
DNA Methylation of Fibroblast Phenotypes and Contributions to Lung Fibrosis


Abstract
:1. Introduction
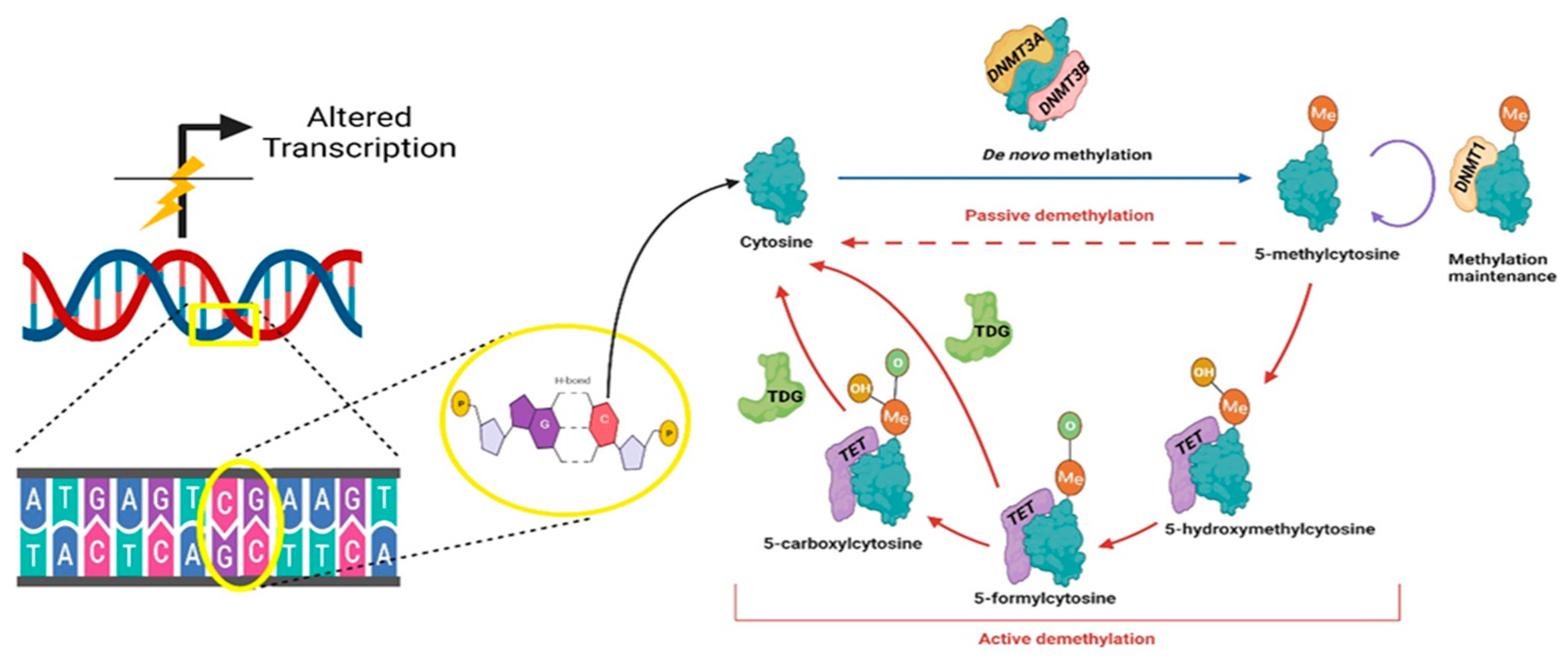
2. DNA Methylation
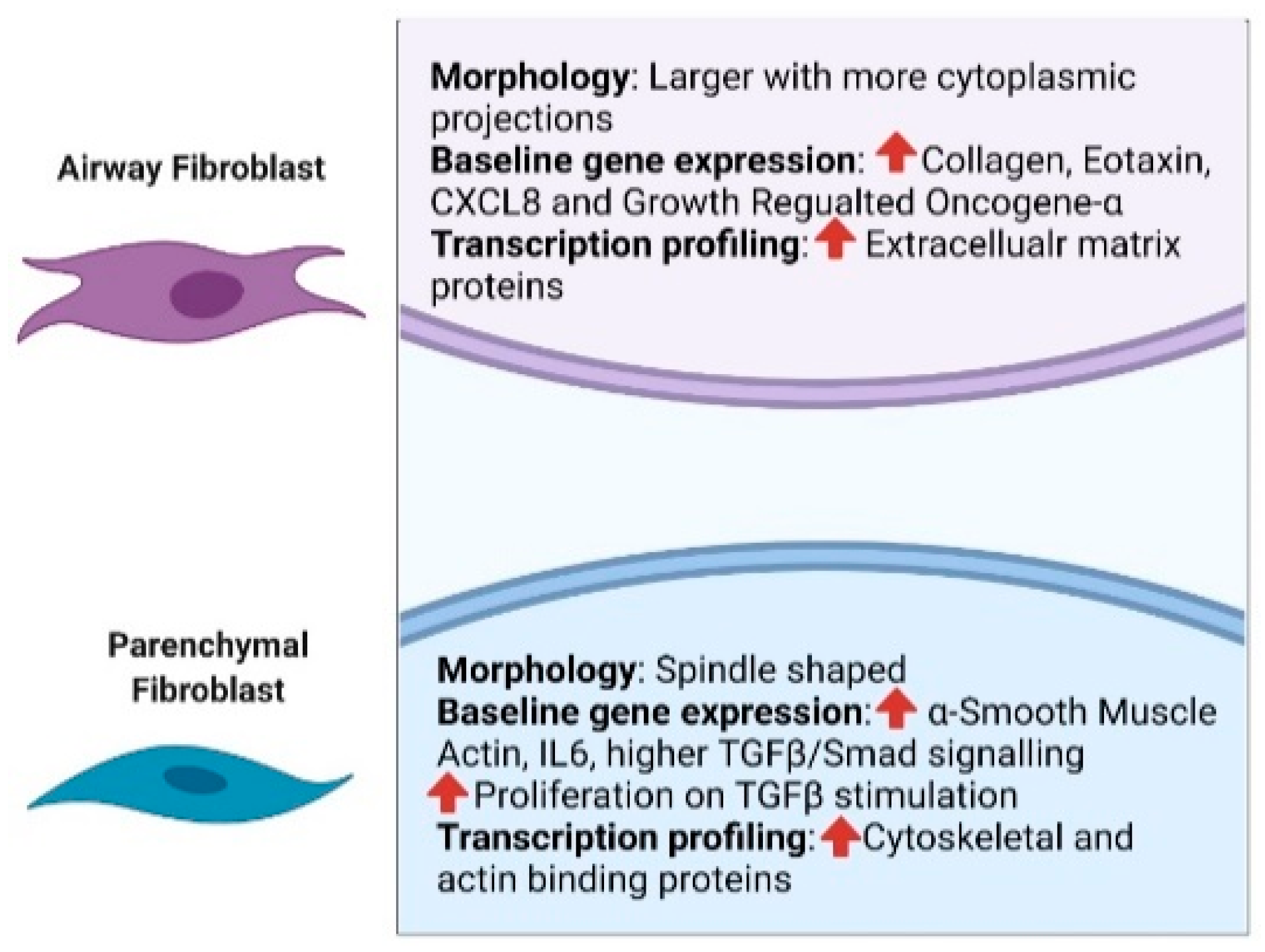
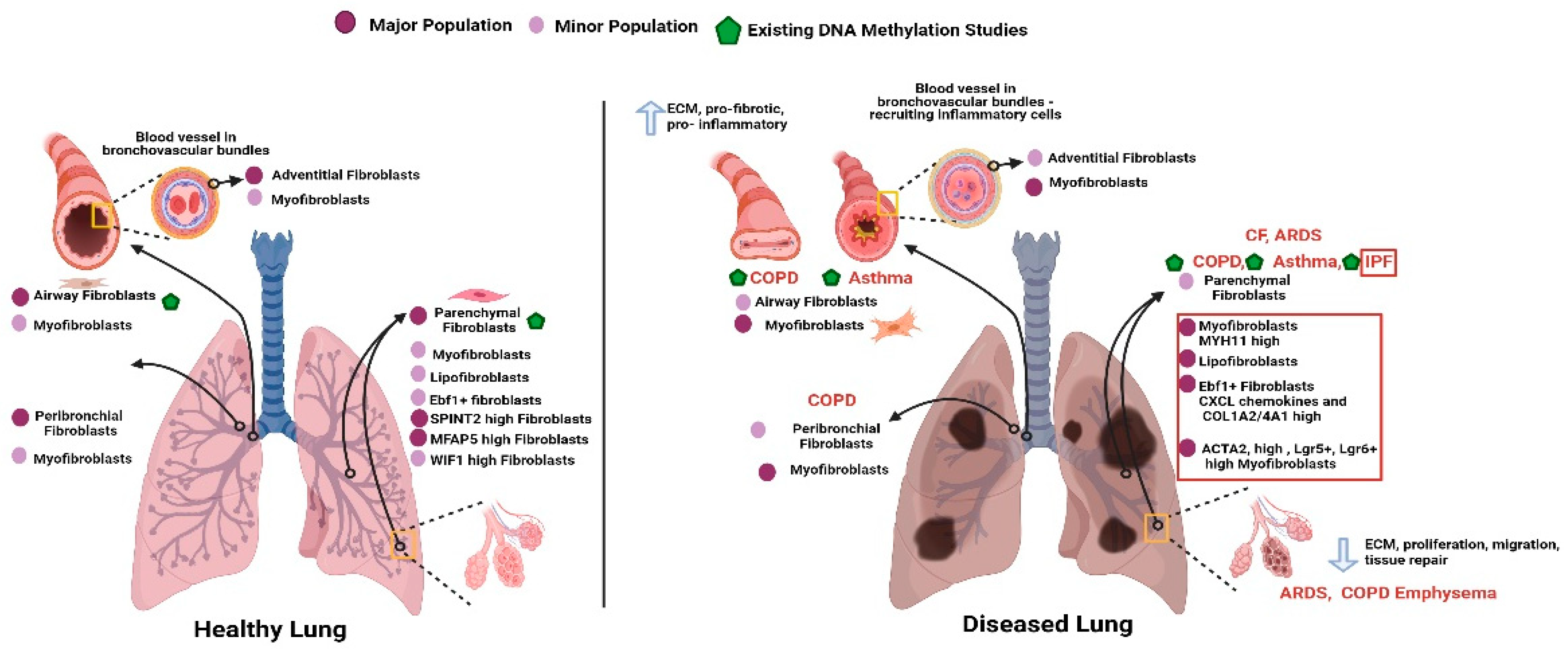
3. DNA Methylation and Fibroblast Heterogeneity
4. DNA Methylation and Fibrosis in Lung Disease
4.1. Idiopathic Pulmonary Fibrosis
4.2. Asthma
4.3. Chronic Obstructive Pulmonary Disease (COPD)
4.4. Acute Respiratory Distress Syndrome (ARDS)
4.5. Cystic Fibrosis
5. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lynch, M.D.; Watt, F.M. Fibroblast heterogeneity: Implications for human disease. J. Clin. Investig. 2018, 128, 26–35. [Google Scholar] [CrossRef] [Green Version]
- Shaw, T.J.; Rognoni, E. Dissecting Fibroblast Heterogeneity in Health and Fibrotic Disease. Curr. Rheumatol. Rep. 2020, 22, 33. [Google Scholar] [CrossRef] [PubMed]
- LeBleu, V.S.; Neilson, E.G. Origin and functional heterogeneity of fibroblasts. FASEB J. 2020, 34, 3519–3536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rinn, J.L.; Bondre, C.; Gladstone, H.B.; Brown, P.O.; Chang, H.Y. Anatomic demarcation by positional variation in fibroblast gene expression programs. PLoS Genet. 2006, 2, e119. [Google Scholar] [CrossRef] [PubMed]
- Fries, K.M.; Blieden, T.; Looney, R.J.; Sempowski, G.D.; Silvera, M.R.; Willis, R.A.; Phipps, R.P. Evidence of fibroblast heterogeneity and the role of fibroblast subpopulations in fibrosis. Clin. Immunol. Immunopathol. 1994, 72, 283–292. [Google Scholar] [CrossRef]
- Jordana, M.; Schulman, J.; McSharry, C.; Irving, L.B.; Newhouse, M.T.; Jordana, G.; Gauldie, J. Heterogeneous proliferative characteristics of human adult lung fibroblast lines and clonally derived fibroblasts from control and fibrotic tissue. Am. Rev. Respir. Dis. 1988, 137, 579–584. [Google Scholar] [CrossRef] [PubMed]
- Ko, S.D.; Page, R.C.; Narayanan, A.S. Fibroblast heterogeneity and prostaglandin regulation of subpopulations. Proc. Natl. Acad. Sci. USA 1977, 74, 3429–3432. [Google Scholar] [CrossRef] [Green Version]
- Derdak, S.; Penney, D.P.; Keng, P.; Felch, M.E.; Brown, D.; Phipps, R.P. Differential collagen and fibronectin production by thy1+ and thy1- lung fibroblast subpopulations. Am. J. Physiol. 1992, 263, L283–L290. [Google Scholar]
- Foote, A.G.; Wang, Z.; Kendziorski, C.; Thibeault, S.L. Tissue specific human fibroblast differential expression based on RNAsequencing analysis. BMC Genom. 2019, 20, 308. [Google Scholar] [CrossRef] [PubMed]
- Dessalle, K.; Narayanan, V.; Kyoh, S.; Mogas, A.; Halayko, A.J.; Nair, P.; Baglole, C.J.; Eidelman, D.H.; Ludwig, M.S.; Hamid, Q. Human bronchial and parenchymal fibroblasts display differences in basal inflammatory phenotype and response to IL-17A. Clin. Exp. Allergy 2016, 46, 945–956. [Google Scholar] [CrossRef] [PubMed]
- Kotaru, C.; Schoonover, K.J.; Trudeau, J.B.; Huynh, M.L.; Zhou, X.; Hu, H.; Wenzel, S.E. Regional fibroblast heterogeneity in the lung: Implications for remodeling. Am. J. Respir. Crit. Care Med. 2006, 173, 1208–1215. [Google Scholar] [CrossRef] [Green Version]
- Pechkovsky, D.V.; Hackett, T.L.; An, S.S.; Shaheen, F.; Murray, L.A.; Knight, D.A. Human Lung Parenchyma but Not Proximal Bronchi Produces Fibroblasts with Enhanced TGF-beta Signaling and alpha-SMA Expression. Am. J. Respir. Cell Mol. Biol. 2010, 43, 641–651. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.X.; Wu, W.; Hu, H.Z.; Milosevic, J.; Konishi, K.; Kaminski, N.; Wenzel, S.E. Genomic Differences Distinguish the Myofibroblast Phenotype of Distal Lung Fibroblasts from Airway Fibroblasts. Am. J. Respir. Cell Mol. Biol. 2011, 45, 1256–1262. [Google Scholar] [CrossRef] [Green Version]
- Raredon, M.S.B.; Adams, T.S.; Suhail, Y.; Schupp, J.C.; Poli, S.; Neumark, N.; Leiby, K.L.; Greaney, A.M.; Yuan, Y.F.; Horien, C.; et al. Single-cell connectomic analysis of adult mammalian lungs. Sci. Adv. 2019, 5, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reyfman, P.A.; Walter, J.M.; Joshi, N.; Anekalla, K.R.; McQuattie-Pimentel, A.C.; Chiu, S.; Fernandez, R.; Akbarpour, M.; Chen, C.I.; Ren, Z.Y.; et al. Single-Cell Transcriptomic Analysis of Human Lung Provides Insights into the Pathobiology of Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2019, 199, 1517–1536. [Google Scholar] [CrossRef]
- Liu, X.; Rowan, S.; Liang, C.J.; Yao, C.; Deng, N.; Huang, G.; Xie, T.; Wang, Y.; Stripp, B.R.; Noble, P.W.; et al. Definition and Signatures of Lung Fibroblast Populations in Development and Fibrosis in Mice and Men. Am. J. Respir. Crit. Care Med. 2020, 201. [Google Scholar] [CrossRef]
- Tsukui, T.; Sun, K.H.; Wetter, J.B.; Wilson-Kanamori, J.R.; Hazelwood, L.A.; Henderson, N.C.; Adams, T.S.; Schupp, J.C.; Poli, S.D.; Rosas, I.O.; et al. Collagen-producing lung cell atlas identifies multiple subsets with distinct localization and relevance to fibrosis. Nat. Commun. 2020, 11, 1920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valenzi, E.; Bulik, M.; Tabib, T.; Morse, C.; Sembrat, J.; Bittar, H.T.; Rojas, M.; Lafyatis, R. Single-cell analysis reveals fibroblast heterogeneity and myofibroblasts in systemic sclerosis-associated interstitial lung disease. Ann. Rheum. Dis. 2019, 78, 1379–1387. [Google Scholar] [CrossRef] [PubMed]
- Sucre, J.M.S.; Hagood, J. Single cell analysis of human lung development: Knowing what mesenchymal cells are and what they may be. Eur. Respir. J. 2020, 55, 1902327. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Costello, J. DNA methylation: An epigenetic mark of cellular memory. Exp. Mol. Med. 2017, 49, e322. [Google Scholar] [CrossRef] [Green Version]
- Chen, P.Y.; Feng, S.H.; Joo, J.W.J.; Jacobsen, S.E.; Pellegrini, M. A comparative analysis of DNA methylation across human embryonic stem cell lines. Genome Biol. 2011, 12. [Google Scholar] [CrossRef] [Green Version]
- Lyko, F. The DNA methyltransferase family: A versatile toolkit for epigenetic regulation. Nat. Rev. Genet. 2018, 19, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Okano, M.; Bell, D.W.; Haber, D.A.; Li, E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 1999, 99, 247–257. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.J.; Zhang, Y. TET-mediated active DNA demethylation: Mechanism, function and beyond. Nat. Rev. Genet. 2017, 18, 517–534. [Google Scholar] [CrossRef]
- Suzuki, M.M.; Bird, A. DNA methylation landscapes: Provocative insights from epigenomics. Nat. Rev. Genet. 2008, 9, 465–476. [Google Scholar] [CrossRef] [PubMed]
- Aran, D.; Toperoff, G.; Rosenberg, M.; Hellman, A. Replication timing-related and gene body-specific methylation of active human genes. Hum. Mol. Genet. 2011, 20, 670–680. [Google Scholar] [CrossRef]
- Zemach, A.; McDaniel, I.E.; Silva, P.; Zilberman, D. Genome-Wide Evolutionary Analysis of Eukaryotic DNA Methylation. Science 2010, 328, 916–919. [Google Scholar] [CrossRef] [Green Version]
- Weber, M.; Davies, J.J.; Wittig, D.; Oakeley, E.J.; Haase, M.; Lam, W.L.; Schubeler, D. Chromosome-wide and promoter-specific analyses identify sites of differential DNA methylation in normal and transformed human cells. Nat. Genet. 2005, 37, 853–862. [Google Scholar] [CrossRef]
- Harris, R.A.; Wang, T.; Coarfa, C.; Nagarajan, R.P.; Hong, C.B.; Downey, S.L.; Johnson, B.E.; Fouse, S.D.; Delaney, A.; Zhao, Y.J.; et al. Comparison of sequencing-based methods to profile DNA methylation and identification of monoallelic epigenetic modifications. Nat. Biotechnol. 2010, 28, 1097–1194. [Google Scholar] [CrossRef]
- Meissner, A.; Gnirke, A.; Bell, G.W.; Ramsahoye, B.; Lander, E.S.; Jaenisch, R. Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis. Nucleic Acids Res. 2005, 33, 5868–5877. [Google Scholar] [CrossRef] [Green Version]
- Gupta, R.; Nagarajan, A.; Wajapeyee, N. Advances in genome-wide DNA methylation analysis. BioTechniques 2010, 49, III–XI. [Google Scholar] [CrossRef] [Green Version]
- Bock, C. Analysing and interpreting DNA methylation data. Nat. Rev. Genet. 2012, 13, 705–719. [Google Scholar] [CrossRef]
- Li, Y.; Tollefsbol, T.O. DNA methylation detection: Bisulfite genomic sequencing analysis. Methods Mol. Biol. 2011, 791, 11–21. [Google Scholar] [CrossRef] [Green Version]
- Bibikova, M.; Lin, Z.W.; Zhou, L.X.; Chudin, E.; Garcia, E.W.; Wu, B.; Doucet, D.; Thomas, N.J.; Wang, Y.H.; Vollmer, E.; et al. High-throughput DNA methylation profiling using universal bead arrays. Genome Res. 2006, 16, 383–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bibikova, M.; Le, J.; Barnes, B.; Saedinia-Melnyk, S.; Zhou, L.X.; Shen, R.; Gunderson, K.L. Genome-wide DNA methylation profiling using Infinium (R) assay. Epigenomics 2009, 1, 177–200. [Google Scholar] [CrossRef]
- Pidsley, R.; Zotenko, E.; Peters, T.J.; Lawrence, M.G.; Risbridger, G.P.; Molloy, P.; Van Djik, S.; Muhlhausler, B.; Stirzaker, C.; Clark, S.J. Critical evaluation of the Illumina MethylationEPIC BeadChip microarray for whole-genome DNA methylation profiling. Genome Biol. 2016, 17, 208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bibikova, M.; Barnes, B.; Tsan, C.; Ho, V.; Klotzle, B.; Le, J.M.; Delano, D.; Zhang, L.; Schroth, G.P.; Gunderson, K.L.; et al. High density DNA methylation array with single CpG site resolution. Genomics 2011, 98, 288–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irizarry, R.A.; Ladd-Acosta, C.; Carvalho, B.; Wu, H.; Brandenburg, S.A.; Jeddeloh, J.A.; Wen, B.; Feinberg, A.P. Comprehensive high-throughput arrays for relative methylation (CHARM). Genome Res. 2008, 18, 780–790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dupont, J.M.; Tost, J.; Jammes, H.; Gut, N.G. De novo quantitative bisulfite sequencing using the pyrosequencing technology. Anal. Biochem. 2004, 333, 119–127. [Google Scholar] [CrossRef]
- Sestakova, S.; Salek, C.; Remesova, H. DNA Methylation Validation Methods: A Coherent Review with Practical Comparison. Biol. Proced. Online 2019, 21, 1–11. [Google Scholar] [CrossRef]
- Karemaker, I.D.; Vermeulen, M. Single-Cell DNA Methylation Profiling: Technologies and Biological Applications. Trends Biotechnol. 2018, 36, 952–965. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, N.A.; Tao, R.; Chenoweth, J.G.; Brandtjen, A.; Mighdoll, M.I.; Genova, J.D.; McKay, R.D.; Jia, Y.; Weinberger, D.R.; Kleinman, J.E.; et al. Strong Components of Epigenetic Memory in Cultured Human Fibroblasts Related to Site of Origin and Donor Age. PLoS Genet. 2016, 12, e1005819. [Google Scholar] [CrossRef] [PubMed]
- Varley, K.E.; Gertz, J.; Bowling, K.M.; Parker, S.L.; Reddy, T.E.; Pauli-Behn, F.; Cross, M.K.; Williams, B.A.; Stamatoyannopoulos, J.A.; Crawford, G.E.; et al. Dynamic DNA methylation across diverse human cell lines and tissues. Genome Res. 2013, 23, 555–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blake, L.E.; Roux, J.; Hernando-Herraez, I.; Banovich, N.E.; Perez, R.G.; Hsiao, C.J.; Eres, I.; Cuevas, C.; Marques-Bonet, T.; Gilad, Y. A comparison of gene expression and DNA methylation patterns across tissues and species. Genome Res. 2020, 30, 250–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irizarry, R.A.; Ladd-Acosta, C.; Wen, B.; Wu, Z.; Montano, C.; Onyango, P.; Cui, H.; Gabo, K.; Rongione, M.; Webster, M.; et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat. Genet. 2009, 41, 178–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koch, C.M.; Suschek, C.V.; Lin, Q.; Bork, S.; Goergens, M.; Joussen, S.; Pallua, N.; Ho, A.D.; Zenke, M.; Wagner, W. Specific age-associated DNA methylation changes in human dermal fibroblasts. PLoS ONE 2011, 6, e16679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clifford, R.L.; Yang, C.X.; Fishbane, N.; Patel, J.; MacIsaac, J.L.; McEwen, L.M.; May, S.T.; Castellanos-Uribe, M.; Nair, P.; Obeidat, M.; et al. TWIST1 DNA methylation is a cell marker of airway and parenchymal lung fibroblasts that are differentially methylated in asthma. Clin. Epigenet. 2020, 12, 145. [Google Scholar] [CrossRef]
- Selman, M.; Pardo, A. Idiopathic pulmonary fibrosis: An epithelial/fibroblastic cross-talk disorder. Respir. Res. 2001, 3, 8. [Google Scholar] [CrossRef]
- Brody, A.R.; Soler, P.; Basset, F.; Haschek, W.M.; Witschi, H. Epithelial-mesenchymal associations of cells in human pulmonary fibrosis and in bht-oxygen-induced fibrosis in mice. Exp. Lung Res. 1981, 2, 207–220. [Google Scholar] [CrossRef]
- John, A.E.; Joseph, C.; Jenkins, G.; Tatler, A.L. COVID-19 and pulmonary fibrosis: A potential role for lung epithelial cells and fibroblasts. Immunol. Rev. 2021, 302, 228–240. [Google Scholar] [CrossRef]
- Nemeth, J.; Schundner, A.; Frick, M. Insights Into Development and Progression of Idiopathic Pulmonary Fibrosis From Single Cell RNA Studies. Front. Med. 2020, 7, 611728. [Google Scholar] [CrossRef] [PubMed]
- Adams, T.S.; Schupp, J.C.; Poli, S.; Ayaub, E.A.; Neumark, N.; Ahangari, F.; Chu, S.G.; Raby, B.A.; DeIuliis, G.; Januszyk, M.; et al. Single-cell RNA-seq reveals ectopic and aberrant lung-resident cell populations in idiopathic pulmonary fibrosis. Sci. Adv. 2020, 6, eaba1983. [Google Scholar] [CrossRef] [PubMed]
- Habermann, A.C.; Gutierrez, A.J.; Bui, L.T.; Yahn, S.L.; Winters, N.I.; Calvi, C.L.; Peter, L.; Chung, M.I.; Taylor, C.J.; Jetter, C.; et al. Single-cell RNA sequencing reveals profibrotic roles of distinct epithelial and mesenchymal lineages in pulmonary fibrosis. Sci. Adv. 2020, 6, eaba1972. [Google Scholar] [CrossRef] [PubMed]
- Sanders, Y.Y.; Ambalavanan, N.; Halloran, B.; Zhang, X.; Liu, H.; Crossman, D.K.; Bray, M.; Zhang, K.; Thannickal, V.J.; Hagood, J.S. Altered DNA methylation profile in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2012, 186, 525–535. [Google Scholar] [CrossRef]
- Yang, I.V.; Pedersen, B.S.; Rabinovich, E.; Hennessy, C.E.; Davidson, E.J.; Murphy, E.; Guardela, B.J.; Tedrow, J.R.; Zhang, Y.; Singh, M.K.; et al. Relationship of DNA methylation and gene expression in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2014, 190, 1263–1272. [Google Scholar] [CrossRef]
- Huang, S.K.; Scruggs, A.M.; McEachin, R.C.; White, E.S.; Peters-Golden, M. Lung fibroblasts from patients with idiopathic pulmonary fibrosis exhibit genome-wide differences in DNA methylation compared to fibroblasts from nonfibrotic lung. PLoS ONE 2014, 9, e107055. [Google Scholar] [CrossRef] [Green Version]
- Negreros, M.; Hagood, J.S.; Espinoza, C.R.; Balderas-Martinez, Y.I.; Selman, M.; Pardo, A. Transforming growth factor beta 1 induces methylation changes in lung fibroblasts. PLoS ONE 2019, 14, e0223512. [Google Scholar] [CrossRef] [Green Version]
- Hough, K.P.; Curtiss, M.L.; Blain, T.J.; Liu, R.M.; Trevor, J.; Deshane, J.S.; Thannickal, V.J. Airway Remodeling in Asthma. Front. Med. 2020, 7, 191. [Google Scholar] [CrossRef]
- Royce, S.G.; Cheng, V.; Samuel, C.S.; Tang, M.L. The regulation of fibrosis in airway remodeling in asthma. Mol. Cell. Endocrinol. 2012, 351, 167–175. [Google Scholar] [CrossRef]
- Mautino, G.; Henriquet, C.; Gougat, C.; Le Cam, A.; Dayer, J.M.; Bousquet, J.; Capony, F. Increased expression of tissue inhibitor of metalloproteinase-1 and loss of correlation with matrix metalloproteinase-9 by macrophages in asthma. Lab. Investig. 1999, 79, 39–47. [Google Scholar]
- Vignola, A.M.; Riccobono, L.; Mirabella, A.; Profita, M.; Chanez, P.; Bellia, V.; Mautino, G.; D’Accardi, P.; Bousquet, J.; Bonsignore, G. Sputum metalloproteinase-9 tissue inhibitor of metalloproteinase-1 ratio correlates with airflow obstruction in asthma and chronic bronchitis. Am. J. Respir. Crit. Care Med. 1998, 158, 1945–1950. [Google Scholar] [CrossRef] [Green Version]
- Hayakawa, T.; Yamashita, K.; Tanzawa, K.; Uchijima, E.; Iwata, K. Growth-promoting activity of tissue inhibitor of metalloproteinases-1 (timp-1) for a wide-range of cells—A possible new growth-factor in serum. FEBS Lett. 1992, 298, 29–32. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, M.; Sun, G.; Stacey, M.A.; Mori, L.; Mattoli, S. Identification of circulating fibrocytes as precursors of bronchial myofibroblasts in asthma. J. Immunol. 2003, 171, 380–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gizycki, M.J.; Adelroth, E.; Rogers, A.V.; Obyrne, P.M.; Jeffery, P.K. Myofibroblast involvement in the allergen-induced late response in mild atopic asthma. Am. J. Respir. Cell Mol. Biol. 1997, 16, 664–673. [Google Scholar] [CrossRef]
- Postma, D.S.; Timens, W. Remodeling in asthma and chronic obstructive pulmonary disease. Proc. Am. Thorac. Soc. 2006, 3, 434–439. [Google Scholar] [CrossRef]
- Minshall, E.M.; Leung, D.Y.M.; Martin, R.J.; Song, Y.L.; Cameron, L.; Ernst, P.; Hamid, Q. Eosinophil-associated TGF-beta(1) mRNA expression and airways fibrosis in bronchial asthma. Am. J. Respir. Cell Mol. Biol. 1997, 17, 326–333. [Google Scholar] [CrossRef] [PubMed]
- Chetta, A.; Foresi, A.; DelDonno, M.; Bertorelli, G.; Pesci, A.; Olivieri, D. Airways remodeling is a distinctive feature of asthma and is related to severity of disease. Chest 1997, 111, 852–857. [Google Scholar] [CrossRef] [Green Version]
- Boser, S.R.; Mauad, T.; Araujo-Paulino, B.B.; Mitchell, I.; Shrestha, G.; Chiu, A.; Butt, J.; Kelly, M.M.; Caldini, E.; James, A.; et al. Myofibroblasts are increased in the lung parenchyma in asthma. PLoS ONE 2017, 12, e0182378. [Google Scholar] [CrossRef] [Green Version]
- Weitoft, M.; Andersson, C.; Andersson-Sjoland, A.; Tufvesson, E.; Bjermer, L.; Erjefalt, J.; Westergren-Thorsson, G. Controlled and uncontrolled asthma display distinct alveolar tissue matrix compositions. Respir. Res. 2014, 15, 67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnes, P.J. Small airway fibrosis in COPD. Int. J. Biochem. Cell Biol. 2019, 116, 105598. [Google Scholar] [CrossRef] [PubMed]
- Jeffery, P.K. Remodeling and inflammation of bronchi in asthma and chronic obstructive pulmonary disease. Proc. Am. Thorac. Soc. 2004, 1, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Hogg, J.C.; Chu, F.; Utokaparch, S.; Woods, R.; Elliott, W.M.; Buzatu, L.; Cherniack, R.M.; Rogers, R.M.; Sciurba, F.C.; Coxson, H.O.; et al. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N. Engl. J. Med. 2004, 350, 2645–2653. [Google Scholar] [CrossRef] [PubMed]
- Wrench, C.L.; Baker, J.R.; Fenwick, P.S.; Donnelly, L.E.; Barnes, P.J. Chylous cardiac tapenade with chylothoracies secondary to Hodgkin’s lymphoma: The first recorded case of successful octreotide treatment. In D42. Pleural Disease Case Reports II; American Thoracic Society: New York, NY, USA, 2019; p. A6422. [Google Scholar]
- Wrench, C.; Baker, J.; Fenwick, P.; Donnelly, L.; Barnes, P. Small airway fibroblasts from COPD patients are senescent and pro-fibrotic. Eur. Respir. J. 2018, 52. [Google Scholar] [CrossRef]
- Holz, O.; Zuhlke, I.; Jaksztat, E.; Muller, K.C.; Welker, L.; Nakashima, M.; Diemel, K.D.; Branscheid, D.; Magnussen, H.; Jorres, R.A. Lung fibroblasts from patients with emphysema show a reduced proliferation rate in culture. Eur. Respir. J. 2004, 24, 575–579. [Google Scholar] [CrossRef]
- Togo, S.; Holz, O.; Liu, X.; Sugiura, H.; Kamio, K.; Wang, X.; Kawasaki, S.; Ahn, Y.; Fredriksson, K.; Skold, C.M.; et al. Lung fibroblast repair functions in patients with chronic obstructive pulmonary disease are altered by multiple mechanisms. Am. J. Respir. Crit. Care Med. 2008, 178, 248–260. [Google Scholar] [CrossRef]
- Campbell, J.D.; McDonough, J.E.; Zeskind, J.E.; Hackett, T.L.; Pechkovsky, D.V.; Brandsma, C.A.; Suzuki, M.; Gosselink, J.V.; Liu, G.; Alekseyev, Y.O.; et al. A gene expression signature of emphysema-related lung destruction and its reversal by the tripeptide GHK. Genome Med. 2012, 4, 67. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Wu, L.; Qu, J.M.; Bai, C.X.; Merrilees, M.J.; Black, P.N. Pro-inflammatory phenotype of COPD fibroblasts not compatible with repair in COPD lung. J. Cell Mol. Med. 2012, 16, 1522–1532. [Google Scholar] [CrossRef] [PubMed]
- Li, X.Y.; Noell, G.; Tabib, T.; Gregory, A.D.; Bittar, H.T.E.; Vats, R.; Kaminski, T.W.; Sembrat, J.; Snyder, M.E.; Chandra, D.; et al. Single cell RNA sequencing identifies IGFBP5 and QKI as ciliated epithelial cell genes associated with severe COPD. Respir. Res. 2021, 22. [Google Scholar] [CrossRef] [PubMed]
- Qiu, W.; Baccarelli, A.; Carey, V.J.; Boutaoui, N.; Bacherman, H.; Klanderman, B.; Rennard, S.; Agusti, A.; Anderson, W.; Lomas, D.A.; et al. Variable DNA methylation is associated with chronic obstructive pulmonary disease and lung function. Am. J. Respir. Crit. Care Med. 2012, 185, 373–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoo, S.; Takikawa, S.; Geraghty, P.; Argmann, C.; Campbell, J.; Lin, L.; Huang, T.; Tu, Z.; Foronjy, R.F.; Spira, A.; et al. Integrative analysis of DNA methylation and gene expression data identifies EPAS1 as a key regulator of COPD. PLoS Genet. 2015, 11, e1004898. [Google Scholar] [CrossRef] [Green Version]
- Sundar, I.K.; Yin, Q.; Baier, B.S.; Yan, L.; Mazur, W.; Li, D.; Susiarjo, M.; Rahman, I. DNA methylation profiling in peripheral lung tissues of smokers and patients with COPD. Clin. Epigenet. 2017, 9, 38. [Google Scholar] [CrossRef]
- Morrow, J.D.; Cho, M.H.; Hersh, C.P.; Pinto-Plata, V.; Celli, B.; Marchetti, N.; Criner, G.; Bueno, R.; Washko, G.; Glass, K.; et al. DNA methylation profiling in human lung tissue identifies genes associated with COPD. Epigenetics 2016, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Vucic, E.A.; Chari, R.; Thu, K.L.; Wilson, I.M.; Cotton, A.M.; Kennett, J.Y.; Zhang, M.; Lonergan, K.M.; Steiling, K.; Brown, C.J.; et al. DNA methylation is globally disrupted and associated with expression changes in chronic obstructive pulmonary disease small airways. Am. J. Respir. Cell Mol. Biol. 2014, 50, 912–922. [Google Scholar] [CrossRef] [Green Version]
- Clifford, R.L.; Fishbane, N.; Patel, J.; MacIsaac, J.L.; McEwen, L.M.; Fisher, A.J.; Brandsma, C.A.; Nair, P.; Kobor, M.S.; Hackett, T.L.; et al. Altered DNA methylation is associated with aberrant gene expression in parenchymal but not airway fibroblasts isolated from individuals with COPD. Clin. Epigenet. 2018, 10, 32. [Google Scholar] [CrossRef] [Green Version]
- Paul, D.S.; Teschendorff, A.E.; Dang, M.A.; Lowe, R.; Hawa, M.I.; Ecker, S.; Beyan, H.; Cunningham, S.; Fouts, A.R.; Ramelius, A.; et al. Increased DNA methylation variability in type 1 diabetes across three immune effector cell types. Nat. Commun. 2016, 7, 13555. [Google Scholar] [CrossRef] [PubMed]
- Matthay, M.A.; Zemans, R.L.; Zimmerman, G.A.; Arabi, Y.M.; Beitler, J.R.; Mercat, A.; Herridge, M.; Randolph, A.G.; Calfee, C.S. Acute respiratory distress syndrome. Nat. Rev. Dis. Primers 2019, 5. [Google Scholar] [CrossRef] [PubMed]
- Soares, M.P.; Teixeira, L.; Moita, L.F. Disease tolerance and immunity in host protection against infection. Nat. Rev. Immunol. 2017, 17, 83–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donnelly, S.C.; Haslett, C. Cellular mechanisms of acute lung injury—Implications for future treatment in the adult respiratory-distress syndrome. Thorax 1992, 47, 260–263. [Google Scholar] [CrossRef] [Green Version]
- Dawes, K.E.; Gray, A.J.; Laurent, G.J. Thrombin stimulates fibroblast chemotaxis and replication. Eur. J. Cell Biol. 1993, 61, 126–130. [Google Scholar]
- Bachofen, M.; Weibel, E.R. Structural alterations of lung parenchyma in the adult respiratory-distress syndrome. Clin. Chest Med. 1982, 3, 35–56. [Google Scholar] [CrossRef]
- Marshall, R.; Bellingan, G.; Laurent, G. The acute respiratory distress syndrome: Fibrosis in the fast lane. Thorax 1998, 53, 815–817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szilagyi, K.L.; Lw, C.; Zhang, X.; Wang, T.; Fortman, J.D.; Zhang, W.; Garcia, J.G.N. Epigenetic contribution of the myosin light chain kinase gene to the risk for acute respiratory distress syndrome. Transl. Res. 2017, 180, 12–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Y.C.; Zhang, R.Y.; Zhu, Z.Z.; Shen, S.P.; Su, L.; Christiani, D.C. Epigenome-wide association study for 28-day survival of acute respiratory distress syndrome. Intens. Care Med. 2018, 44, 1182–1184. [Google Scholar] [CrossRef] [PubMed]
- Davies, J.C.; Alton, E.; Bush, A. Cystic fibrosis. BMJ Br. Med. J. 2007, 335, 1255–1259. [Google Scholar] [CrossRef] [PubMed]
- Cutting, G.R. Cystic fibrosis genetics: From molecular understanding to clinical application. Nat. Rev. Genet. 2015, 16, 45–56. [Google Scholar] [CrossRef] [Green Version]
- Rout-Pitt, N.; Farrow, N.; Parsons, D.; Donnelley, M. Epithelial mesenchymal transition (EMT): A universal process in lung diseases with implications for cystic fibrosis pathophysiology. Respir. Res. 2018, 19. [Google Scholar] [CrossRef] [PubMed]
- Mazio, C.; Scognamiglio, L.S.; De Cegli, R.; Galietta, L.J.V.; Di Bernardo, D.; Casale, C.; Urciuolo, F.; Imparato, G.; Netti, P.A. Intrinsic Abnormalities of Cystic Fibrosis Airway Connective Tissue Revealed by an In Vitro 3D Stromal Model. Cells 2020, 9, 1371. [Google Scholar] [CrossRef]
- Magalhaes, M.; Rivals, I.; Claustres, M.; Varilh, J.; Thomasset, M.; Bergougnoux, A.; Mely, L.; Leroy, S.; Corvol, H.; Guillot, L.; et al. DNA methylation at modifier genes of lung disease severity is altered in cystic fibrosis. Clin. Epigenet. 2017, 9. [Google Scholar] [CrossRef] [Green Version]
- Magalhaes, M.; Tost, J.; Pineau, F.; Rivals, I.; Busato, F.; Alary, N.; Mely, L.; Leroy, S.; Murris, M.; Caimmi, D.; et al. Dynamic changes of DNA methylation and lung disease in cystic fibrosis: Lessons from a monogenic disease. Epigenomics 2018, 10, 1131–1145. [Google Scholar] [CrossRef]
- Chen, Y.D.H.; Armstrong, D.A.; Salas, L.A.; Hazlett, H.F.; Nymon, A.B.; Dessaint, J.A.; Aridgides, D.S.; Mellinger, D.L.; Liu, X.Y.; Christensen, B.C.; et al. Genome-wide DNA methylation profiling shows a distinct epigenetic signature associated with lung macrophages in cystic fibrosis. Clin. Epigenet. 2018, 10. [Google Scholar] [CrossRef] [Green Version]
- Pineau, F.; Caimmi, D.; Taviaux, S.; Reveil, M.; Brosseau, L.; Rivals, I.; Drevait, M.; Vachier, I.; Claustres, M.; Chiron, R.; et al. DNA Methylation at ATP11A cg11702988 Is a Biomarker of Lung Disease Severity in Cystic Fibrosis: A Longitudinal Study. Genes 2021, 12, 441. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Disease | Findings | Cell/Tissue Type | Array Platform 1 | Ref |
|---|---|---|---|---|
| IPF | 870 differentially methylated genes including IPF linked genes COL3A1 and MMP7 | Whole Lung Tissue (n = 12 IPF; 7 Controls) | Illumina 27 k | [54] |
| IPF | 2130 differentially methylated regions | Whole Lung Tissue (n = 94 IPF; 67 Controls) | CHARM | [55] |
| IPF | 125 differentially methylated CpGs | Parenchymal fibroblasts (n = 6 IPF; 3 non-fibrotic Controls; 3 normal lung fibroblast cell lines | Illumina 27 k | [56] |
| Asthma | 17 and 112 differentially methylated regions in airway and parenchymal fibroblast respectively. TWIST1 identified as airway vs. parenchymal fibroblast marker | Airway fibroblasts (n = 9 Asthma; 8 Controls) and Parenchymal fibroblasts (n = 8 Asthma; 9 Controls) | Illumina EPIC | [47] |
| COPD | 349 disease severity associated differentially methylated CpGs | Whole Blood (2 cohorts with n = 620 COPD; 325 Controls and 181 COPD; 109 Controls) | Illumina 27 k | [81] |
| COPD | Identified EPAS1 as key regulator of COPD | Whole Lung Tissue (n = 176 COPD; 76 Controls) | Nimblegen 2.1 | [82] |
| COPD | 280 and 10 differentially methylated CpGs in COPD compared to non-smokers and smokers respectively. | Lung Tissue (n = 8 non-smokers; 8 smokers with normal lung function; 8 COPD) | Illumina 450 k | [83] |
| COPD | 535 differentially methylated CpGs | Whole Lung Tissue (n = 114 COPD; 46 Controls) | Illumina 450 k | [84] |
| COPD | 1260 differentially methylated CpGs | Small airway epithelial cells (n = 15 COPD; 23 Controls) | Illumina 27 k | [85] |
| COPD | 887 and 44 differentially methylated regions in airway and parenchymal fibroblasts from COPD patients; 359 differentially variable CpGs in COPD parenchymal fibroblasts | Airway fibroblasts (n = 7COPD; 8 Controls) and Parenchymal fibroblasts (n = 29 COPD; 17 Controls) | Illumina 450 k | [86] |
| ARDS | 2 differentially methylated CpGs on myosin light chain kinase gene associated with ARDS | Whole Blood (n = 39 ARDS; 75 ICU Controls) | Illumina 450 k | [94] |
| ARDS | 2 differentially methylated CpGs located within prostaglandin D2 receptor and integral membrane ATPase genes | Whole Blood (185 moderate-to-severe ARDS) | Illumina 450 k | [95] |
| CF | Methylation changes in 3 genes (Heme Oxygenase 1, Glutathione S-Transferase Mu 3, Endothelin Receptor Type A) associated with disease severity | Nasal epithelial cells and whole blood (n = 48 CF; 24 Controls) | Targeted sequencing for CFTR and 13 lung disease modifier genes | [100] |
| CF | Differential methylation at 50 CpGs correlated with lung function in CF patients | Nasal epithelial cells (n = 32 CF; 16 Controls) | Illumina 450 k | [101] |
| CF | 109 differentially methylated CpGs | Bronchoalveolar Lavage (n = 4 CF; 4 Controls) | Illumina EPIC | [102] |
| CF | Differential methylation at CpG (cg11702988) showed negative correlation with disease severity. Validated in sputum as a biomarker. | Nasal epithelial cells (n = 51 CF; 24 Controls) | Illumina 450 k | [101] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rajasekar, P.; Patel, J.; Clifford, R.L. DNA Methylation of Fibroblast Phenotypes and Contributions to Lung Fibrosis. Cells 2021, 10, 1977. https://doi.org/10.3390/cells10081977
Rajasekar P, Patel J, Clifford RL. DNA Methylation of Fibroblast Phenotypes and Contributions to Lung Fibrosis. Cells. 2021; 10(8):1977. https://doi.org/10.3390/cells10081977
Chicago/Turabian StyleRajasekar, Poojitha, Jamie Patel, and Rachel L. Clifford. 2021. "DNA Methylation of Fibroblast Phenotypes and Contributions to Lung Fibrosis" Cells 10, no. 8: 1977. https://doi.org/10.3390/cells10081977
APA StyleRajasekar, P., Patel, J., & Clifford, R. L. (2021). DNA Methylation of Fibroblast Phenotypes and Contributions to Lung Fibrosis. Cells, 10(8), 1977. https://doi.org/10.3390/cells10081977