee, G.H.; Kazdoba, T.M.; Crowell, B.; Domogauer, J.; Mahoney, H.M.; Franco, S.J.; Müller, U.; Weeber, E.J.; D’Arcangelo, G. Dab1 is required for synaptic plasticity and associative learning. J. Neurosci. 2013, 33, 15652–15668. [Google Scholar] [CrossRef]
- Dibattista, A.; Dumanis, S.B.; Song, J.M.; Bu, G.; Weeber, E.; Rebeck, G.W.; Hoe, H.-S. Very low density lipoprotein receptor regulates dendritic spine formation in a RasGRF1/CaMKII dependent manner. Biochim. et Biophys. Acta (BBA) - Bioenergy 2015, 1853, 904–917. [Google Scholar] [CrossRef][Green Version]
- Kim, M.; Jeong, Y.; Chang, Y.-C. Extracellular matrix protein reelin regulate dendritic spine density through CaMKIIβ. Neurosci. Lett. 2015, 599, 97–101. [Google Scholar] [CrossRef]
- Qiu, S.; Korwek, K.M.; Pratt-Davis, A.R.; Peters, M.; Bergman, M.Y.; Weeber, E.J. Cognitive disruption and altered hippocampus synaptic function in Reelin haploinsufficient mice. Neurobiol. Learn. Mem. 2006, 85, 228–242. [Google Scholar] [CrossRef]
- Weeber, E.J.; Beffert, U.; Jones, C.; Christian, J.M.; Förster, E.; Sweatt, J.D.; Herz, J. Reelin and ApoE Receptors Cooperate to Enhance Hippocampal Synaptic Plasticity and Learning. J. Biol. Chem. 2002, 277, 39944–39952. [Google Scholar] [CrossRef]
- Pujadas, L.; Gruart, A.; Bosch, C.; Delgado, L.; Teixeira, C.; Rossi, D.; De Lecea, L.; Martínez, A.; Delgado-García, J.M.; Soriano, E. Reelin Regulates Postnatal Neurogenesis and Enhances Spine Hypertrophy and Long-Term Potentiation. J. Neurosci. 2010, 30, 4636–4649. [Google Scholar] [CrossRef] [PubMed]
- Lane-Donovan, C.; Philips, G.T.; Wasser, C.R.; Durakoglugil, M.S.; Masiulis, I.; Upadhaya, A.; Pohlkamp, T.; Coskun, C.; Kotti, T.; Steller, L.; et al. Reelin protects against amyloid β toxicity in vivo. Sci. Signal. 2015, 8, ra67. [Google Scholar] [CrossRef] [PubMed]
- Rogers, J.T.; Rusiana, I.; Trotter, J.; Zhao, L.; Donaldson, E.; Pak, D.T.; Babus, L.; Peters, M.; Banko, J.L.; Chavis, P.; et al. Reelin supplementation enhances cognitive ability, synaptic plasticity, and dendritic spine density. Learn. Mem. 2011, 18, 558–564. [Google Scholar] [CrossRef] [PubMed]
- Lüscher, C.; Malenka, R.C. NMDA Receptor-Dependent Long-Term Potentiation and Long-Term Depression (LTP/LTD). Cold Spring Harb. Perspect. Biol. 2012, 4, a005710. [Google Scholar] [CrossRef] [PubMed]
- Chater, T.E.; Goda, Y. The role of AMPA receptors in postsynaptic mechanisms of synaptic plasticity. Front. Cell. Neurosci. 2014, 8, 401. [Google Scholar] [CrossRef]
- Chen, Y.; Beffert, U.; Ertunc, M.; Tang, T.-S.; Kavalali, E.T.; Bezprozvanny, I.B.; Herz, J. Reelin Modulates NMDA Receptor Activity in Cortical Neurons. J. Neurosci. 2005, 25, 8209–8216. [Google Scholar] [CrossRef]
- Brai, E.; Marathe, S.; Astori, S.; Ben Fredj, N.; Perry, E.; Lamy, C.; Scotti, A.; Alberi, L. Notch1 Regulates Hippocampal Plasticity Through Interaction with the Reelin Pathway, Glutamatergic Transmission and CREB Signaling. Front. Cell. Neurosci. 2015, 9, 1217. [Google Scholar] [CrossRef]
- Qiu, S.; Weeber, E.J. Reelin Signaling Facilitates Maturation of CA1 Glutamatergic Synapses. J. Neurophysiol. 2007, 97, 2312–2321. [Google Scholar] [CrossRef]
- Sinagra, M.; Verrier, D.; Franková, D.; Korwek, K.M.; Blahoš, J.; Weeber, E.J.; Manzoni, O.J.J.; Chavis, P. Reelin, Very-Low-Density Lipoprotein Receptor, and Apolipoprotein E Receptor 2 Control Somatic NMDA Receptor Composition during Hippocampal Maturation In Vitro. J. Neurosci. 2005, 25, 6127–6136. [Google Scholar] [CrossRef]
- Ventruti, A.; Kazdoba, T.; Niu, S.; D’Arcangelo, G. Reelin deficiency causes specific defects in the molecular composition of the synapses in the adult brain. Neurosci. 2011, 189, 32–42. [Google Scholar] [CrossRef]
- Qiu, S.; Zhao, L.F.; Korwek, K.M.; Weeber, E.J. Differential Reelin-Induced Enhancement of NMDA and AMPA Receptor Activity in the Adult Hippocampus. J. Neurosci. 2006, 26, 12943–12955. [Google Scholar] [CrossRef]
- Pfennig, S.; Foss, F.; Bissen, D.; Harde, E.; Treeck, J.C.; Segarra, M.; Acker-Palmer, A. GRIP1 Binds to ApoER2 and EphrinB2 to Induce Activity-Dependent AMPA Receptor Insertion at the Synapse. Cell Rep. 2017, 21, 84–96. [Google Scholar] [CrossRef] [PubMed]
- Bal, M.; Leitz, J.; Reese, A.L.; Ramirez, D.M.; Durakoglugil, M.; Herz, J.; Monteggia, L.M.; Kavalali, E.T. Reelin mobilizes a VAMP7-dependent synaptic vesicle pool and selectively augments spontaneous neurotransmission. Neuron 2013, 80, 934–946. [Google Scholar] [CrossRef] [PubMed]
- Hellwig, S.; Hack, I.; Kowalski, J.; Brunne, B.; Jarowyj, J.; Unger, A.; Bock, H.H.; Junghans, D.; Frotscher, M. Role for Reelin in Neurotransmitter Release. J. Neurosci. 2011, 31, 2352–2360. [Google Scholar] [CrossRef] [PubMed]
- Boycott, K.M.; Bonnemann, C.; Herz, J.; Neuert, S.; Beaulieu, C.; Scott, J.N.; Venkatasubramanian, A.; Parboosingh, J.S. Mutations in VLDLR as a cause for autosomal recessive cerebellar ataxia with mental retardation (dysequilibrium syndrome). J. Child Neurol. 2009, 24, 1310–1315. [Google Scholar] [CrossRef]
- Micalizzi, A.; Moroni, I.; Ginevrino, M.; Biagini, T.; Mazza, T.; Romani, M.; Valente, E.M. Very mild features of dysequilibrium syndrome associated with a novel VLDLR missense mutation. Neurogenetics 2016, 17, 191–195. [Google Scholar] [CrossRef]
- Seixas, A.I.; Loureiro, J.; Costa, C.; Ordóñez-Ugalde, A.; Marcelino, H.; Oliveira, C.; Loureiro, J.L.; Dhingra, A.; Brandão, E.; Cruz, V.T.; et al. A Pentanucleotide ATTTC Repeat Insertion in the Non-coding Region of DAB1, Mapping to SCA37, Causes Spinocerebellar Ataxia. Am. J. Hum. Genet. 2017, 101, 87–103. [Google Scholar] [CrossRef]
- Persico, A.; Napolioni, V. Autism genetics. Behav. Brain Res. 2013, 251, 95–112. [Google Scholar] [CrossRef]
- A Skaar, D.; Shao, Y.; Haines, J.L.; E Stenger, J.; Jaworski, J.; Martin, E.R.; Delong, G.R.; Moore, J.H.; McCauley, J.L.; Sutcliffe, J.S.; et al. Analysis of the RELN gene as a genetic risk factor for autism. Mol. Psychiatry 2004, 10, 563–571. [Google Scholar] [CrossRef]
- Lammert, D.; Howell, B.W. RELN Mutations in Autism Spectrum Disorder. Front. Cell. Neurosci. 2016, 10, 81. [Google Scholar] [CrossRef]
- Sánchez-Sánchez, S.M.; Magdalon, J.; Griesi-Oliveira, K.; Yamamoto, G.L.; Santacruz-Perez, C.; Fogo, M.; Passos-Bueno, M.R.; Sertie, A. Rare RELN variants affect Reelin-DAB1 signal transduction in autism spectrum disorder. Hum. Mutat. 2018, 39, 1372–1383. [Google Scholar] [CrossRef] [PubMed]
- Impagnatiello, F.; Guidotti, A.R.; Pesold, C.; Dwivedi, Y.; Caruncho, H.; Pisu, M.G.; Uzunov, D.P.; Smalheiser, N.R.; Davis, J.M.; Pandey, G.N.; et al. A decrease of reelin expression as a putative vulnerability factor in schizophrenia. Proc. Natl. Acad. Sci. USA 1998, 95, 15718–15723. [Google Scholar] [CrossRef] [PubMed]
- Fatemi, S.H. The role of Reelin in pathology of autism. Mol. Psychiatry 2002, 7, 919–920. [Google Scholar] [CrossRef] [PubMed]
- Zhubi, A.; Chen, Y.; Guidotti, A.; Grayson, D. Epigenetic regulation of RELN and GAD1 in the frontal cortex (FC) of autism spectrum disorder (ASD) subjects. Int. J. Dev. Neurosci. 2017, 62, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Grayson, D.; Jia, X.; Chen, Y.; Sharma, R.P.; Mitchell, C.P.; Guidotti, A.; Costa, E. Reelin promoter hypermethylation in schizophrenia. Proc. Natl. Acad. Sci. USA 2005, 102, 9341–9346. [Google Scholar] [CrossRef] [PubMed]
- Guidotti, A.; Auta, J.; Davis, J.M.; Dwivedi, Y.; Grayson, D.R.; Impagnatiello, F.; Pandey, G.; Pesold, C.; Sharma, R.; Uzunov, D.; et al. Decrease in reelin and glutamic acid decarboxylase67 (GAD67) expression in schizophrenia and bipolar disorder: A postmortem brain study. Arch. Gen. Psychiatry 2000, 57, 1061–1069. [Google Scholar] [CrossRef]
- Fikri, R.M.N.; Norlelawati, A.T.; El-Huda, A.R.N.; Hanisah, M.N.; Kartini, A.; Norsidah, K.; Zamzila, A.N. Reelin (RELN) DNA methylation in the peripheral blood of schizophrenia. J. Psychiatr. Res. 2017, 88, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Tueting, P.; Costa, E.; Dwivedi, Y.; Guidotti, A.; Impagnatiello, F.; Manev, R.; Pesold, C. The phenotypic characteristics of heterozygous reeler mouse. NeuroReport 1999, 10, 1329–1334. [Google Scholar] [CrossRef] [PubMed]
- Sobue, A.; Kushima, I.; Nagai, T.; Shan, W.; Kohno, T.; Aleksic, B.; Aoyama, Y.; Mori, D.; Arioka, Y.; Kawano, N.; et al. Genetic and animal model analyses reveal the pathogenic role of a novel deletion of RELN in schizophrenia. Sci. Rep. 2018, 8, 13046. [Google Scholar] [CrossRef] [PubMed]
- Dazzo, E.; Fanciulli, M.; Serioli, E.; Minervini, G.; Pulitano, P.; Binelli, S.; Di Bonaventura, C.; Luisi, C.; Pasini, E.; Striano, S.; et al. Heterozygous Reelin Mutations Cause Autosomal-Dominant Lateral Temporal Epilepsy. Am. J. Hum. Genet. 2015, 96, 992–1000. [Google Scholar] [CrossRef] [PubMed]
- Jackson, J.S.; Jambrina, E.; Li, J.; Marston, H.; Menzies, F.; Phillips, K.; Gilmour, G. Targeting the Synapse in Alzheimer’s Disease. Front. Mol. Neurosci. 2019, 13, 735. [Google Scholar] [CrossRef] [PubMed]
- Parameshwaran, K.; Dhanasekaran, M.; Suppiramaniam, V. Amyloid beta peptides and glutamatergic synaptic dysregulation. Exp. Neurol. 2008, 210, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Antoniades, D.; Katopodi, T.; Pappa, S.; Lampropoulos, A.; Konsta, V.; Frydas, E.; Mpalogiannis, S.; Hatzistilianou, M. The role of reelin gene polymorphisms in the pathogenesis of Alzheimer’s disease in a Greek population. J. Biol. Regul. Homeost. Agents 2011, 25, 351–358. [Google Scholar] [PubMed]
- Kramer, P.L.; Xu, H.; Woltjer, R.L.; Westaway, S.K.; Clark, D.; Erten-Lyons, D.; Kaye, J.; Welsh-Bohmer, K.A.; Troncoso, J.C.; Markesbery, W.R.; et al. Alzheimer disease pathology in cognitively healthy elderly: A genome-wide study. Neurobiol. Aging 2010, 32, 2113–2122. [Google Scholar] [CrossRef] [PubMed]
- Karim, S.; Mirza, Z.; Ansari, S.; Rasool, M.; Iqbal, Z.; Sohrab, S.; Kamal, M.A.; Abuzenadah, A.M.; Al-Qahtani, M. Transcriptomics Study of Neurodegenerative Disease: Emphasis on Synaptic Dysfunction Mechanism in Alzheimer’s Disease. CNS Neurol. Disord. - Drug Targets 2014, 13, 1202–1212. [Google Scholar] [CrossRef] [PubMed]
- Herring, A.; Donath, A.; Steiner, K.M.; Widera, M.P.; Hamzehian, S.; Kanakis, D.; Kölble, K.; ElAli, A.; Hermann, D.M.; Paulus, W.; et al. Reelin Depletion is an Early Phenomenon of Alzheimer’s Pathology. J. Alzheimer’s Dis. 2012, 30, 963–979. [Google Scholar] [CrossRef]
- Ohkubo, N.; Lee, Y.-D.; Morishima, A.; Terashima, T.; Kikkawa, S.; Tohyama, M.; Sakanaka, M.; Tanaka, J.; Maeda, N.; Vitek, M.P.; et al. Apolipoprotein E and Reelin ligands modulate tau phosphorylation through an Apolipoprotein E receptor/disabled-1/glycogen synthase kinase-3β cascade. FASEB J. 2002, 17, 295–297. [Google Scholar] [CrossRef] [PubMed]
- Hoe, H.-S.; Tran, T.S.; Matsuoka, Y.; Howell, B.W.; Rebeck, G.W. DAB1 and Reelin Effects on Amyloid Precursor Protein and ApoE Receptor 2 Trafficking and Processing. J. Biol. Chem. 2006, 281, 35176–35185. [Google Scholar] [CrossRef]
- Durakoglugil, M.S.; Chen, Y.; White, C.L.; Kavalali, E.T.; Herz, J. Reelin signaling antagonizes β-amyloid at the synapse. Proc. Natl. Acad. Sci. USA 2009, 106, 15938–15943. [Google Scholar] [CrossRef]
- Kocherhans, S.; Madhusudan, A.; Doehner, J.; Breu, K.S.; Nitsch, R.M.; Fritschy, J.-M.; Knuesel, I. Reduced Reelin Expression Accelerates Amyloid-β Plaque Formation and Tau Pathology in Transgenic Alzheimer’s Disease Mice. J. Neurosci. 2010, 30, 9228–9240. [Google Scholar] [CrossRef]
- Pujadas, L.; Rossi, D.; Andres, R.; Teixeira, C.; Serra-Vidal, B.; Parcerisas, A.; Maldonado, R.; Giralt, E.; Carulla, N.; Soriano, E. Reelin delays amyloid-beta fibril formation and rescues cognitive deficits in a model of Alzheimer’s disease. Nat. Commun. 2014, 5, 3443. [Google Scholar] [CrossRef] [PubMed]
- Rossi, D.; Gruart, A.; Contreras-Murillo, G.; Muhaisen, A.; Ávila, J.; Delgado-García, J.M.; Pujadas, L.; Soriano, E. Reelin reverts biochemical, physiological and cognitive alterations in mouse models of Tauopathy. Prog. Neurobiol. 2020, 186, 101743. [Google Scholar] [CrossRef] [PubMed]
- Homayouni, R.; Rice, D.S.; Sheldon, M.; Curran, T. Disabled-1 Binds to the Cytoplasmic Domain of Amyloid Precursor-Like Protein 1. J. Neurosci. 1999, 19, 7507–7515. [Google Scholar] [CrossRef] [PubMed]
- Hoareau, C.; Borrell, V.; Soriano, E.; Krebs, M.-O.; Prochiantz, A.; Allinquant, B. Amyloid precursor protein cytoplasmic domain antagonizes reelin neurite outgrowth inhibition of hippocampal neurons. Neurobiol. Aging 2008, 29, 542–553. [Google Scholar] [CrossRef] [PubMed]
- Hoe, H.-S.; Wessner, D.; Beffert, U.; Becker, A.G.; Matsuoka, Y.; Rebeck, G.W. F-Spondin Interaction with the Apolipoprotein E Receptor ApoEr2 Affects Processing of Amyloid Precursor Protein. Mol. Cell. Biol. 2005, 25, 9259–9268. [Google Scholar] [CrossRef]
- Helbecque, N.; Cottel, D.; Amouyel, P. Low-density lipoprotein receptor-related protein 8 gene polymorphisms and dementia. Neurobiol. Aging 2009, 30, 266–271. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, H.; Kamimura, K.; Tanahashi, H.; Takahashi, K.; Asada, T.; Tabira, T. Genetic Risk Factors in Japanese Alzheimer’s Disease Patients: α1-ACT, VLDLR, and ApoE. Neurobiol. Aging 1998, 19, S43–S46. [Google Scholar] [CrossRef]
- Corder, E.; Saunders, A.; Strittmatter, W.; Schmechel, D.; Gaskell, P.; Small, G.; Roses, A.; Haines, J.; Pericak-Vance, M. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 1993, 261, 921–923. [Google Scholar] [CrossRef]
- Chen, Y.; Durakoglugil, M.S.; Xian, X.; Herz, J. ApoE4 reduces glutamate receptor function and synaptic plasticity by selectively impairing ApoE receptor recycling. Proc. Natl. Acad. Sci. USA 2010, 107, 12011–12016. [Google Scholar] [CrossRef]


© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jossin, Y. Reelin Functions, Mechanisms of Action and Signaling Pathways During Brain Development and Maturation. Biomolecules 2020, 10, 964. https://doi.org/10.3390/biom10060964
Jossin Y. Reelin Functions, Mechanisms of Action and Signaling Pathways During Brain Development and Maturation. Biomolecules. 2020; 10(6):964. https://doi.org/10.3390/biom10060964
Chicago/Turabian StyleJossin, Yves. 2020. "Reelin Functions, Mechanisms of Action and Signaling Pathways During Brain Development and Maturation" Biomolecules 10, no. 6: 964. https://doi.org/10.3390/biom10060964
APA StyleJossin, Y. (2020). Reelin Functions, Mechanisms of Action and Signaling Pathways During Brain Development and Maturation. Biomolecules, 10(6), 964. https://doi.org/10.3390/biom10060964