The Intersection between Oral Microbiota, Host Gene Methylation and Patient Outcomes in Head and Neck Squamous Cell Carcinoma

 , , , ,
, , , ,  , , , and
, , , and  add
Show full author list
add
Show full author list
Abstract
:Simple Summary
Abstract

1. Background
2. Methods
2.1. Ethical Approval
2.2. HNSCC Tissue Collection and Processing
2.3. Collection of Oral Rinse Samples
2.4. DNA Extraction of Tissues and Oral Rinses
2.5. HPV Genotyping
2.6. 16S rRNA Gene PCR Amplification and Sequencing
2.7. 16S Sequence Data Processing and Community Composition Statistical Analyses
2.8. Microbial Rank Correlation and Identifying Indicator Markers
2.9. Functional Prediction Based on 16S rRNA Gene Community Composition
2.10. Fusobacterium nucleatum in Situ Hybridization (ISH) Analysis
2.11. Quantitative PCR (qPCR) for Fusobacterium nucleatum
2.12. Human DNA CpG Methylation from HNSCC Tissue
3. Results
3.1. Study Subjects
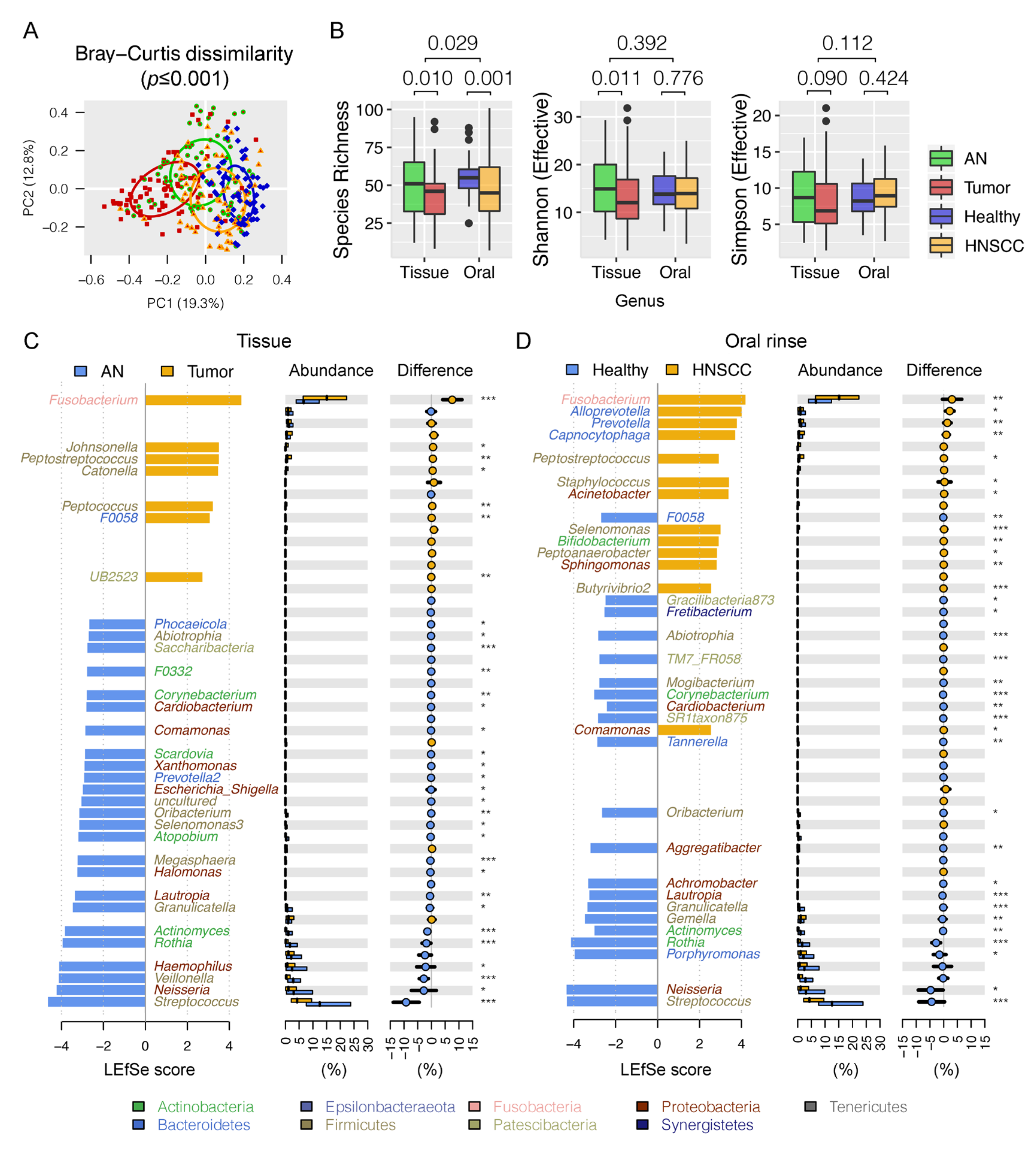
3.2. Differential Oral Microbial Community in HNSCCs Compared to Controls
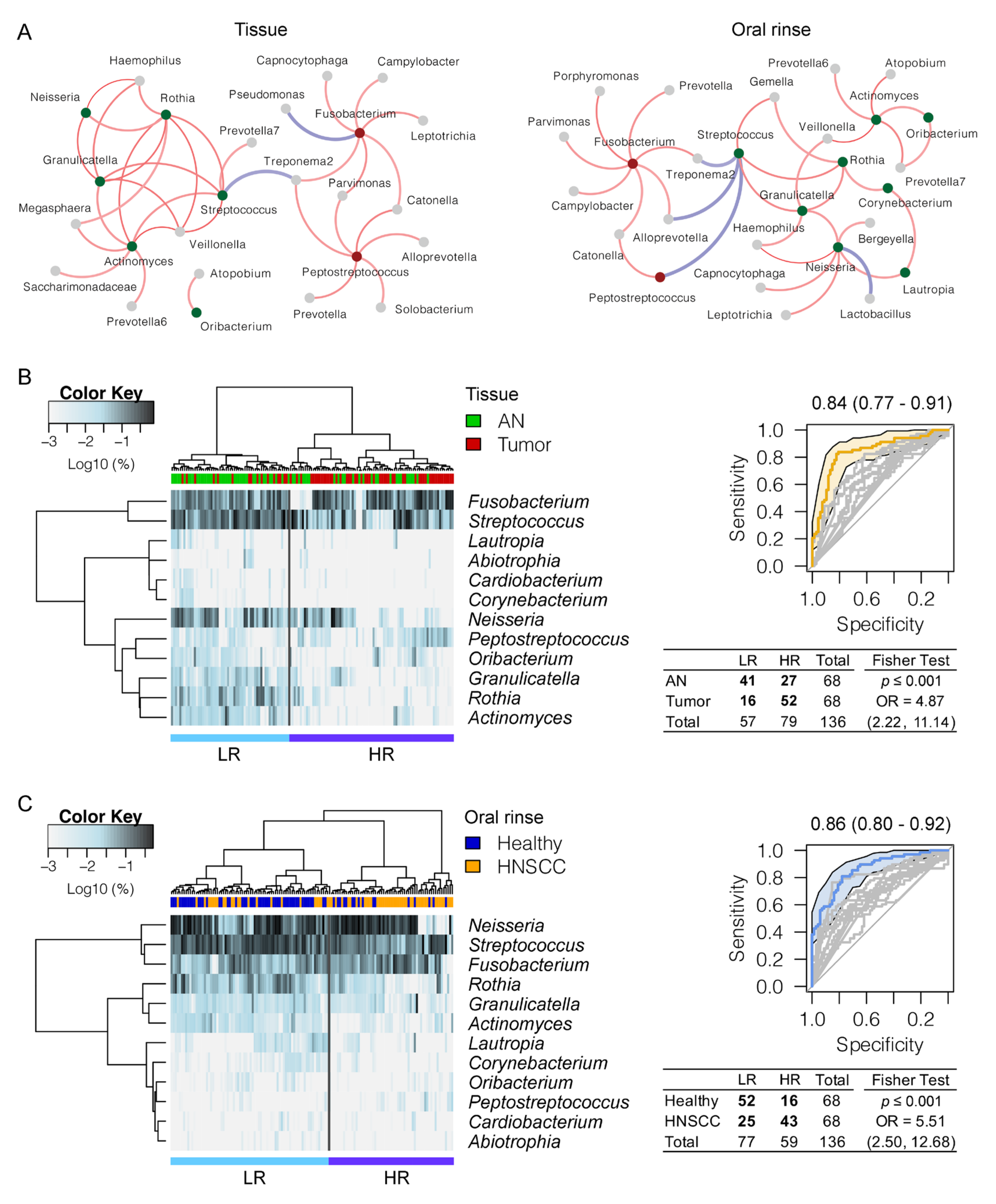
3.3. Panel of the Bacterial Microbiome Useful in Differentiating HNSCC from Controls
3.4. Pathways Related to Bacterial Adaptation and Infectious Disease Enriched in HNSCC
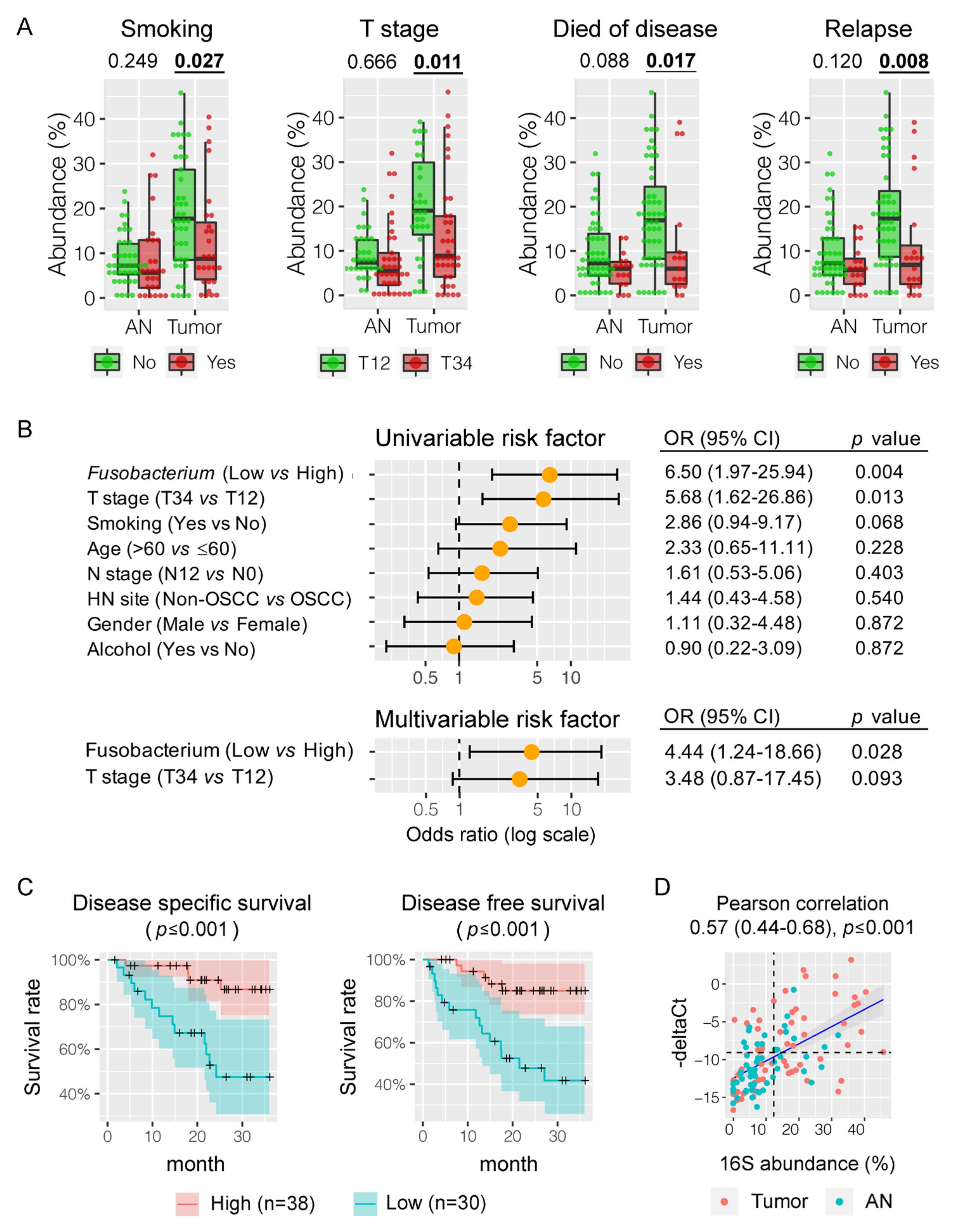
3.5. Fusobacterium nucleatum is Associated with HNSCC Patient Outcomes
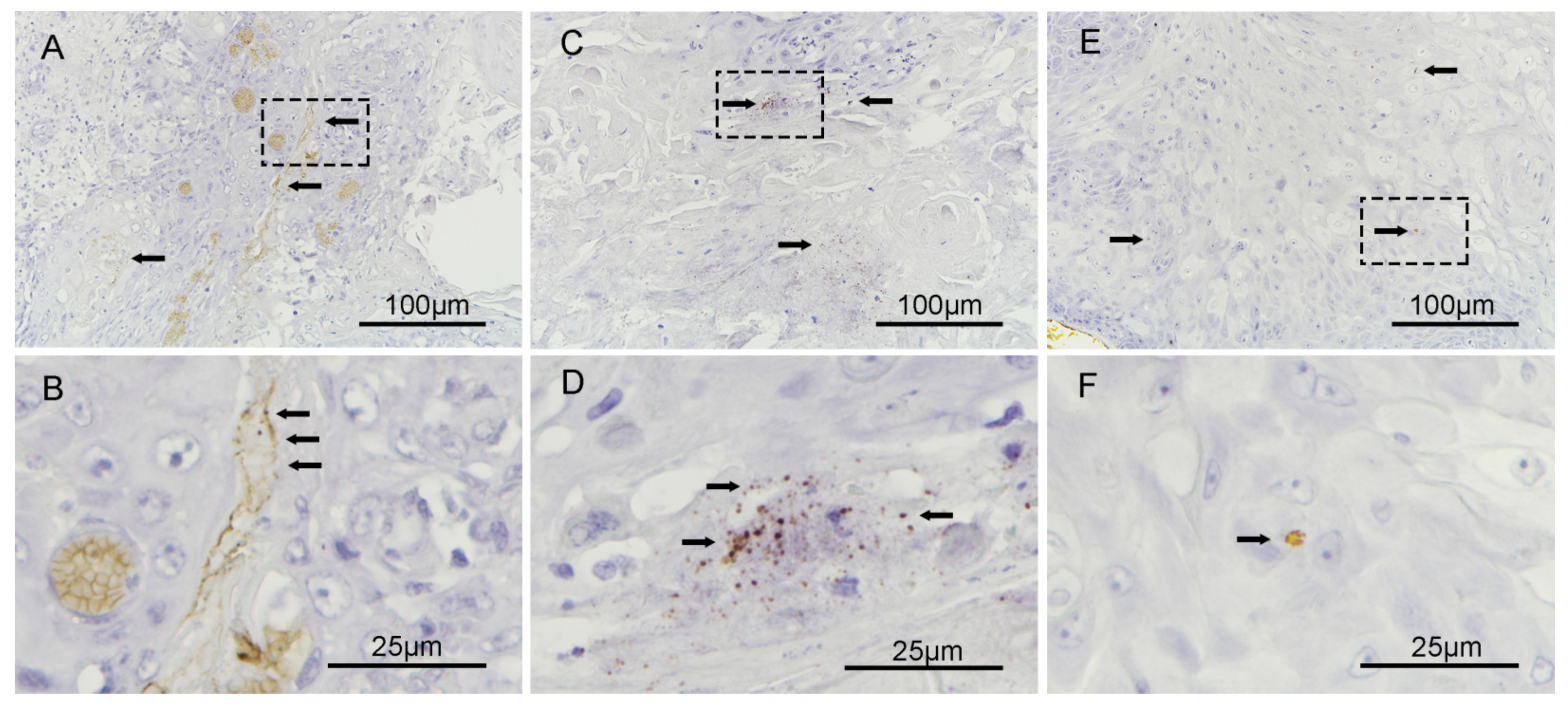
3.6. Fusobacterium nucleatum Localizes to the Intracellular Compartments of Tumor Cells
3.7. Enrichment of Fusobacterium is Associated with Host Gene Promoter Methylation in HNSCC
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| HNSCC | Head and neck squamous cell carcinoma |
| AUC | Area under the receiver operating curve |
| HPV | Human papillomavirus |
| AN | Adjacent normal |
| ORF | Open reading frame |
| OUT | Operational taxonomic unit |
| ASV | amplicon sequence variant |
| PERMANOVA | Permutational multivariate analysis of variance |
| MWU | Mann–Whitney Wilcoxon rank sum test |
| WSR | Wilcoxon signed rank test |
| KW | Kruskal–Wallis test |
| Tukey HSD | Tukey’s honest significant difference |
| ROC | Receiver operating characteristic |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| KO | Kyoto Encyclopedia of Genes and Genomes Orthology |
| FFPE | Formalin-fixed paraffin-embedded |
| DMRs | Differentially methylated regions |
| TCGA | The cancer genome atlas |
| HR | High risk |
| PCoA | Principal coordinate analysis |
| LefSe | Linear discriminant analysis effect size |
| LDA | Linear discriminant analysis |
| PICRUSt2 | Phylogenetic Investigation of Communities by Reconstruction of Unobserved States 2 |
| DSS | Disease specific survival |
| DFS | Disease free survival |
| ISH | In situ hybridization |
| Glm nb | generalized linear regression model test |
| TSG | tumor suppressor gene |
| LXN | Latexin |
| qPCR | Quantitative PCR |
| F-Low | Fusobacterium-low |
| F-High | Fusobacterium-high |
References
- Hsiao, J.-R.; Chang, C.-C.; Lee, W.-T.; Huang, C.-C.; Ou, C.-Y.; Tsai, S.-T.; Chen, K.-C.; Huang, J.-S.; Wong, T.-Y.; Lai, Y.-H.; et al. The interplay between oral microbiome, lifestyle factors and genetic polymorphisms in the risk of oral squamous cell carcinoma. Carcinogens 2018, 39, 778–787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, A. Radiation therapy for oral cavity and oropharyngeal cancers. Dent. Clin. North. Am. 2018, 62, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Isayeva, T.; Li, Y.; Maswahu, D.; Brandwein-Gensler, M. Human Papillomavirus in non-oropharyngeal head and neck cancers: A systematic literature review. Head Neck Pathol. 2012, 6, 104–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahn, J.; Sinha, R.; Pei, Z.; Dominianni, C.; Wu, J.; Shi, J.; Goedert, J.J.; Hayes, R.B.; Yang, L. Human gut microbiome and risk for colorectal cancer. J. Natl. Cancer Inst. 2013, 105, 1907–1911. [Google Scholar] [CrossRef] [Green Version]
- Guevarra, L.A., Jr.; Afable, A.C.F.; Belza, P.J.O.; Dy, K.J.S.; Lee, S.J.Q.; Sy-Ortin, T.T.; Albano, P.M.S.P. Immunogenicity of a Fap2 peptide mimotope of Fusobacterium nucleatum and its potential use in the diagnosis of colorectal cancer. Infect. Agents Cancer 2018, 13, 1–6. [Google Scholar] [CrossRef]
- Forman, D.; Newell, D.G.; Fullerton, F.; Yarnell, J.W.; Stacey, A.R.; Wald, N.; Sitas, F. Association between infection with Helicobacter pylori and risk of gastric cancer: Evidence from a prospective investigation. BMJ 1991, 302, 1302–1305. [Google Scholar] [CrossRef] [Green Version]
- Hansson, L.R.; Engstrand, L.; Nyren, O.; Lindgren, A. Prevalence of helicobacter pylori infection in subtypes of gastric cancer. Gastroenterology 1995, 109, 885–888. [Google Scholar] [CrossRef]
- Mima, K.; Nishihara, R.; Qian, Z.R.; Cao, Y.; Sukawa, Y.; Nowak, J.A.; Yang, J.; Dou, R.; Masugi, Y.; Song, M.; et al. Fusobacterium nucleatum in colorectal carcinoma tissue and patient prognosis. Gut 2016, 65, 1973–1980. [Google Scholar] [CrossRef] [Green Version]
- Tahara, T.; Hirata, I.; Nakano, N.; Tahara, S.; Horiguchi, N.; Kawamura, T.; Okubo, M.; Ishizuka, T.; Yamada, H.; Yoshida, D.; et al. Potential link between Fusobacterium enrichment and DNA methylation accumulation in the inflammatory colonic mucosa in ulcerative colitis. Oncotarget 2017, 8, 61917–61926. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Ye, J.; Fang, D.; Lv, L.; Wu, W.; Shi, D.; Li, Y.; Yang, L.; Bian, X.; Wu, J.; et al. Multi-omic profiling reveals associations between the gut mucosal microbiome, the metabolome, and host DNA methylation associated gene expression in patients with colorectal cancer. BMC Microbiol. 2020, 20, 1–13. [Google Scholar] [CrossRef]
- Peterson, S.N.; Snesrud, E.; Liu, J.; Ong, A.C.; Kilian, M.; Schork, N.J.; Bretz, W. The dental plaque microbiome in health and disease. PLoS ONE 2013, 8, e58487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tezal, M.; Sullivan, M.A.; Hyland, A.; Marshall, J.R.; Stoler, D.; Reid, M.E.; Loree, T.R.; Rigual, N.R.; Merzianu, M.; Hauck, L.; et al. Chronic periodontitis and the incidence of head and neck squamous cell carcinoma. Cancer Epidemiol. Biomark. Prev. 2009, 18, 2406–2412. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Jia, J.W.H. Metagenome-wide association studies: Fine-mining the microbiome. Nat. Rev. Genet. 2016, 14, 508–522. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Ganly, I. The oral microbiome and oral cancer. Clin. Lab. Med. 2014, 34, 711–719. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Yang, J.; Feng, Q.; Chen, B.; Li, M.; Liang, C.; Li, M.; Li, Z.; Xu, Q.; Zhang, L.; et al. Compositional and functional analysis of the microbiome in tissue and saliva of oral squamous cell carcinoma. Front. Microbiol. 2019, 10, 1439. [Google Scholar] [CrossRef]
- Yang, C.-Y.; Yeh, Y.-M.; Yu, H.-Y.; Chin, C.-Y.; Hsu, C.-W.; Liu, H.; Huang, P.-J.; Hu, S.-N.; Liao, C.-T.; Chang, K.-P.; et al. Oral microbiota community dynamics associated with oral squamous cell carcinoma staging. Front. Microbiol. 2018, 9, 862. [Google Scholar] [CrossRef] [Green Version]
- Ganly, I.; Yang, L.; Giese, R.A.; Hao, Y.; Nossa, C.W.; Morris, L.G.T.; Rosenthal, M.; Migliacci, J.; Kelly, D.; Tseng, W.; et al. Periodontal pathogens are a risk factor of oral cavity squamous cell carcinoma, independent of tobacco and alcohol and human papillomavirus. Int. J. Cancer 2019, 145, 775–784. [Google Scholar] [CrossRef]
- Guerrero-Preston, R.; White, J.R.; Godoy-Vitorino, F.; Rodríguez-Hilario, A.; Navarro, K.; González, H.; Michailidi, C.; Jedlicka, A.; Canapp, S.; Bondy, J.; et al. High-resolution microbiome profiling uncovers Fusobacterium nucleatum, Lactobacillus gasseri/johnsonii, and Lactobacillus vaginalis associated to oral and oropharyngeal cancer in saliva from HPV positive and HPV negative patients treated with surgery and chemo-radiation. Oncotarget 2017, 8, 110931–110948. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Funchain, P.; Bebek, G.; Altemus, J.; Zhang, H.; Niazi, F.; Peterson, C.; Lee, W.T.; Burkey, B.B.; Eng, C. Microbiomic differences in tumor and paired-normal tissue in head and neck squamous cell carcinomas. Genome Med. 2017, 9, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Guerrero-Preston, R.; Godoy-Vitorino, F.; Jedlicka, A.; Rodríguez-Hilario, A.; González, H.; Bondy, J.; Lawson, F.; Folawiyo, O.; Michailidi, C.; Dziedzic, A.; et al. 16S rRNA amplicon sequencing identifies microbiota associated with oral cancer, human papilloma virus infection and surgical treatment. Oncotarget 2016, 7, 51320–51334. [Google Scholar] [CrossRef] [Green Version]
- Carvalho, A.L.; Jerónimo, C.; Kim, M.M.; Henrique, R.; Zhang, Z.; Hoque, M.O.; Chang, S.; Brait, M.; Nayak, C.S.; Jiang, W.-W.; et al. Evaluation of promoter hypermethylation detection in body fluids as a screening/diagnosis tool for head and neck squamous cell carcinoma. Clin. Cancer Res. 2008, 14, 97–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosas, S.L.; Koch, W.; Carvalho, M.G.D.C.; Wu, L.; Califano, J.; Westra, W.; Jen, J.; Sidransky, D. Promoter hypermethylation patterns of p16, O6-methylguanine-DNA-methyltransferase, and death-associated protein kinase in tumors and saliva of head and neck cancer patients. Cancer Res. 2001, 61, 939–942. [Google Scholar] [PubMed]
- Wong, M.C.S.; Vlantis, A.C.; Liang, M.; Wong, P.Y.; Ho, W.C.S.; Boon, S.S.; Sze, R.K.H.; Leung, C.; Chan, P.K.S.; Chen, Z. Prevalence and epidemiologic profile of oral infection with alpha, beta, and gamma Papillomaviruses in an Asian Chinese population. J. Infect. Dis. 2018, 218, 388–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef]
- Chen, Z.; Hui, P.C.; Hui, M.; Yeoh, Y.K.; Wong, P.Y.; Chan, M.C.W.; Wong, M.C.S.; Ng, S.C.; Chan, F.K.L.; Chan, P.K.S. Impact of preservation method and 16S rRNA hypervariable region on gut microbiota profiling. mSystems 2019, 4, e00271-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilbert, J.A.; Jansson, J.K.; Knight, R. The Earth microbiome project: Successes and aspirations. BMC Biol. 2014, 12, 1–4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Matsen, F.A.; Kodner, R.B.; Armbrust, E.V. pplacer: Linear time maximum-likelihood and Bayesian phylogenetic placement of sequences onto a fixed reference tree. BMC Bioinform. 2010, 11, 538. [Google Scholar] [CrossRef] [Green Version]
- Friedman, J.; Alm, E.J. Inferring correlation networks from genomic survey data. PLoS Comput. Biol. 2012, 8, e1002687. [Google Scholar] [CrossRef] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Rohart, F.; Eslami, A.; Matigian, N.; Bougeard, S.; Cao, K.-A.L. MINT: A multivariate integrative method to identify reproducible molecular signatures across independent experiments and platforms. BMC Bioinform. 2017, 18, 128. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 002832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castellarin, M.; Warren, R.L.; Freeman, J.D.; Dreolini, L.; Krzywinski, M.; Strauss, J.; Barnes, R.; Watson, P.; Allen-Vercoe, E.; Moore, R.A.; et al. Fusobacterium nucleatum infection is prevalent in human colorectal carcinoma. Genome Res. 2012, 22, 299–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krueger, F.; Andrews, S.R. Bismark: A flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 2011, 27, 1571–1572. [Google Scholar] [CrossRef] [PubMed]
- Akalin, A.; Kormaksson, M.; Li, S.; Garrett-Bakelman, F.E.; Figueroa, M.E.M.; Melnick, A.; Mason, C.E. methylKit: A comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol. 2012, 13, R87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seed, R.I.; Taurozzi, A.J.; Wilcock, D.J.; Nappo, G.; Erb, H.H.H.; Read, M.L.; Gurney, M.; Archer, L.K.; Ito, S.; Rumsby, M.G.; et al. The putative tumour suppressor protein Latexin is secreted by prostate luminal cells and is downregulated in malignancy. Sci. Rep. 2019, 9, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Osisami, M.; Dai, J.; Keller, J.M.; Escara-Wilke, J.; Mizokami, A.; Keller, E.T. Bone microenvironment changes in latexin expression promote chemoresistance. Mol. Cancer Res. 2017, 15, 457–466. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Sun, C.; Tan, Y.; Li, L.; Zhang, H.; Liang, Y.; Zeng, J.; Zou, H. Transcription levels and prognostic significance of the NFI family members in human cancers. PeerJ 2020, 8, e8816. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, C.; Li, Z.; Wang, C.; Jia, J.; Gao, T.; Hildebrandt, G.; Zhou, D.; Bondada, S.; Ji, P.; et al. Latexin inactivation enhances survival and long-term engraftment of hematopoietic stem cells and expands the entire hematopoietic system in mice. Stem Cell Rep. 2017, 8, 991–1004. [Google Scholar] [CrossRef]
- Shaw, R.; Beasley, N. Aetiology and risk factors for head and neck cancer: United Kingdom National multidisciplinary guidelines. J. Laryngol. Otol. 2016, 130, S9–S12. [Google Scholar] [CrossRef] [PubMed]
- Mitsunaga, K.; Kikuchi, J.; Wada, T.; Furukawa, Y. Latexin regulates the abundance of multiple cellular proteins in hematopoietic stem cells. J. Cell. Physiol. 2011, 227, 1138–1147. [Google Scholar] [CrossRef]
- You, Y.; Wen, R.; Pathak, R.D.; Li, A.; Li, W.; Clair, D.S.; Hauerjensen, M.; Zhou, D.; Liang, Y. Latexin sensitizes leukemogenic cells to gamma-irradiation-induced cell-cycle arrest and cell death through Rps3 pathway. Cell Death Dis. 2014, 5, e1493. [Google Scholar] [CrossRef] [PubMed]
- Geng, F.; Zhang, Y.; Lu, Z.; Zhang, S.; Pan, Y. Fusobacterium nucleatum Caused DNA damage and promoted cell proliferation by the Ku70/p53 pathway in oral cancer cells. DNA Cell Biol. 2020, 39, 144–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubinstein, M.R.; Wang, X.; Liu, W.; Hao, Y.; Cai, G.; Han, Y.W. Fusobacterium nucleatum promotes colorectal carcinogenesis by modulating E-cadherin/β-catenin signaling via its FadA adhesin. Cell Host Microbe 2013, 14, 195–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, Z.; Zhou, Y.; Wang, C.; Zheng, J.; Zhang, P.; Zhou, L.; Wu, L.; Shan, Y.; Ye, M.; He, Y.; et al. Latexin exhibits tumor-suppressor potential in pancreatic ductal adenocarcinoma. Oncol. Rep. 2015, 35, 50–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehrenhöfer-Wölfer, K.; Puchner, T.; Schwarz, C.; Rippka, J.; Blaha-Ostermann, S.; Strobl, U.; Hörmann, A.; Bader, G.; Kornigg, S.; Zahn, S.; et al. SMARCA2-deficiency confers sensitivity to targeted inhibition of SMARCA4 in esophageal squamous cell carcinoma cell lines. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; He, K.; Zhang, Y.; Song, J.; Shi, Z.; Chen, W.; Shao, Y. Inactivation of SMARCA2 by promoter hypermethylation drives lung cancer development. Gene 2019, 687, 193–199. [Google Scholar] [CrossRef]
- Glaros, S.; Cirrincione, G.M.; Muchardt, C.; Kleer, C.G.; Michael, C.W.; Reisman, D. The reversible epigenetic silencing of BRM: Implications for clinical targeted therapy. Oncogene 2007, 26, 7058–7066. [Google Scholar] [CrossRef] [Green Version]
- Salem, N.; Kamal, I.; Al-Maghrabi, J.; Abuzenadah, A.M.; Peer-Zada, A.A.; Qari, Y.; Al-Ahwal, M.S.; Al-Qahtani, M.; Buhmeida, A. High expression of matrix metalloproteinases: MMP-2 and MMP-9 predicts poor survival outcome in colorectal carcinoma. Futur. Oncol. 2016, 12, 323–331. [Google Scholar] [CrossRef]
- Lee, H.-Y.; Yeh, B.-W.; Chan, T.-C.; Yang, K.-F.; Li, W.-M.; Huang, C.-N.; Ke, H.-L.; Li, C.-C.; Yeh, H.-C.; Liang, P.-I.; et al. Sulfatase-1 overexpression indicates poor prognosis in urothelial carcinoma of the urinary bladder and upper tract. Oncotarget 2017, 8, 47216–47229. [Google Scholar] [CrossRef] [PubMed]
- Walker, C.; Mojares, E.; Hernández, A.D.R. Role of extracellular matrix in development and cancer progression. Int. J. Mol. Sci. 2018, 19, 3028. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| AN | Tumor | Healthy | HNSCC | Tumor Tissue Compared to AN | HNSCC Oral Rinse Compared to Healthy Subjects | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Taxonomy (Phylum; Genus) | Abundance (Mean ± smd) | Abundance (Mean ± smd) | Abundance (Mean ± smd) | Abundance (Mean ± smd) | Difference in Abundance | p Value (mwu Test) | LEfSe assign | LEfSe p adj | AUC Value | Difference in Abundance | p value (mwu test) | LEfSe Assign | LEfSe p adj | AUC Value |
| Fusobacteria; Fusobacterium | 8.60 ± 0.87 | 16.25 ± 1.51 | 4.60 ± 0.36 | 7.69 ± 0.72 | 7.65 (4.14,11.15) | 0.0000 | Tumor | 0.0002 | 0.69 (0.60, 0.78) | 3.09 (−0.42, 6.60) | 0.0027 | HNSCC | 0.0025 | 0.65 (0.55, 0.74) |
| Firmicutes; Peptostreptococcus | 0.79 ± 0.19 | 1.33 ± 0.19 | 0.06 ± 0.01 | 0.22 ± 0.06 | 0.54 (0.03, 1.05) | 0.0042 | Tumor | 0.0095 | 0.63 (0.53, 0.72) | 0.16 (−0.35, 0.67) | 0.0132 | HNSCC | 0.0130 | 0.62 (0.52, 0.72) |
| Firmicutes; Streptococcus | 17.16 ± 1.71 | 8.00 ± 1.33 | 15.00 ± 0.92 | 10.64 ± 1.02 | −9.17 (−13.86, −4.48) | 0.0000 | AN | 0.0000 | 0.75 (0.67, 0.83) | −4.36 (−9.05, 0.33) | 0.0001 | Healthy | 0.0001 | 0.70 (0.61, 0.79) |
| Proteobacteria; Neisseria | 6.80 ± 1.12 | 4.07 ± 0.96 | 18.82 ± 1.51 | 14.10 ± 1.28 | −2.73 (−7.25, 1.79) | 0.0076 | AN | 0.0400 | 0.60 (0.51, 0.70) | −4.72 (−9.24, −0.21) | 0.0222 | Healthy | 0.0228 | 0.61 (0.52, 0.71) |
| Actinobacteria; Rothia | 2.99 ± 0.48 | 1.21 ± 0.37 | 4.47 ± 0.57 | 1.77 ± 0.28 | −1.78 (−3.39, −0.18) | 0.0000 | AN | 0.0003 | 0.68 (0.59, 0.77) | −2.70 (−4.31, −1.10) | 0.0000 | Healthy | 0.0000 | 0.75 (0.67, 0.83) |
| Actinobacteria; Actinomyces | 1.99 ± 0.39 | 0.57 ± 0.16 | 0.87 ± 0.11 | 0.69 ± 0.13 | −1.42 (−2.24, −0.60) | 0.0000 | AN | 0.0000 | 0.73 (0.65, 0.82) | −0.18 (−1.00, 0.64) | 0.0100 | Healthy | 0.0097 | 0.63 (0.53, 0.72) |
| Firmicutes; Granulicatella | 1.42 ± 0.20 | 0.91 ± 0.25 | 1.20 ± 0.14 | 1.10 ± 0.40 | −0.52 (−1.48, 0.45) | 0.0126 | AN | 0.0127 | 0.62 (0.53, 0.72) | −0.10 (−1.07, 0.87) | 0.0002 | Healthy | 0.0002 | 0.68 (0.59, 0.77) |
| Proteobacteria; Lautropia | 0.47 ± 0.14 | 0.05 ± 0.02 | 0.64 ± 0.14 | 0.27 ± 0.07 | −0.42 (−0.80, −0.04) | 0.0008 | AN | 0.0096 | 0.60 (0.53, 0.67) | −0.37 (−0.75, 0.00) | 0.0000 | Healthy | 0.0000 | 0.71 (0.62, 0.80) |
| Proteobacteria; Cardiobacterium | 0.13 ± 0.04 | 0.05 ± 0.02 | 0.09 ± 0.01 | 0.09 ± 0.03 | −0.09 ( −0.18, 0.01) | 0.0192 | AN | 0.0410 | 0.58 (0.50, 0.65) | −0.00 (−0.10, 0.10) | 0.0020 | Healthy | 0.0020 | 0.65 (0.56, 0.73) |
| Actinobacteria; Corynebacterium | 0.15 ± 0.04 | 0.08 ± 0.05 | 0.27 ± 0.03 | 0.09 ± 0.03 | −0.07 (−0.21, 0.08) | 0.0008 | AN | 0.0091 | 0.60 (0.53, 0.68) | −0.18 (−0.32, −0.04) | 0.0000 | Healthy | 0.0000 | 0.80 (0.72, 0.87) |
| Firmicutes; Abiotrophia | 0.14 ± 0.08 | 0.08 ± 0.04 | 0.08 ± 0.02 | 0.02 ± 0.01 | −0.06 (−0.22, 0.11) | 0.1402 | AN | 0.0106 | 0.58 (0.52, 0.65) | −0.06 (−0.23, 0.10) | 0.0001 | Healthy | 0.0001 | 0.66 (0.59, 0.73) |
| Firmicutes; Oribacterium | 0.77 ± 0.16 | 0.72 ± 0.25 | 0.15 ± 0.02 | 0.11 ± 0.02 | −0.05 (−0.59, 0.49) | 0.0383 | AN | 0.0050 | 0.64 (0.54, 0.73) | −0.04 (−0.58, 0.49) | 0.0123 | Healthy | 0.0121 | 0.62 (0.53, 0.71) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Z.; Wong, P.Y.; Ng, C.W.K.; Lan, L.; Fung, S.; Li, J.W.; Cai, L.; Lei, P.; Mou, Q.; Wong, S.H.; et al. The Intersection between Oral Microbiota, Host Gene Methylation and Patient Outcomes in Head and Neck Squamous Cell Carcinoma. Cancers 2020, 12, 3425. https://doi.org/10.3390/cancers12113425
Chen Z, Wong PY, Ng CWK, Lan L, Fung S, Li JW, Cai L, Lei P, Mou Q, Wong SH, et al. The Intersection between Oral Microbiota, Host Gene Methylation and Patient Outcomes in Head and Neck Squamous Cell Carcinoma. Cancers. 2020; 12(11):3425. https://doi.org/10.3390/cancers12113425
Chicago/Turabian StyleChen, Zigui, Po Yee Wong, Cherrie W. K. Ng, Linlin Lan, Sherwood Fung, Jing W. Li, Liuyang Cai, Pu Lei, Qianqian Mou, Sunny H. Wong, and et al. 2020. "The Intersection between Oral Microbiota, Host Gene Methylation and Patient Outcomes in Head and Neck Squamous Cell Carcinoma" Cancers 12, no. 11: 3425. https://doi.org/10.3390/cancers12113425