
Inhibition of Non-Small Cell Lung Cancer Proliferation and Survival by Rosemary Extract Is Associated with Activation of ERK and AMPK



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Rosemary Extract Preparation
2.3. Cell Culture and Treatment
2.4. Cell Proliferation Assay
2.5. Clonogenic Survival Assay
2.6. Immunoblotting
2.7. Wound Healing Assay
2.8. Statistical Analysis
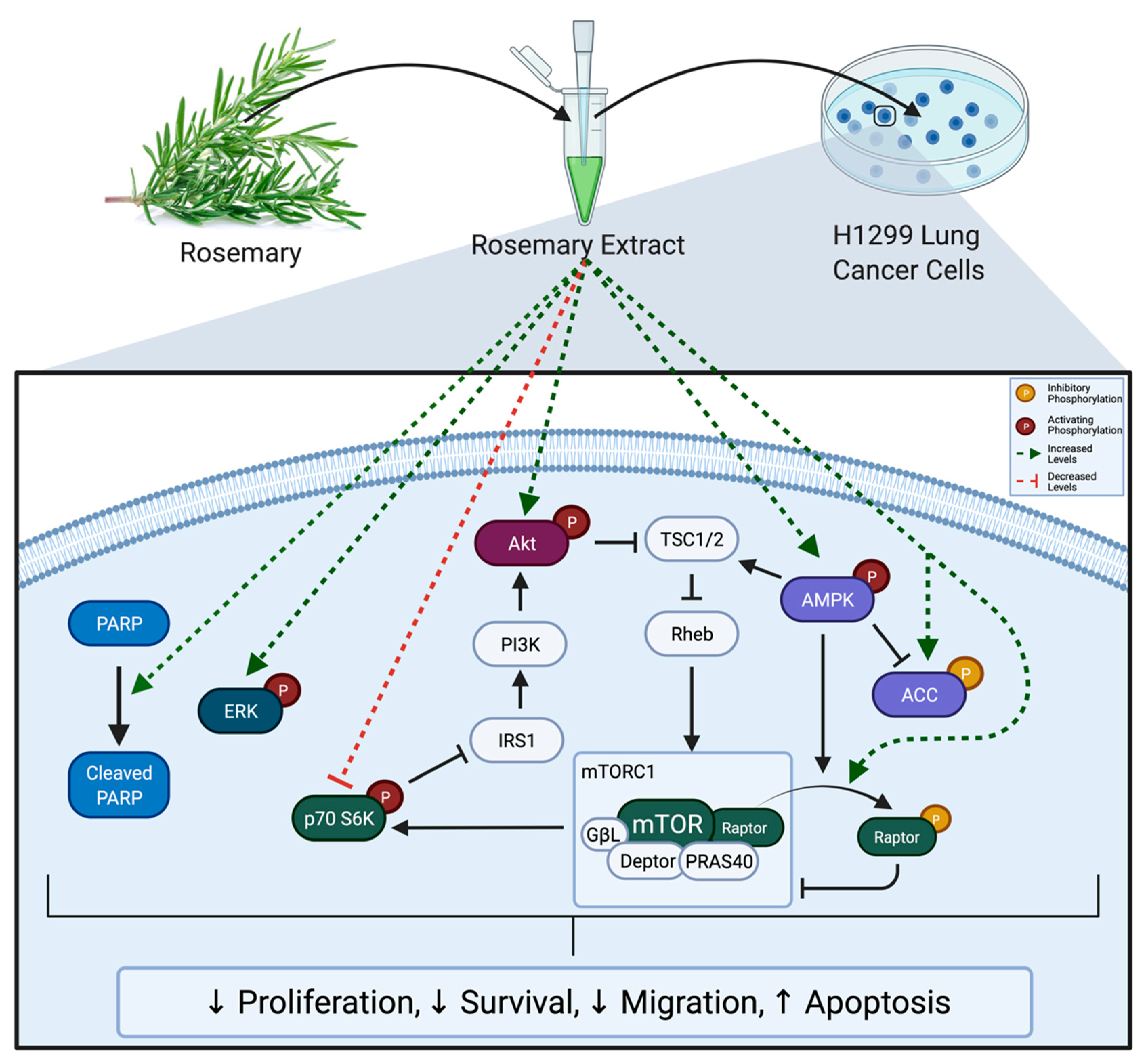
3. Results
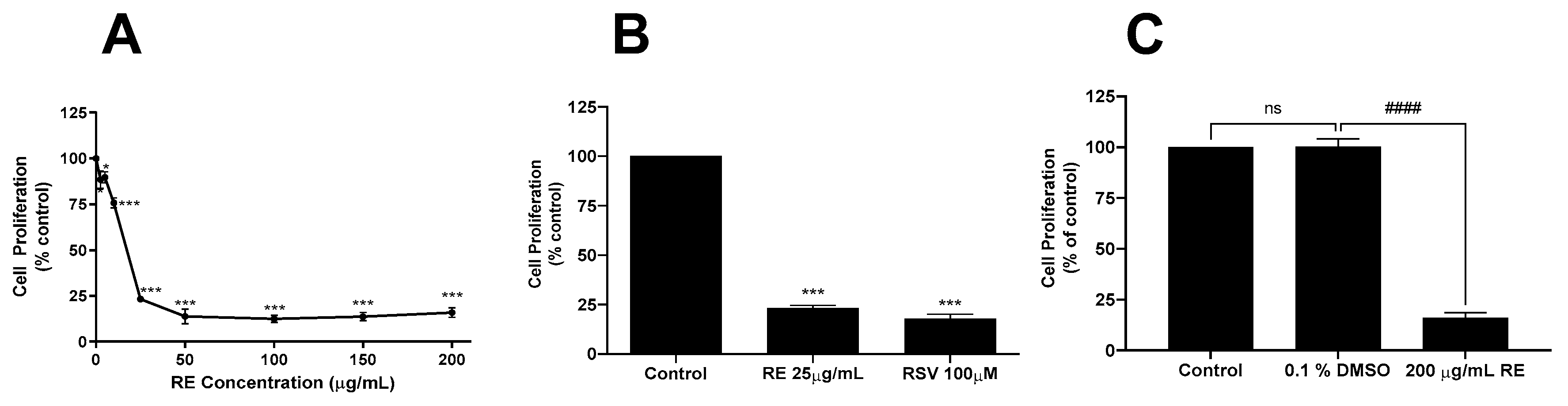
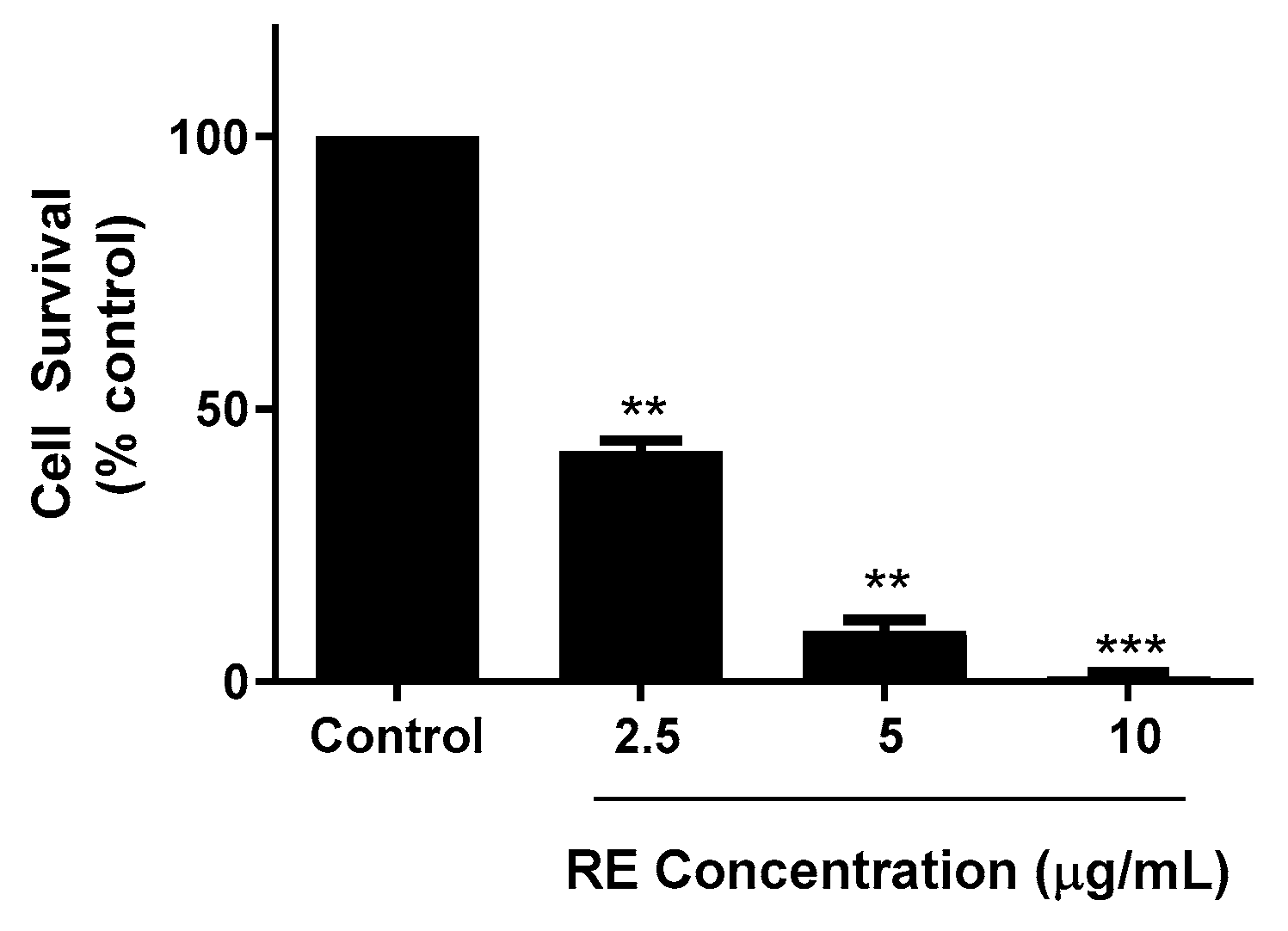
3.1. RE Inhibits Proliferation and Survival and Promotes Apoptosis of H1299 Lung Cancer Cells
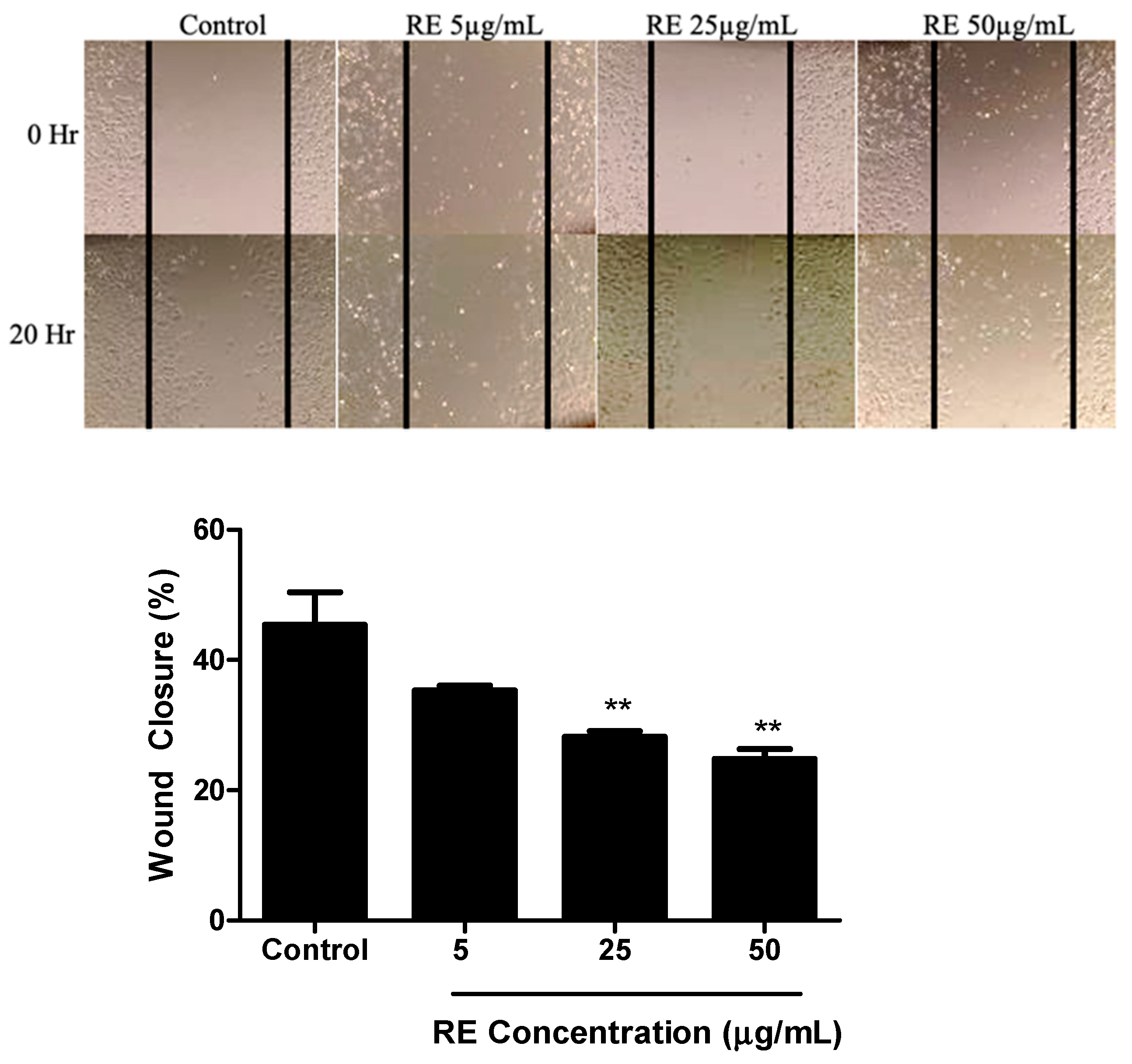
3.2. RE Inhibits Migration of H1299 Lung Cancer Cells
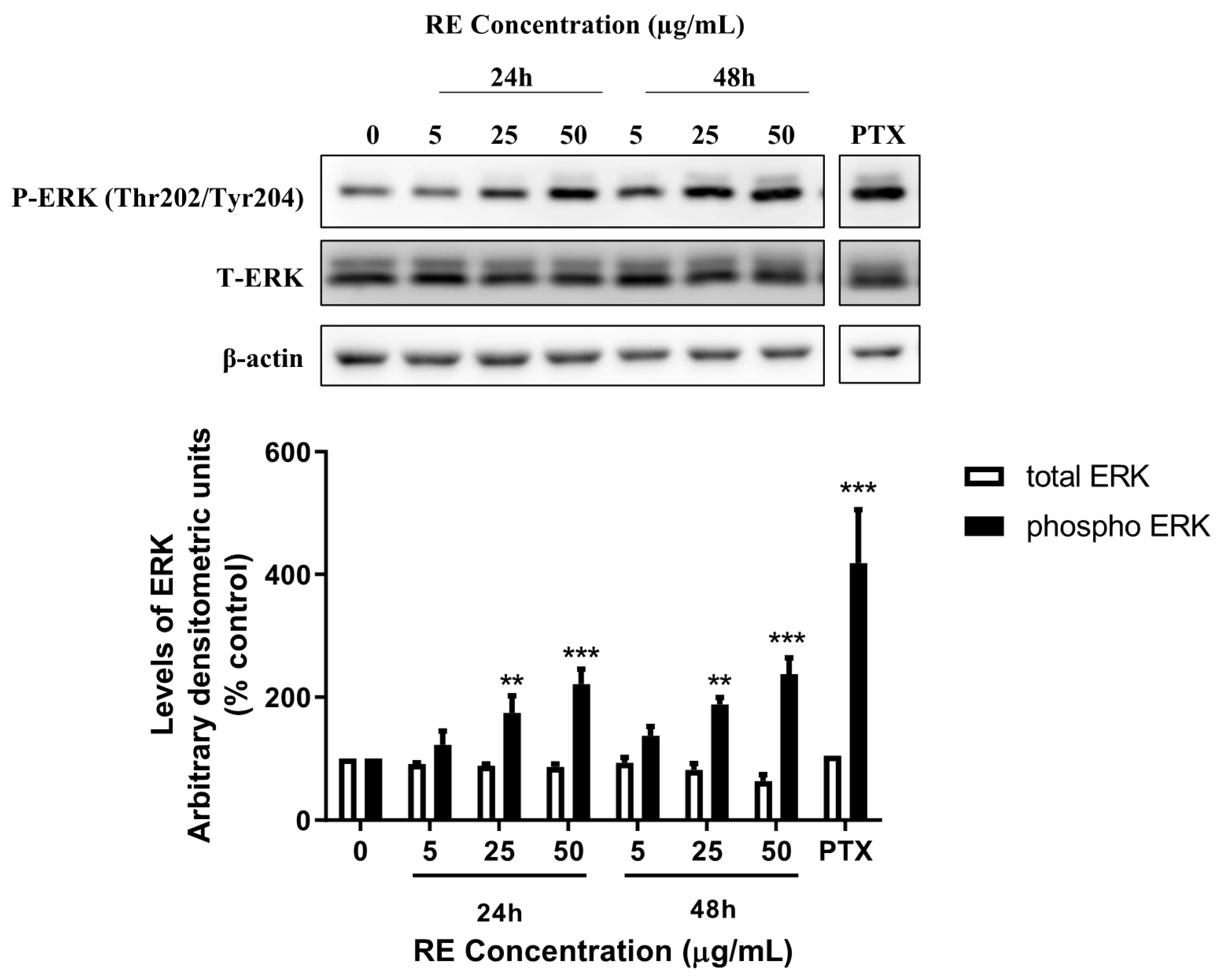
3.3. RE Increases ERK Phosphorylation in H1299 Lung Cancer Cells
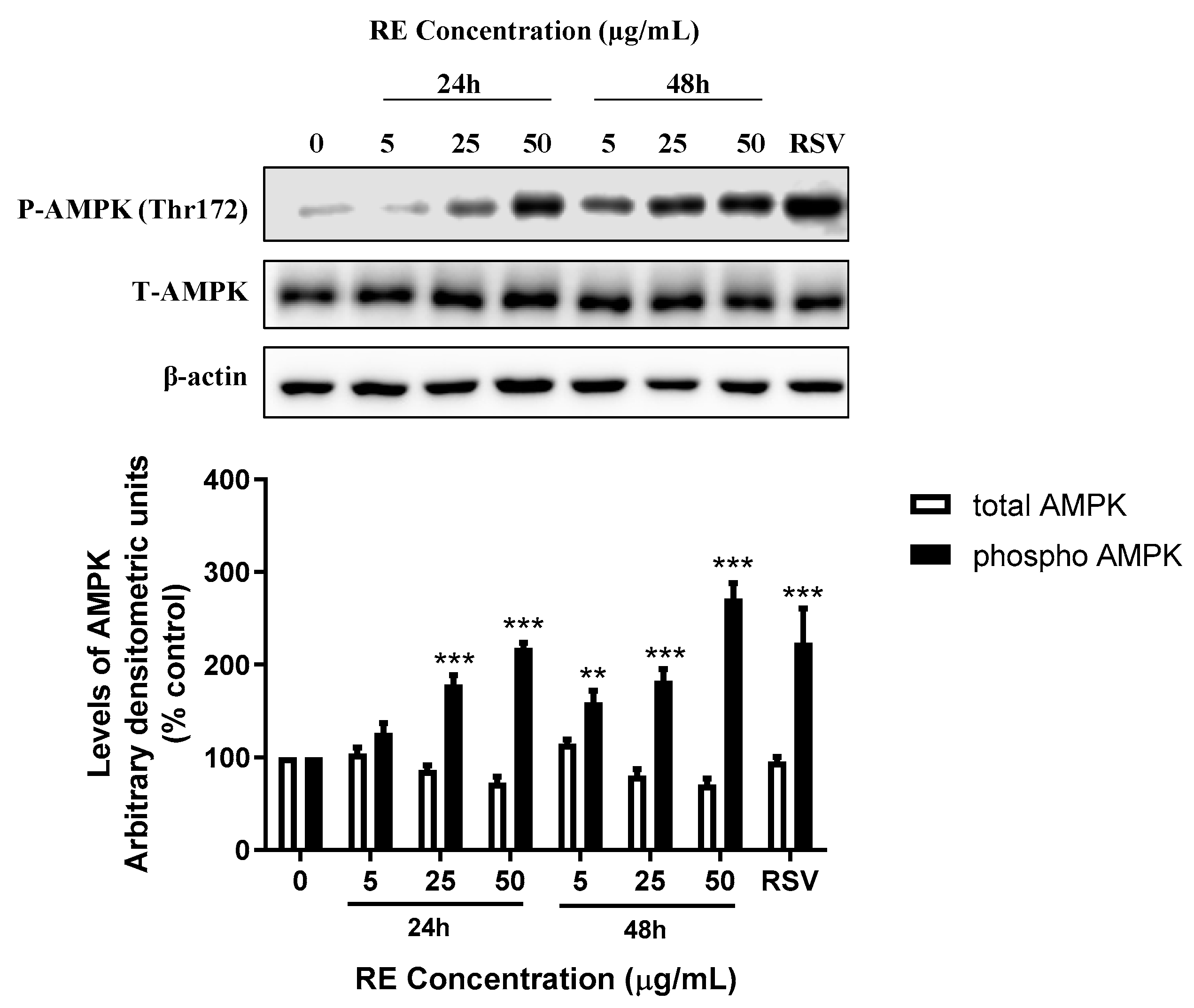
3.4. RE Increases AMPK Phosphorylation in H1299 Lung Cancer Cells
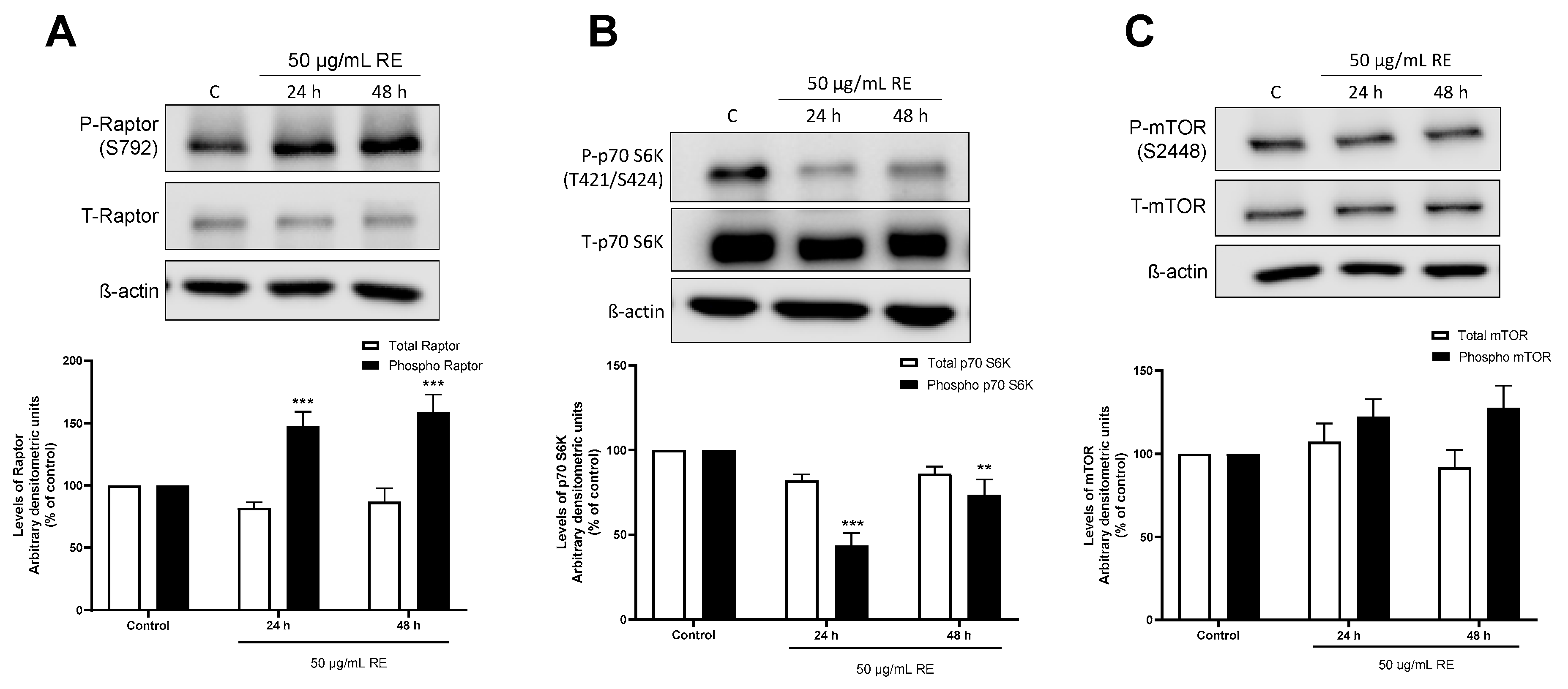
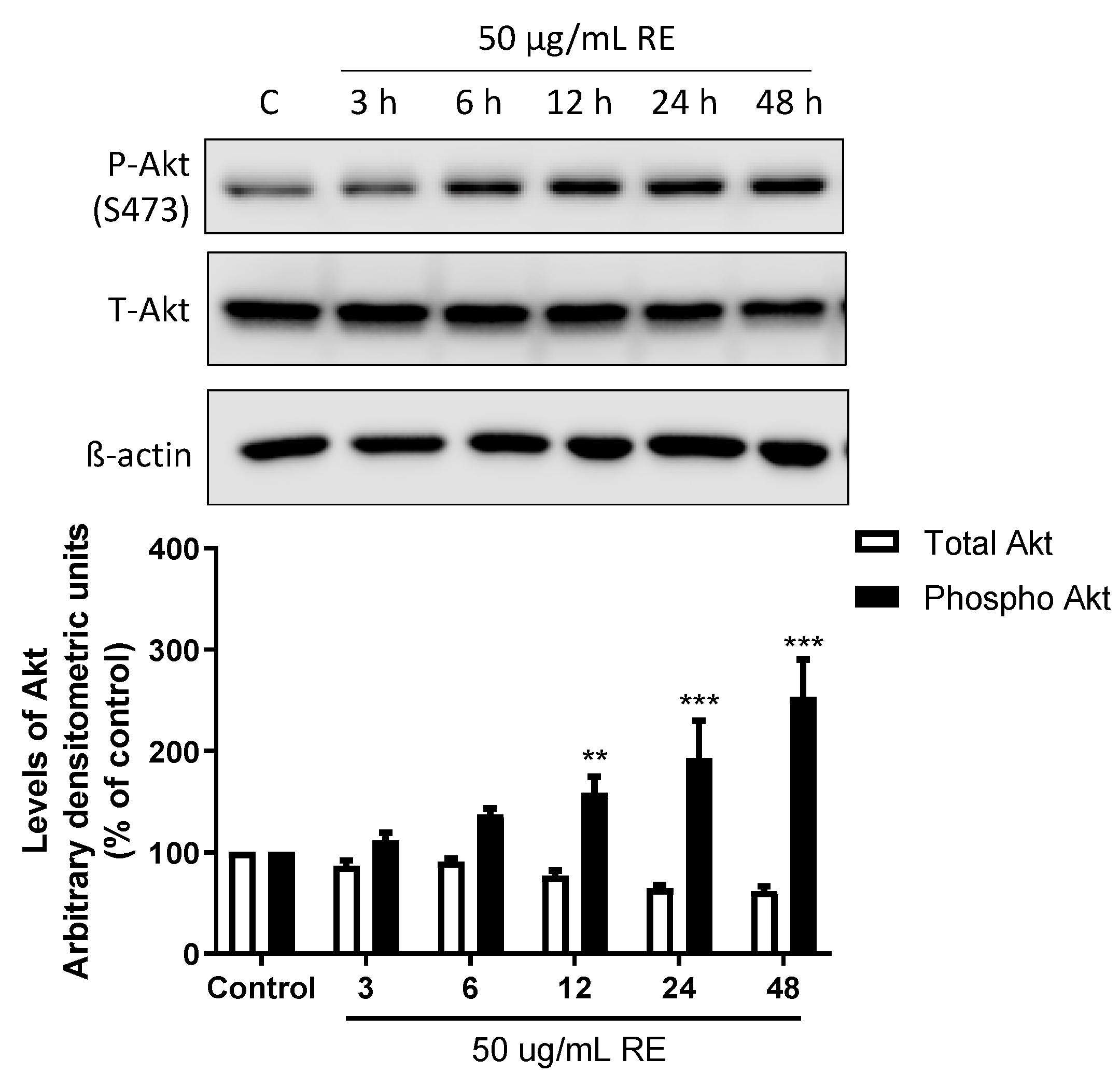
3.5. RE phosphorylates Raptor in H1299 NSCLC Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Araujo, L.H.; Horn, L.; Merritt, R.E.; Shilo, K.; Meng, X.-W.; Carbone, D.P. Cancer of the Lung: Non-Small Cell Lung Cancer and Small Cell Lung Cancer. In Abeloff’s Clinical Oncology; Elsevier: Philadelphia, PA, USA, 2019; pp. 1108–1158. ISBN 978-0-323-47674-4. [Google Scholar]
- Weaver, B.A. How Taxol/Paclitaxel Kills Cancer Cells. Mol. Biol. Cell 2014, 25, 2677–2681. [Google Scholar] [CrossRef] [PubMed]
- van Oosterom, A.T.; Schrijvers, D.; Schrijvers, D. Docetaxel (Taxotere), a Review of Preclinical and Clinical Experience. Part II: Clinical Experience. Anticancer Drugs 1995, 6, 356–368. [Google Scholar] [CrossRef] [PubMed]
- Cheung, S.; Tai, J. Anti-Proliferative and Antioxidant Properties of Rosemary Rosmarinus Officinalis. Oncol. Rep. 2007, 17, 1525–1531. [Google Scholar] [CrossRef] [Green Version]
- Moore, J.; Yousef, M.; Tsiani, E. Anticancer Effects of Rosemary (Rosmarinus officinalis L.) Extract and Rosemary Extract Polyphenols. Nutrients 2016, 8, 731. [Google Scholar] [CrossRef] [PubMed]
- Mena, P.; Cirlini, M.; Tassotti, M.; Herrlinger, K.; Dall’Asta, C.; Del Rio, D. Phytochemical Profiling of Flavonoids, Phenolic Acids, Terpenoids, and Volatile Fraction of a Rosemary (Rosmarinus officinalis L.) Extract. Molecules 2016, 21, 1576. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, F.; Yang, X.; Geng, M.; Huang, M. Targeting ERK, an Achilles’ Heel of the MAPK Pathway, in Cancer Therapy. Acta Pharm. Sin. B 2018, 8, 552–562. [Google Scholar] [CrossRef] [PubMed]
- Chambard, J.-C.; Lefloch, R.; Pouysségur, J.; Lenormand, P. ERK Implication in Cell Cycle Regulation. Biochim. Biophys. Acta BBA-Mol. Cell Res. 2007, 1773, 1299–1310. [Google Scholar] [CrossRef] [PubMed]
- Alessi, D.R.; Sakamoto, K.; Bayascas, J.R. LKB1-Dependent Signaling Pathways. Annu. Rev. Biochem. 2006, 75, 137–163. [Google Scholar] [CrossRef]
- Zhang, Y.; Meng, Q.; Sun, Q.; Xu, Z.-X.; Zhou, H.; Wang, Y. LKB1 Deficiency-Induced Metabolic Reprogramming in Tumorigenesis and Non-Neoplastic Diseases. Mol. Metab. 2021, 44, 101131. [Google Scholar] [CrossRef] [PubMed]
- Mihaylova, M.M.; Shaw, R.J. The AMPK Signalling Pathway Coordinates Cell Growth, Autophagy and Metabolism. Nat. Cell Biol. 2011, 13, 1016–1023. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Wang, X.; Chen, L.; Huang, L.; Dong, R. Belinostat-Induced Apoptosis and Growth Inhibition in Pancreatic Cancer Cells Involve Activation of TAK1-AMPK Signaling Axis. Biochem. Biophys. Res. Commun. 2013, 437, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Isebaert, S.F.; Swinnen, J.V.; McBride, W.H.; Begg, A.C.; Haustermans, K.M. 5-Aminoimidazole-4-Carboxamide Riboside Enhances Effect of Ionizing Radiation in PC3 Prostate Cancer Cells. Int. J. Radiat. Oncol. Biol. Phys. 2011, 81, 1515–1523. [Google Scholar] [CrossRef] [PubMed]
- Vazquez-Martin, A.; Oliveras-Ferraros, C.; Menendez, J. The Antidiabetic Drug Metformin Suppresses HER2 (ErbB-2) Oncoprotein Overexpression via Inhibition of the MTOR Effector P70S6K1 in Human Breast Carcinoma Cells. Cell Cycle Georget. Tex 2009, 8, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Storozhuk, Y.; Hopmans, S.N.; Sanli, T.; Barron, C.; Tsiani, E.; Cutz, J.-C.; Pond, G.; Wright, J.; Singh, G.; Tsakiridis, T. Metformin Inhibits Growth and Enhances Radiation Response of Non-Small Cell Lung Cancer (NSCLC) through ATM and AMPK. Br. J. Cancer 2013, 108, 2021–2032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marín-Aguilar, F.; Pavillard, L.; Giampieri, F.; Bullón, P.; Cordero, M. Adenosine Monophosphate (AMP)-Activated Protein Kinase: A New Target for Nutraceutical Compounds. Int. J. Mol. Sci. 2017, 18, 288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Ma, J.; Zhang, N.; Yang, Q.; Jin, Y.; Wang, Y. The Acetyl-CoA Carboxylase Enzyme: A Target for Cancer Therapy? Expert Rev. Anticancer Ther. 2015, 15, 667–676. [Google Scholar] [CrossRef]
- Kim, D.-H.; Sarbassov, D.D.; Ali, S.M.; Latek, R.R.; Guntur, K.V.P.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. GbetaL, a Positive Regulator of the Rapamycin-Sensitive Pathway Required for the Nutrient-Sensitive Interaction between Raptor and MTOR. Mol. Cell 2003, 11, 895–904. [Google Scholar] [CrossRef]
- Inoki, K.; Li, Y.; Zhu, T.; Wu, J.; Guan, K.-L. TSC2 Is Phosphorylated and Inhibited by Akt and Suppresses MTOR Signalling. Nat. Cell Biol. 2002, 4, 648–657. [Google Scholar] [CrossRef]
- Navé, B.T.; Ouwens, M.; Withers, D.J.; Alessi, D.R.; Shepherd, P.R. Mammalian Target of Rapamycin Is a Direct Target for Protein Kinase B: Identification of a Convergence Point for Opposing Effects of Insulin and Amino-Acid Deficiency on Protein Translation. Biochem. J. 1999, 344 Pt 2, 427–431. [Google Scholar] [CrossRef] [PubMed]
- Bond, P. Regulation of MTORC1 by Growth Factors, Energy Status, Amino Acids and Mechanical Stimuli at a Glance. J. Int. Soc. Sports Nutr. 2016, 13, 8. [Google Scholar] [CrossRef] [Green Version]
- Hara, K.; Maruki, Y.; Long, X.; Yoshino, K.; Oshiro, N.; Hidayat, S.; Tokunaga, C.; Avruch, J.; Yonezawa, K. Raptor, a Binding Partner of Target of Rapamycin (TOR), Mediates TOR Action. Cell 2002, 110, 177–189. [Google Scholar] [CrossRef] [Green Version]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK Phosphorylation of Raptor Mediates a Metabolic Checkpoint. Mol. Cell 2008, 30, 214–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hresko, R.C.; Mueckler, M. MTOR.RICTOR Is the Ser473 Kinase for Akt/Protein Kinase B in 3T3-L1 Adipocytes. J. Biol. Chem. 2005, 280, 40406–40416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murugan, A.K. MTOR: Role in Cancer, Metastasis and Drug Resistance. Semin. Cancer Biol. 2019, 59, 92–111. [Google Scholar] [CrossRef] [PubMed]
- Tian, T.; Li, X.; Zhang, J. MTOR Signaling in Cancer and MTOR Inhibitors in Solid Tumor Targeting Therapy. Int. J. Mol. Sci. 2019, 20, 755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, J.; Megaly, M.; MacNeil, A.J.; Klentrou, P.; Tsiani, E. Rosemary Extract Reduces Akt/MTOR/P70S6K Activation and Inhibits Proliferation and Survival of A549 Human Lung Cancer Cells. Biomed. Pharmacother. 2016, 83, 725–732. [Google Scholar] [CrossRef] [PubMed]
- Naimi, M.; Tsakiridis, T.; Stamatatos, T.C.; Alexandropoulos, D.I.; Tsiani, E. Increased Skeletal Muscle Glucose Uptake by Rosemary Extract through AMPK Activation. Appl. Physiol. Nutr. Metab. 2014, 40, 407–413. [Google Scholar] [CrossRef] [PubMed]
- Moore, J.; Pickering, G.; Gaudette, N.J.; Tsiani, E. Resveratrol-Fortification of Red Wine Does Not Provide Greater Inhibition of Human Lung Cancer Cell Survival Compared to Non-Fortified Wine. J. Mol. Biochem. 2015, 4. Available online: http://jmolbiochem.com/index.php/JmolBiochem/article/view/148 (accessed on 20 July 2021).
- Jaglanian, A.; Termini, D.; Tsiani, E. Rosemary (Rosmarinus officinalis L.) Extract Inhibits Prostate Cancer Cell Proliferation and Survival by Targeting Akt and MTOR. Biomed. Pharmacother. 2020, 131, 110717. [Google Scholar] [CrossRef]
- Yousef, M.; Vlachogiannis, I.A.; Tsiani, E. Effects of Resveratrol against Lung Cancer: In Vitro and In Vivo Studies. Nutrients 2017, 9, 1231. [Google Scholar] [CrossRef] [Green Version]
- Jaglanian, A.; Tsiani, E. Rosemary Extract Inhibits Proliferation, Survival, Akt, and MTOR Signaling in Triple-Negative Breast Cancer Cells. Int. J. Mol. Sci. 2020, 21, 810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bacus, S.S.; Gudkov, A.V.; Lowe, M.; Lyass, L.; Yung, Y.; Komarov, A.P.; Keyomarsi, K.; Yarden, Y.; Seger, R. Taxol-Induced Apoptosis Depends on MAP Kinase Pathways (ERK and P38) and Is Independent of P53. Oncogene 2001, 20, 147–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lan, F.; Weikel, K.A.; Cacicedo, J.M.; Ido, Y. Resveratrol-Induced AMP-Activated Protein Kinase Activation Is Cell-Type Dependent: Lessons from Basic Research for Clinical Application. Nutrients 2017, 9, 751. [Google Scholar] [CrossRef] [Green Version]
- Rashid, A.; Liu, C.; Sanli, T.; Tsiani, E.; Singh, G.; Bristow, R.G.; Dayes, I.; Lukka, H.; Wright, J.; Tsakiridis, T. Resveratrol Enhances Prostate Cancer Cell Response to Ionizing Radiation. Modulation of the AMPK, Akt and MTOR Pathways. Radiat. Oncol. 2011, 6, 144. [Google Scholar] [CrossRef] [Green Version]
- Jose, C.; Hébert-Chatelain, E.; Bellance, N.; Larendra, A.; Su, M.; Nouette-Gaulain, K.; Rossignol, R. AICAR inhibits cancer cell growth and triggers cell-type distinct effects on OXPHOS biogenesis, oxidative stress and Akt activation. Biochim. Biophys. Acta BBA Bioenerget. 2011, 1807, 707–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, X.; Harkavy, B.; Shen, N.; Grohar, P.; Helman, L.J. Rapamycin Induces Feedback Activation of Akt Signaling through an IGF-1R-Dependent Mechanism. Oncogene 2007, 26, 1932–1940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breuleux, M.; Klopfenstein, M.; Stephan, C.; Doughty, C.A.; Barys, L.; Maira, S.-M.; Kwiatkowski, D.; Lane, H.A. Increased AKT S473 Phosphorylation after MTORC1 Inhibition Is Rictor Dependent and Does Not Predict Tumor Cell Response to PI3K/MTOR Inhibition. Mol. Cancer Ther. 2009, 8, 742–753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phelps, R.M.; Johnson, B.E.; Ihde, D.C.; Gazdar, A.F.; Carbone, D.P.; McClintock, P.R.; Linnoila, R.I.; Matthews, M.J.; Bunn, P.A.; Carney, D.; et al. NCI-Navy Medical Oncology Branch Cell Line Data Base. J. Cell. Biochem. 1996, 63, 32–91. [Google Scholar] [CrossRef] [PubMed]
- Yesil-Celiktas, O.; Sevimli, C.; Bedir, E.; Vardar-Sukan, F. Inhibitory Effects of Rosemary Extracts, Carnosic Acid and Rosmarinic Acid on the Growth of Various Human Cancer Cell Lines. Plant Foods Hum. Nutr. 2010, 65, 158–163. [Google Scholar] [CrossRef]
- Cagnol, S.; Chambard, J.-C. ERK and cell death: Mechanisms of ERK-induced cell death—Apoptosis, autophagy and senescence: Erk and cell death. FEBS J. 2010, 277, 2–21. [Google Scholar] [CrossRef] [PubMed]
- Reddy, K.B.; Nabha, S.M.; Atanaskova, N. Role of MAP Kinase in Tumor Progression and Invasion. Cancer Metastasis Rev. 2003, 22, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Yan, M.; Li, G.; Petiwala, S.M.; Householter, E.; Johnson, J.J. Standardized Rosemary (Rosmarinus officinalis) Extract Induces Nrf2/Sestrin-2 Pathway in Colon Cancer Cells. J. Funct. Foods 2015, 13, 137–147. [Google Scholar] [CrossRef]
- Sheridan, C.; Brumatti, G.; Elgendy, M.; Brunet, M.; Martin, S.J. An ERK-Dependent Pathway to Noxa Expression Regulates Apoptosis by Platinum-Based Chemotherapeutic Drugs. Oncogene 2010, 29, 6428–6441. [Google Scholar] [CrossRef] [Green Version]
- Bouzidi, A.; Magnifico, M.C.; Paiardini, A.; Macone, A.; Boumis, G.; Giardina, G.; Rinaldo, S.; Liberati, F.R.; Lauro, C.; Limatola, C.; et al. Cytosolic Serine Hydroxymethyltransferase Controls Lung Adenocarcinoma Cells Migratory Ability by Modulating AMP Kinase Activity. Cell Death Dis. 2020, 11. [Google Scholar] [CrossRef]
- Meisse, D.; Van de Casteele, M.; Beauloye, C.; Hainault, I.; Kefas, B.A.; Rider, M.H.; Foufelle, F.; Hue, L. Sustained Activation of AMP-Activated Protein Kinase Induces c-Jun N-Terminal Kinase Activation and Apoptosis in Liver Cells. FEBS Lett. 2002, 526, 38–42. [Google Scholar] [CrossRef]
- Hwang, J.-T.; Ha, J.; Park, O.J. Combination of 5-Fluorouracil and Genistein Induces Apoptosis Synergistically in Chemo-Resistant Cancer Cells through the Modulation of AMPK and COX-2 Signaling Pathways. Biochem. Biophys. Res. Commun. 2005, 332, 433–440. [Google Scholar] [CrossRef]
- Song, X.; Kim, S.-Y.; Zhang, L.; Tang, D.; Bartlett, D.L.; Kwon, Y.T.; Lee, Y.J. Role of AMP-Activated Protein Kinase in Cross-Talk between Apoptosis and Autophagy in Human Colon Cancer. Cell Death Dis. 2014, 5, e1504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, J.; Li, Y.; Gao, H.; Yang, D.; He, X.; Fang, Y.; Zhou, G. Phenolic Compound Ellagic Acid Inhibits Mitochondrial Respiration and Tumor Growth in Lung Cancer. Food Funct. 2020, 11, 6332–6339. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.-Z.; Guo, S.-S.; Liu, D.-X.; Zhang, L.; Sun, G.-C. Antiproliferative Effect and Autophagy Induction of Curcumin Derivative ZYX02-Na on the Human Lung Cancer Cells A549. J. Biochem. Mol. Toxicol. 2020, 34, e22592. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Zhang, Y.; Zhang, Y.; Chen, L.; Xu, X.; Dang, Y.; Ti, X. Phillygenin regulates proliferation and apoptosis of non-small cell lung cancer through by AMPK/ERK/NF-ΚB axis. Pharm. Int. J. Pharm. Sci. 2020, 75, 512–515. [Google Scholar] [CrossRef]
- Barber, M.C.; Price, N.T.; Travers, M.T. Structure and regulation of acetyl-CoA carboxylase genes of metazoa. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2005, 1733, 1–28. [Google Scholar] [CrossRef]
- Cho, Y.S.; Lee, J.I.; Shin, D.; Kim, H.T.; Jung, H.Y.; Lee, T.G.; Kang, L.-W.; Ahn, Y.-J.; Cho, H.-S.; Heo, Y.-S. Molecular Mechanism for the Regulation of Human ACC2 through Phosphorylation by AMPK. Biochem. Biophys. Res. Commun. 2010, 391, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Coughlan, K.A.; Valentine, R.J.; Ruderman, N.B.; Saha, A.K. AMPK Activation: A Therapeutic Target for Type 2 Diabetes? Diabetes Metab. Syndr. Obes. Targets Ther. 2014, 7, 241–253. [Google Scholar] [CrossRef] [Green Version]
- Svensson, R.U.; Parker, S.J.; Eichner, L.J.; Kolar, M.J.; Wallace, M.; Brun, S.N.; Lombardo, P.S.; Van Nostrand, J.L.; Hutchins, A.; Vera, L.; et al. Inhibition of Acetyl-CoA Carboxylase Suppresses Fatty Acid Synthesis and Tumor Growth of Non-Small-Cell Lung Cancer in Preclinical Models. Nat. Med. 2016, 22, 1108–1119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Saud, S.M.; Young, M.R.; Chen, G.; Hua, B. Targeting AMPK for Cancer Prevention and Treatment. Oncotarget 2015, 6, 7365–7378. [Google Scholar] [CrossRef] [Green Version]
- Bhaskar, P.T.; Hay, N. The Two TORCs and Akt. Dev. Cell 2007, 12, 487–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nogueira, V.; Park, Y.; Chen, C.-C.; Xu, P.-Z.; Chen, M.-L.; Tonic, I.; Unterman, T.; Hay, N. Akt Determines Replicative Senescence and Oxidative or Oncogenic Premature Senescence and Sensitizes Cells to Oxidative Apoptosis. Cancer Cell 2008, 14, 458–470. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Hu, X.; Liu, Y.; Dong, S.; Wen, Z.; He, W.; Zhang, S.; Huang, Q.; Shi, M. ROS Signaling under Metabolic Stress: Cross-Talk between AMPK and AKT Pathway. Mol. Cancer 2017, 16, 79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valdés, A.; Sullini, G.; Ibáñez, E.; Cifuentes, A.; García-Cañas, V. Rosemary Polyphenols Induce Unfolded Protein Response and Changes in Cholesterol Metabolism in Colon Cancer Cells. J. Funct. Foods 2015, 15, 429–439. [Google Scholar] [CrossRef]
- Min, K.-J.; Jung, K.-J.; Kwon, T.K. Carnosic Acid Induces Apoptosis through Reactive Oxygen Species-Mediated Endoplasmic Reticulum Stress Induction in Human Renal Carcinoma Caki Cells. J. Cancer Prev. 2014, 19, 170–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.-H.; Park, K.-W.; Chae, I.G.; Kundu, J.; Kim, E.-H.; Kundu, J.K.; Chun, K.-S. Carnosic Acid Inhibits STAT3 Signaling and Induces Apoptosis through Generation of ROS in Human Colon Cancer HCT116 Cells. Mol. Carcinog. 2016, 55, 1096–1110. [Google Scholar] [CrossRef]
- Valdés, A.; García-Cañas, V.; Koçak, E.; Simó, C.; Cifuentes, A. Foodomics Study on the Effects of Extracellular Production of Hydrogen Peroxide by Rosemary Polyphenols on the Anti-Proliferative Activity of Rosemary Polyphenols against HT-29 Cells. Electrophoresis 2016, 37, 1795–1804. [Google Scholar] [CrossRef] [Green Version]
- Liao, X.; Gao, Y.; Sun, L.; Liu, J.; Chen, H.; Yu, L.; Chen, Z.; Chen, W.; Lin, L. Rosmarinic Acid Reverses Non-small Cell Lung Cancer Cisplatin Resistance by Activating the MAPK Signaling Pathway. Phytother. Res. 2020, 34, 1142–1153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, L.; Zhang, J.; Fan, Y.; Li, Y. Antiproliferative Activity of Carnosic Acid Is Mediated via Inhibition of Cell Migration and Invasion, and Suppression of Phosphatidylinositol 3-Kinases (PI3K)/AKT/Mammalian Target of Rapamycin (MTOR) Signaling Pathway. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2019, 25, 7864–7871. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
O’Neill, E.J.; Moore, J.; Song, J.; Tsiani, E.L. Inhibition of Non-Small Cell Lung Cancer Proliferation and Survival by Rosemary Extract Is Associated with Activation of ERK and AMPK. Life 2022, 12, 52. https://doi.org/10.3390/life12010052
O’Neill EJ, Moore J, Song J, Tsiani EL. Inhibition of Non-Small Cell Lung Cancer Proliferation and Survival by Rosemary Extract Is Associated with Activation of ERK and AMPK. Life. 2022; 12(1):52. https://doi.org/10.3390/life12010052
Chicago/Turabian StyleO’Neill, Eric J., Jessy Moore, Joon Song, and Evangelia Litsa Tsiani. 2022. "Inhibition of Non-Small Cell Lung Cancer Proliferation and Survival by Rosemary Extract Is Associated with Activation of ERK and AMPK" Life 12, no. 1: 52. https://doi.org/10.3390/life12010052
APA StyleO’Neill, E. J., Moore, J., Song, J., & Tsiani, E. L. (2022). Inhibition of Non-Small Cell Lung Cancer Proliferation and Survival by Rosemary Extract Is Associated with Activation of ERK and AMPK. Life, 12(1), 52. https://doi.org/10.3390/life12010052