The Effect of Glyceraldehyde-Derived Advanced Glycation End Products on β-Tubulin-Inhibited Neurite Outgrowth in SH-SY5Y Human Neuroblastoma Cells


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Cultures
2.2. MTT Assay
2.3. Preparation of Anti-TAGE Antibody
2.4. Slot Blotting Analysis for TAGE
2.5. Detection of TAGE Proteins by 2-Dimensional Gel Electrophoresis (2-DE) and Western Blotting
2.6. Protein Identification
2.7. Western Blot Analysis
2.8. Neurite Outgrowth
2.9. Immunocytochemistry
2.10. Statistics
3. Results
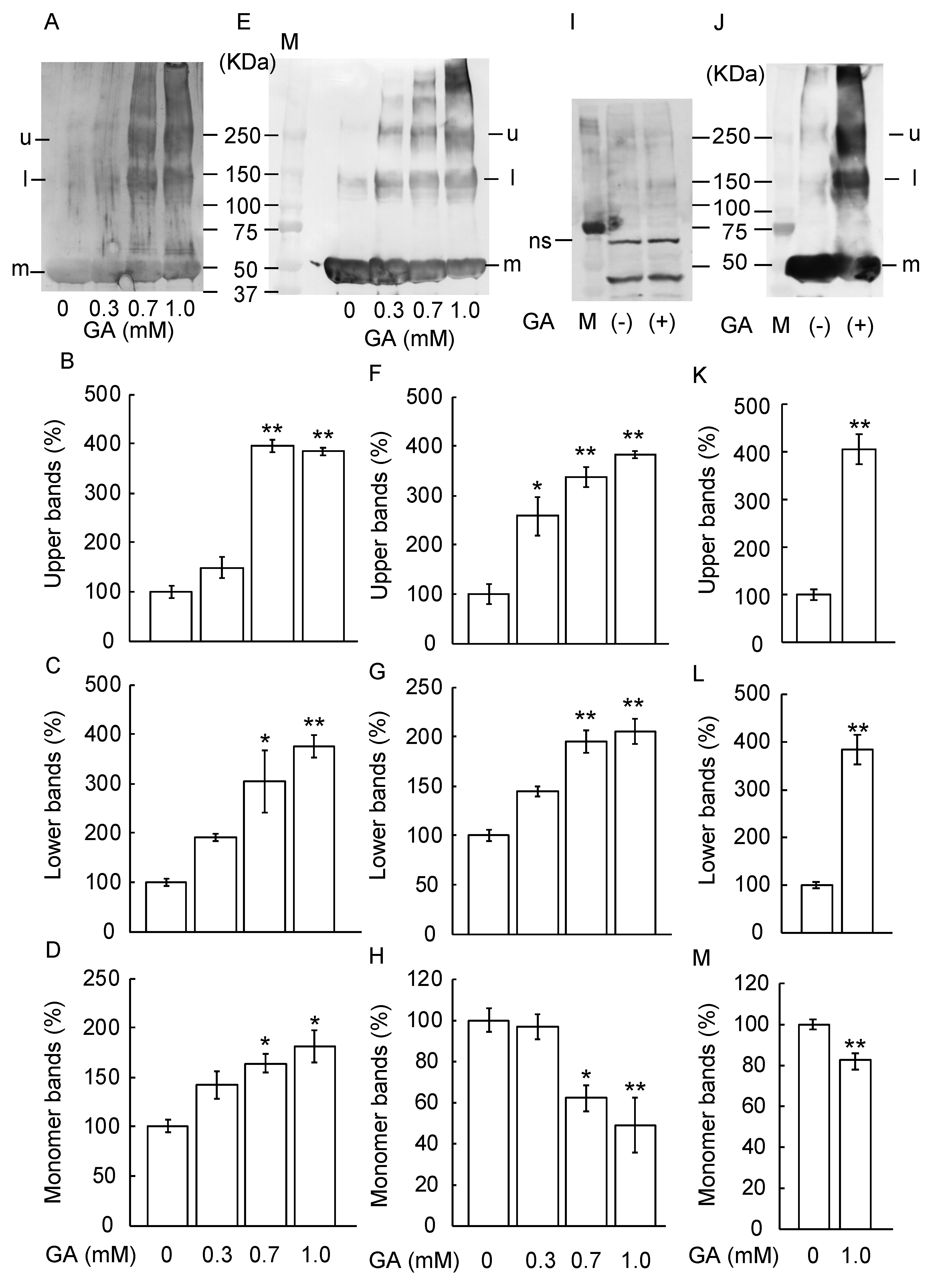
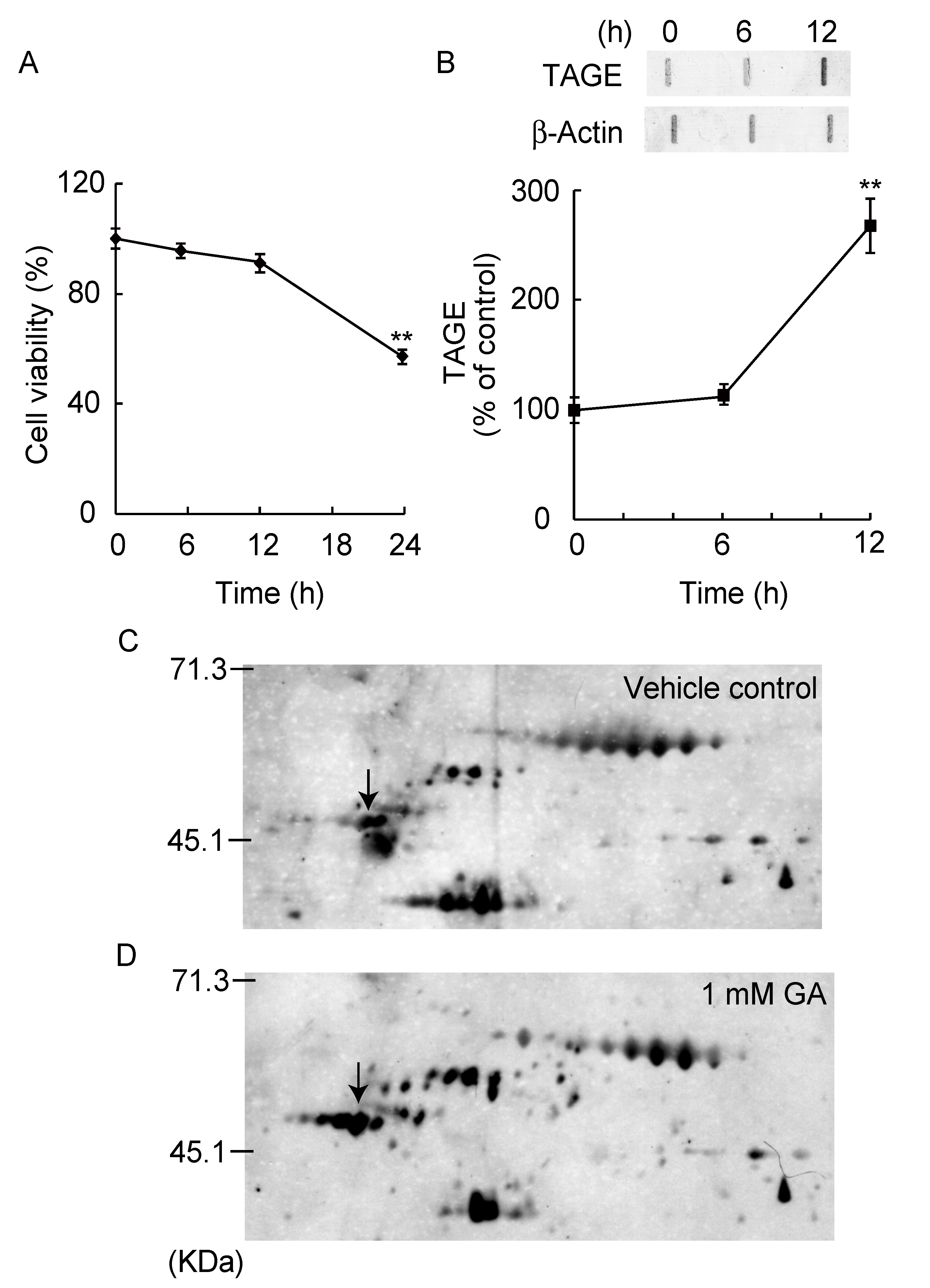
3.1. Detection and Identification of TAGE Proteins
3.2. TAGE Formation and Abnormal Aggregation of β-Tubulin by GA
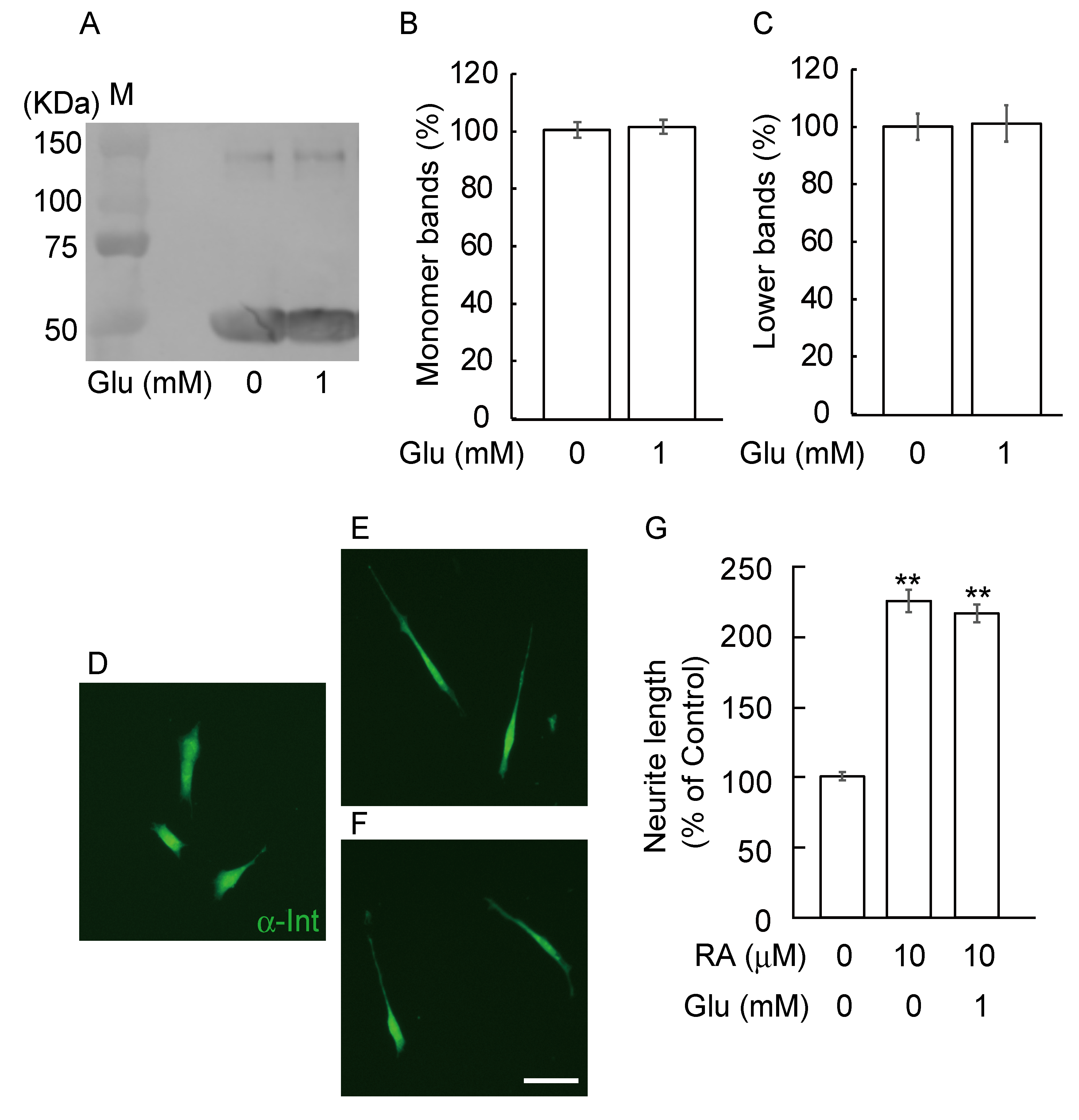
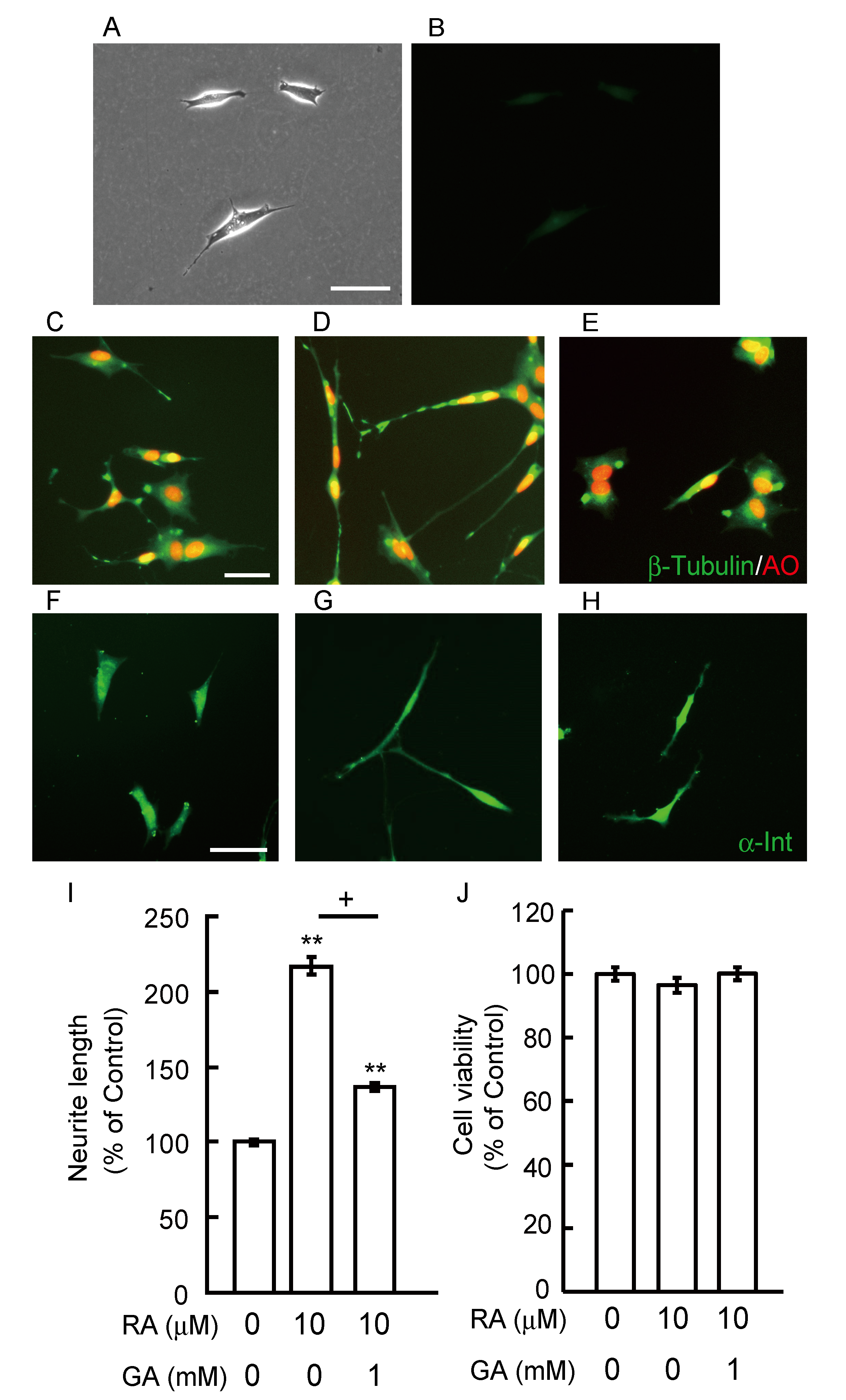
3.3. GA Inhibits Neurite Outgrowth From SH-SY5Y Cells
3.4. Glucose Did Not Induce Abnormal β-Tubulin Aggregation and Did Not Inhibit Neurite Outgrowth
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Sato, T.; Shimogaito, N.; Wu, X.; Kikuchi, S.; Yamagishi, S.; Takeuchi, M. Toxic advanced glycation end products (TAGE) theory in Alzheimer’s disease. Am. J. Alzheimers Dis. Other Demen. 2006, 21, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, M.; Makita, Z. Alternative routes for the formation of immunochemically distinct advanced glycation end-products in vivo. Curr. Mol. Med. 2001, 1, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, M.; Yamagishi, S. TAGE (toxic AGEs) hypothesis in various chronic diseases. Med. Hypotheses. 2004, 63, 449–452. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, M.; Takino, J.; Yamagishi, S. Involvement of the toxic AGEs (TAGE)-RAGE system in the pathogenesis of diabetic vascular complications: A novel therapeutic strategy. Curr. Drug Targets 2010, 11, 1468–1482. [Google Scholar] [CrossRef] [Green Version]
- Smith, M.A.; Taneda, S.; Richey, P.L.; Miyata, S.; Yan, S.D.; Stern, D.; Sayre, L.M.; Monnier, V.M.; Perry, G. Advanced maillard reaction end products are associated with Alzheimer disease pathology. Proc. Natl. Acad. Sci. USA 1994, 91, 5710–5714. [Google Scholar] [CrossRef] [Green Version]
- Yan, S.D.; Yan, S.F.; Chen, X.; Fu, J.; Chen, M.; Kuppusamy, P.; Smith, M.A.; Perry, G.; Godman, G.C.; Nawroth, P. Non-enzymatically glycated tau in Alzheimer’s disease induces neuronal oxidant stress resulting in cytokine gene expression and release of amyloid beta-peptide. Nat. Med. 1995, 1, 693–699. [Google Scholar] [CrossRef]
- Takeuchi, M. Serum Levels of toxic AGEs (TAGE) may be a promising novel biomarker for the onset/progression of lifestyle-related diseases. Diagnostics (Basel) 2016, 6, 23. [Google Scholar] [CrossRef]
- Takeuchi, M.; Bucala, R.; Suzuki, T.; Ohkubo, T.; Yamazaki, M.; Koike, T.; Kameda, Y.; Makita, Z. Neurotoxicity of advanced glycation end-products for cultured cortical neurons. J. Neuropathol. Exp. Neurol. 2000, 59, 1094–1105. [Google Scholar] [CrossRef] [Green Version]
- Koriyama, Y.; Furukawa, A.; Muramatsu, M.; Takino, J.; Takeuchi, M. Glyceraldehyde caused Alzheimer’s disease-like alterations in diagnostic marker levels in SH-SY5Y human neuroblastoma cells. Sci. Rep. 2015, 5, 13313. [Google Scholar] [CrossRef] [Green Version]
- Takata, T.; Sakasai-Sakai, A.; Ueda, T.; Takeuchi, M. Intracellular toxic advanced glycation end-products in cardiomyocytes may cause cardiovascular disease. Sci. Rep. 2019, 9, 2121. [Google Scholar] [CrossRef] [Green Version]
- Takino, J.; Nagamine, K.; Takeuchi, M.; Hori, T. In vitro identification of nonalcoholic fatty liver disease-related protein hnRNPM. World J. Gastroenterol. 2015, 21, 1784–1793. [Google Scholar] [CrossRef] [PubMed]
- Lee, V.M.; Goedert, M.; Trojanowski, J.Q. Neurodegenerative tauopathies. Annu. Rev. Neurosci. 2001, 24, 1121–1159. [Google Scholar] [CrossRef] [PubMed]
- Kirschner, M.; Mitchison, T. Beyond self-assembly: From microtubules to morphogenesis. Cell 1986, 45, 329–342. [Google Scholar] [CrossRef]
- Choei, H.; Sasaki, N.; Takeuchi, M.; Yoshida, T.; Ukai, W.; Yamagishi, S.; Kikuchi, S.; Saito, T. Glyceraldehyde-derived advanced glycation end products in Alzheimer’s disease. Acta. Neuropathol. 2004, 108, 189–193. [Google Scholar] [CrossRef]
- Koriyama, Y.; Chiba, K.; Mohri, T. Propentofylline protects beta-amyloid protein-induced apoptosis in cultured rat hippocampal neurons. Eur. J. Pharmacol. 2003, 458, 235–241. [Google Scholar] [CrossRef]
- Takeuchi, M.; Makita, Z.; Bucala, R.; Suzuki, T.; Koike, T.; Kameda, Y. Immunological evidence that non-carboxymethyllysine advanced glycation end-products are produced from short chain sugars and dicarbonyl compounds in vivo. Mol. Med. 2000, 6, 114–125. [Google Scholar] [CrossRef] [Green Version]
- Tahara, N.; Yamagishi, S.; Takeuchi, M.; Honda, A.; Tahara, A.; Nitta, Y.; Kodama, N.; Mizoguchi, M.; Kaida, H.; Ishibashi, M.; et al. Positive association between serum level of glyceraldehyde-derived advanced glycation end products and vascular inflammation evaluated by [(18)F] fluorodeoxyglucose positron emission tomography. Diabetes Care 2012, 35, 2618–2625. [Google Scholar] [CrossRef] [Green Version]
- Furukawa, A.; Kawamoto, Y.; Chiba, Y.; Takei, S.; Hasegawa-Ishii, S.; Kawamura, N.; Yoshikawa, K.; Hosokawa, M.; Oikawa, S.; Kato, M.; et al. Proteomic identification of hippocampal proteins vulnerable to oxidative stress in excitotoxin-induced acute neuronal injury. Neurobiol. Dis. 2011, 43, 706–714. [Google Scholar] [CrossRef]
- Iwasaki, S.; Iguchi, M.; Watanabe, K.; Hoshino, R.; Tsujimoto, M.; Kohno, M. Specific activation of the p38 mitogen-activated protein kinase signaling pathway and induction of neurite outgrowth in PC12 cells by bone morphogenetic protein-2. J. Biol. Chem. 1999, 274, 26503–26510. [Google Scholar] [CrossRef] [Green Version]
- Yuan, A.; Rao, M.V.; Sasaki, T.; Chen, Y.; Kumar, A.; Veeranna; Liem, R.K.H.; Eyer, J.; Peterson, A.C.; Julien, J.P.; et al. Alpha-internexin is structurally and functionally associated with the neurofilament triplet proteins in the mature CNS. J. Neurosci. 2006, 26, 10006–10019. [Google Scholar] [CrossRef]
- Ott, A.; Stolk, R.P.; van Harskamp, F.; Pols, H.A.; Hofman, A.; Breteler, M.M. Diabetes mellitus and the risk of dementia: The Rotterdam Study. Neurology 1999, 53, 1937–1942. [Google Scholar] [CrossRef] [PubMed]
- Gasparini, L.; Netzer, W.J.; Greengard, P.; Xu, H. Does insulin dysfunction play a role in Alzheimer’s disease? Trends Pharmacol. Sci. 2002, 23, 288–293. [Google Scholar] [CrossRef]
- Watson, G.S.; Craft, S. The role of insulin resistance in the pathogenesis of Alzheimer’s disease: Implications for treatment. CNS Drugs 2003, 17, 27–45. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.D.; Chen, X.; Fu, J.; Chen, M.; Zhu, H.; Roher, A.; Slattery, T.; Zhao, L.; Nagashima, M.; Morser, J.; et al. RAGE and amyloid-beta peptide neurotoxicity in Alzheimer’s disease. Nature 1996, 382, 685–691. [Google Scholar] [CrossRef]
- Vitek, M.P.; Bhattacharya, K.; Glendening, J.M.; Stopa, E.; Vlassara, H.; Bucala, R.; Manogue, K.; Cerami, A. Advanced glycation end products contribute to amyloidosis in Alzheimer disease. Proc. Natl. Acad. Sci. USA 1994, 91, 4766–4770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arriba, S.G.D.; Loske, C.; Meiners, I.; Fleischer, G.; Lobisch, M.; Wessel, K.; Tritschler, H.; Schinzel, R.; Münch, G. Advanced glycation endproducts induce changes in glucose consumption, lactate production, and ATP levels in SH-SY5Y neuroblastoma cells by a redox-sensitive mechanism. J. Cereb. Blood Flow Metab. 2003, 23, 1307–1313. [Google Scholar] [CrossRef] [Green Version]
- Kuhla, B.; Loske, C.; Arriba, S.G.D.; Schinzel, R.; Huber, J.; Münch, G. Differential effects of “advanced glycation endproducts”and beta-amyloid peptide on glucose utilization and ATP levels in the neuronal cell line SH-SY5Y. J. Neural Transm. 2004, 111, 427–439. [Google Scholar] [CrossRef]
- Takeuchi, M.; Makita, Z.; Yanagisawa, K.; Kameda, Y.; Koike, T. Detection of noncarboxymethyllysine and carboxymethyllysine advanced glycation end products (AGE) in serum of diabetic patients. Mol. Med. 1999, 5, 393–405. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, M.; Kikuchi, S.; Sasaki, N.; Suzuki, T.; Watai, T.; Iwaki, M.; Bucala, R.; Yamagishi, S. Involvement of advanced glycation end-products (AGEs) in Alzheimer’s disease. Curr. Alzheimer Res. 2004, 1, 39–46. [Google Scholar] [CrossRef]
- Cullum, N.A.; Mahon, J.; Stringer, K.; McLean, W.G. Glycation of rat sciatic nerve tubulin in experimental diabetes mellitus. Diabetologia 1991, 34, 387–389. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, M.; Yamagishi, S. Possible involvement of advanced glycation end-products (AGEs) in the pathogenesis of Alzheimer’s disease. Curr. Pharm. Des. 2008, 14, 973–978. [Google Scholar] [CrossRef] [PubMed]
- Oates, P.J. Polyol pathway and diabetic peripheral neuropathy. Int. Rev. Neurobiol. 2002, 50, 325–392. [Google Scholar] [PubMed]
- Hallfrisch, J. Metabolic effects of dietary fructose. FASEB J. 1990, 4, 2652–2660. [Google Scholar] [CrossRef] [PubMed]
- Hassel, B.; Elsais, A.; Frøland, A.S.; Taubøll, E.; Gjerstad, L.; Quan, Y.; Dingledine, R.; Rise, F. Uptake and metabolism of fructose by rat neocortical cells in vivo and by isolated nerve terminals in vitro. J. Neurochem. 2015, 133, 572–581. [Google Scholar] [CrossRef] [Green Version]
- Bais, R.; James, H.M.; Rofe, A.M.; Conyers, R.A. The purification and properties of human liver ketohexokinase. A role for ketohexokinase and fructose-bisphosphate aldolase in the metabolic production of oxalate from xylitol. Biochem. J. 1985, 230, 53–60. [Google Scholar] [CrossRef] [Green Version]
- Ihara, Y.; Nukina, N.; Miura, R.; Ogawara, M. Phosphorylated tau protein is integrated into paired helical filaments in Alzheimer’s disease. J. Biochem. 1986, 99, 1807–1810. [Google Scholar] [CrossRef]
- Iqbal, K.; Grundke-Iqbal, I.; Smith, A.J.; George, L.; Tung, Y.C.; Zaidi, T. Identification and localization of a tau peptide to paired helical filaments of Alzheimer disease. Proc. Natl. Acad. Sci. U.S.A. 1989, 86, 5646–5650. [Google Scholar] [CrossRef] [Green Version]
- Ledesma, M.D.; Bonay, P.; Vila, J. Tau protein from Alzheimer’s disease patients is glycated at its tubulin-binding domain. J. Neurochem. 1995, 65, 1658–1664. [Google Scholar] [CrossRef]
- Ando, K.; Otake, A.M.; Ohtake, Y.; Hayashishita, M.; Sekiya, M.; Iijima, K.M. Stabilization of microtubule-unbound tau via tau phosphorylation at Ser262/356 by Par-1/MARK contributes to augmentation of AD-related phosphorylation and Aβ42-induced tau toxicity. PLoS Genet. 2016, 12, e1005917. [Google Scholar] [CrossRef] [Green Version]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nasu, R.; Furukawa, A.; Suzuki, K.; Takeuchi, M.; Koriyama, Y. The Effect of Glyceraldehyde-Derived Advanced Glycation End Products on β-Tubulin-Inhibited Neurite Outgrowth in SH-SY5Y Human Neuroblastoma Cells. Nutrients 2020, 12, 2958. https://doi.org/10.3390/nu12102958
Nasu R, Furukawa A, Suzuki K, Takeuchi M, Koriyama Y. The Effect of Glyceraldehyde-Derived Advanced Glycation End Products on β-Tubulin-Inhibited Neurite Outgrowth in SH-SY5Y Human Neuroblastoma Cells. Nutrients. 2020; 12(10):2958. https://doi.org/10.3390/nu12102958
Chicago/Turabian StyleNasu, Ryuto, Ayako Furukawa, Keita Suzuki, Masayoshi Takeuchi, and Yoshiki Koriyama. 2020. "The Effect of Glyceraldehyde-Derived Advanced Glycation End Products on β-Tubulin-Inhibited Neurite Outgrowth in SH-SY5Y Human Neuroblastoma Cells" Nutrients 12, no. 10: 2958. https://doi.org/10.3390/nu12102958