
Patients with Adult-Onset Still’s Disease in Germany: A Retrospective Analysis of Clinical Characteristics and Treatment Practices Ahead of the Release of the German Recommendations


 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Design and Setting
2.2. Outcomes
2.3. Statistical Analysis
3. Results
3.1. Symptoms and Laboratory Findings: Initial Diagnosis vs. Current Status
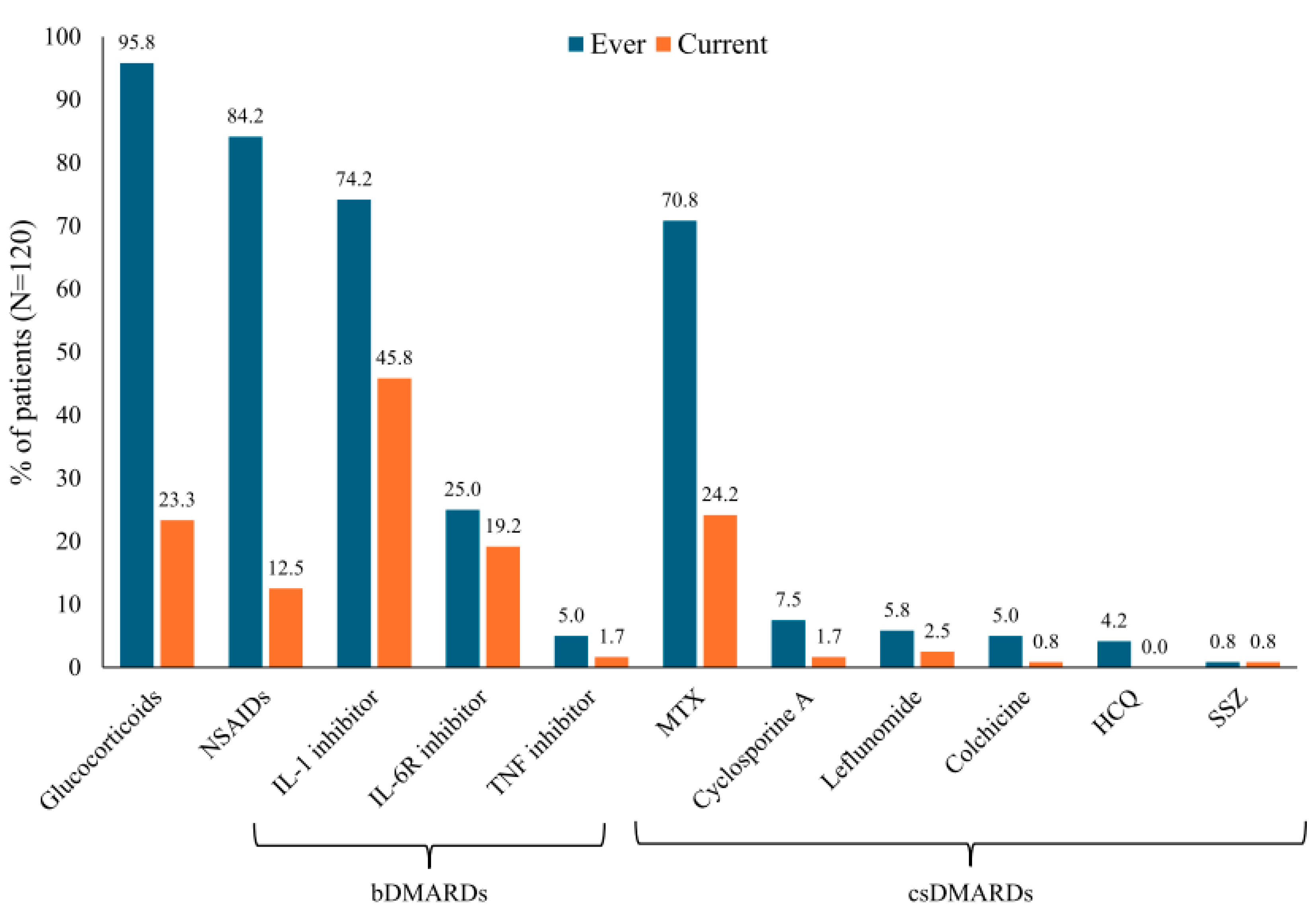
3.2. Drug Therapies and Satisfaction with Treatment
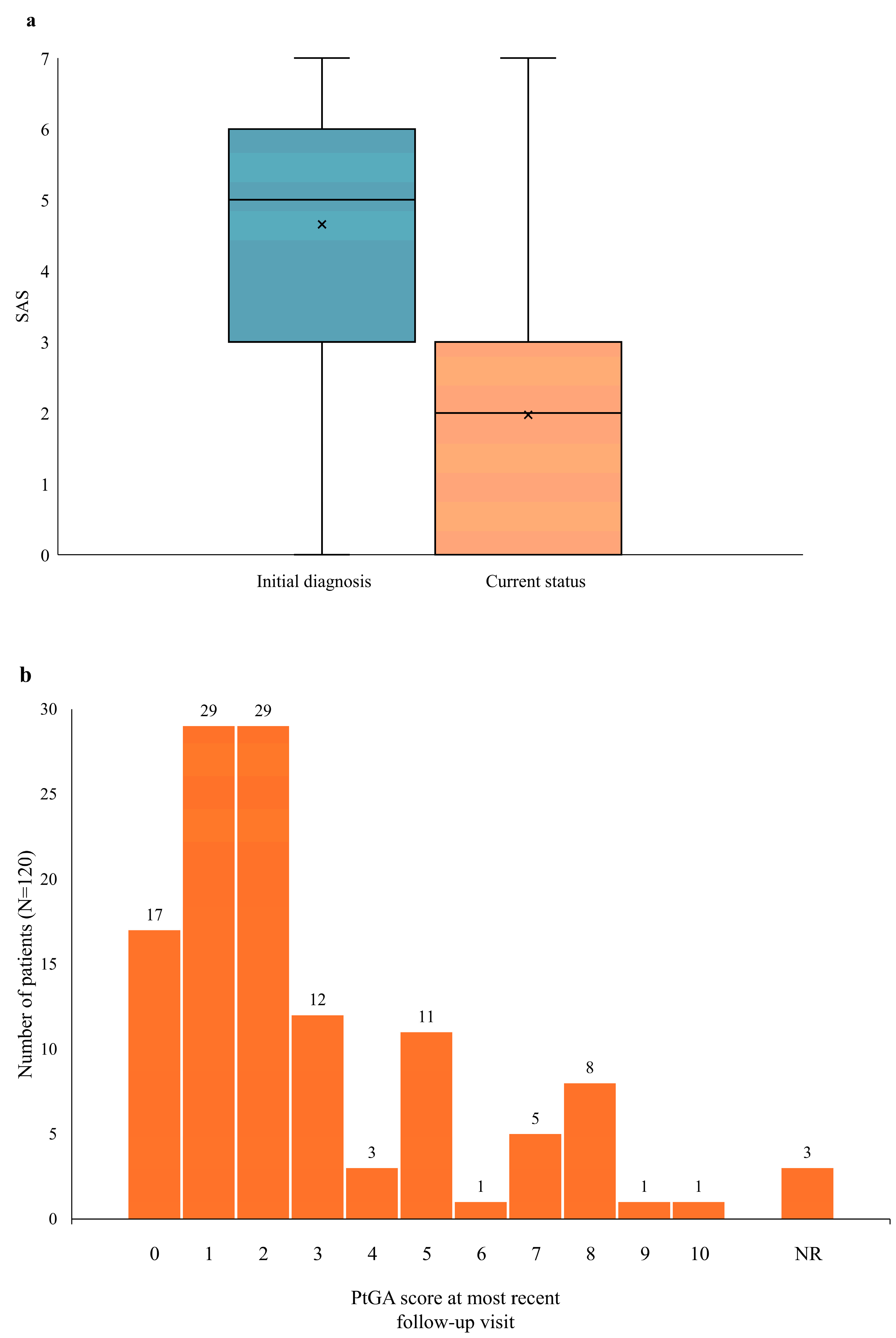
3.3. Disease Activity: Initial Diagnosis vs. Current Status
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fautrel, B.; Mitrovic, S.; De Matteis, A.; Bindoli, S.; Antón, J.; Belot, A.; Bracaglia, C.; Constantin, T.; Dagna, L.; Di Bartolo, A.; et al. EULAR/PReS recommendations for the diagnosis and management of Still’s disease, comprising systemic juvenile idiopathic arthritis and adult-onset Still’s disease. Ann. Rheum. Dis. 2024, 83, 1614–1627. [Google Scholar] [CrossRef]
- De Matteis, A.; Bindoli, S.; De Benedetti, F.; Carmona, L.; Fautrel, B.; Mitrovic, S. Systemic juvenile idiopathic arthritis and adult-onset Still’s disease are the same disease: Evidence from systematic reviews and meta-analyses informing the 2023 EULAR/PReS recommendations for the diagnosis and management of Still’s disease. Ann. Rheum. Dis. 2024, 83, 1748–1761. [Google Scholar] [CrossRef] [PubMed]
- Gerfaud-Valentin, M.; Jamilloux, Y.; Iwaz, J.; Seve, P. Adult-onset Still’s disease. Autoimmun. Rev. 2014, 13, 708–722. [Google Scholar] [CrossRef] [PubMed]
- Suda, T.; Zoshima, T.; Takeji, A.; Suzuki, Y.; Mizushima, I.; Yamada, K.; Nakashima, A.; Yachie, A.; Kawano, M. Elderly-onset Still’s disease complicated by macrophage activation syndrome: A case report and review of the literature. Intern. Med. 2020, 59, 721–728. [Google Scholar] [CrossRef] [PubMed]
- Ruscitti, P.; Feist, E.; Canon-Garcia, V.; Rabijns, H.; Toennessen, K.; Bartlett, C.; Gregg, E.; Miller, P.; McGonagle, D. Burden of adult-onset Still’s disease: A systematic review of health-related quality of life, utilities, costs and resource use. Semin. Arthritis Rheum. 2023, 63, 152264. [Google Scholar] [CrossRef] [PubMed]
- Mittal, S.; Schroeder, B.; Alfaki, M. Mortality, length of stay and cost of hospitalization among patients with adult-onset Still’s disease: Results from the National Inpatient Sample 2016-2019. Diseases 2024, 12, 166. [Google Scholar] [CrossRef]
- Bywaters, E.G. Still’s disease in the adult. Ann. Rheum. Dis. 1971, 30, 121–133. [Google Scholar] [CrossRef] [PubMed]
- Tomaras, S.; Goetzke, C.C.; Kallinich, T.; Feist, E. Adult-onset Still’s disease: Clinical aspects and therapeutic approach. J. Clin. Med. 2021, 10, 733. [Google Scholar] [CrossRef] [PubMed]
- Priori, R.; Colafrancesco, S.; Alessandri, C.; Minniti, A.; Perricone, C.; Iaiani, G.; Palazzo, D.; Valesini, G. Interleukin 18: A biomarker for differential diagnosis between adult-onset Still’s disease and sepsis. J. Rheumatol. 2014, 41, 1118–1123. [Google Scholar] [CrossRef] [PubMed]
- Feist, E.; Mitrovic, S.; Fautrel, B. Mechanisms, biomarkers and targets for adult-onset Still’s disease. Nat. Rev. Rheumatol. 2018, 14, 603–618. [Google Scholar] [CrossRef]
- Föll, D.; Wittkowski, H.; Hinze, C. Das Still-Syndrom als biphasische Erkrankung: Aktuelle Erkenntnisse zur Pathogenese und neue therapeutische Ansätze [Still’s disease as biphasic disorder: Current knowledge on pathogenesis and novel treatment approaches]. Z. Rheumatol. 2020, 79, 639–648. [Google Scholar] [CrossRef]
- Cush, J.J.; Medsger, T.A., Jr.; Christy, W.C.; Herbert, D.C.; Cooperstein, L.A. Adult-onset Still’s disease. Clinical course and outcome. Arthritis Rheum. 1987, 30, 186–194. [Google Scholar] [CrossRef]
- Efthimiou, P.; Kontzias, A.; Hur, P.; Rodha, K.; Ramakrishna, G.S.; Nakasato, P. Adult-onset Still’s disease in focus: Clinical manifestations, diagnosis, treatment, and unmet needs in the rra of targeted therapies. Semin. Arthritis Rheum. 2021, 51, 858–874. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.Y.; Kim, J.W.; Suh, C.H.; Kim, H.A. Roles of interactions between toll-like receptors and their endogenous ligands in the pathogenesis of systemic juvenile idiopathic arthritis and adult-onset Still’s disease. Front. Immunol. 2020, 11, 583513. [Google Scholar] [CrossRef] [PubMed]
- Nigrovic, P.A. Review: Is there a window of opportunity for treatment of systemic juvenile idiopathic arthritis? Arthritis Rheumatol. 2014, 66, 1405–1413. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, R.; Kernder, A.; Blank, N.; Ernst, D.; Henes, J.; Keyßer, G.; Klemm, P.; Krusche, M.; Meinecke, A.; Rech, J.; et al. Implementation of the new DGRh S2e guideline on diagnostics and treatment of adult-onset Still’s disease in Germany. Implications for clinical practice in rheumatology. Z. Rheumatol. 2024; Epub ahead of print. [Google Scholar] [CrossRef]
- Crispin, J.C.; Martinez-Banos, D.; Alcocer-Varela, J. Adult-onset Still disease as the cause of fever of unknown origin. Medicine 2005, 84, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Sautner, J. Makrophagenaktivierungssyndrom—Eine seltene Komplikation bei adultem Morbus Still. Rheuma Plus 2020, 19, 28–31. [Google Scholar] [CrossRef]
- Baerlecken, N.T.; Schmidt, R.E. Adulter Morbus Still, Fieber, Diagnose und Therapie [Adult onset Still’s disease, fever, diagnosis and therapy]. Z. Rheumatol. 2012, 71, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, M. Macrophage activation syndrome associated with adult-onset Still’s disease. Nihon Rinsho Meneki Gakkai Kaishi 2007, 30, 428–431. [Google Scholar] [CrossRef]
- Rigante, D.; Emmi, G.; Fastiggi, M.; Silvestri, E.; Cantarini, L. Macrophage activation syndrome in the course of monogenic autoinflammatory disorders. Clin. Rheumatol. 2015, 34, 1333–1339. [Google Scholar] [CrossRef] [PubMed]
- Saper, V.E.; Chen, G.; Deutsch, G.H.; Guillerman, R.P.; Birgmeier, J.; Jagadeesh, K.; Canna, S.; Schulert, G.; Deterding, R.; Xu, J.; et al. Emergent high fatality lung disease in systemic juvenile arthritis. Ann. Rheum. Dis. 2019, 78, 1722–1731. [Google Scholar] [CrossRef] [PubMed]
- Ruscitti, P.; Bruno, F.; Berardicurti, O.; Acanfora, C.; Pavlych, V.; Palumbo, P.; Conforti, A.; Carubbi, F.; Di Cola, I.; Di Benedetto, P.; et al. Lung involvement in macrophage activation syndrome and severe COVID-19: Results from a cross-sectional study to assess clinical, laboratory and artificial intelligence-radiological differences. Ann. Rheum. Dis. 2020, 79, 1152–1155. [Google Scholar] [CrossRef] [PubMed]
- Chantarogh, S.; Vilaiyuk, S.; Tim-Aroon, T.; Worawichawong, S. Clinical improvement of renal amyloidosis in a patient with systemic-onset juvenile idiopathic arthritis who received tocilizumab treatment: A case report and literature review. BMC Nephrol. 2017, 18, 159. [Google Scholar] [CrossRef] [PubMed]
- Pouchot, J.; Sampalis, J.S.; Beaudet, F.; Carette, S.; Décary, F.; Salusinsky-Sternbach, M.; Hill, R.O.; Gutkowski, A.; Harth, M.; Myhal, D.; et al. Adult Still’s disease: Manifestations, disease course, and outcome in 62 patients. Medicine 1991, 70, 118–136. [Google Scholar] [CrossRef] [PubMed]
- Rau, M.; Schiller, M.; Krienke, S.; Heyder, P.; Lorenz, H.; Blank, N. Clinical manifestations but not cytokine profiles differentiate adult-onset Still’s disease and sepsis. J. Rheumatol. 2010, 37, 2369–2376. [Google Scholar] [CrossRef]
- Kalyoncu, U.; Kasifoglu, T.; Omma, A.; Bes, C.; Cinar, M.; Emmungil, H.; Kucuksahin, O.; Akar, S.; Aksu, K.; Yildiz, F.; et al. Derivation and validation of adult Still Activity Score (SAS). Jt. Bone Spine 2023, 90, 105499. [Google Scholar] [CrossRef]
- Bindoli, S.; De Matteis, A.; Mitrovic, S.; Fautrel, B.; Carmona, L.; De Benedetti, F. Efficacy and safety of therapies for Still’s disease and macrophage activation syndrome (MAS): A systematic review informing the EULAR/PReS guidelines for the management of Still’s disease. Ann. Rheum. Dis. 2024, 83, 1731–1747. [Google Scholar] [CrossRef] [PubMed]
- Correia Marques, M.; Balay-Dustrude, E.; Bracaglia, C.; Twilt, M.; Onel, K.; Appenzeller, S.; Dedeoglu, F.; Eloseily, E.; Martinez Jimenez, P.; Trachtman, R.; et al. Reporting of clinical features and outcome measures in Still’s disease: A systematic literature review of sJIA and AOSD cohorts [abstract]. Arthritis Rheumatol. 2024, 76 (Suppl. S9), 0402. Available online: https://acrabstracts.org/abstract/reporting-of-clinical-features-and-outcome-measures-in-stills-disease-a-systematic-literature-review-of-sjia-and-aosd-cohorts/ (accessed on 2 December 2024).
- Ruscitti, P.; Stamm, T.; Ritschl, V.; Mitrovic, S.; Girard-Guyonvarc’h, C.; Alexanderson, H.; Barten, B.; Bostrøm, C.; Fell, D.; Gattorno, M.; et al. The development of the EULAR Score for the definition of disease activity in adult-onset Still’s sisease; The “DAVID” Project [abstract]. Arthritis Rheumatol. 2024, 76 (Suppl. S9), 2030. Available online: https://acrabstracts.org/abstract/the-development-of-the-eular-score-for-the-definition-of-disease-activity-in-adult-onset-stills-disease-the-david-project/ (accessed on 2 December 2024).
- Leavis, H.L.; van Daele, P.L.A.; Mulders-Manders, C.; Michels, R.; Rutgers, A.; Legger, E.; Bijl, M.; Hak, E.A.; Lam-Tse, W.K.; Bonte-Mineur, F.; et al. Management of adult-onset Still’s disease: Evidence- and consensus-based recommendations by experts. Rheumatology 2024, 63, 1656–1663. [Google Scholar] [CrossRef]
- Vordenbäumen, S.; Feist, E.; Rech, J.; Fleck, M.; Blank, N.; Haas, J.-P.; Kötter, I.; Krusche, M.; Chehab, G.; Hoyer, B.; et al. DGRh-S2e-Leitlinie: Diagnostik und Therapie des adulten Still-Syndroms (AOSD). Z. Rheumatol. 2022, 81, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, M.; Ohta, A.; Tsunematsu, T.; Kasukawa, R.; Mizushima, Y.; Kashiwagi, H.; Kashiwazaki, S.; Tanimoto, K.; Matsumoto, Y.; Ota, T.; et al. Preliminary criteria for classification of adult Still’s disease. J. Rheumatol. 1992, 19, 424–430. [Google Scholar] [PubMed]
- Lebrun, D.; Mestrallet, S.; Dehoux, M.; Golmard, J.L.; Granger, B.; Georgin-Lavialle, S.; Arnaud, L.; Grateau, G.; Pouchot, J.; Fautrel, B. Validation of the Fautrel Classification Criteria for Adult-Onset Still’s Disease. Semin. Arthritis Rheum. 2018, 47, 578–585. [Google Scholar] [CrossRef] [PubMed]
- Franchini, S.; Dagna, L.; Salvo, F.; Aiello, P.; Baldissera, E.; Sabbadini, M.G. Adult onset Still’s disease: Clinical presentation in a large cohort of Italian patients. Clin. Exp. Rheumatol. 2010, 28, 41–48. [Google Scholar] [PubMed]
- Kalyoncu, U.; Solmaz, D.; Emmungil, H.; Yazici, A.; Kasifoglu, T.; Kimyon, G.; Balkarli, A.; Bes, C.; Ozmen, M.; Alibaz-Oner, F.; et al. Response rate of initial conventional treatments, disease course, and related factors of patients with adult-onset Still’s disease: Data from a large multicenter cohort. J. Autoimmun. 2016, 69, 59–63. [Google Scholar] [CrossRef] [PubMed]
- Ruscitti, P.; Natoli, V.; Consolaro, A.; Caorsi, R.; Rosina, S.; Giancane, G.; Naddei, R.; Di Cola, I.; Di Muzio, C.; Berardicurti, O.; et al. Disparities in the prevalence of clinical reatures between systemic juvenile idiopathic arthritis and adult-onset Still’s disease. Rheumatology 2022, 61, 4124–4129. [Google Scholar] [CrossRef] [PubMed]
- Fautrel, B.; Patterson, J.; Bowe, C.; Arber, M.; Glanville, J.; Mealing, S.; Canon-Garcia, V.; Fagerhed, L.; Rabijns, H.; Giacomelli, R. Systematic review on the use of biologics in adult-onset Still’s disease. Semin. Arthritis Rheum. 2023, 58, 152139. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency. Summary of Opinion (Post Authorization): Ilaris (Canakinumab). 23 June 2016. Available online: https://www.ema.europa.eu/en/documents/smop/chmp-post-authorisation-summary-positive-opinion-ilaris_en.pdf-1 (accessed on 21 November 2024).
- Sobi. Kineret® (anakinra) approved in the EU for the treatment of Still’s disease. 11 April 2018. Available online: https://www.sobi.com/sites/sobi/files/pr/201804107653-1.pdf (accessed on 21 November 2024).
- Kernder, A.; Filla, T.; Friedrich, R.; Blank, N.; Ernst, D.; Henes, J.; Keyßer, G.; Klemm, P.; Krusche, M.; Meinecke, A.; et al. First-line biological vs. conventional therapy in adult-onset Still’s disease: A multicentre, retrospective, propensity weighted analysis. Lancet Rheum. 2025; Manuscript accepted. [Google Scholar] [CrossRef]
- Ursini, F.; Gregg, E.; Canon-Garcia, V.; Rabijns, H.; Toennessen, K.; Bartlett, K.; Graziadio, S. Care pathway analysis and evidence gaps in adult-onset Still’s disease: Interviews with experts from the UK, France, Italy, and Germany. Front. Med. 2023, 10, 1257413. [Google Scholar] [CrossRef]
- Hinze, C.H.; Holzinger, D.; Lainka, E.; Haas, J.P.; Speth, F.; Kallinich, T.; Rieber, N.; Hufnagel, M.; Jansson, A.F.; Hedrich, C.; et al. PRO-KIND SJIA project collaborators. Practice and consensus-based strategies in diagnosing and managing systemic juvenile idiopathic arthritis in Germany. Pediatr. Rheumatol. Online J. 2018, 16, 7. [Google Scholar] [CrossRef] [PubMed]
- Ravelli, A.; Consolaro, A.; Horneff, G.; Laxer, R.M.; Lovell, D.J.; Wulffraat, N.M.; Akikusa, J.D.; Al-Mayouf, S.M.; Antón, J.; Avcin, T.; et al. Treating juvenile idiopathic arthritis to target: Recommendations of an international task force. Ann. Rheum. Dis. 2018, 77, 819–828. [Google Scholar] [CrossRef] [PubMed]
- El Tal, T.; Ryan, M.E.; Feldman, B.M.; Bingham, C.A.; Burnham, J.M.; Batthish, M.; Bullock, D.; Ferraro, K.; Gilbert, M.; Gillispie-Taylor, M.; et al. Consensus approach to a treat-to-target strategy in juvenile idiopathic arthritis care: Report From the 2020 PR-COIN Consensus Conference. J. Rheumatol. 2022, 49, 497–503. [Google Scholar] [CrossRef] [PubMed]
- Onel, K.B.; Horton, D.B.; Lovell, D.J.; Shenoi, S.; Cuello, C.A.; Angeles-Han, S.T.; Becker, M.L.; Cron, R.Q.; Feldman, B.M.; Ferguson, P.J.; et al. 2021 American College of Rheumatology guideline for the treatment of juvenile idiopathic arthritis: Therapeutic approaches for oligoarthritis, temporomandibular joint arthritis, and systemic juvenile idiopathic arthritis. Arthritis Rheumatol. 2022, 74, 553–569. [Google Scholar] [CrossRef] [PubMed]
- ter Haar, N.M.; van Dijkhuizen, E.H.P.; Swart, J.F.; van Royen-Kerkhof, A.; El Idrissi, A.; Leek, A.P.; de Jager, W.; de Groot, M.C.H.; Haitjema, S.; Holzinger, D.; et al. Treatment to target using recombinant interleukin-1 receptor antagonist as first-line monotherapy in new-onset systemic juvenile idiopathic arthritis: Results from a five-year follow-up study. Arthritis Rheumatol. 2019, 71, 1163–1173. [Google Scholar] [CrossRef]
- Klein, A.; Minden, K.; Hospach, A.; Foeldvari, I.; Weller-Heinemann, F.; Trauzeddel, R.; Huppertz, H.I.; Horneff, G. Treat-to-target study for improved outcome in polyarticular juvenile idiopathic arthritis. Ann. Rheum. Dis. 2020, 79, 969–974. [Google Scholar] [CrossRef] [PubMed]
- Rezaei, E.; Hogan, D.; Trost, B.; Kusalik, A.J.; Boire, G.; Cabral, D.A.; Campillo, S.; Chédeville, G.; Chetaille, A.L.; Dancey, P.; et al. Clinical and associated inflammatory biomarker features predictive of short-term outcomes in non-systemic juvenile idiopathic arthritis. Rheumatology 2020, 59, 2402–2411. [Google Scholar] [CrossRef] [PubMed]
- Glerup, M.; Kessel, C.; Foell, D.; Berntson, L.; Fasth, A.; Myrup, C.; Nordal, E.; Rypdal, V.; Rygg, M.; Arnstad, E.D.; et al. Nordic Study Group of Paediatric Rheumatology (NoSPeR). Inflammatory biomarkers predicting long-term remission and active disease in juvenile idiopathic arthritis: A population-based study of the Nordic JIA cohort. RMD Open 2024, 10, e004317. [Google Scholar] [CrossRef] [PubMed]
- Vastert, S.J.; Jamilloux, Y.; Quartier, P.; Ohlman, S.; Osterling Koskinen, L.; Kullenberg, T.; Franck-Larsson, K.; Fautrel, B.; de Benedetti, F. Anakinra in children and adults with Still’s disease. Rheumatology 2019, 58 (Suppl. S6), vi9–vi22. [Google Scholar] [CrossRef] [PubMed]
- Bindoli, S.; Baggio, C.; Doria, A.; Sfriso, P. Adult-onset Still’s disease (AOSD): Advances in understanding pathophysiology, genetics and emerging treatment options. Drugs 2024, 84, 257–274. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Characteristic | Value |
|---|---|
| N | 120 |
| Age at initial diagnosis, years a | |
| Mean (SD) | 41.4 (16.8) |
| Median (Q1, Q3) | 40 (27, 55) |
| Age at most recent visit, years a | |
| Mean (SD) | 50.7 (16.2) |
| Median (Q1, Q3) | 51 (36, 62) |
| Sex (as categorized by physician) | |
| Female | 67 (55.8%) |
| Male | 53 (44.2%) |
| Reason for visit | |
| Regular follow-up following earlier AOSD diagnosis | 86 (71.7%) |
| Disease recurrence | 34 (28.3%) |
| Disease course | |
| Monocyclic | 25 (20.8%) |
| Polycyclic | 66 (55.0%) |
| Chronic | 29 (24.1%) |
| Current complications and comorbidities | |
| Hypertension | 20 (16.7%) |
| MAS | 12 (10.0%) |
| Osteoporosis | 10 (8.3%) |
| Diabetes mellitus | 8 (6.7%) |
| Time since diagnosis for most recent visit, years a | |
| Mean (SD) | 9.4 (6.9) |
| Median (Q1, Q3) | 9 (4, 11) |
| Pouchot score at initial diagnosis a | |
| Mean (SD) | 5.1 (2.0) |
| Median (IQR) | 5 (4, 6.5) |
| Outcome | p Value * |
|---|---|
| Signs and symptoms | |
| Abdominal pain | <0.001 |
| Arthralgia | 0.129 |
| Cough | <0.001 |
| Fever | 0.178 |
| Hepatomegaly | <0.001 |
| Large-joint involvement | <0.001 |
| Lymphadenopathy | <0.001 |
| Shortness of breath | <0.001 |
| Small-joint involvement | <0.001 |
| Sore throat | <0.001 |
| Splenomegaly | <0.001 |
| Swollen joints | <0.001 |
| Thoracic pain | <0.001 |
| Measures of disease activity | |
| Ferritin > 350 ng/mL | 0.416 |
| Neutrophils > 65% | 0.002 |
| Still’s Activity Score | <0.001 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schoenau, V.; Wendel, S.; Tascilar, K.; Henes, J.; Feist, E.; Baerlecken, N.T.; Popp, F.; Schmidt-Haendle, M.; Hellmich, B.; Kötter, I.; et al. Patients with Adult-Onset Still’s Disease in Germany: A Retrospective Analysis of Clinical Characteristics and Treatment Practices Ahead of the Release of the German Recommendations. J. Clin. Med. 2025, 14, 981. https://doi.org/10.3390/jcm14030981
Schoenau V, Wendel S, Tascilar K, Henes J, Feist E, Baerlecken NT, Popp F, Schmidt-Haendle M, Hellmich B, Kötter I, et al. Patients with Adult-Onset Still’s Disease in Germany: A Retrospective Analysis of Clinical Characteristics and Treatment Practices Ahead of the Release of the German Recommendations. Journal of Clinical Medicine. 2025; 14(3):981. https://doi.org/10.3390/jcm14030981
Chicago/Turabian StyleSchoenau, Verena, Sarah Wendel, Koray Tascilar, Joerg Henes, Eugen Feist, Niklas Thomas Baerlecken, Florian Popp, Matthias Schmidt-Haendle, Bernhard Hellmich, Ina Kötter, and et al. 2025. "Patients with Adult-Onset Still’s Disease in Germany: A Retrospective Analysis of Clinical Characteristics and Treatment Practices Ahead of the Release of the German Recommendations" Journal of Clinical Medicine 14, no. 3: 981. https://doi.org/10.3390/jcm14030981
APA StyleSchoenau, V., Wendel, S., Tascilar, K., Henes, J., Feist, E., Baerlecken, N. T., Popp, F., Schmidt-Haendle, M., Hellmich, B., Kötter, I., Andreica, I., & Rech, J. (2025). Patients with Adult-Onset Still’s Disease in Germany: A Retrospective Analysis of Clinical Characteristics and Treatment Practices Ahead of the Release of the German Recommendations. Journal of Clinical Medicine, 14(3), 981. https://doi.org/10.3390/jcm14030981