Leptin Signaling Contributes to Aromatase Inhibitor Resistant Breast Cancer Cell Growth and Activation of Macrophages

 , , ,
, , ,  , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents, Antibodies, and Plasmids
2.2. Cell Culture
2.3. Cell Proliferation Assays
2.4. ERE-Luciferase Reporter Assay
2.5. Aromatase Activity Assays
2.6. Real-Time PCR Assays
2.7. Immunoblot Analysis
2.8. Leptin Measurement by Enzyme-Linked Immunosorbent Assay (ELISA)
2.9. Wound-Healing Assays
2.10. Migration/Chemiotaxis Assays
2.11. Conditioned Medium and Coculture Systems
2.12. Leptin-Immunodepleted Conditioned Media
2.13. RNA Sequencing Analysis
2.14. Statistical Analysis
3. Results
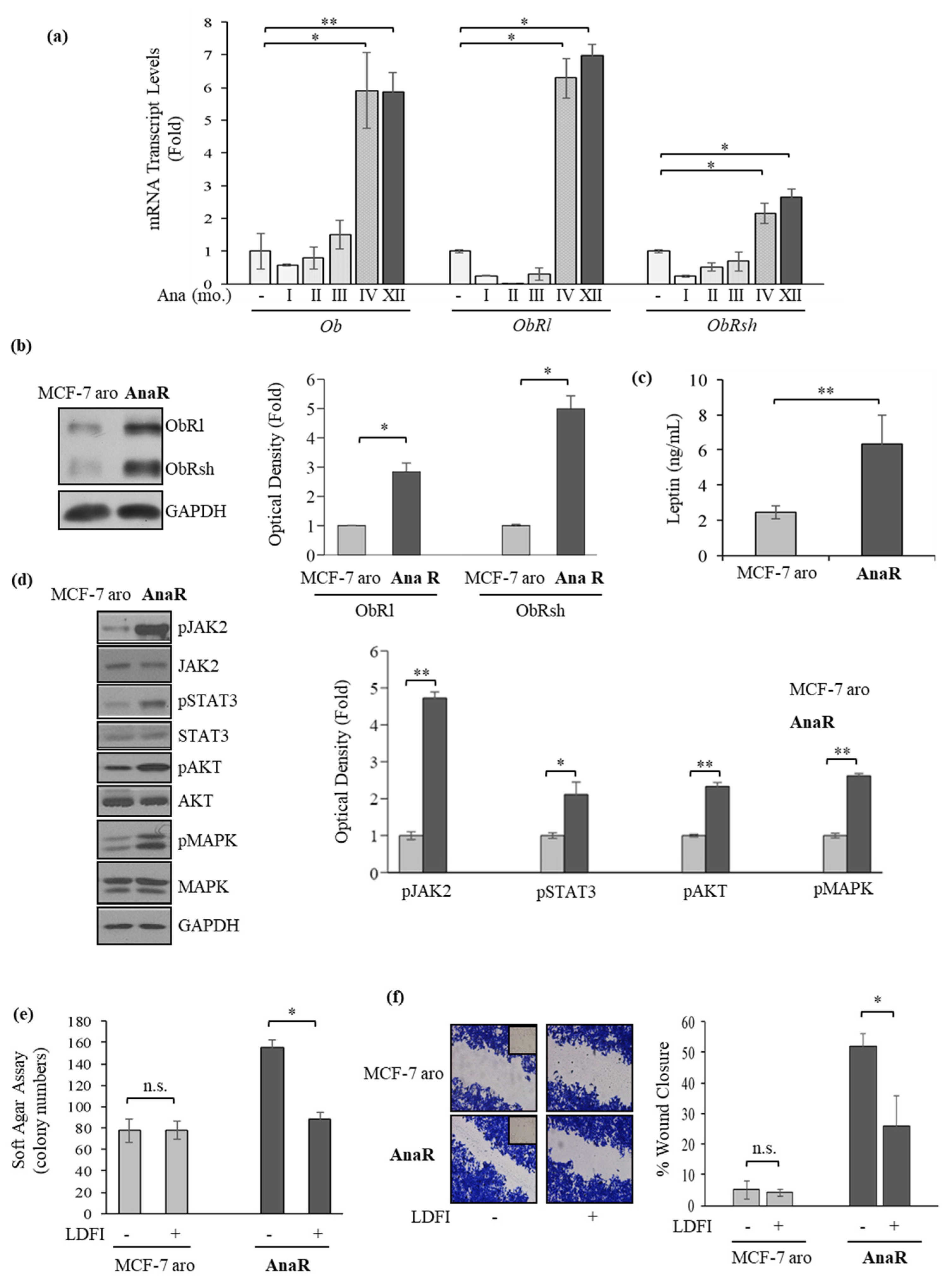
3.1. Anastrozole-Resistant (AnaR) Cells Exhibit Increased Activation of Leptin Signaling Pathway
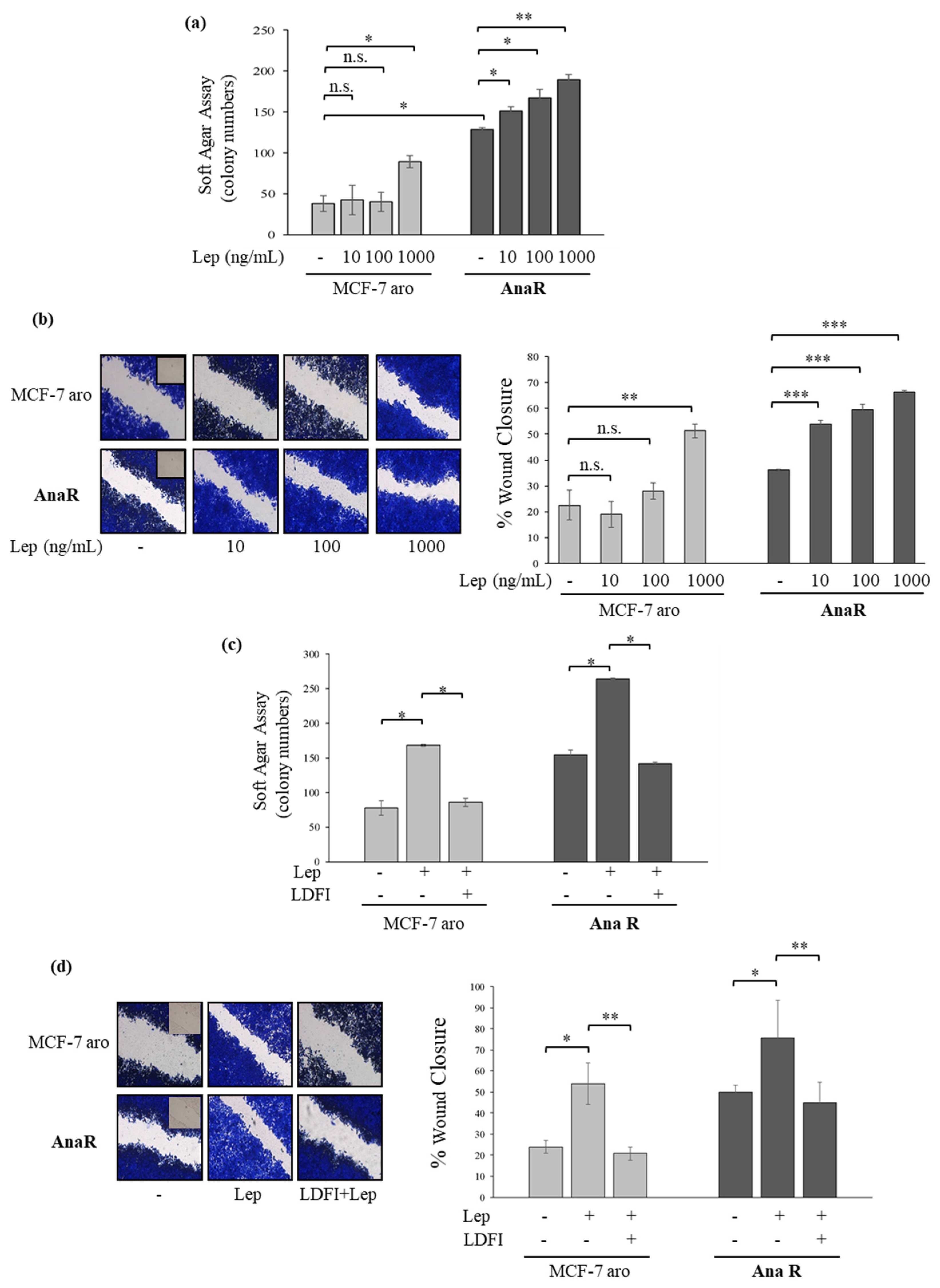
3.2. Anastrozole-Resistant Breast Cancer Cells Show Leptin Hypersensitivity
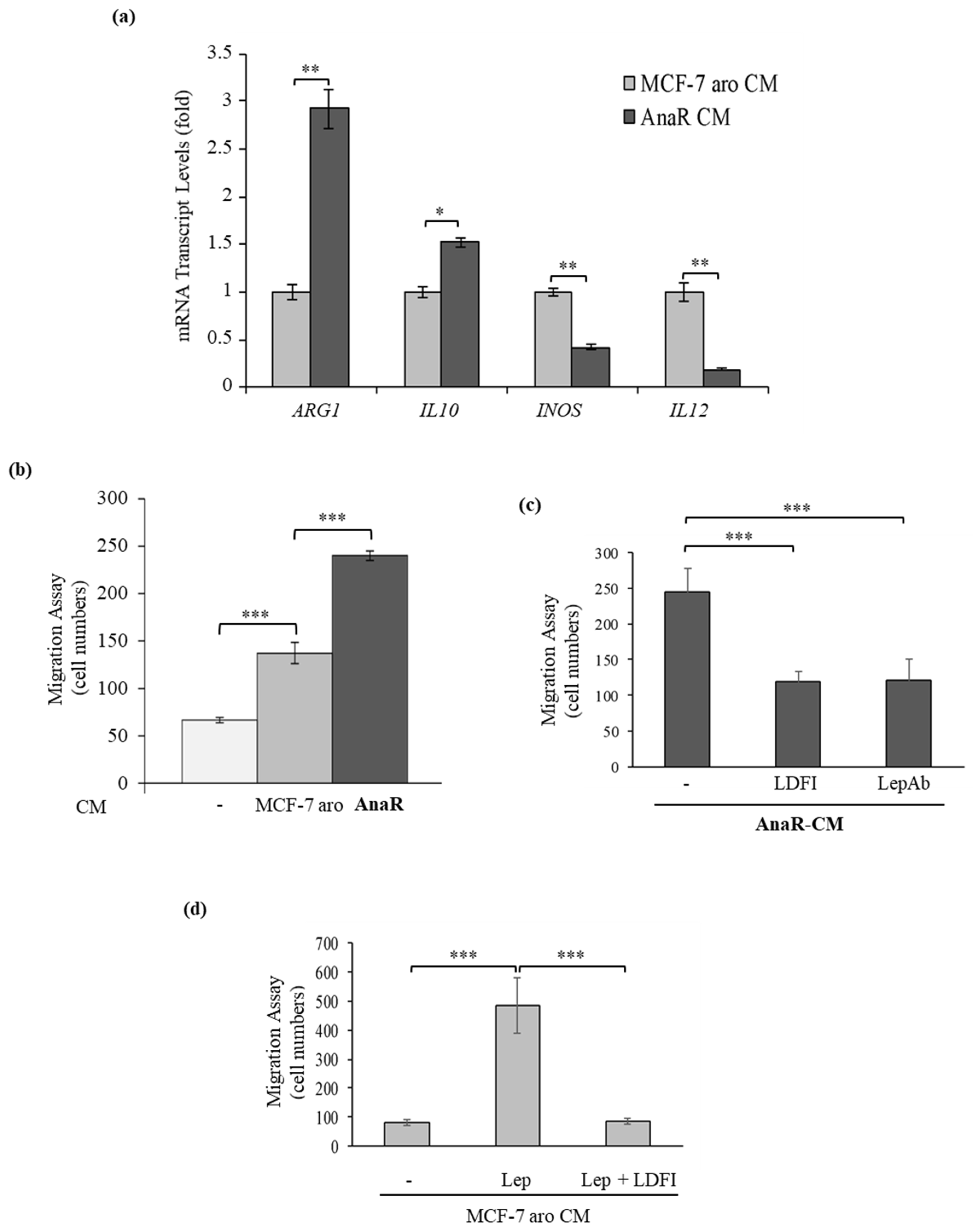
3.3. Enhanced Recruitment and Protumor Activation of Macrophages from AnaR Cells throughLeptin Signaling
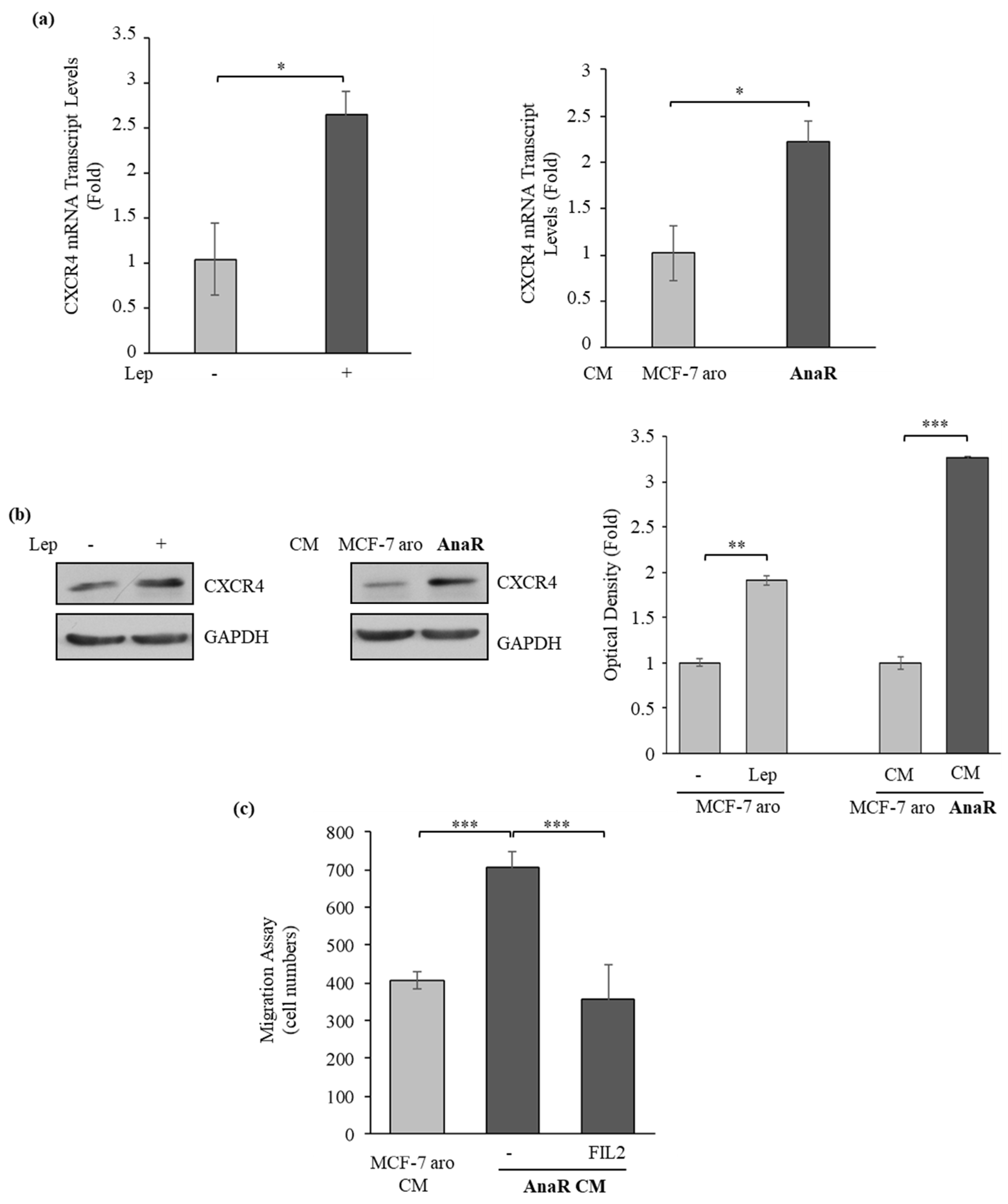
3.4. AnaR Cells Affect Motility of Macrophage through an Increase in CXCR4 expression
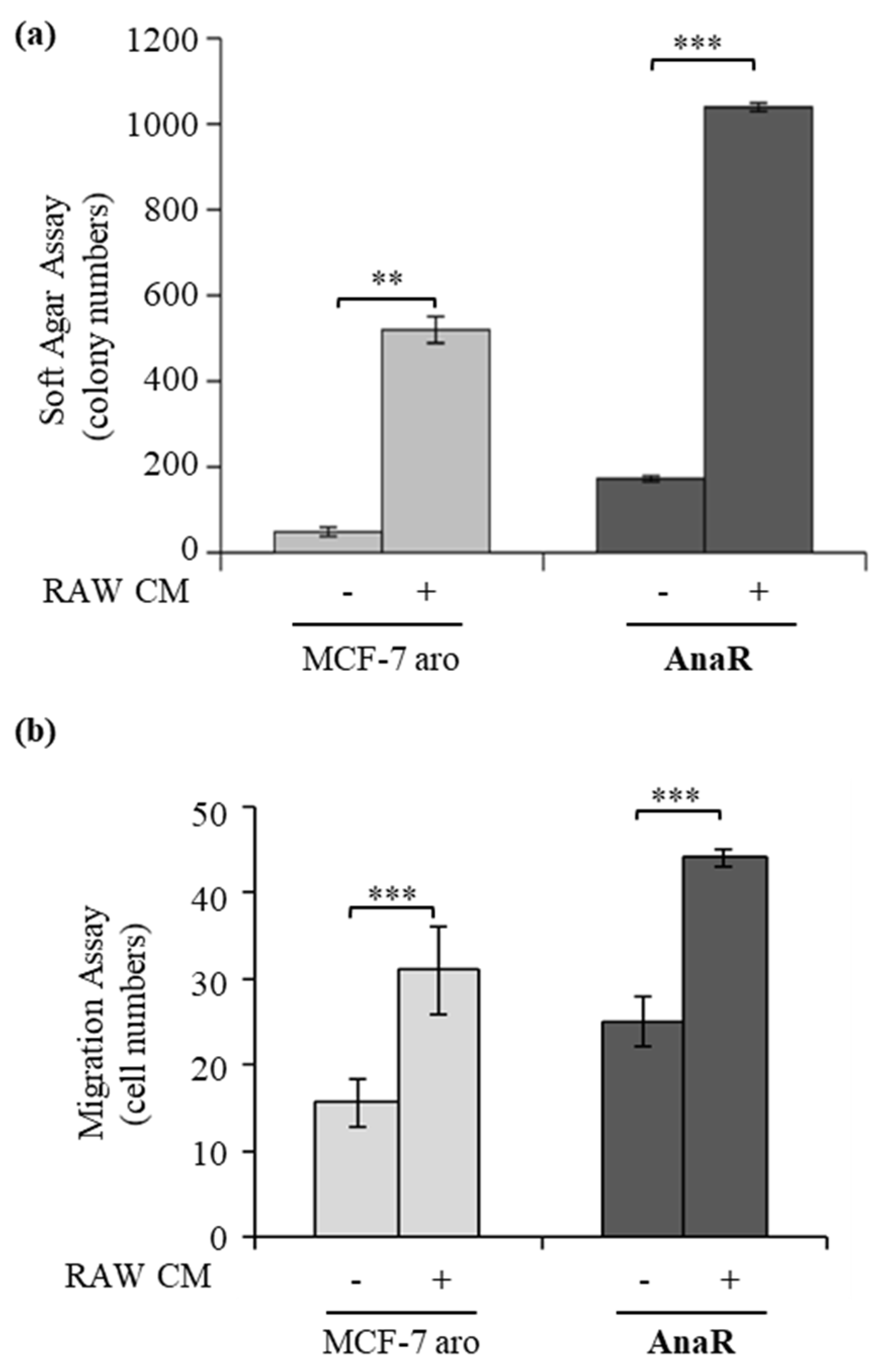
3.5. Activated Macrophages Promote Growth and Migration of AnaR Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Martin, L.A.; Farmer, I.; Johnston, S.R.; Ali, S.; Dowsett, M. Elevated ERK1/ERK2/estrogen receptor cross-talk enhances estrogen-mediated signaling during long-term estrogen deprivation. Endocr.-Relat. Cancer 2005, 12 (Suppl. 1), S75–S84. [Google Scholar] [CrossRef] [PubMed]
- Sabnis, G.J.; Jelovac, D.; Long, B.; Brodie, A. The role of growth factor receptor pathways in human breast cancer cells adapted to long-term estrogen deprivation. Cancer Res. 2005, 65, 3903–3910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santen, R.J.; Song, R.X.; Zhang, Z.; Kumar, R.; Jeng, M.H.; Masamura, A.; Lawrence, J., Jr.; Berstein, L.; Yue, W. Long-term estradiol deprivation in breast cancer cells up-regulates growth factor signaling and enhances estrogen sensitivity. Endocr.-Relat. Cancer 2005, 12 (Suppl. 1), S61–S73. [Google Scholar] [CrossRef] [PubMed]
- Chen, S. An “omics” approach to determine the mechanisms of acquired aromatase inhibitor resistance. Omics J. Integr. Biol. 2011, 15, 347–352. [Google Scholar] [CrossRef] [Green Version]
- Wong, C.; Chen, S. The development, application and limitations of breast cancer cell lines to study tamoxifen and aromatase inhibitor resistance. J. Steroid Biochem. Mol. Biol. 2012, 131, 83–92. [Google Scholar] [CrossRef] [Green Version]
- Murphy, C.G.; Dickler, M.N. Endocrine resistance in hormone-responsive breast cancer: Mechanisms and therapeutic strategies. Endocr.-Relat. Cancer 2016, 23, R337–R352. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.Y.; Wu, C.Y.; Petrossian, K.; Huang, T.T.; Tseng, L.M.; Chen, S. Treatment for the endocrine resistant breast cancer: Current options and future perspectives. J. Steroid Biochem. Mol. Biol. 2017, 172, 166–175. [Google Scholar] [CrossRef]
- Osborne, C.K.; Schiff, R. Mechanisms of endocrine resistance in breast cancer. Annu. Rev. Med. 2011, 62, 233–247. [Google Scholar] [CrossRef] [Green Version]
- Ford, N.A.; Devlin, K.L.; Lashinger, L.M.; Hursting, S.D. Deconvoluting the obesity and breast cancer link: Secretome, soil and seed interactions. J. Mammary Gland Biol. Neoplasia 2013, 18, 267–275. [Google Scholar] [CrossRef] [Green Version]
- Calle, E.E.; Kaaks, R. Overweight, obesity and cancer: Epidemiological evidence and proposed mechanisms. Nat. Rev. Cancer 2004, 4, 579–591. [Google Scholar] [CrossRef]
- Ando, S.; Gelsomino, L.; Panza, S.; Giordano, C.; Bonofiglio, D.; Barone, I.; Catalano, S. Obesity, Leptin and Breast Cancer: Epidemiological Evidence and Proposed Mechanisms. Cancers 2019, 11, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiralerspong, S.; Goodwin, P.J. Obesity and Breast Cancer Prognosis: Evidence, Challenges, and Opportunities. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2016, 34, 4203–4216. [Google Scholar] [CrossRef] [PubMed]
- Picon-Ruiz, M.; Morata-Tarifa, C.; Valle-Goffin, J.J.; Friedman, E.R.; Slingerland, J.M. Obesity and adverse breast cancer risk and outcome: Mechanistic insights and strategies for intervention. CA Cancer J. Clin. 2017, 67, 378–397. [Google Scholar] [CrossRef] [PubMed]
- Ando, S.; Barone, I.; Giordano, C.; Bonofiglio, D.; Catalano, S. The Multifaceted Mechanism of Leptin Signaling within Tumor Microenvironment in Driving Breast Cancer Growth and Progression. Front. Oncol. 2014, 4, 340. [Google Scholar] [CrossRef] [Green Version]
- Giordano, C.; Chemi, F.; Panza, S.; Barone, I.; Bonofiglio, D.; Lanzino, M.; Cordella, A.; Campana, A.; Hashim, A.; Rizza, P.; et al. Leptin as a mediator of tumor-stromal interactions promotes breast cancer stem cell activity. Oncotarget 2016, 7, 1262–1275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiorio, E.; Mercanti, A.; Terrasi, M.; Micciolo, R.; Remo, A.; Auriemma, A.; Molino, A.; Parolin, V.; Di Stefano, B.; Bonetti, F.; et al. Leptin/HER2 crosstalk in breast cancer: In vitro study and preliminary in vivo analysis. BMC Cancer 2008, 8, 305. [Google Scholar] [CrossRef] [Green Version]
- Garofalo, C.; Koda, M.; Cascio, S.; Sulkowska, M.; Kanczuga-Koda, L.; Golaszewska, J.; Russo, A.; Sulkowski, S.; Surmacz, E. Increased expression of leptin and the leptin receptor as a marker of breast cancer progression: Possible role of obesity-related stimuli. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2006, 12, 1447–1453. [Google Scholar] [CrossRef] [Green Version]
- Ishikawa, M.; Kitayama, J.; Nagawa, H. Enhanced expression of leptin and leptin receptor (OB-R) in human breast cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2004, 10, 4325–4331. [Google Scholar] [CrossRef] [Green Version]
- Jarde, T.; Caldefie-Chezet, F.; Damez, M.; Mishellany, F.; Penault-Llorca, F.; Guillot, J.; Vasson, M.P. Leptin and leptin receptor involvement in cancer development: A study on human primary breast carcinoma. Oncol. Rep. 2008, 19, 905–911. [Google Scholar] [CrossRef] [Green Version]
- Miyoshi, Y.; Funahashi, T.; Tanaka, S.; Taguchi, T.; Tamaki, Y.; Shimomura, I.; Noguchi, S. High expression of leptin receptor mRNA in breast cancer tissue predicts poor prognosis for patients with high, but not low, serum leptin levels. Int. J. Cancer 2006, 118, 1414–1419. [Google Scholar] [CrossRef]
- Ando, S.; Catalano, S. The multifactorial role of leptin in driving the breast cancer microenvironment. Nat. Rev. Endocrinol. 2011, 8, 263–275. [Google Scholar] [CrossRef] [PubMed]
- Barone, I.; Giordano, C.; Bonofiglio, D.; Ando, S.; Catalano, S. The weight of obesity in breast cancer progression and metastasis: Clinical and molecular perspectives. Semin. Cancer Biol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Catalano, S.; Marsico, S.; Giordano, C.; Mauro, L.; Rizza, P.; Panno, M.L.; Ando, S. Leptin enhances, via AP-1, expression of aromatase in the MCF-7 cell line. J. Biol. Chem. 2003, 278, 28668–28676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catalano, S.; Mauro, L.; Marsico, S.; Giordano, C.; Rizza, P.; Rago, V.; Montanaro, D.; Maggiolini, M.; Panno, M.L.; Ando, S. Leptin induces, via ERK1/ERK2 signal, functional activation of estrogen receptor alpha in MCF-7 cells. J. Biol. Chem. 2004, 279, 19908–19915. [Google Scholar] [CrossRef] [Green Version]
- Barone, I.; Catalano, S.; Gelsomino, L.; Marsico, S.; Giordano, C.; Panza, S.; Bonofiglio, D.; Bossi, G.; Covington, K.R.; Fuqua, S.A.; et al. Leptin mediates tumor-stromal interactions that promote the invasive growth of breast cancer cells. Cancer Res. 2012, 72, 1416–1427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giordano, C.; Vizza, D.; Panza, S.; Barone, I.; Bonofiglio, D.; Lanzino, M.; Sisci, D.; De Amicis, F.; Fuqua, S.A.; Catalano, S.; et al. Leptin increases HER2 protein levels through a STAT3-mediated up-regulation of Hsp90 in breast cancer cells. Mol. Oncol. 2013, 7, 379–391. [Google Scholar] [CrossRef]
- Saxena, N.K.; Taliaferro-Smith, L.; Knight, B.B.; Merlin, D.; Anania, F.A.; O’Regan, R.M.; Sharma, D. Bidirectional crosstalk between leptin and insulin-like growth factor-I signaling promotes invasion and migration of breast cancer cells via transactivation of epidermal growth factor receptor. Cancer Res. 2008, 68, 9712–9722. [Google Scholar] [CrossRef] [Green Version]
- Guo, S.; Gonzalez-Perez, R.R. Notch, IL-1 and leptin crosstalk outcome (NILCO) is critical for leptin-induced proliferation, migration and VEGF/VEGFR-2 expression in breast cancer. PLoS ONE 2011, 6, e21467. [Google Scholar] [CrossRef] [Green Version]
- Cao, R.; Brakenhielm, E.; Wahlestedt, C.; Thyberg, J.; Cao, Y. Leptin induces vascular permeability and synergistically stimulates angiogenesis with FGF-2 and VEGF. Proc. Natl. Acad. Sci. USA 2001, 98, 6390–6395. [Google Scholar] [CrossRef] [Green Version]
- Zarkesh-Esfahani, H.; Pockley, A.G.; Wu, Z.; Hellewell, P.G.; Weetman, A.P.; Ross, R.J. Leptin indirectly activates human neutrophils via induction of TNF-alpha. J. Immunol. 2004, 172, 1809–1814. [Google Scholar] [CrossRef] [Green Version]
- Santander, A.M.; Lopez-Ocejo, O.; Casas, O.; Agostini, T.; Sanchez, L.; Lamas-Basulto, E.; Carrio, R.; Cleary, M.P.; Gonzalez-Perez, R.R.; Torroella-Kouri, M. Paracrine Interactions between Adipocytes and Tumor Cells Recruit and Modify Macrophages to the Mammary Tumor Microenvironment: The Role of Obesity and Inflammation in Breast Adipose Tissue. Cancers 2015, 7, 143–178. [Google Scholar] [CrossRef] [PubMed]
- Yom, C.K.; Lee, K.M.; Han, W.; Kim, S.W.; Kim, H.S.; Moon, B.I.; Jeong, K.Y.; Im, S.A.; Noh, D.Y. Leptin as a potential target for estrogen receptor-positive breast cancer. J. Breast Cancer 2013, 16, 138–145. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Zha, X.; Chen, W.; Zhu, T.; Qiu, J.; Roe, O.D.; Li, J.; Wang, Z.; Yin, Y. Leptin attenuates the anti-estrogen effect of tamoxifen in breast cancer. Biomed. Pharmacother. Biomed. Pharmacother. 2013, 67, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.; Shi, D.; Qiu, J.; Zhu, F.; Qian, J.; He, S.; Shu, Y.; Yin, Y.; Chen, X. ObRb downregulation increases breast cancer cell sensitivity to tamoxifen. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2015, 36, 6813–6821. [Google Scholar] [CrossRef]
- Papanikolaou, V.; Stefanou, N.; Dubos, S.; Papathanasiou, I.; Palianopoulou, M.; Valiakou, V.; Tsezou, A. Synergy of leptin/STAT3 with HER2 receptor induces tamoxifen resistance in breast cancer cells through regulation of apoptosis-related genes. Cell. Oncol. 2015, 38, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Bougaret, L.; Delort, L.; Billard, H.; Le Huede, C.; Boby, C.; De la Foye, A.; Rossary, A.; Mojallal, A.; Damour, O.; Auxenfans, C.; et al. Adipocyte/breast cancer cell crosstalk in obesity interferes with the anti-proliferative efficacy of tamoxifen. PLoS ONE 2018, 13, e0191571. [Google Scholar] [CrossRef] [Green Version]
- Delort, L.; Bougaret, L.; Cholet, J.; Vermerie, M.; Billard, H.; Decombat, C.; Bourgne, C.; Berger, M.; Dumontet, C.; Caldefie-Chezet, F. Hormonal Therapy Resistance and Breast Cancer: Involvement of Adipocytes and Leptin. Nutrients 2019, 11, 2839. [Google Scholar] [CrossRef] [Green Version]
- Barone, I.; Cui, Y.; Herynk, M.H.; Corona-Rodriguez, A.; Giordano, C.; Selever, J.; Beyer, A.; Ando, S.; Fuqua, S.A. Expression of the K303R estrogen receptor-alpha breast cancer mutation induces resistance to an aromatase inhibitor via addiction to the PI3K/Akt kinase pathway. Cancer Res. 2009, 69, 4724–4732. [Google Scholar] [CrossRef] [Green Version]
- Rovito, D.; Giordano, C.; Plastina, P.; Barone, I.; De Amicis, F.; Mauro, L.; Rizza, P.; Lanzino, M.; Catalano, S.; Bonofiglio, D.; et al. Omega-3 DHA- and EPA-dopamine conjugates induce PPARgamma-dependent breast cancer cell death through autophagy and apoptosis. Biochim. Biophys. Acta 2015, 1850, 2185–2195. [Google Scholar] [CrossRef]
- Mancuso, R.; Raut, D.S.; Marino, N.; De Luca, G.; Giordano, C.; Catalano, S.; Barone, I.; Ando, S.; Gabriele, B. A Palladium-Catalyzed Carbonylation Approach to Eight-Membered Lactam Derivatives with Antitumor Activity. Chemistry 2016, 22, 3053–3064. [Google Scholar] [CrossRef]
- Mauro, L.; Naimo, G.D.; Gelsomino, L.; Malivindi, R.; Bruno, L.; Pellegrino, M.; Tarallo, R.; Memoli, D.; Weisz, A.; Panno, M.L.; et al. Uncoupling effects of estrogen receptor alpha on LKB1/AMPK interaction upon adiponectin exposure in breast cancer. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2018, 32, 4343–4355. [Google Scholar] [CrossRef] [Green Version]
- Bartella, V.; Rizza, P.; Barone, I.; Zito, D.; Giordano, F.; Giordano, C.; Catalano, S.; Mauro, L.; Sisci, D.; Panno, M.L.; et al. Estrogen receptor beta binds Sp1 and recruits a corepressor complex to the estrogen receptor alpha gene promoter. Breast Cancer Res. Treat. 2012, 134, 569–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mauro, L.; Sisci, D.; Bartucci, M.; Salerno, M.; Kim, J.; Tam, T.; Guvakova, M.A.; Ando, S.; Surmacz, E. SHC-alpha5beta1 integrin interactions regulate breast cancer cell adhesion and motility. Exp. Cell Res. 1999, 252, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Catalano, S.; Campana, A.; Giordano, C.; Gyorffy, B.; Tarallo, R.; Rinaldi, A.; Bruno, G.; Ferraro, A.; Romeo, F.; Lanzino, M.; et al. Expression and Function of Phosphodiesterase Type 5 in Human Breast Cancer Cell Lines and Tissues: Implications for Targeted Therapy. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 2271–2282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panza, S.; Gelsomino, L.; Malivindi, R.; Rago, V.; Barone, I.; Giordano, C.; Giordano, F.; Leggio, A.; Comande, A.; Liguori, A.; et al. Leptin Receptor as a Potential Target to Inhibit Human Testicular Seminoma Growth. Am. J. Pathol. 2019, 189, 687–698. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Mihaly, Z.; Kormos, M.; Lanczky, A.; Dank, M.; Budczies, J.; Szasz, M.A.; Gyorffy, B. A meta-analysis of gene expression-based biomarkers predicting outcome after tamoxifen treatment in breast cancer. Breast Cancer Res. Treat. 2013, 140, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Catalano, S.; Leggio, A.; Barone, I.; De Marco, R.; Gelsomino, L.; Campana, A.; Malivindi, R.; Panza, S.; Giordano, C.; Liguori, A.; et al. A novel leptin antagonist peptide inhibits breast cancer growth in vitro and in vivo. J. Cell. Mol. Med. 2015, 19, 1122–1132. [Google Scholar] [CrossRef]
- Gruen, M.L.; Hao, M.; Piston, D.W.; Hasty, A.H. Leptin requires canonical migratory signaling pathways for induction of monocyte and macrophage chemotaxis. Am. J. Physiol. Cell Physiol. 2007, 293, C1481–C1488. [Google Scholar] [CrossRef]
- Li, K.; Wei, L.; Huang, Y.; Wu, Y.; Su, M.; Pang, X.; Wang, N.; Ji, F.; Zhong, C.; Chen, T. Leptin promotes breast cancer cell migration and invasion via IL-18 expression and secretion. Int. J. Oncol. 2016, 48, 2479–2487. [Google Scholar] [CrossRef] [Green Version]
- Cao, H.; Huang, Y.; Wang, L.; Wang, H.; Pang, X.; Li, K.; Dang, W.; Tang, H.; Wei, L.; Su, M.; et al. Leptin promotes migration and invasion of breast cancer cells by stimulating IL-8 production in M2 macrophages. Oncotarget 2016, 7, 65441–65453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandis, A.Z.; Cherla, R.P.; Ganju, R.K. Differential regulation of CXCR4-mediated T-cell chemotaxis and mitogen-activated protein kinase activation by the membrane tyrosine phosphatase, CD45. J. Biol. Chem. 2003, 278, 9536–9543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajasekaran, D.; Groning, S.; Schmitz, C.; Zierow, S.; Drucker, N.; Bakou, M.; Kohl, K.; Mertens, A.; Lue, H.; Weber, C.; et al. Macrophage Migration Inhibitory Factor-CXCR4 Receptor Interactions: EVIDENCE FOR PARTIAL ALLOSTERIC AGONISM IN COMPARISON WITH CXCL12 CHEMOKINE. J. Biol. Chem. 2016, 291, 15881–15895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruytinx, P.; Proost, P.; Van Damme, J.; Struyf, S. Chemokine-Induced Macrophage Polarization in Inflammatory Conditions. Front. Immunol. 2018, 9, 1930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tulotta, C.; Stefanescu, C.; Chen, Q.; Torraca, V.; Meijer, A.H.; Snaar-Jagalska, B.E. CXCR4 signaling regulates metastatic onset by controlling neutrophil motility and response to malignant cells. Sci. Rep. 2019, 9, 2399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grande, F.; Barone, I.; Aiello, F.; Brancale, A.; Cancellieri, M.; Badolato, M.; Chemi, F.; Giordano, C.; Vircillo, V.; Bonofiglio, D.; et al. Identification of novel 2-(1H-indol-1-yl)-benzohydrazides CXCR4 ligands impairing breast cancer growth and motility. Future Med. Chem. 2016, 8, 93–106. [Google Scholar] [CrossRef]
- Cassetta, L.; Pollard, J.W. Targeting macrophages: Therapeutic approaches in cancer. Nat. Rev. Drug Discov. 2018, 17, 887–904. [Google Scholar] [CrossRef]
- Chanmee, T.; Ontong, P.; Konno, K.; Itano, N. Tumor-associated macrophages as major players in the tumor microenvironment. Cancers 2014, 6, 1670–1690. [Google Scholar] [CrossRef] [Green Version]
- Acedo, S.C.; Gambero, S.; Cunha, F.G.; Lorand-Metze, I.; Gambero, A. Participation of leptin in the determination of the macrophage phenotype: An additional role in adipocyte and macrophage crosstalk. In Vitro Cell. Dev. Biol. Anim. 2013, 49, 473–478. [Google Scholar] [CrossRef]
- Nagasawa, T.; Hirota, S.; Tachibana, K.; Takakura, N.; Nishikawa, S.; Kitamura, Y.; Yoshida, N.; Kikutani, H.; Kishimoto, T. Defects of B-cell lymphopoiesis and bone-marrow myelopoiesis in mice lacking the CXC chemokine PBSF/SDF-1. Nature 1996, 382, 635–638. [Google Scholar] [CrossRef] [PubMed]
- Ye, J. Emerging role of adipose tissue hypoxia in obesity and insulin resistance. Int. J. obes. 2009, 33, 54–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schioppa, T.; Uranchimeg, B.; Saccani, A.; Biswas, S.K.; Doni, A.; Rapisarda, A.; Bernasconi, S.; Saccani, S.; Nebuloni, M.; Vago, L.; et al. Regulation of the chemokine receptor CXCR4 by hypoxia. J. Exp. Med. 2003, 198, 1391–1402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romeo, E.; Caserta, C.A.; Rumio, C.; Marcucci, F. The Vicious Cross-Talk between Tumor Cells with an EMT Phenotype and Cells of the Immune System. Cells 2019, 8, 460. [Google Scholar] [CrossRef] [Green Version]
- Pfeiler, G.; Konigsberg, R.; Fesl, C.; Mlineritsch, B.; Stoeger, H.; Singer, C.F.; Postlberger, S.; Steger, G.G.; Seifert, M.; Dubsky, P.; et al. Impact of body mass index on the efficacy of endocrine therapy in premenopausal patients with breast cancer: An analysis of the prospective ABCSG-12 trial. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2011, 29, 2653–2659. [Google Scholar] [CrossRef] [Green Version]
- Coates, A.S.; Winer, E.P.; Goldhirsch, A.; Gelber, R.D.; Gnant, M.; Piccart-Gebhart, M.; Thurlimann, B.; Senn, H.J.; Panel, M. Tailoring therapies--improving the management of early breast cancer: St Gallen International Expert Consensus on the Primary Therapy of Early Breast Cancer 2015. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2015, 26, 1533–1546. [Google Scholar] [CrossRef]
- Goodwin, P.J. Obesity, insulin resistance and breast cancer outcomes. Breast 2015, 24 (Suppl. 2), S56–S59. [Google Scholar] [CrossRef]
- Morgan, M.M.; Arendt, L.M.; Alarid, E.T.; Beebe, D.J.; Johnson, B.P. Mammary adipose stromal cells derived from obese women reduce sensitivity to the aromatase inhibitor anastrazole in an organotypic breast model. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2019, 33, 8623–8633. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Categories | Diseases or Functions Annotation | p-Value | Activation z-score | Molecules | #Molecules |
|---|---|---|---|---|---|
| Cellular Movement | Invasion of tumor cell lines | 1.97 × 10−3 | 1.797 | ARRDC3, BCAR3, CXCR4, EDN1, FABP5, HIPK2, IL13RA2, LCN2, NDRG2, NOS2, SPP1, VEGFB | 12 |
| Cellular Movement, Hematological System Development and Function, Immune Cell Trafficking, Inflammatory Response | Chemotaxis of phagocytes | 7.26 × 10−4 | 1.616 | CSF1, CXCR4, IL1B, S100A8, SPP1, VEGFB | 6 |
| Cardiovascular System Development and Function, Cellular Development, Cellular Growth and Proliferation, Organismal Development, Tissue Development | Proliferation of endothelial cells | 1.33 × 10−5 | 1.562 | CSF1, EDN1, FABP4, GAS6, HIPK2, IL1B, PROCR, TNFSF15, VEGFB | 9 |
| Cellular Movement | Invasion of cells | 1.67 × 10−4 | 1.487 | ARRDC3, BCAR3, CLCA2, CXCR4, EDN1, FABP5, HIPK2, IER3, IL13RA2, IL1B, LCN2, NDRG2, NOS2, SPP1, VEGFB | 15 |
| Cellular Movement, Hematological System Development and Function, Immune Cell Trafficking, Inflammatory Response | Chemotaxis of myeloid cells | 9.32 × 10−4 | 1.387 | CSF1, CXCR4, IL1B, S100A8, SPP1, VEGFB | 6 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gelsomino, L.; Giordano, C.; La Camera, G.; Sisci, D.; Marsico, S.; Campana, A.; Tarallo, R.; Rinaldi, A.; Fuqua, S.; Leggio, A.; et al. Leptin Signaling Contributes to Aromatase Inhibitor Resistant Breast Cancer Cell Growth and Activation of Macrophages. Biomolecules 2020, 10, 543. https://doi.org/10.3390/biom10040543
Gelsomino L, Giordano C, La Camera G, Sisci D, Marsico S, Campana A, Tarallo R, Rinaldi A, Fuqua S, Leggio A, et al. Leptin Signaling Contributes to Aromatase Inhibitor Resistant Breast Cancer Cell Growth and Activation of Macrophages. Biomolecules. 2020; 10(4):543. https://doi.org/10.3390/biom10040543
Chicago/Turabian StyleGelsomino, Luca, Cinzia Giordano, Giusi La Camera, Diego Sisci, Stefania Marsico, Antonella Campana, Roberta Tarallo, Antonio Rinaldi, Suzanne Fuqua, Antonella Leggio, and et al. 2020. "Leptin Signaling Contributes to Aromatase Inhibitor Resistant Breast Cancer Cell Growth and Activation of Macrophages" Biomolecules 10, no. 4: 543. https://doi.org/10.3390/biom10040543