
Glycoconjugation of Betulin Derivatives Using Copper-Catalyzed 1,3-Dipolar Azido-Alkyne Cycloaddition Reaction and a Preliminary Assay of Cytotoxicity of the Obtained Compounds

 , , , , and
, , , , and
Abstract
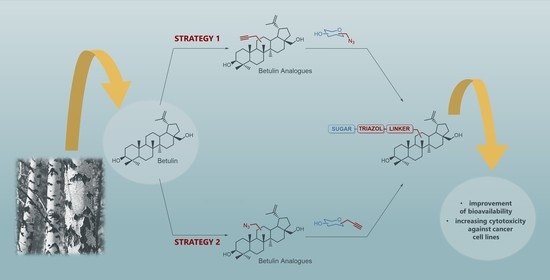
1. Introduction
2. Results and Discussion
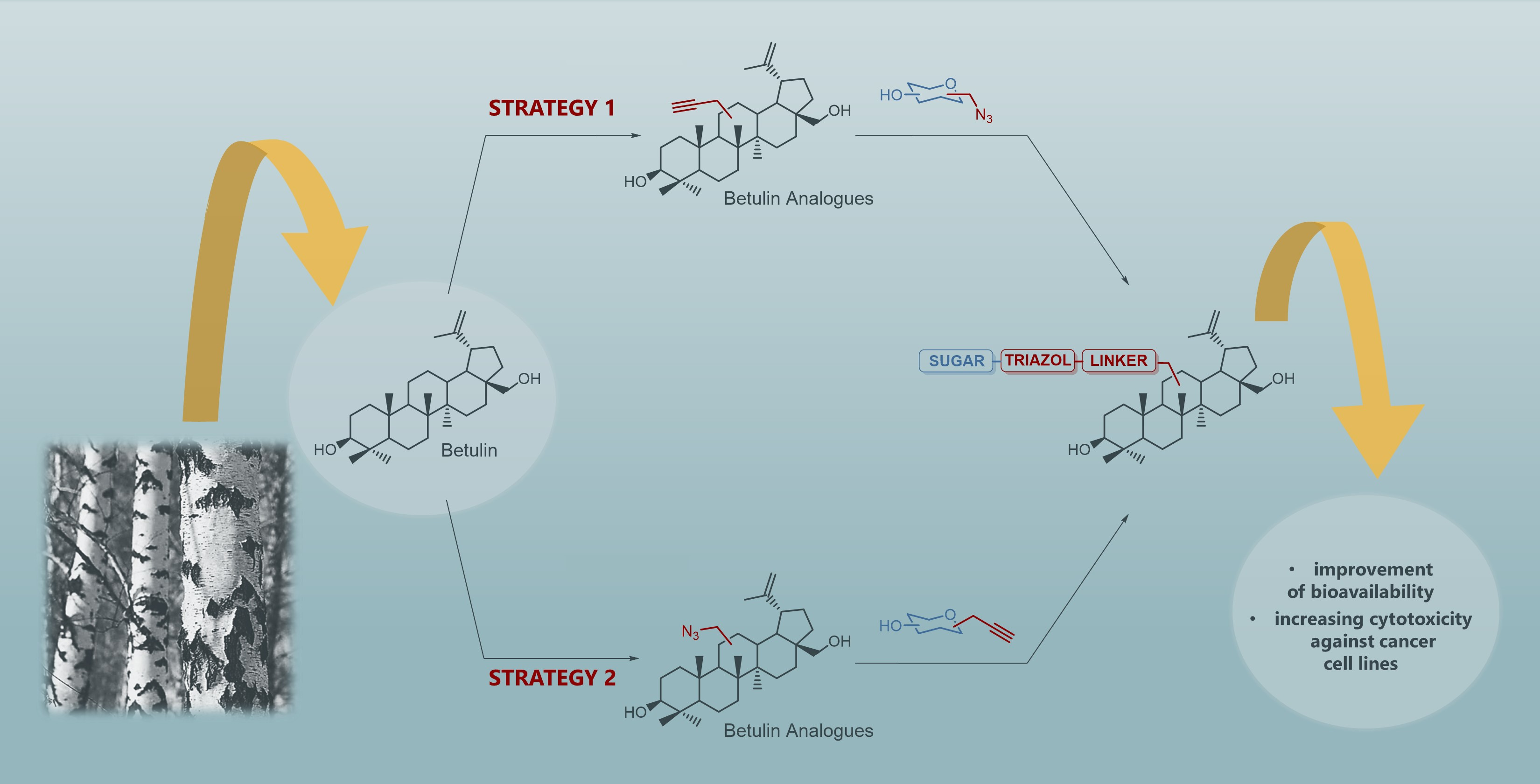
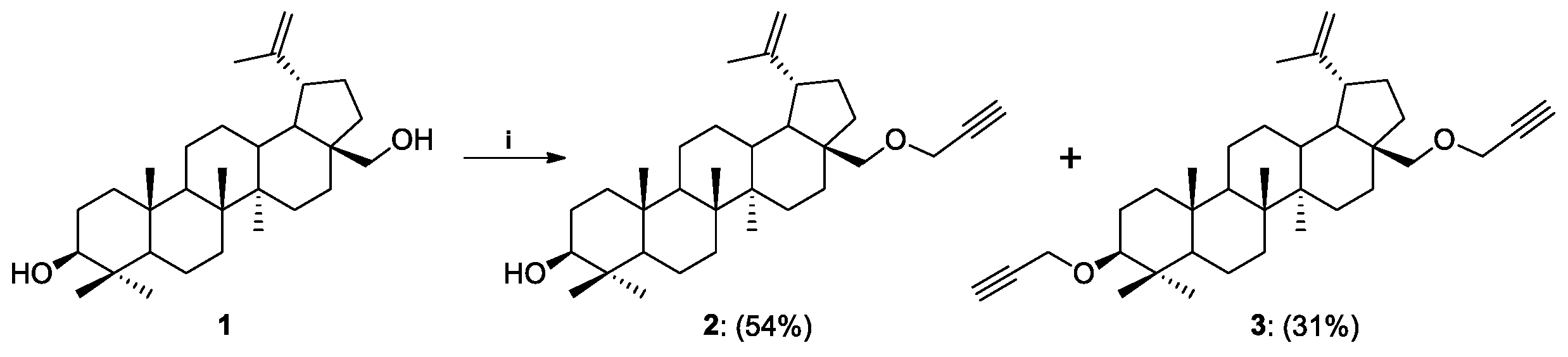
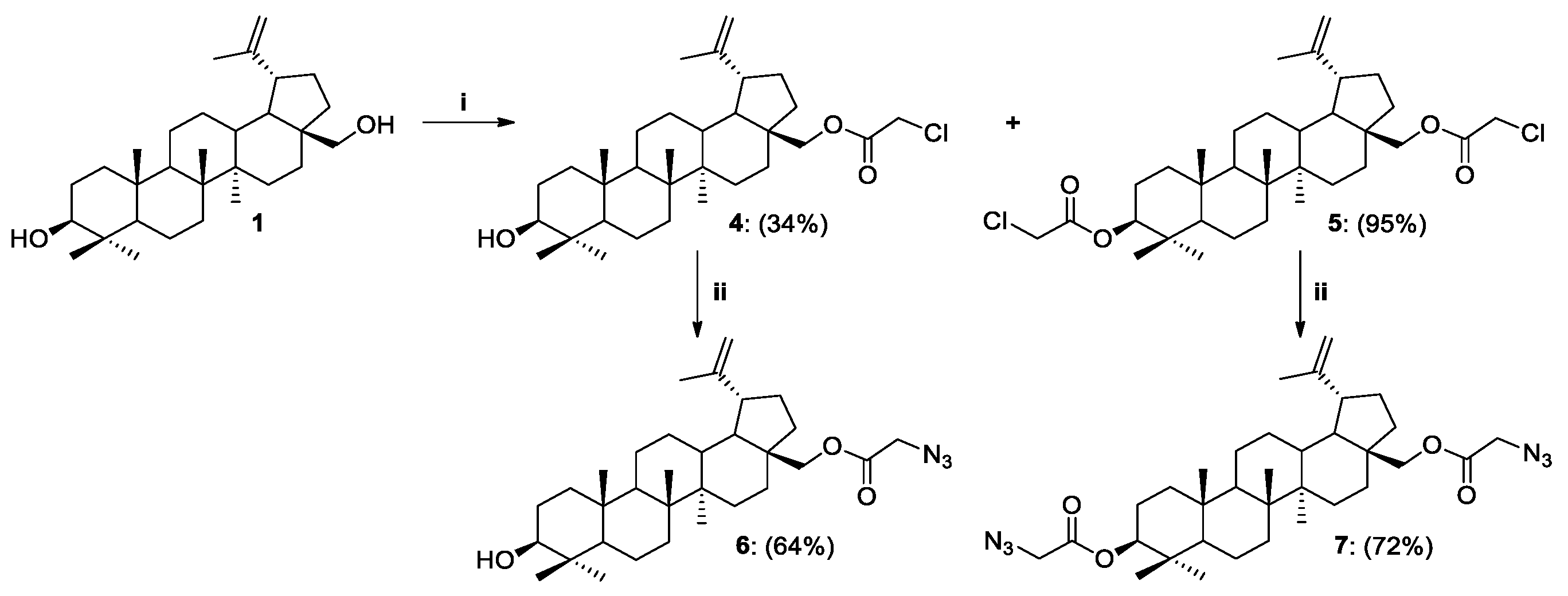
2.1. Synthesis of Betulin Analogs
2.2. Synthesis of Sugar Derivatives
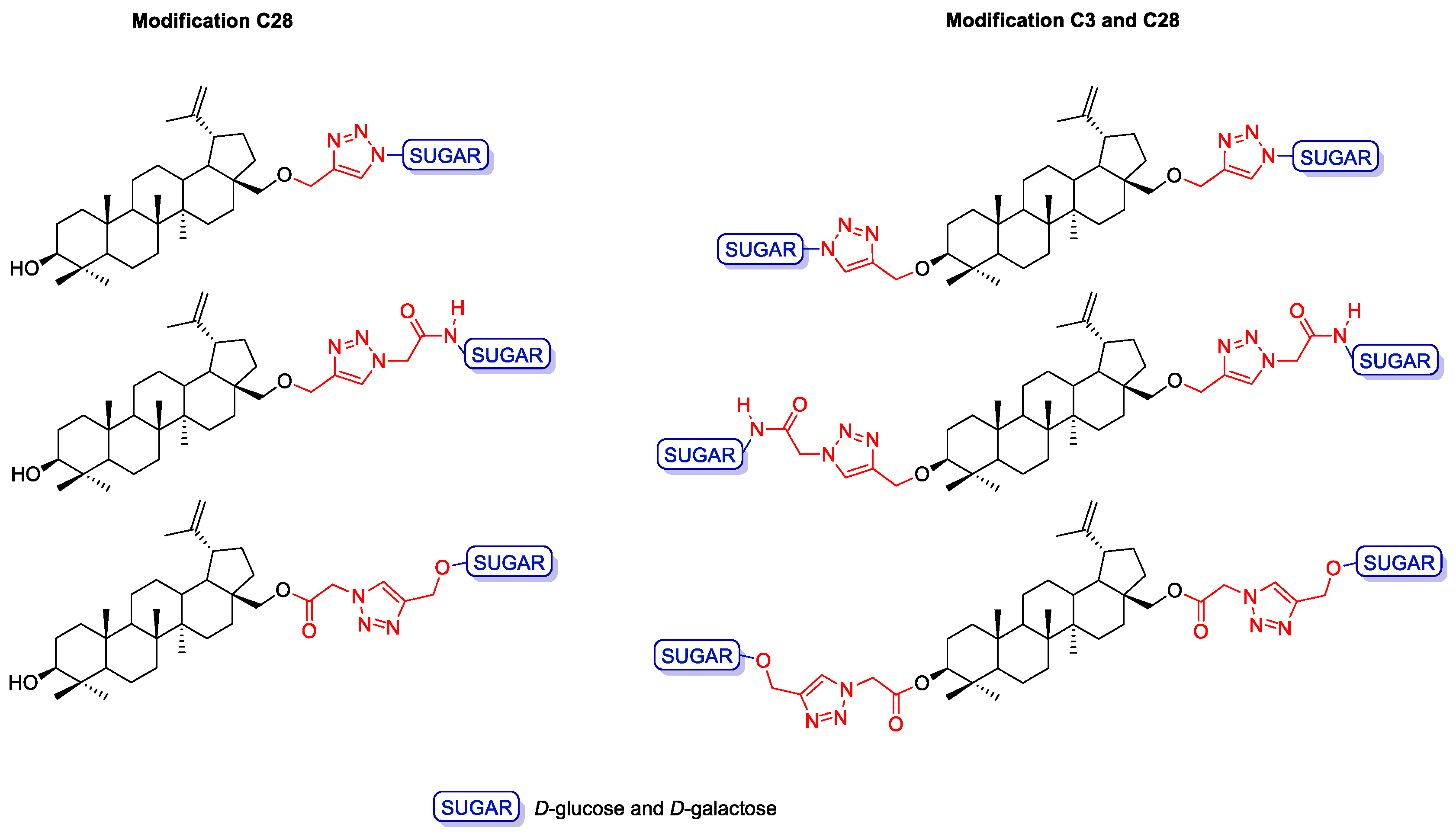
2.3. Modifications of the Natural Betulin Skeleton via Click Chemistry
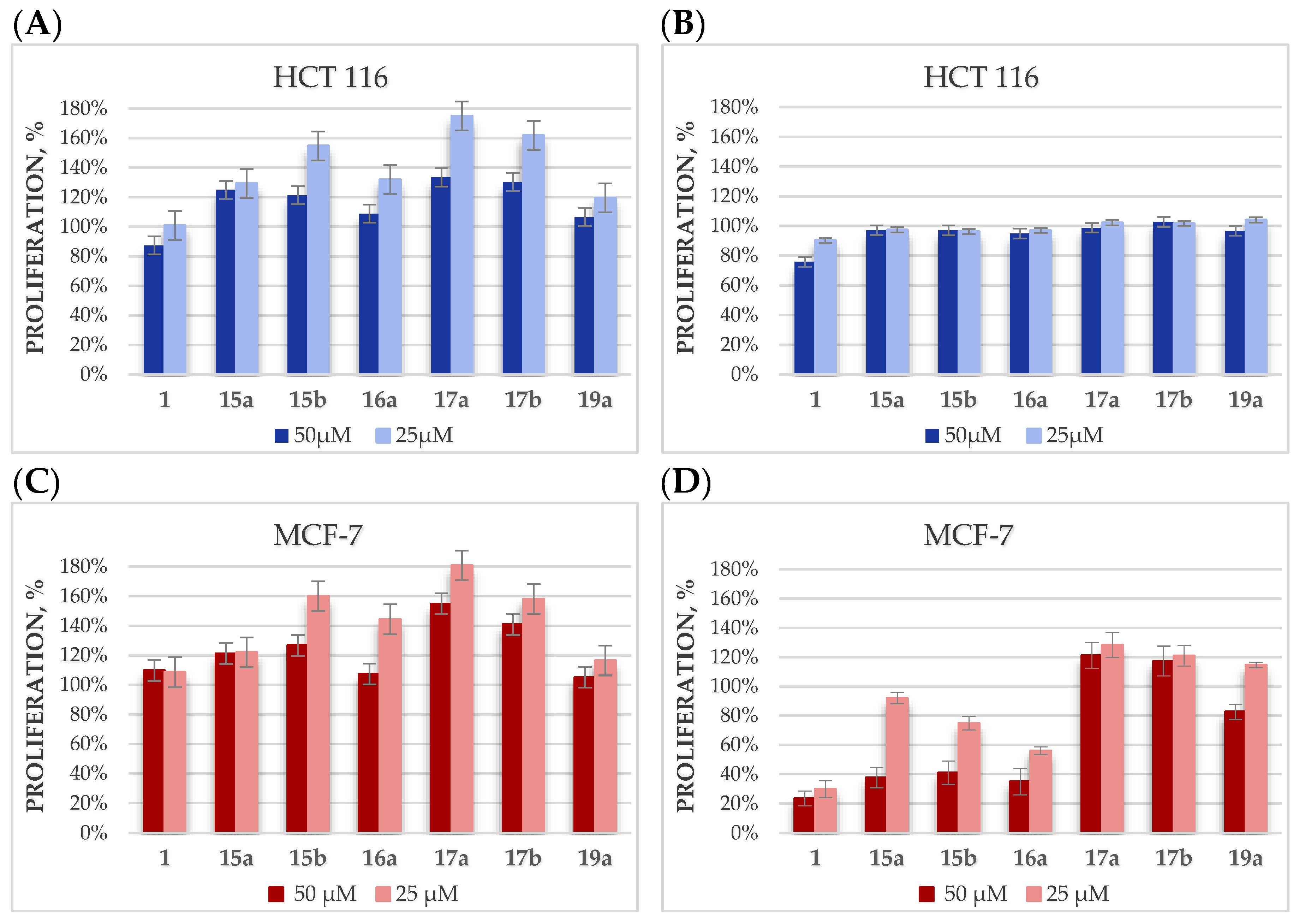
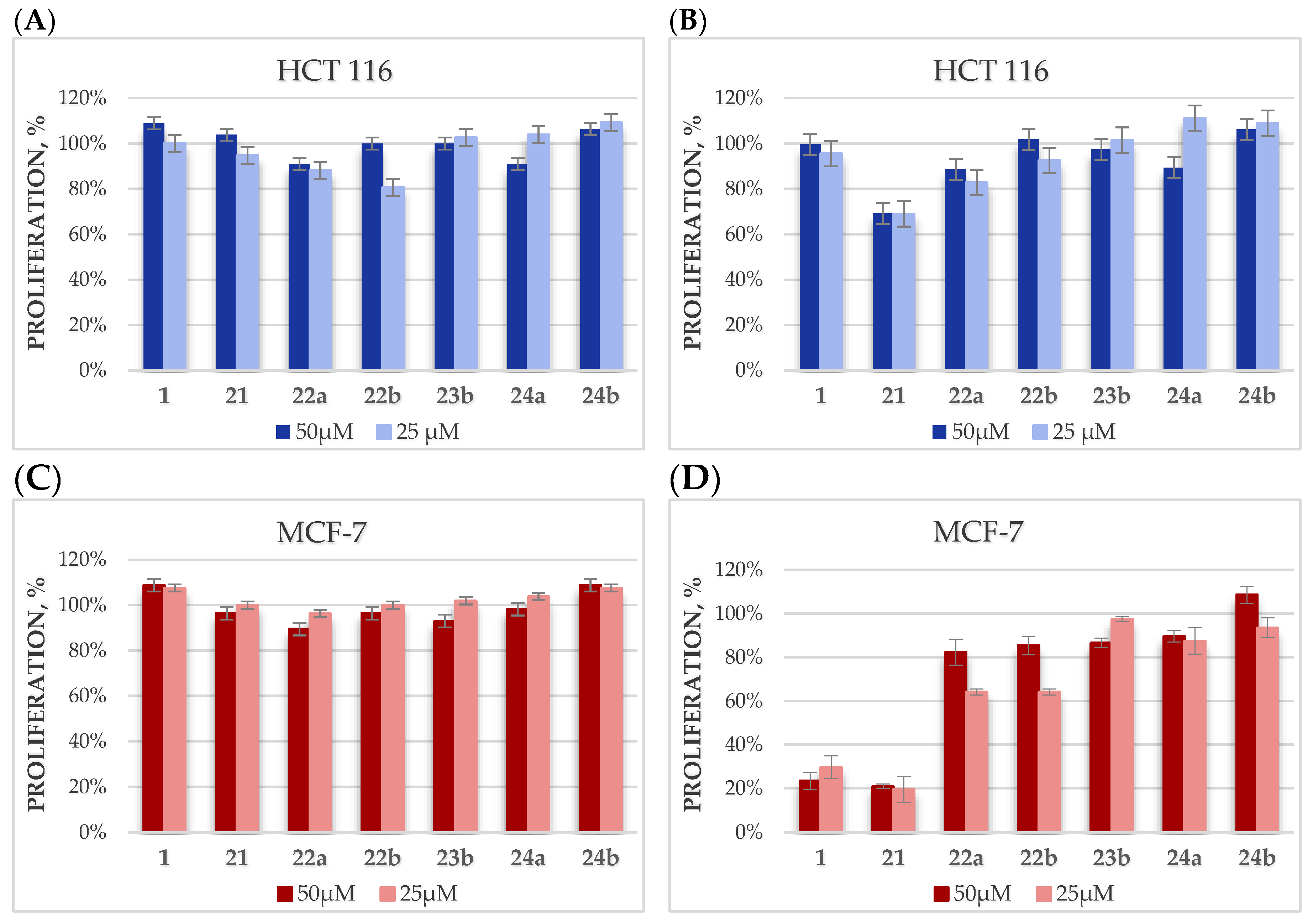
2.4. Cytotoxicity Studies
2.5. Cytotoxic Activity Predicted Lipophilicity
3. Materials and Methods
3.1. General Information
3.2. Chemistry
3.2.1. Procedure for the Synthesis of Betulin Analogs
Procedure for the Synthesis of 28-O-propargylbetulin 2 and 3,28-O,O’-di(propargyl)betulin 3
Procedure for the Synthesis of 28-O-(2-azidoacetyl)betulin 6
Procedure for the Synthesis of 3,28-O,O’-di(2-azidoacetyl)betulin 7
3.2.2. General Procedure for the Synthesis of Betulin Glycoconjugates 15–16 according to Strategy I (Modification C28)
3.2.3. General Procedure for the Synthesis of Betulin Glycoconjugates 17–18 according to Strategy I (Modification C3 and C28)
3.2.4. Procedure for the Synthesis of Betulin Glycoconjugates 19a according to Strategy II (Modification C28)
3.2.5. Procedure for the Synthesis of Betulin Glycoconjugates 20a according to Strategy II (Modification C3 and C28)
3.2.6. Procedure for the Synthesis of 3,28-O,O’-di(2-(4-(hydroxymethyl-1H-1,2,3-triazol-1-yl)acetyl)betulin 21
3.2.7. Procedure for the Deprotection of Betulin Glycoconjugates 15–17
3.3. Biological Assays
3.3.1. Cell Lines
3.3.2. CCK-8 Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Krasutsky, P.A. Birch bark research and development. Nat. Prod. Rep. 2006, 23, 919–942. [Google Scholar] [CrossRef] [PubMed]
- Safe, S.; Kasiappan, R. Natural Products as Mechanism-based Anticancer Agents: Sp Transcription Factors as Targets. Phytother. Res. 2016, 30, 1723–1732. [Google Scholar] [CrossRef] [PubMed]
- Hordyjewska, A.; Ostapiuk, A.; Horecka, A. Betulin and betulinic acid in cancer research. J. Pre-Clin. Clin. Res. 2018, 12, 72–75. [Google Scholar] [CrossRef]
- Dutta, D.; Chakraborty, B.; Sarkar, A.; Chowdhury, C.; Das, P. A potent betulinic acid analogue ascertains an antagonistic mechanism between autophagy and proteasomal degradation pathway in HT-29 cells. BMC Cancer 2016, 16, 23. [Google Scholar] [CrossRef] [PubMed]
- Boryczka, S.; Bębenek, E.; Wietrzyk, J.; Kempińska, K.; Jastrzębska, M.; Kusz, J.; Nowak, M. Synthesis, Structure and Cytotoxic Activity of New Acetylenic Derivatives of Betulin. Molecules 2013, 18, 4526–4543. [Google Scholar] [CrossRef]
- Baratto, L.C.; Porsani, M.V.; Pimentel, I.C.; Netto, A.B.P.; Paschke, R.; Oliveira, B.H. Preparation of betulinic acid derivatives by chemical and biotransformation methods and determination of cytotoxicity against selected cancer cell lines. Eur. J. Med. Chem. 2013, 68, 121–131. [Google Scholar] [CrossRef]
- Kommera, H.; Kaluđerović, G.N.; Kalbitz, J.; Dräger, B.; Paschke, R. Small structural changes of pentacyclic lupane type triterpenoid derivatives lead to significant differences in their anticancer properties. Eur. J. Med. Chem. 2010, 45, 3346–3353. [Google Scholar] [CrossRef]
- Kommera, H.; Kaluđerović, G.N.; Dittrich, S.; Kalbitz, J.; Dräger, B.; Mueller, T.; Paschke, R. Carbamate derivatives of betulinic acid and betulin with selective cytotoxic activity. Bioorg. Med. Chem. Lett. 2010, 20, 3409–3412. [Google Scholar] [CrossRef]
- Dangroo, N.A.; Singh, J.; Rath, S.K.; Gupta, N.; Qayum, A.; Singh, S.; Sangwan, P.L. A convergent synthesis of novel alkyne–azide cycloaddition congeners of betulinic acid as potent cytotoxic agent. Steroids 2017, 123, 1–12. [Google Scholar] [CrossRef]
- Gauthier, C.; Legault, J.; Lebrun, M.; Dufour, P.; Pichette, A. Glycosidation of lupane-type triterpenoids as potent in vitro cytotoxic agents. Bioorg. Med. Chem. 2006, 14, 6713–6725. [Google Scholar] [CrossRef]
- Drąg-Zalesińska, M.; Kulbacka, J.; Saczko, J.; Wysocka, T.; Zabel, M.; Surowiak, P.; Drag, M. Esters of betulin and betulinic acid with amino acids have improved water solubility and are selectively cytotoxic toward cancer cells. Bioorg. Med. Chem. Lett. 2009, 19, 4814–4817. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.-J.; Liu, M.-C.; Xiang, H.-M.; Zhao, Q.; Xue, W.; Yang, S. Synthesis and in vitro antitumor evaluation of betulin acid ester derivatives as novel apoptosis inducers. Eur. J. Med. Chem. 2015, 102, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.-M.; Xu, H.-G.; Wang, L.; Li, Y.-J.; Sun, P.; Wu, X.-M.; Wang, G.; Chen, W.-M.; Ye, W.-C. Betulinic Acid and its Derivatives as Potential Antitumor Agents. Med. Res. Rev. 2015, 35, 1127–1155. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Liu, Z.; Liu, W.; Han, X.; Zhao, M. Betulin attenuates lung and liver injuries in sepsis. Int. Immunopharmacol. 2016, 30, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Moghaddam, M.G.; Ahmad, F.B.H.; Samzadeh-Kermani, A. Biological Activity of Betulinic Acid: A Review. Pharmacol. Pharm. 2012, 3, 119–123. [Google Scholar] [CrossRef]
- Zhao, H.; Zheng, Q.; Hu, X.; Shen, H.; Li, F. Betulin attenuates kidney injury in septic rats through inhibiting TLR4/NF-κB signaling pathway. Life Sci. 2016, 144, 185–193. [Google Scholar] [CrossRef]
- Fulda, S.; Kroemer, G. Targeting mitochondrial apoptosis by betulinic acid in human cancers. Drug Discov. Today 2009, 14, 885–890. [Google Scholar] [CrossRef]
- Suman, P.; Patel, A.; Solano, L.; Jampana, G.; Gardner, Z.S.; Holt, C.M.; Jonnalagadda, S.C. Synthesis and cytotoxicity of Baylis-Hillman template derived betulinic acid-triazole conjugates. Tetrahedron 2017, 73, 4214–4226. [Google Scholar] [CrossRef]
- Jonnalagadda, S.; Suman, P.; Morgan, D.; Seay, J. Recent Developments on the Synthesis and Applications of Betulin and Betulinic Acid Derivatives as Therapeutic Agents. Bioact. Nat. Prod. (Part O) 2017, 53, 45–84. [Google Scholar] [CrossRef]
- Baltina, L.A.; Kazakova, O.B.; Nigmatullina, L.; Boreko, E.I.; Pavlova, N.; Nikolaeva, S.; Savinova, O.; Tolstikov, G. Lupane triterpenes and derivatives with antiviral activity. Bioorg. Med. Chem. Lett. 2003, 13, 3549–3552. [Google Scholar] [CrossRef]
- Bori, I.D.; Hung, H.-Y.; Qian, K.; Chen, C.-H.; Morris-Natschke, S.L.; Lee, K.-H. Anti-AIDS agents 88. Anti-HIV conjugates of betulin and betulinic acid with AZT prepared via click chemistry. Tetrahedron Lett. 2012, 53, 1987–1989. [Google Scholar] [CrossRef]
- Amiri, S.; Dastghaib, S.; Ahmadi, M.; Mehrbod, P.; Khadem, F.; Behrouj, H.; Aghanoori, M.-R.; Machaj, F.; Ghamsari, M.; Rosik, J.; et al. Betulin and its derivatives as novel compounds with different pharmacological effects. Biotechnol. Adv. 2020, 38, 107409. [Google Scholar] [CrossRef]
- Zdzisińska, B. Właściwości lecznicze betuliny i kwasu betulinowego, składników ekstraktu z kory brzozy. Farm. Przegl. Nauk. 2010, 3, 33–39. [Google Scholar]
- Grymel, M.; Zawojak, M.; Adamek, J. Triphenylphosphonium Analogues of Betulin and Betulinic Acid with Biological Activity: A Comprehensive Review. J. Nat. Prod. 2019, 82, 1719–1730. [Google Scholar] [CrossRef] [PubMed]
- Pokorný, J.; Horka, V.; Sidova, V.; Urban, M. Synthesis and characterization of new conjugates of betulin diacetate and bis(triphenysilyl)betulin with substituted triazoles. Mon. Für Chem.—Chem. Mon. 2018, 149, 839–845. [Google Scholar] [CrossRef]
- Bhunia, D.; Pallavi, P.M.C.; Bonam, S.R.; Reddy, S.A.; Verma, Y.K.; Halmuthur, S.K.M. Design, Synthesis, and Evaluation of Novel 1,2,3-Triazole-Tethered Glycolipids as Vaccine Adjuvants. Arch. Pharm. 2015, 348, 689–703. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Zhang, T.; Yuan, H.; Li, D.; Lou, H.; Fan, P. Mitochondria-Targeted Lupane Triterpenoid Derivatives and Their Selective Apoptosis-Inducing Anticancer Mechanisms. J. Med. Chem. 2017, 60, 6353–6363. [Google Scholar] [CrossRef] [PubMed]
- Dalvie, D.K.; Kalgutkar, A.S.; Khojasteh-Bakht, S.C.; Obach, R.S.; O’Donnell, J.P. Biotransformation Reactions of Five-Membered Aromatic Heterocyclic Rings. Chem. Res. Toxicol. 2002, 15, 269–299. [Google Scholar] [CrossRef]
- Valverde, I.E.; Bauman, A.; Kluba, C.A.; Vomstein, S.; Walter, M.A.; Mindt, T.L. 1,2,3-Triazoles as Amide Bond Mimics: Triazole Scan Yields Protease-Resistant Peptidomimetics for Tumor Targeting. Angew. Chem. Int. Ed. 2013, 52, 8957–8960. [Google Scholar] [CrossRef]
- Dheer, D.; Singh, V.; Shankar, R. Medicinal attributes of 1,2,3-triazoles: Current developments. Bioorganic Chem. 2017, 71, 30–54. [Google Scholar] [CrossRef]
- Thirumurugan, P.; Matosiuk, D.; Jozwiak, K. Click Chemistry for Drug Development and Diverse Chemical–Biology Applications. Chem. Rev. 2013, 113, 4905–4979. [Google Scholar] [CrossRef] [PubMed]
- Pokorný, L.B.A.M.U.J.; Borkova, L.; Urban, M. Click Reactions in Chemistry of Triterpenes—Advances Towards Development of Potential Therapeutics. Curr. Med. Chem. 2018, 25, 636–658. [Google Scholar] [CrossRef] [PubMed]
- Antimonova, A.N.; Petrenko, N.I.; Shakirov, M.M.; Rybalova, T.V.; Frolova, T.S.; Shul’Ts, E.E.; Kukina, T.P.; Sinitsyna, O.I.; Tolstikov, G.A. Synthesis and study of mutagenic properties of lupane triterpenoids containing 1,2,3-triazole fragments in the C-30 position. Chem. Nat. Compd. 2013, 49, 657–664. [Google Scholar] [CrossRef]
- Bębenek, E.; Jastrzębska, M.; Kadela-Tomanek, M.; Chrobak, E.; Orzechowska, B.; Zwolińska, K.; Latocha, M.; Mertas, A.; Czuba, Z.P.; Boryczka, S. Novel Triazole Hybrids of Betulin: Synthesis and Biological Activity Profile. Molecules 2017, 22, 1876. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.; Tang, N.; Yan, W. Synthesis and cytotoxicity of triterpenoids derived from betulin and betulinic acid via click chemistry. J. Asian Nat. Prod. Res. 2015, 17, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Krawczyk, M.; Pastuch-Gawołek, G.; Mrozek-Wilczkiewicz, A.; Kuczak, M.; Skonieczna, M.; Musiol, R. Synthesis of 8-hydroxyquinoline glycoconjugates and preliminary assay of their β1,4-GalT inhibitory and anti-cancer properties. Bioorg. Chem. 2019, 84, 326–338. [Google Scholar] [CrossRef] [PubMed]
- Krawczyk, M.; Pastuch-Gawołek, G.; Pluta, A.; Erfurt, K.; Domiński, A.; Kurcok, P. 8-Hydroxyquinoline Glycoconjugates: Modifications in the Linker Structure and Their Effect on the Cytotoxicity of the Obtained Compounds. Molecules 2019, 24, 4181. [Google Scholar] [CrossRef]
- Krawczyk, M.; Pastuch-Gawołek, G.; Hadasik, A.; Erfurt, K. 8-Hydroxyquinoline Glycoconjugates Containing Sulfur at the Sugar Anomeric Position—Synthesis and Preliminary Evaluation of Their Cytotoxicity. Molecules 2020, 25, 4174. [Google Scholar] [CrossRef]
- Domiński, A.; Krawczyk, M.; Konieczny, T.; Kasprów, M.; Foryś, A.; Pastuch-Gawołek, G.; Kurcok, P. Biodegradable pH-responsive micelles loaded with 8-hydroxyquinoline glycoconjugates for Warburg effect based tumor targeting. Eur. J. Pharm. Biopharm. 2020, 154, 317–329. [Google Scholar] [CrossRef]
- Křen, V.; Martinkova, L. Glycosides in Medicine: “The Role of Glycosidic Residue in Biological Activity”. Curr. Med. Chem. 2001, 8, 1303–1328. [Google Scholar] [CrossRef]
- A Smith, T. Facilitative glucose transporter expression in human cancer tissue. Br. J. Biomed. Sci. 1999, 56, 285–292. [Google Scholar] [PubMed]
- Heiden, M.G.V.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Calvaresi, E.C.; Hergenrother, P.J. Glucose conjugation for the specific targeting and treatment of cancer. Chem. Sci. 2013, 4, 2319–2333. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.-Q. Functional Properties and Genomics of Glucose Transporters. Curr. Genom. 2007, 8, 113–128. [Google Scholar] [CrossRef]
- Barnett, J.E.G.; Holman, G.D.; Munday, K.A. Structural requirements for binding to the sugar-transport system of the human erythrocyte. Biochem. J. 1973, 131, 211–221. [Google Scholar] [CrossRef]
- Ma, J.; Liu, H.; Xi, Z.; Hou, J.; Li, Y.; Niu, J.; Liu, T.; Bi, S.; Wang, X.; Wang, C.; et al. Protected and De-protected Platinum(IV) Glycoconjugates With GLUT1 and OCT2-Mediated Selective Cancer Targeting: Demonstrated Enhanced Transporter-Mediated Cytotoxic Properties in vitro and in vivo. Front. Chem. 2018, 6, 386. [Google Scholar] [CrossRef]
- Khan, I.; Guru, S.K.; Rath, S.K.; Chinthakindi, P.K.; Singh, B.; Koul, S.; Bhushan, S.; Sangwan, P.L. A novel triazole derivative of betulinic acid induces extrinsic and intrinsic apoptosis in human leukemia HL-60 cells. Eur. J. Med. Chem. 2016, 108, 104–116. [Google Scholar] [CrossRef]
- Komissarova, N.G.; Dubovitskii, S.N.; Shitikova, O.V.; Vyrypaev, E.M.; Spirikhin, L.V.; Eropkina, E.M.; Lobova, T.G.; Eropkin, M.; Yunusov, M.S. Synthesis of Conjugates of Lupane-Type Pentacyclic Triterpenoids with 2-Aminoethane- and N-Methyl-2-Aminoethanesulfonic Acids. Assessment of in vitro Toxicity. Chem. Nat. Compd. 2017, 53, 907–914. [Google Scholar] [CrossRef]
- Yang, Q.-R.; Qiao, W.-H.; Zhang, S.-M.; Qu, J.-P.; Liu, D.-L. Synthesis and Characterization of a New Cationic Galactolipid with Carbamate for Gene Delivery. Tenside Surfactants Deterg. 2010, 47, 294–299. [Google Scholar] [CrossRef]
- Rajaram, H.; Palanivelu, M.K.; Arumugam, T.V.; Rao, V.M.; Shaw, P.N.; McGeary, R.P.; Ross, B.P. ‘Click’ assembly of glycoclusters and discovery of a trehalose analogue that retards Aβ40 aggregation and inhibits Aβ40-induced neurotoxicity. Bioorg. Med. Chem. Lett. 2014, 24, 4523–4528. [Google Scholar] [CrossRef]
- Lancuški, A.; Bossard, F.; Fort, S. Carbohydrate-Decorated PCL Fibers for Specific Protein Adhesion. Biomacromolecules 2013, 14, 1877–1884. [Google Scholar] [CrossRef] [PubMed]
- Zemplén, G.; Pacsu, E. Über die Verseifung acetylierter Zucker und verwandter Substanzen. Berichte Dtsch. Chem. Ges. (A B Ser.) 1929, 62, 1613–1614. [Google Scholar] [CrossRef]
- Brown, R.S.; Wahl, R.L. Overexpression of Glut-1 glucose transporter in human breast cancer. An im-munohistochemical study. Cancer 1993, 72, 2979–2985. [Google Scholar] [CrossRef]
- Haber, R.S.; Rathan, A.; Weiser, K.R.; Pritsker, A.; Itzkowitz, S.H.; Bodian, C.; Slater, G.; Weiss, A.; Burstein, D.E. GLUT1 glucose transporter expression in colorectal carcinoma: A marker for poor progno-sis. Cancer 1998, 83, 34–40. [Google Scholar] [CrossRef]
- Kumamoto, K.; Goto, Y.; Sekikawa, K.; Takenoshita, S.; Ishida, N.; Kawakita, M.; Kannagi, R. Increased expression of UDP-galactose transporter messenger RNA in human colon cancer tissues and its implica-tion in synthesis of Thomsen-Friedenreich antigen and sialyl Lewis A/X determinants. Cancer Res. 2001, 61, 4620–4627. [Google Scholar]
- Lutter, A.-H.; Scholka, J.; Richter, H.; Anderer, U. Applying XTT, WST-1, and WST-8 to human chondrocytes: A comparison of membrane-impermeable tetrazolium salts in 2D and 3D cultures. Clin. Hemorheol. Microcirc. 2017, 67, 327–342. [Google Scholar] [CrossRef]
- Van De Waterbeemd, H.; Smith, D.A.; Jones, B.C. Lipophilicity in PK design: Methyl, ethyl, futile. J. Comput. Mol. Des. 2001, 15, 273–286. [Google Scholar] [CrossRef]
- Lombardo, F.; Obach, R.; Shalaeva, M.Y.; Gao, F. Prediction of Volume of Distribution Values in Hu-mans for Neutral and Basic Drugs Using Physicochemical Measurements and Plasma Protein Binding Data. J. Med. Chem. 2002, 45, 2867–2876. [Google Scholar] [CrossRef]
- Deng, D.; Xu, C.; Sun, P.; Wu, J.; Yan, C.; Hu, M.; Yan, N. Crystal structure of the human glucose transporter GLUT1. Nat. Cell Biol. 2014, 510, 121–125. [Google Scholar] [CrossRef]
- Fernández, C.; Nieto, O.; Rivas, E.; Montenegro, G.; Fontenla, J.A.; Fernández-Mayoralas, A. Synthesis and biological studies of glycosyl dopamine derivatives as potential antiparkinsonian agents. Carbohydr. Res. 2000, 327, 353–365. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Substrate | Product | ||||||
|---|---|---|---|---|---|---|---|---|
| No. | Solvent | Temp., °C | Time, h | No. | R (C3) | R1 (C28) | Yield, % | |
| 1 | 1 | THF | r.t. | 24 | 2 | OH | OCH2C≡CH | 54 |
| 3 | OCH2C≡CH | OCH2C≡CH | 31 | |||||
| 2 | 1 | THF | r.t. | 24 | 4 | OH | O(CO)CH2Cl | 34 |
| 3 | 1 | THF | r.t. | 24 | 5 | O(CO)CH2Cl | O(CO)CH2Cl | 95 |
| 4 | 4 | DMF | 90 | 3 | 6 | OH | O(CO)CH2N3 | 64 |
| 5 | 5 | DMF | 90 | 3 | 7 | O(CO)CH2N3 | O(CO)CH2N3 | 72 |
| Entry | Substrate | Product | |||||
|---|---|---|---|---|---|---|---|
| Analog BN | No. | Sugar | R1, R2, R3 | No. | Linker | Yield, % | |
| STRATEGY I | |||||||
| 1 | 2 | 12a | Glc | OAc | 15a | 28-OCH2Triaz | 99 |
| 2 | 2 | 12b | Gal | OAc | 15b | 28-OCH2Triaz | 97 |
| 3 | 2 | 14a | Glc | OAc | 16a | 28-OCH2TriazCH2(CO)NH | 98 |
| 4 | 2 | 14b | Gal | OAc | 16b | 28-OCH2TriazCH2(CO)NH | 97 |
| 5 | 3 | 12a | Glc | OAc | 17a | 3,28-di-OCH2Triaz | 99 |
| 6 | 3 | 12b | Gal | OAc | 17b | 3,28-di-OCH2Triaz | 99 |
| 7 | 3 | 14b | Gal | OAc | 18b | 3,28-di-OCH2TriazCH2(CO)NH | 82 |
| STRATEGY II | |||||||
| 8 | 6 | 10a | Glc | OAc | 19a | 28-O(CO)CH2TriazCH2O | 97 |
| 9 | 7 | 10a | Glc | OAc | 20a | 3,28-di-O(CO)CH2TriazCH2O | 87 |
| 10 | 7 | − | − | − | 21 | 3,28-di-O(CO)CH2TriazOH | 77 |
| DEPROTECTION | |||||||
| 11 | 15a | − | Glc | OH | 22a | 28-OCH2Triaz | 87 |
| 12 | 15b | − | Gal | OH | 22b | 28-OCH2Triaz | 99 |
| 13 | 16b | − | Gal | OH | 23b | 28-OCH2TriazCH2(CO)NH | 96 |
| 14 | 17a | − | Glc | OH | 24a | 3,28-di-OCH2Triaz | 87 |
| 15 | 17b | − | Gal | OH | 24b | 3,28-di-OCH2Triaz | 69 |
| Compound | Activity IC50 [µM] a,b | |
|---|---|---|
| MCF-7 | NHDF | |
| 1 | 12.40 ± 0.14 | 11.60 ± 0.22 |
| 15a | 65.28 ± 0.68 | 60.62 ± 0. 64 |
| 15b | 47.70 ± 0.72 | 47.61 ± 0.86 |
| 16a | 29.77 ± 0.52 | 29.82 ± 0.45 |
 | |||
|---|---|---|---|
| No. | R (C3) | R1 (C28) | logP |
| 1 | OH | OH | 7.16 |
| 2 | OH | OCH2C≡CH | 7.94 |
| 3 | OCH2C≡CH | OCH2C≡CH | 8.54 |
| 4 | OH | O(CO)CH2Cl | 8.23 |
| 5 | O(CO)CH2Cl | O(CO)CH2Cl | 8.86 |
| 6 | OH | O(CO)CH2N3 | 8.31 |
| 7 | O(CO)CH2N3 | O(CO)CH2N3 | 8.94 |
| 15a | OH | OCH2TriazGlc(OAc) | 7.59 |
| 16a | OH | OCH2TriazCH2(CO)NHGlc(OAc) | 6.69 |
| 17a | OCH2TriazGlc(OAc) | OCH2TriazGlc(OAc) | 8.01 |
| 18b | OCH2TriazCH2(CO)NHGal(OAc) | OCH2TriazCH2(CO)NHGal(OAc) | 6.22 |
| 19a | OH | O(CO)CH2TriazCH2OGlc(OAc) | 7.45 |
| 20a | O(CO)CH2TriazCH2OGlc(OAc) | O(CO)CH2TriazCH2OGlc(OAc) | 7.74 |
| 21 | O(CO)CH2TriazOH | O(CO)CH2TriazOH | 6.38 |
| 22a | OH | OCH2TriazGlc(OH) | 5.21 |
| 23b | OH | OCH2TriazCH2(CO)NHGal(OH) | 4.31 |
| 24a | OCH2TriazGlc(OH) | OCH2TriazGlc(OH) | 3.25 |
Sample Availability: Samples of the compounds 15–24 are available from the authors. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grymel, M.; Pastuch-Gawołek, G.; Lalik, A.; Zawojak, M.; Boczek, S.; Krawczyk, M.; Erfurt, K. Glycoconjugation of Betulin Derivatives Using Copper-Catalyzed 1,3-Dipolar Azido-Alkyne Cycloaddition Reaction and a Preliminary Assay of Cytotoxicity of the Obtained Compounds. Molecules 2020, 25, 6019. https://doi.org/10.3390/molecules25246019
Grymel M, Pastuch-Gawołek G, Lalik A, Zawojak M, Boczek S, Krawczyk M, Erfurt K. Glycoconjugation of Betulin Derivatives Using Copper-Catalyzed 1,3-Dipolar Azido-Alkyne Cycloaddition Reaction and a Preliminary Assay of Cytotoxicity of the Obtained Compounds. Molecules. 2020; 25(24):6019. https://doi.org/10.3390/molecules25246019
Chicago/Turabian StyleGrymel, Mirosława, Gabriela Pastuch-Gawołek, Anna Lalik, Mateusz Zawojak, Seweryn Boczek, Monika Krawczyk, and Karol Erfurt. 2020. "Glycoconjugation of Betulin Derivatives Using Copper-Catalyzed 1,3-Dipolar Azido-Alkyne Cycloaddition Reaction and a Preliminary Assay of Cytotoxicity of the Obtained Compounds" Molecules 25, no. 24: 6019. https://doi.org/10.3390/molecules25246019
APA StyleGrymel, M., Pastuch-Gawołek, G., Lalik, A., Zawojak, M., Boczek, S., Krawczyk, M., & Erfurt, K. (2020). Glycoconjugation of Betulin Derivatives Using Copper-Catalyzed 1,3-Dipolar Azido-Alkyne Cycloaddition Reaction and a Preliminary Assay of Cytotoxicity of the Obtained Compounds. Molecules, 25(24), 6019. https://doi.org/10.3390/molecules25246019