
Alkylaminophenol and GPR17 Agonist for Glioblastoma Therapy: A Combinational Approach for Enhanced Cell Death Activity

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
3. Results
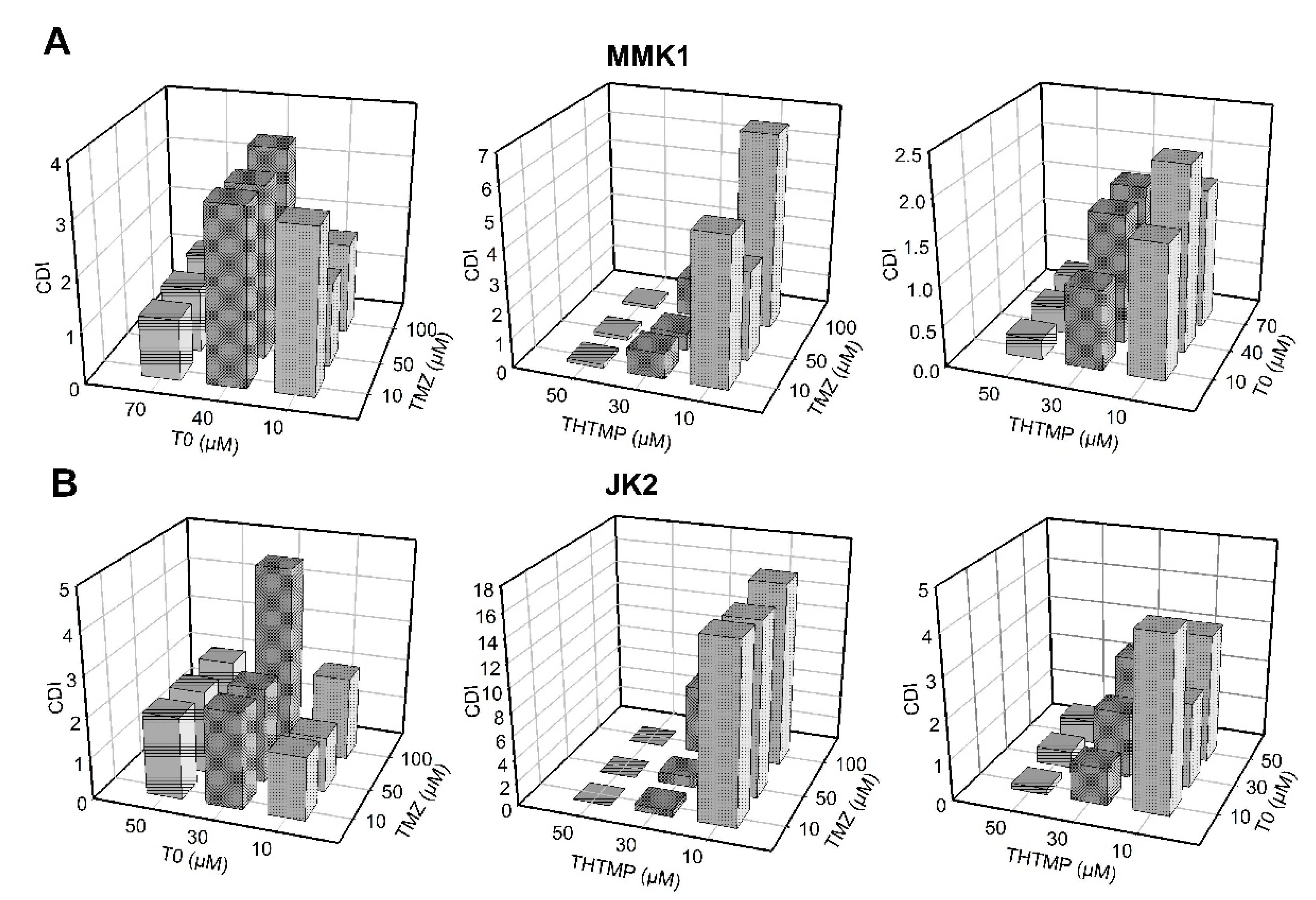
3.1. Synergic Effect of THTMP + T0 against Patient Derived GBM Cells
3.2. Evaluation of Drug Synergy by Combination Therapy
3.3. Combinatorial Drug THTMP + T0 Attenuates Migration and Invasion of GBM Cells
3.4. Clonogenicity of THTMP + T0 in Primary GBM Tumor Cells
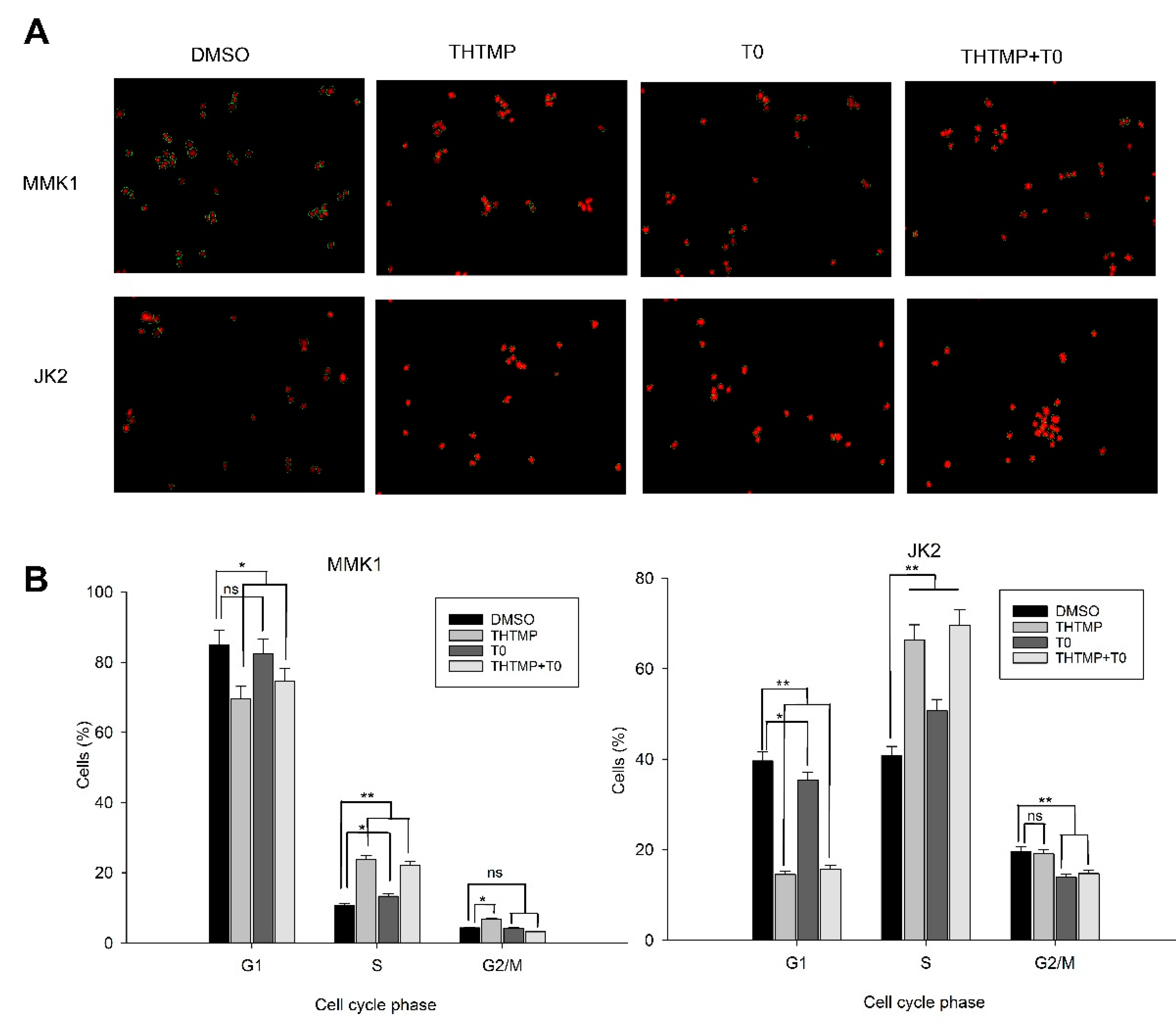
3.5. THTMP + T0 Unveils the Cell Cycle Checkpoints in GBM
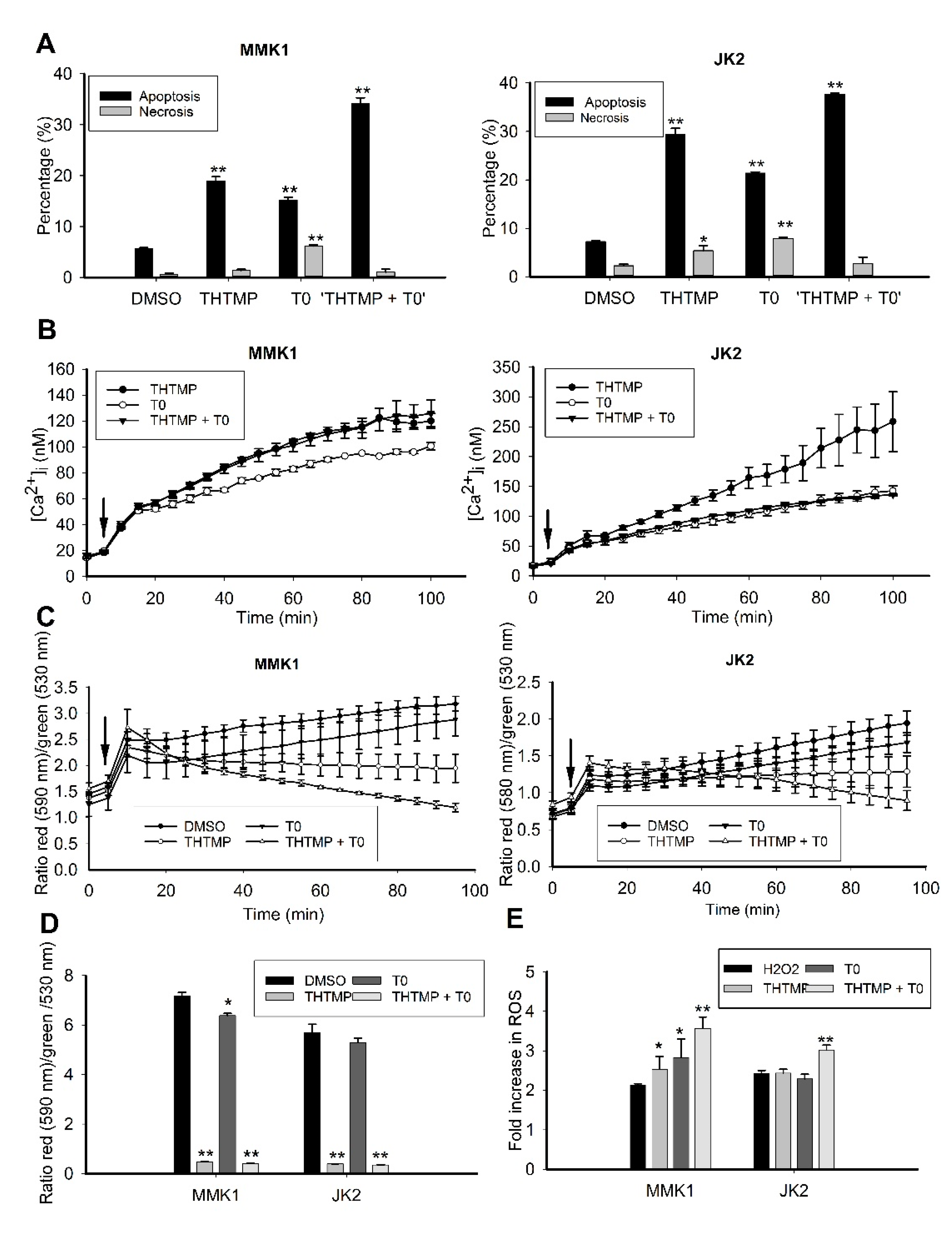
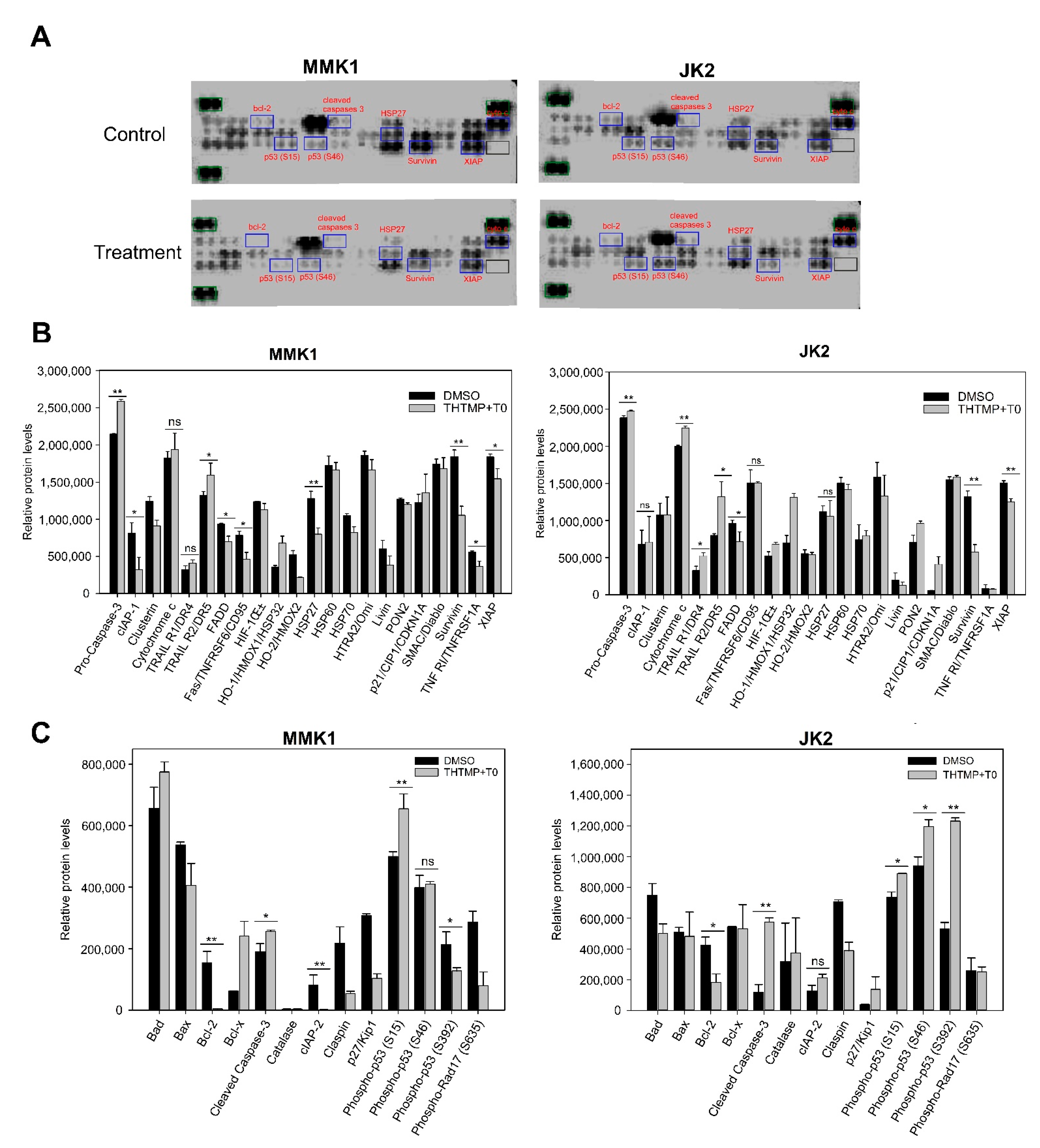
3.6. THTMP + T0 Activates Intrinsic Pathways of Apoptosis Induction
3.7. Combination of THTMP/T0 Induces Intrinsic Apoptotic Pathway Rather Than Extrinsic Apoptotic Pathway in GBM Cells
3.8. In Vivo Anti-Tumor Efficacy of Combinatorial Drug, THTMP + T0
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ostrom, Q.T.; Cote, D.J.; Ascha, M.; Kruchko, C.; Barnholtz-Sloan, J.S. Adult Glioma Incidence and Survival by Race or Ethnicity in the United States from 2000 to 2014. JAMA Oncol. 2018, 4, 1254–1262. [Google Scholar] [CrossRef] [Green Version]
- Stupp, R.; Mason, W.; van den Bent, M.J.; Weller, M.; Fisher, B.M.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant\nand Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Koshy, M.; Villano, J.L.; Dolecek, T.A.; Howard, A.; Mahmood, U.; Chmura, S.J.; Weichselbaum, R.R.; McCarthy, B.J. Improved survival time trends for glioblastoma using the SEER 17 population-based registries. J. Neurooncol. 2012, 107, 207–212. [Google Scholar] [CrossRef] [Green Version]
- Zhao, M.; van Straten, D.; Broekman, M.L.D.; Préat, V.; Schiffelers, R.M. Nanocarrier-based drug combination therapy for glioblastoma. Theranostics 2020, 10, 1355. [Google Scholar] [CrossRef]
- Hegi, M.E.; Diserens, A.-C.; Gorlia, T.; Hamou, M.-F.; de Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT Gene Silencing and Benefit from Temozolomide in Glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef] [Green Version]
- McLendon, R.; Friedman, A.; Bigner, D.; Van Meir, E.G.; Brat, D.J.; Mastrogianakis, G.M.; Olson, J.J.; Mikkelsen, T.; Lehman, N.; Aldape, K.; et al. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061. [Google Scholar]
- Qazi, M.A.; Vora, P.; Venugopal, C.; Sidhu, S.S.; Moffat, J.; Swanton, C.; Singh, S.K. Intratumoral heterogeneity: Pathways to treatment resistance and relapse in human glioblastoma. Ann. Oncol. 2017, 28, 1448–1456. [Google Scholar] [CrossRef]
- Karjalainen, A.; Yli-Harja, O.; Kandhavelu, M.; Doan, P.; Candeias, N.R.; Sandberg, O.; Chandraseelan, J.G. Synthesis of Phenol-derivatives and Biological Screening for Anticancer Activity. Anticancer. Agents Med. Chem. 2017, 17, 1710–1720. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Shnimizu, M.; Moriwaki, H. Cancer chemoprevention by carotenoids. Molecules 2012, 17, 3202–3242. [Google Scholar] [CrossRef] [PubMed]
- Doan, P.; Karjalainen, A.; Chandraseelan, J.G.; Sandberg, O.; Yli-Harja, O.; Rosholm, T.; Franzen, R.; Candeias, N.R.; Kandhavelu, M. Synthesis and biological screening for cytotoxic activity of N-substituted indolines and morpholines. Eur. J. Med. Chem. 2016, 120, 296–303. [Google Scholar] [CrossRef] [PubMed]
- Von Pawel, J.; Schiller, J.H.; Shepherd, F.A.; Fields, S.Z.; Kleisbauer, J.P.; Chrysson, N.G.; Stewart, D.J.; Clark, P.I.; Palmer, M.C.; Depierre, A.; et al. Topotecan versus cyclophosphamide, doxorubicin, and vincristine for the treatment of recurrent small-cell lung cancer. J. Clin. Oncol. 1999, 17, 658. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.; Wang, M.; Gralow, J.; Dickler, M.; Cobleigh, M.; Perez, E.A.; Shenkier, T.; Cella, D.; Davidson, N.E. Paclitaxel plus Bevacizumab versus Paclitaxel Alone for Metastatic Breast Cancer. N. Engl. J. Med. 2007, 357, 2666–2676. [Google Scholar] [CrossRef] [Green Version]
- Alvandi, F.; Kwitkowski, V.E.; Ko, C.-W.; Rothmann, M.D.; Ricci, S.; Saber, H.; Ghosh, D.; Brown, J.; Pfeiler, E.; Chikhale, E.; et al. U.S. Food and Drug Administration Approval Summary: Omacetaxine Mepesuccinate as Treatment for Chronic Myeloid Leukemia. Oncologist 2013, 19, 94. [Google Scholar] [CrossRef] [Green Version]
- Doan, P.; Musa, A.; Candeias, N.R.; Emmert-Streib, F.; Yli-Harja, O.; Kandhavelu, M. Alkylaminophenol induces G1/S phase cell cycle arrest in glioblastoma cells through p53 and cyclin-dependent kinase signaling pathway. Front. Pharmacol. 2019, 10, 330. [Google Scholar] [CrossRef] [PubMed]
- Mutharasu, G.; Murugesan, A.; Mani, S.K.; Yli-Harja, O.; Kandhavelu, M. Transcriptomic analysis of glioblastoma multiforme providing new insights into GPR17 signaling communication. J. Biomol. Struct. Dyn. 2020, 1–14. [Google Scholar] [CrossRef]
- Saravanan, K.M.; Palanivel, S.; Yli-Harja, O.; Kandhavelu, M. Identification of novel GPR17-agonists by structural bioinformatics and signaling activation. Int. J. Biol. Macromol. 2018, 106, 901–907. [Google Scholar] [CrossRef]
- Azzabi, A.; Hughes, A.N.; Calvert, P.M.; Plummer, E.R.; Todd, R.; Griffin, M.J.; Lind, M.J.; Maraveyas, A.; Kelly, C.; Fishwick, K.; et al. Phase I study of temozolomide plus paclitaxel in patients with advanced malignant melanoma and associated in vitro investigations. Br. J. Cancer 2005, 92, 1006–1012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ni, S.; Fan, X.; Wang, J.; Qi, H.; Li, X. Biodegradable implants efficiently deliver combination of paclitaxel and temozolomide to glioma C6 cancer cells in vitro. Ann. Biomed. Eng. 2014, 42, 214–221. [Google Scholar] [CrossRef]
- Shchors, K.; Massaras, A.; Hanahan, D. Dual Targeting of the Autophagic Regulatory Circuitry in Gliomas with Repurposed Drugs Elicits Cell-Lethal Autophagy and Therapeutic Benefit. Cancer Cell 2015, 28, 456–471. [Google Scholar] [CrossRef] [Green Version]
- Doan, P.; Nguyen, T.; Yli-Harja, O.; Kandhavelu, M.; Yli-Harja, O.; Doan, P.; Nguyen, T.; Yli-Harja, O.; Candeias, N.R. Effect of alkylaminophenols on growth inhibition and apoptosis of bone cancer cells. Eur. J. Pharm. Sci. 2017, 107, 208–216. [Google Scholar] [CrossRef]
- Day, B.W.; Stringer, B.W.; Al-Ejeh, F.; Ting, M.J.; Wilson, J.; Ensbey, K.S.; Jamieson, P.R.; Bruce, Z.C.; Lim, Y.C.; Offenhäuser, C.; et al. EphA3 Maintains Tumorigenicity and Is a Therapeutic Target in Glioblastoma Multiforme. Cancer Cell 2013, 23, 238–248. [Google Scholar] [CrossRef] [Green Version]
- Pollard, S.M.; Yoshikawa, K.; Clarke, I.D.; Danovi, D.; Stricker, S.; Russell, R.; Bayani, J.; Head, R.; Lee, M.; Bernstein, M.; et al. Glioma Stem Cell Lines Expanded in Adherent Culture Have Tumor-Specific Phenotypes and Are Suitable for Chemical and Genetic Screens. Cell Stem Cell 2009, 4, 568–580. [Google Scholar] [CrossRef] [Green Version]
- Chou, T.C. Drug combination studies and their synergy quantification using the chou-talalay method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franken, N.A.P.; Rodermond, H.M.; Stap, J.; Haveman, J.; van Bree, C. Clonogenic assay of cells in vitro. Nat. Protoc. 2006, 1, 2315–2319. [Google Scholar] [CrossRef]
- Grynkiewicz, G.; Poenie, M.; Tsien, R.Y. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 1985, 260, 3440–3450. [Google Scholar] [CrossRef]
- Vaiyapuri, P.S.; Ali, A.A.; Mohammad, A.A.; Kandhavelu, J.; Kandhavelu, M. Time lapse microscopy observation of cellular structural changes and image analysis of drug treated cancer cells to characterize the cellular heterogeneity. Environ. Toxicol. 2015, 30, 724–734. [Google Scholar] [CrossRef]
- Westermark, B. The deficient density-dependent growth control of human malignant glioma cells and virus-transformed glia-like cells in culture. Int. J. Cancer 1973, 12, 438–451. [Google Scholar] [CrossRef]
- Stringer, B.W.; Day, B.W.; D’Souza, R.C.J.; Jamieson, P.R.; Ensbey, K.S.; Bruce, Z.C.; Lim, Y.C.; Goasdoué, K.; Offenhäuser, C.; Akgül, S.; et al. A reference collection of patient-derived cell line and xenograft models of proneural, classical and mesenchymal glioblastoma. Sci. Rep. 2019, 9, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, S.; Sun, G.; Shen, Y.; Peng, W.; Wang, H.; Wei, W. Synergistic effect of combining paeonol and cisplatin on apoptotic induction of human hepatoma cell lines1. Acta Pharmacol. Sin. 2007, 28, 869–878. [Google Scholar] [CrossRef]
- Chen, L.; Ye, H.-L.; Zhang, G.; Yao, W.-M.; Chen, X.-Z.; Zhang, F.-C.; Liang, G. Autophagy Inhibition Contributes to the Synergistic Interaction between EGCG and Doxorubicin to Kill the Hepatoma Hep3B Cells. PLoS ONE 2014, 9, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Shen, M.; Li, Y.; Sun, Y.; Teng, Y.; Wang, Y.; Duan, Y. The synergic antitumor effects of paclitaxel and temozolomide co-loaded in mPEG-PLGA nanoparticles on glioblastoma cells. Oncotarget 2016, 7, 20890–20901. [Google Scholar] [CrossRef] [Green Version]
- Hajnóczky, G.; Csordás, G.; Das, S.; Garcia-Perez, C.; Saotome, M.; Sinha Roy, S.; Yi, M. Mitochondrial calcium signalling and cell death: Approaches for assessing the role of mitochondrial Ca2+ uptake in apoptosis. Cell Calcium 2006, 40, 553–560. [Google Scholar] [CrossRef] [Green Version]
- Kruman, I.; Guo, Q.; Mattson, M.P. Calcium and reactive oxygen species mediate staurosporine-induced mitochondrial dysfunction and apoptosis in PC12 cells. J. Neurosci. Res. 1998, 51, 293–308. [Google Scholar] [CrossRef]
- Orrenius, S.; Zhivotovsky, B.; Nicotera, P. Calcium: Regulation of cell death: The calcium-apoptosis link. Nat. Rev. Mol. Cell Biol. 2003, 4, 552–565. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Xing, R.; Ou, Z.; Zhao, J.; Hong, G.; Zhao, T.J.; Han, Y.; Chen, Y. G-protein-coupled receptor GPR17 inhibits glioma development by increasing polycomb repressive complex 1-mediated ROS production. Cell Death Dis. 2021, 12, 1–14. [Google Scholar] [CrossRef]
- Quinn, J.A.; Desjardins, A.; Weingart, J.; Brem, H.; Dolan, M.E.; Delaney, S.M.; Vredenburgh, J.; Rich, J.; Friedman, A.H.; Reardon, D.A.; et al. Phase I trial of temozolomide plus O6-benzylguanine for patients with recurrent or progressive malignant glioma. J. Clin. Oncol. 2005, 23, 7178–7187. [Google Scholar] [CrossRef]
- Doan, P.; Musa, A.; Murugesan, A.; Sipilä, V.; Candeias, N.R.; Emmert-Streib, F.; Ruusuvuori, P.; Granberg, K.; Yli-Harja, O.; Kandhavelu, M. Glioblastoma Multiforme Stem Cell Cycle Arrest by Alkylaminophenol through the Modulation of EGFR and CSC Signaling Pathways. Cells 2020, 9, 681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ouyang, L.; Shi, Z.; Zhao, S.; Wang, F.T.; Zhou, T.T.; Liu, B.; Bao, J.K. Programmed cell death pathways in cancer: A review of apoptosis, autophagy and programmed necrosis. Cell Prolif. 2012, 45, 487–498. [Google Scholar] [CrossRef] [PubMed]
- Boucher, M.J.; Duchesne, C.; Rivard, N.; Lainé, J.; Morisset, J. cAMP protection of pancreatic cancer cells against apoptosis induced by ERK inhibition. Biochem. Biophys. Res. Commun. 2001, 285, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Douglas, R.G.; John, C.R. Mitochondria and Apoptosis. Science 1998, 281, 1309–1312. [Google Scholar]
- Tait, S.W.G.; Green, D.R. Mitochondria and cell death: Outer membrane permeabilization and beyond. Nat. Rev. Mol. Cell Biol. 2010, 11, 621–632. [Google Scholar] [CrossRef] [PubMed]
- Mita, A.C.; Mita, M.M.; Nawrocki, S.T.; Giles, F.J. Survivin: Key regulator of mitosis and apoptosis and novel target for cancer therapeutics. Clin. Cancer Res. 2008, 14, 5000–5005. [Google Scholar] [CrossRef] [Green Version]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Aubrey, B.J.; Kelly, G.L.; Janic, A.; Herold, M.J.; Strasser, A. How does p53 induce apoptosis and how does this relate to p53-mediated tumour suppression? Cell Death Differ. 2018, 25, 104–113. [Google Scholar] [CrossRef] [Green Version]
- Berrocal, A.; Perez Segura, P.; Gil, M.; Balaña, C.; Garcia Lopez, J.; Yaya, R.; Rodríguez, J.; Reynes, G.; Gallego, O.; Iglesias, L. Extended-schedule dose-dense temozolomide in refractory gliomas. J. Neurooncol. 2010, 96, 417–422. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.S.; Rait, A.; Kim, E.; DeMarco, J.; Pirollo, K.F.; Chang, E.H. Encapsulation of temozolomide in a tumor-targeting nanocomplex enhances anti-cancer efficacy and reduces toxicity in a mouse model of glioblastoma. Cancer Lett. 2015, 369, 250–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tentori, L.; Graziani, G. Recent Approaches to Improve the Antitumor Efficacy of Temozolomide. Curr. Med. Chem. 2008, 16, 245–257. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Doan, P.; Nguyen, P.; Murugesan, A.; Candeias, N.R.; Yli-Harja, O.; Kandhavelu, M. Alkylaminophenol and GPR17 Agonist for Glioblastoma Therapy: A Combinational Approach for Enhanced Cell Death Activity. Cells 2021, 10, 1975. https://doi.org/10.3390/cells10081975
Doan P, Nguyen P, Murugesan A, Candeias NR, Yli-Harja O, Kandhavelu M. Alkylaminophenol and GPR17 Agonist for Glioblastoma Therapy: A Combinational Approach for Enhanced Cell Death Activity. Cells. 2021; 10(8):1975. https://doi.org/10.3390/cells10081975
Chicago/Turabian StyleDoan, Phuong, Phung Nguyen, Akshaya Murugesan, Nuno R. Candeias, Olli Yli-Harja, and Meenakshisundaram Kandhavelu. 2021. "Alkylaminophenol and GPR17 Agonist for Glioblastoma Therapy: A Combinational Approach for Enhanced Cell Death Activity" Cells 10, no. 8: 1975. https://doi.org/10.3390/cells10081975
APA StyleDoan, P., Nguyen, P., Murugesan, A., Candeias, N. R., Yli-Harja, O., & Kandhavelu, M. (2021). Alkylaminophenol and GPR17 Agonist for Glioblastoma Therapy: A Combinational Approach for Enhanced Cell Death Activity. Cells, 10(8), 1975. https://doi.org/10.3390/cells10081975