Catalpol Ameliorates Insulin Sensitivity and Mitochondrial Respiration in Skeletal Muscle of Type-2 Diabetic Mice Through Insulin Signaling Pathway and AMPK/SIRT1/PGC-1α/PPAR-γ Activation

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Drugs and Reagents
2.2. Animal Studies
2.3. Measurement of FBG, HbA1C, Plasma Insulin, HOMA_IR
2.4. Oral Glucose Tolerance Test (OGTT) and Insulin Tolerance Test (ITT)
2.5. Gene Expression Study
2.6. Protein Expression Study
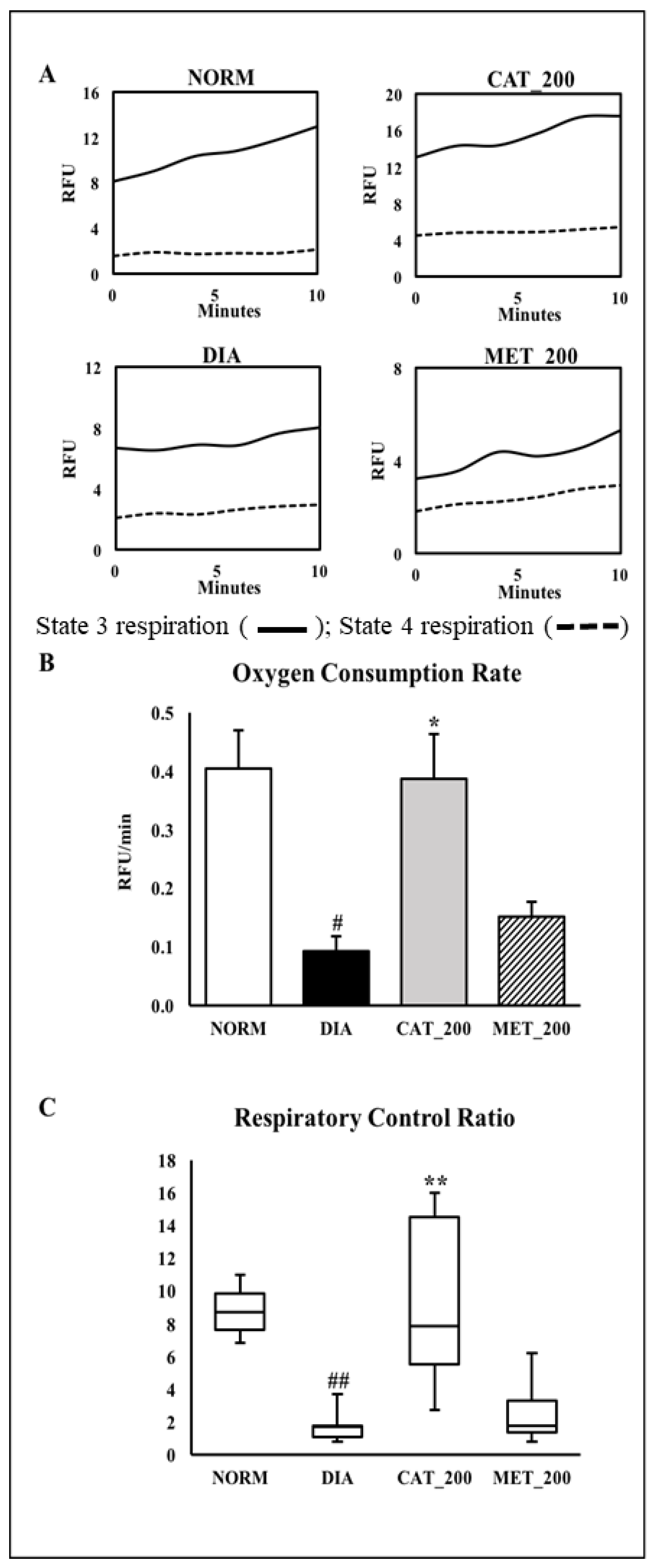
2.7. Mitochondria Function Study
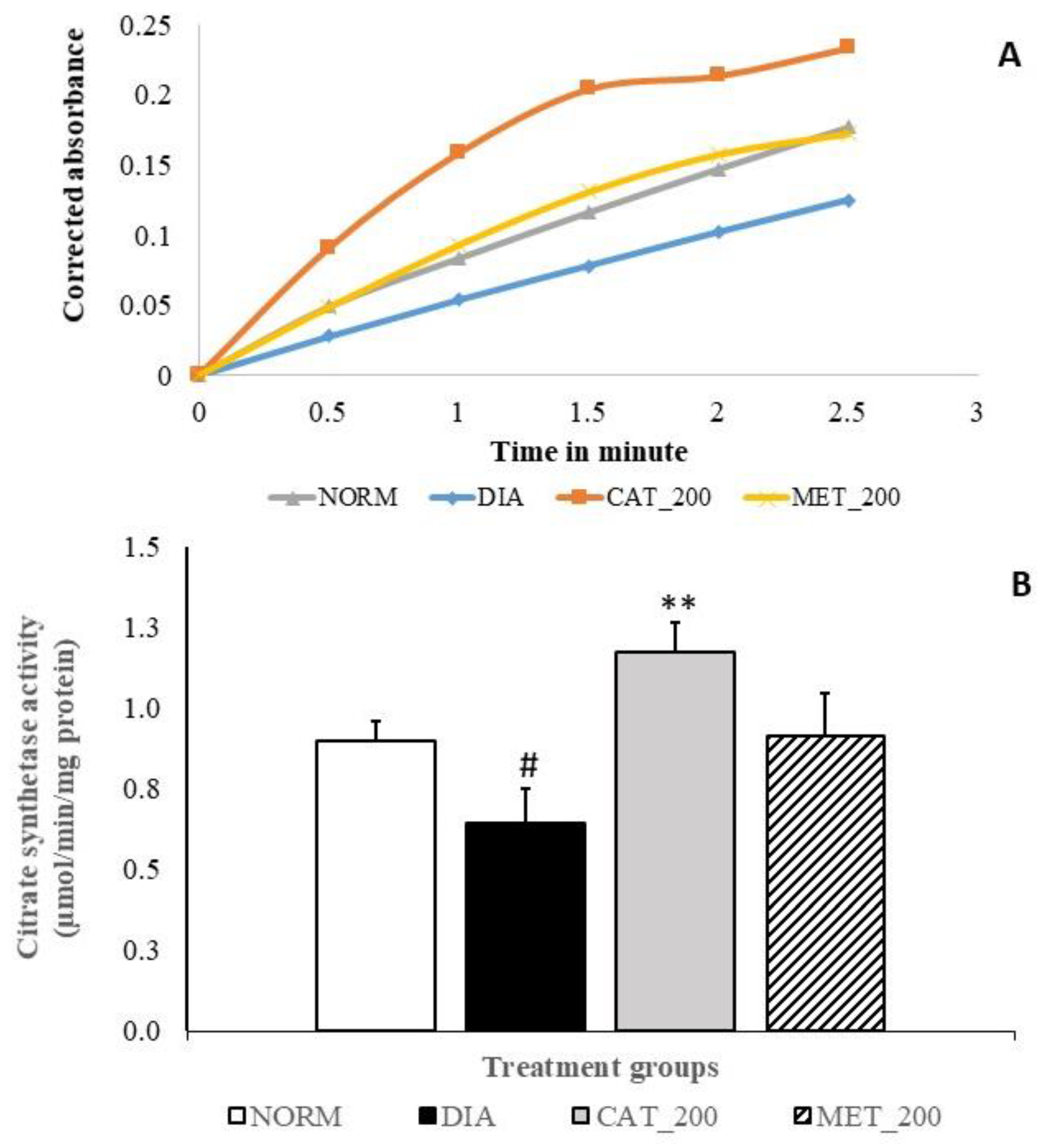
2.8. Citrate Synthase (CS) Activity
2.9. Statistical Analysis
3. Results
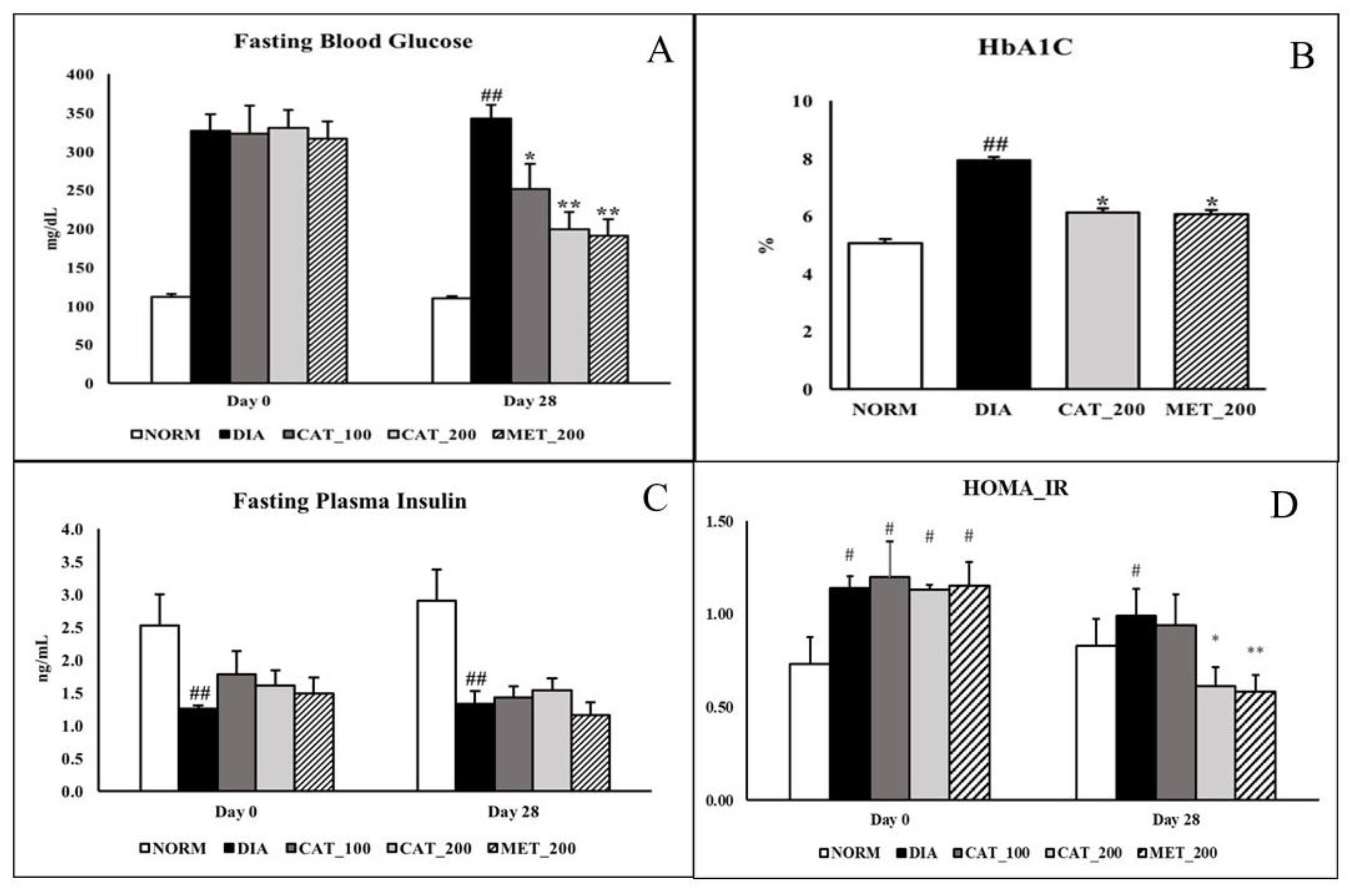
3.1. Measurement of FBG, HbA1C, Plasma Insulin, HOMA_IR
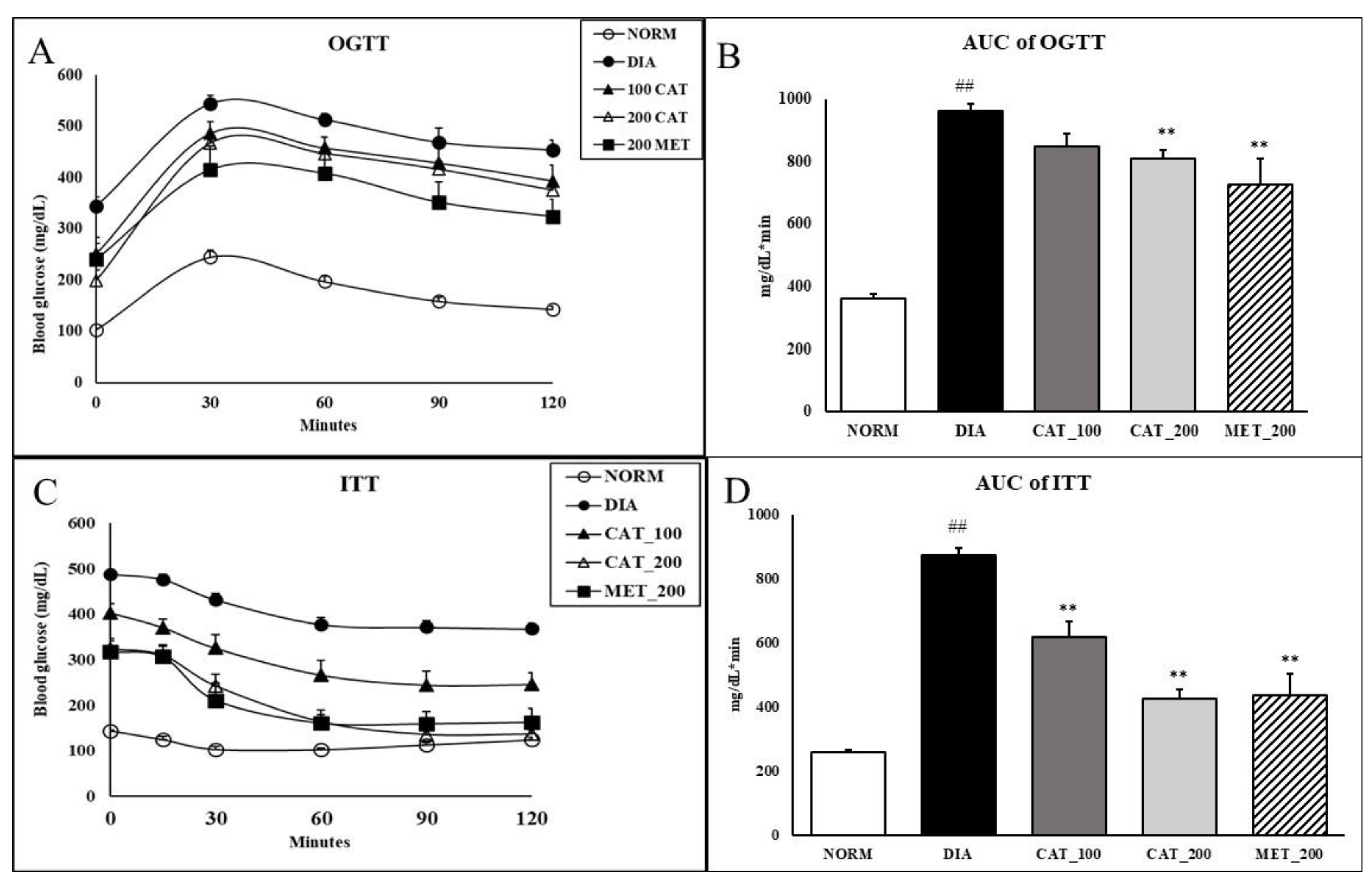
3.2. Oral Glucose Tolerance Test (OGTT) and Insulin Tolerance Test (ITT)
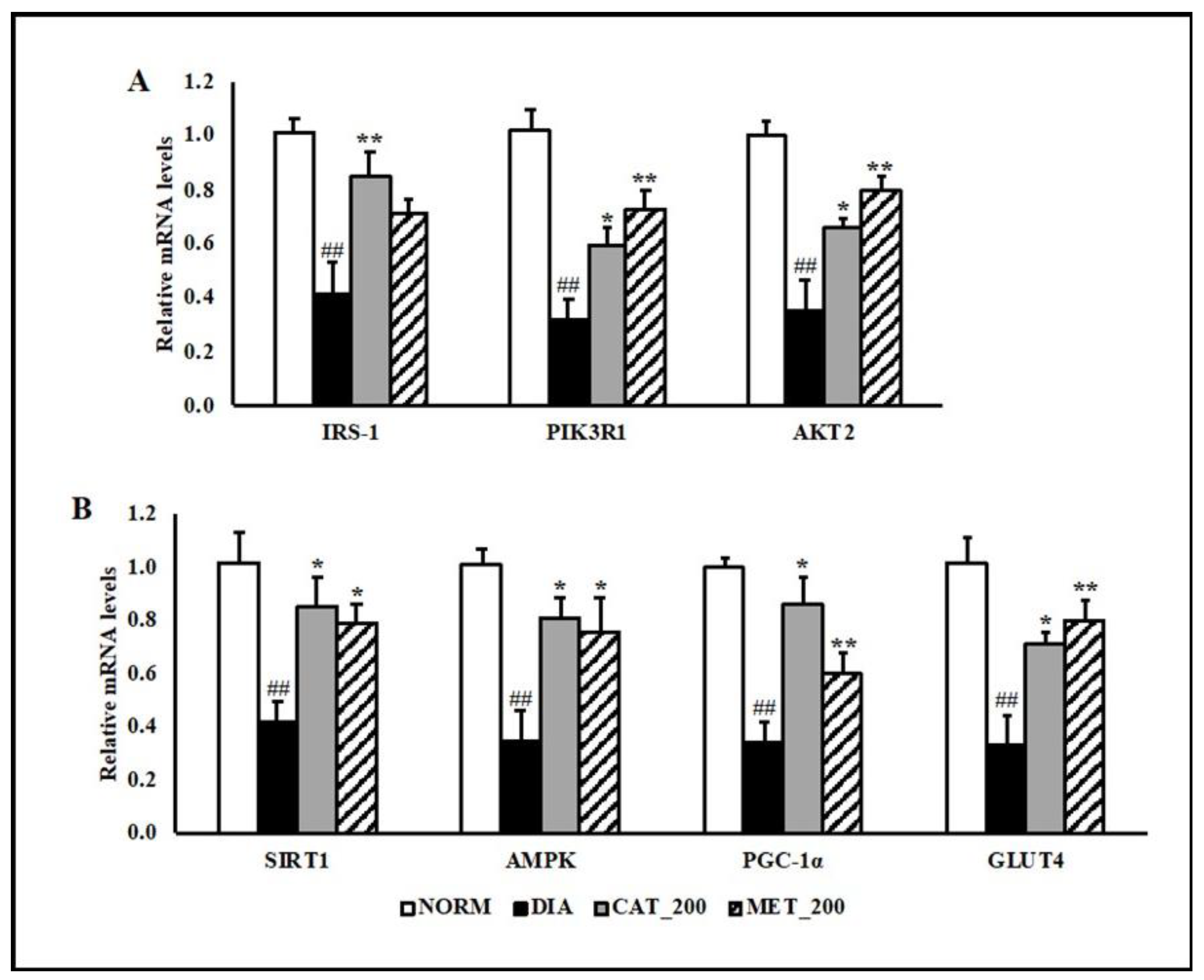
3.3. Gene Expression Study
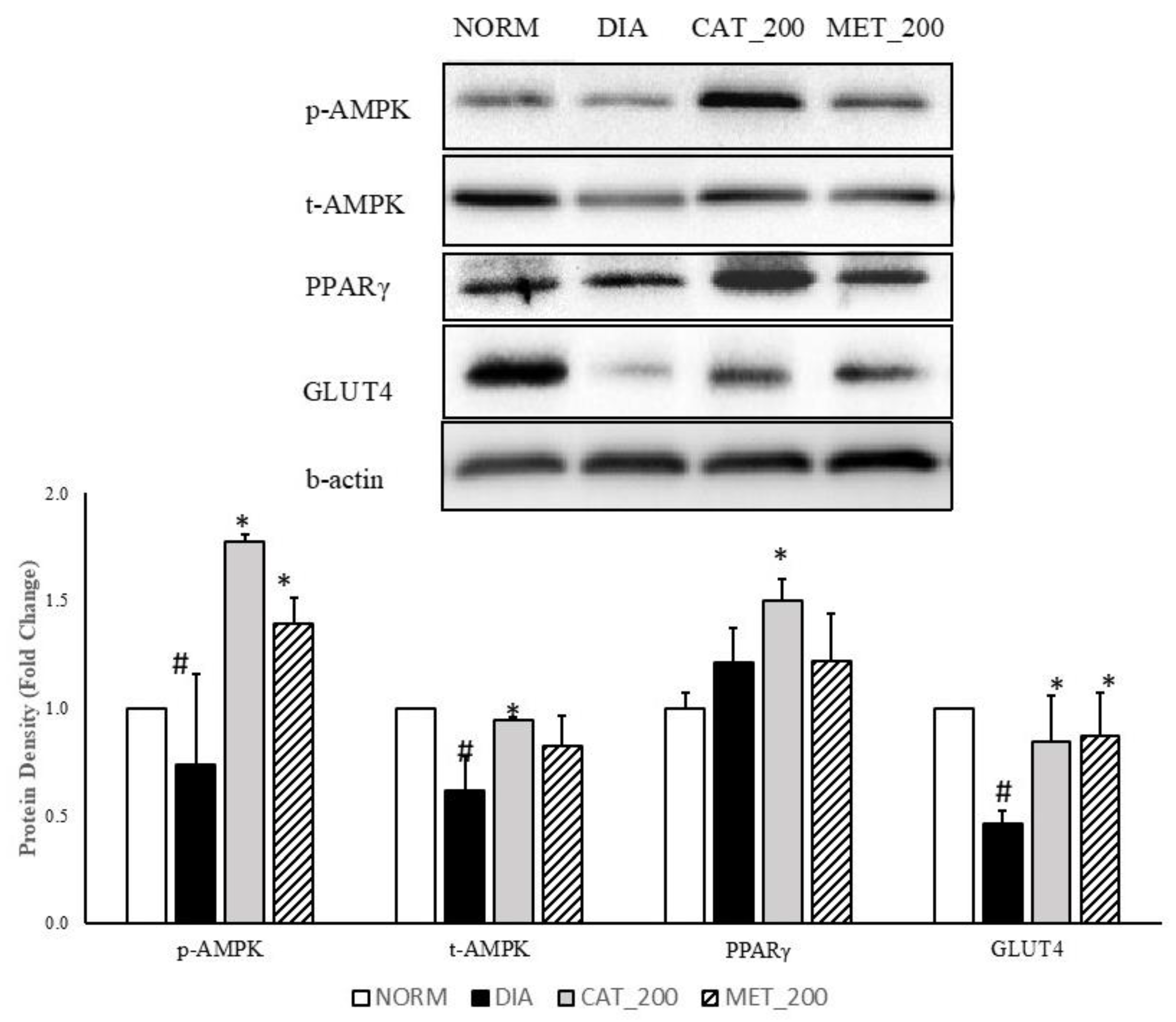
3.4. Protein Expression Study
3.5. Mitochondria Function Study
3.6. Citrate Synthase (CS) Activity
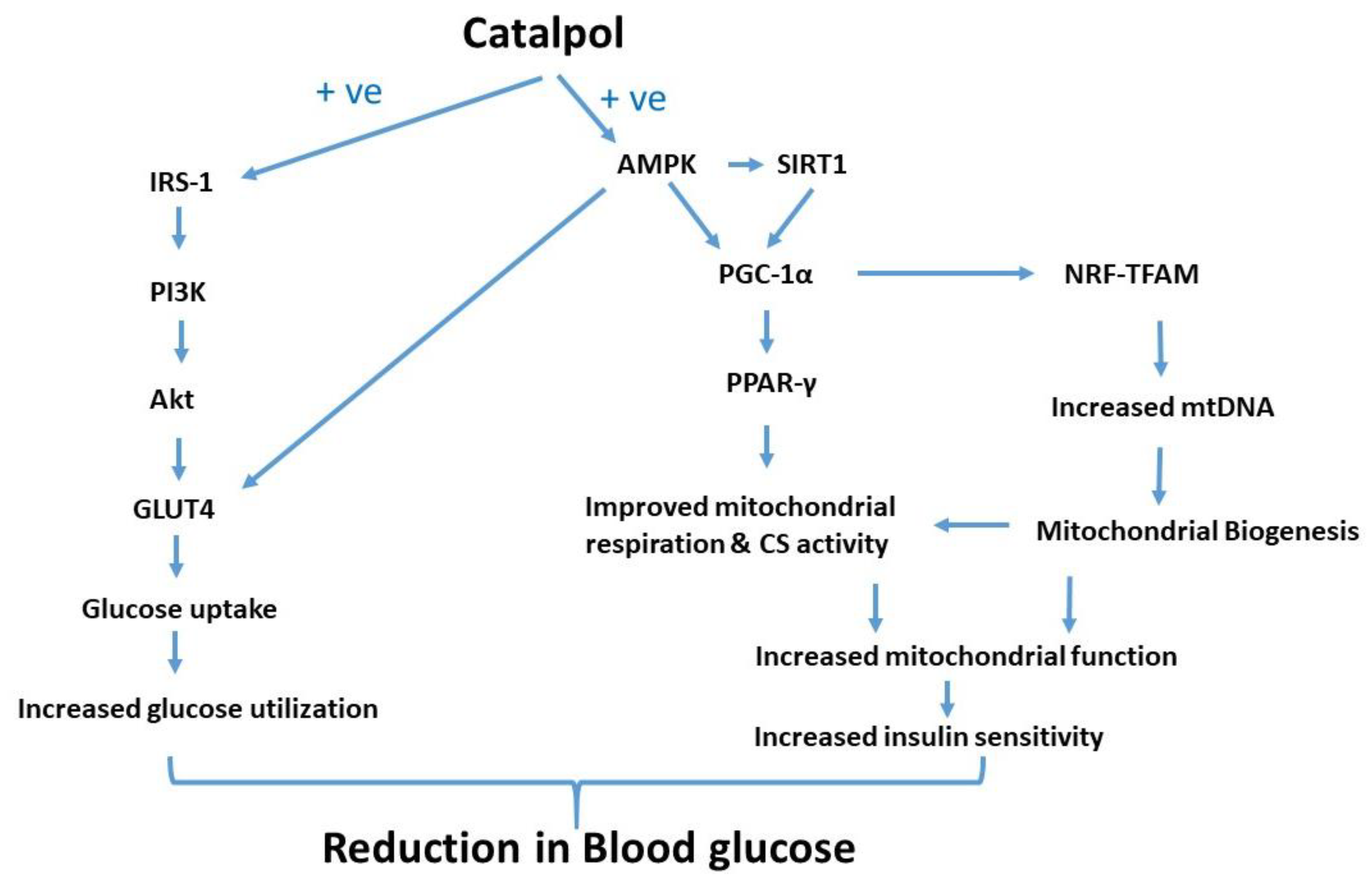
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- World Health Organization. WHO—The Top 10 Causes of Death; WHO: Geneva, Switzerland, 2017. [Google Scholar]
- Reaven, G. Insulin Resistance, Type 2 Diabetes Mellitus, and Cardiovascular Disease. Circulation 2005, 112, 3030–3032. [Google Scholar] [CrossRef]
- Tabák, A.G.; Jokela, M.; Akbaraly, T.N.; Brunner, E.J.; Kivimäki, M.; Witte, D.R. Trajectories of glycaemia, insulin sensitivity, and insulin secretion before diagnosis of type 2 diabetes: An analysis from the Whitehall II study. Lancet 2009, 373, 2215–2221. [Google Scholar] [CrossRef]
- Unwin, N.; Shaw, J.; Zimmet, P.; Alberti, K.G. Impaired glucose tolerance and impaired fasting glycaemia: The current status on definition and intervention. Diabet. Med. 2002, 19, 708–723. [Google Scholar]
- Emerging Risk Factors Collaboration; Sarwar, N.; Gao, P.; Seshasai, S.R.; Gobin, R.; Kaptoge, S.; Di Angelantonio, E.; Ingelsson, E.; Lawlor, D.A.; Selvin, E.; et al. Diabetes mellitus, fasting blood glucose concentration, and risk of vascular disease: A collaborative meta-analysis of 102 prospective studies. Lancet 2010, 375, 2215–2222. [Google Scholar]
- Itani, S.I.; Ruderman, N.B.; Schmieder, F.; Boden, G. Lipid-induced insulin resistance in human muscle is associated with changes in diacylglycerol, protein kinase C, and IkappaB-alpha. Diabetes 2002, 51, 2005–2011. [Google Scholar] [CrossRef]
- Griffin, M.E.; Marcucci, M.J.; Cline, G.W.; Bell, K.; Barucci, N.; Lee, D.; Goodyear, L.J.; Kraegen, E.W.; White, M.F.; Shulman, G.I. Free fatty acid induced insulin resistance is associated with activation of protein kinase C theta and alterations in the insulin signaling cascade. Diabetes 1999, 48, 1270–1274. [Google Scholar] [CrossRef]
- Yu, C.; Chen, Y.; Cline, G.W.; Zhang, D.; Zong, H.; Wang, Y.; Bergeron, R.; Kim, J.K.; Cushman, S.W.; Cooney, G.J.; et al. Mechanism by which fatty acids inhibit insulin activation of insulin receptor substrate-1 (IRS-1)-associated phosphatidylinositol 3-kinase activity in muscle. J. Biol. Chem. 2002, 277, 50230–50236. [Google Scholar] [CrossRef]
- Kim, F.; Pham, M.; Luttrell, I.; Bannerman, D.D.; Tupper, J.; Thaler, J.; Hawn, T.R.; Raines, E.W.; Schwartz, M.W. Toll-like receptor-4 mediates vascular inflammation and insulin resistance in diet-induced obesity. Circ. Res. 2007, 100, 1589–1596. [Google Scholar] [CrossRef]
- Morino, K.; Petersen, K.F.; Dufour, S.; Befroy, D.; Frattini, J.; Shatzkes, N.; Neschen, S.; White, M.F.; Bilz, S.; Sono, S.; et al. Reduced mitochondrial density and increased IRS-1 serine phosphorylation in muscle of insulin-resistant offspring of type 2 diabetic parents. J. Clin. Investig. 2005, 115, 3587–3593. [Google Scholar] [CrossRef]
- Nishikawa, T.; Araki, E. Impact of mitochondrial ROS production in the pathogenesis of diabetes mellitus and its complications. Antioxid. Redox. Signal. 2007, 9, 343–353. [Google Scholar] [CrossRef]
- Shieh, J.P.; Cheng, K.C.; Chung, H.H.; Kerh, Y.F.; Yeh, C.H.; Cheng, J.T. Plasma glucose lowering mechanisms of catalpol, an active principle from roots of Rehmannia glutinosa, in streptozotocin-induced diabetic rats. J. Agric. Food Chem. 2011, 59, 3747–3753. [Google Scholar] [CrossRef] [PubMed]
- Bhattamisra, S.K.; Yap, K.H.; Rao, V.; Choudhury, H. Multiple Biological Effects of an Iridoid Glucoside, Catalpol, and Its Underlying Molecular Mechanisms. Biomolecules 2020, 10, 32. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Zhu, R.; Tian, Y.; Li, R.; Chen, B.; Zhang, H.; Xia, B.; Zhao, D.; Mo, F.; Zhang, D.; et al. Catalpol in Diabetes and its Complications: A Review of Pharmacology, Pharmacokinetics, and Safety. Molecules 2019, 24, 3302. [Google Scholar] [CrossRef]
- Kitagawa, I.; Nishimura, T.; Furubayashi, A.; Yosioka, I. On the constituents of rhizome of Rehmannia glutinosa Libosch. forma hueichingensis Hsiao. Yakugaku Zasshi J. Pharm. Soc. Jpn. 1971, 91, 593–596. [Google Scholar] [CrossRef][Green Version]
- Li, X.; Xu, Z.; Jiang, Z.; Sun, L.; Ji, J.; Miao, J.; Zhang, X.; Li, X.; Huang, S.; Wang, T.; et al. Hypoglycemic effect of catalpol on high-fat diet/streptozotocin-induced diabetic mice by increasing skeletal muscle mitochondrial biogenesis. Acta. Biochim. Biophys. Sin. 2014, 46, 738–748. [Google Scholar] [CrossRef]
- Xu, D.; Wang, L.; Jiang, Z.; Zhao, G.; Hassan, H.M.; Sun, L.; Fan, S.; Zhou, Z.; Zhang, L.; Wang, T. A new hypoglycemic mechanism of catalpol revealed by enhancing MyoD/MyoG-mediated myogenesis. Life Sci. 2018, 209, 313–323. [Google Scholar] [CrossRef]
- Xu, D.Q.; Li, C.J.; Jiang, Z.Z.; Wang, L.; Huang, H.F.; Li, Z.J.; Sun, L.X.; Fan, S.S.; Zhang, L.Y.; Wang, T. The hypoglycemic mechanism of catalpol involves increased AMPK-mediated mitochondrial biogenesis. Acta Pharmacol. Sin. 2020, 41, 791–799. [Google Scholar] [CrossRef] [PubMed]
- Bao, Q.; Shen, X.; Qian, L.; Gong, C.; Nie, M.; Dong, Y. Anti-diabetic activities of catalpol in db/db mice. Korean J. Physiol. Pharmacol. 2016, 20, 153–160. [Google Scholar] [CrossRef]
- Yan, J.; Wang, C.; Jin, Y.; Meng, Q.; Liu, Q.; Liu, Z.; Liu, K.; Sun, H. Catalpol ameliorates hepatic insulin resistance in type 2 diabetes through acting on AMPK/NOX4/PI3K/AKT pathway. Pharmacol. Res. 2018, 130, 466–480. [Google Scholar] [CrossRef]
- Zhou, J.; Xu, G.; Ma, S.; Li, F.; Yuan, M.; Xu, H.; Huang, K. Catalpol ameliorates high-fat diet-induced insulin resistance and adipose tissue inflammation by suppressing the JNK and NF-κB pathways. Biochem. Biophys. Res. Commun. 2015, 467, 853–858. [Google Scholar] [CrossRef]
- Yap, K.; Gan, S.; Candasamy, M.; Md, S.; Majeed, A.A.; Bhattamisra, S. Effect of catalpol on liver glucose homeostasis in high fat diet/low dose streptozotocin-induced type 2 diabetes mellitus. Br. J. Pharmacol. 2019, 176, 3051–3052. [Google Scholar]
- Elhassan, S.A.; Candasamy, M.; Ching, T.S.; Heng, Y.K.; Bhattamisra, S.K. Effect of madecassoside and catalpol in amelioration of insulin sensitivity in pancreatic (INS-1E) β-cell line. Nat. Prod. Res. 2019. [Google Scholar] [CrossRef]
- Rato, L.; Alves, M.G.; Dias, T.R.; Cavaco, J.E.; Oliveira, P.F. Testicular metabolic reprogramming in neonatal streptozotocin-induced type 2 diabetic rats impairs glycolytic flux and promotes glycogen synthesis. J. Diabetes Res. 2015. [Google Scholar] [CrossRef]
- Chen, D.K.; Frizzi, K.E.; Guernsey, L.S.; Ladt, K.; Mizisin, A.P.; Calcutt, N.A. Repeated monitoring of corneal nerves by confocal microscopy as an index of peripheral neuropathy in type-1 diabetic rodents and the effects of topical insulin. J. Peripher. Nerv. Syst. 2013, 18, 306–315. [Google Scholar] [CrossRef] [PubMed]
- Bhattamisra, S.K.; Mohapatra, L.; Panda, B.P.; Parida, S. Effect of isoflavone rich soya seed extract on glucose utilization and endurance capacity in diabetic rat. Diabetol. Croat. 2013, 42, 42–52. [Google Scholar]
- Luo, J.; Quan, J.; Tsai, J.; Hobensack, C.K.; Sullivan, C.; Hector, R.; Reaven, G.M. Nongenetic mouse models of non-insulin-dependent diabetes mellitus. Metabolism 1998, 47, 663–668. [Google Scholar] [CrossRef]
- Manchem, V.P.; Goldfine, I.D.; Kohanski, R.A.; Cristobal, C.P.; Lum, R.T.; Schow, S.R.; Shi, S.; Spevak, W.R.; Laborde, E.; Toavs, D.K.; et al. A novel small molecule that directly sensitizes the insulin receptor in vitro and in vivo. Diabetes 2001, 50, 824–830. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Millar, P.; Pathak, N.; Parthsarathy, V.; Bjourson, A.J.; O’Kane, M.; Pathak, V.; Moffett, R.C.; Flatt, P.R.; Gault, V.A. Metabolic and neuroprotective effects of dapagliflozin and liraglutide in diabetic mice. J. Endocrinol. 2017, 234, 255–267. [Google Scholar] [CrossRef]
- Bonora, E.; Moghetti, P.; Zancanaro, C.; Cigolini, M.; Querena, M.; Cacciatori, V.; Corgnati, A.; Muggeo, M. Estimates of in vivo insulin action in man: Comparison of insulin tolerance tests with euglycemic and hyperglycemic glucose clamp studies. J. Clin. Endocrinol. Metab. 1989, 68, 374–378. [Google Scholar] [CrossRef]
- Akinmokun, A.; Selby, P.L.; Ramaiya, K.; Alberti, K.G. The short insulin tolerance test for determination of insulin sensitivity: A comparison with the euglycaemic clamp. Diabet. Med. 1992, 9, 432–437. [Google Scholar] [CrossRef]
- Gelding, S.V.; Robinson, S.; Lowe, S.; Niththyananthan, R.; Johnston, D.G. Validation of the low dose short insulin tolerance test for evaluation of insulin sensitivity. Clin. Endocrinol. 1994, 40, 611–615. [Google Scholar] [CrossRef]
- Kozawa, J.; Iwahashi, H.; Okita, K.; Okauchi, Y.; Imagawa, A.; Shimomura, I. Insulin tolerance test predicts the effectiveness of insulin sensitizers in Japanese type 2 diabetic patients. Diabetes Ther. 2010, 1, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Wallace, T.M.; Matthews, D.R. The assessment of insulin resistance in man. Diabet Med. 2002, 19, 527–534. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, M.; DeFronzo, R.A. Insulin sensitivity indices obtained from oral glucose tolerance testing. Comparison with the euglycaemic insulin clamp. Diabetes Care. 1999, 22, 1462–1470. [Google Scholar] [CrossRef]
- Inchiostro, S. Measurement of insulin sensitivity in type 2 diabetes mellitus: Comparison between KITT and HOMA-%S indices and evaluation of their relationship with the components of the insulin resistance syndrome. Diabet. Med. 2005, 22, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Wang, Y.; Liu, Z.; Wang, J.; Wan, D.; Feng, S.; Yang, X.; Wang, T. Antidiabetic and antioxidant effects of catalpol extracted from Rehmannia glutinosa (Di Huang) on rat diabetes induced by streptozotocin and high-fat, high-sugar feed. Chin. Med. 2016, 11, 25. [Google Scholar] [CrossRef]
- Morino, K.; Petersen, K.F.; Shulman, G.I. Molecular mechanisms of insulin resistance in humans and their potential links with mitochondrial dysfunction. Diabetes 2006, 55, S9–S15. [Google Scholar] [CrossRef]
- Okada, T.; Kawano, Y.; Sakakibara, T.; Hazeki, O.; Ui, M. Essential role of phosphatidylinositol 3-kinase in insulin-induced glucose transport and antilipolysis in rat adipocytes: Studies with a selective inhibitor wortmannin. J. Biol. Chem. 1994, 269, 3568–3573. [Google Scholar]
- Xu, D.; Huang, X.; Hassan, H.M.; Wang, L.; Li, S.; Jiang, Z.; Zhang, L.; Wang, T. Hypoglycaemic effect of catalpol in a mouse model of high-fat diet-induced pre-diabetes. Appl. Physiol. Nutr. Metab. 2020. [Google Scholar] [CrossRef]
- Asmann, Y.W.; Stump, C.S.; Short, K.R.; Coenen-Schimke, J.M.; Guo, Z.; Bigelow, M.L.; Nair, K.S. Skeletal muscle mitochondrial functions, mitochondrial DNA copy numbers, and gene transcript profiles in type 2 diabetic and nondiabetic subjects at equal levels of low or high insulin and euglycemia. Diabetes 2006, 55, 3309–3319. [Google Scholar] [CrossRef]
- Stump, C.S.; Short, K.R.; Bigelow, M.L.; Schimke, J.M.; Nair, K.S. Effect of insulin on human skeletal muscle mitochondrial ATP production, protein synthesis, and mRNA transcripts. Proc. Natl. Acad. Sci. USA 2003, 100, 7996–8001. [Google Scholar] [CrossRef]
- Yerby, B.; Deacon, R.; Beaulieu, V.; Liang, J.; Gao, J.; Laurent, D. Insulin stimulated mitochondrial adenosine triphosphate synthesis is blunted in skeletal muscles of high-fat-fed rats. Metab. Clin. Exp. 2008, 57, 1584–1590. [Google Scholar] [CrossRef]
- Montgomery, M.K.; Turner, N. Mitochondrial dysfunction and insulin resistance: An update. Endocr. Connect. 2015, 4, R1–R15. [Google Scholar] [CrossRef]
- Montgomery, M.K. Mitochondrial dysfunction and diabetes: Is mitochondrial transfer a friend or foe? Biology 2019, 8, 33. [Google Scholar] [CrossRef]
- Lai, N.; Kummitha, C.; Hoppel, C. Defects in skeletal muscle subsarcolemmal mitochondria in a non-obese model of type 2 diabetes mellitus. PLoS ONE 2017, 12, 183978–183985. [Google Scholar] [CrossRef]
- Cade, W.T. The manifold role of the mitochondria in skeletal muscle insulin resistance. Curr. Opin. Clin. Nutr. Metab. Care 2018, 21, 267–272. [Google Scholar] [CrossRef]
- Newsholme, P.; Haber, E.P.; Hirabara, S.M.; Rebelato, E.L.O.; Procopio, J.; Morgan, D.; Oliveira-Emilio, H.C.; Carpinelli, A.R.; Curi, R. Diabetes associated cell stress and dysfunction: Role of mitochondrial and non-mitochondrial ROS production and activity. J. Physiol. 2007, 583, 9–24. [Google Scholar] [CrossRef]
- Finck, B.N.; Kelly, D.P. PGC-1 coactivators: Inducible regulators of energy metabolism in health and disease. J. Clin Investig. 2006, 116, 615–622. [Google Scholar] [CrossRef]
- Lehman, J.J.; Barger, P.M.; Kovacs, A.; Saffitz, J.E.; Medeiros, D.M.; Kelly, D.P. Peroxisome proliferator-activated receptor gamma coactivator-1 promotes cardiac mitochondrial biogenesis. J. Clin. Investig. 2000, 106, 847–856. [Google Scholar] [CrossRef]
- Baar, K.; Wende, A.R.; Jones, T.E.; Marison, M.; Nolte, L.A.; Chen, M.; Kelly, D.P.; Holloszy, J.O. Adaptations of skeletal muscle to exercise: Rapid increase in the transcriptional coactivator PGC-1. FASEB J. 2002, 16, 1879–1886. [Google Scholar] [CrossRef]
- Yoon, J.C.; Puigserver, P.; Chen, G.; Donovan, J.; Wu, Z.; Rhee, J.; Adelmant, G.; Stafford, J.; Kahn, C.R.; Granner, D.K.; et al. Control of hepatic gluconeogenesis through the transcriptional coactivator PGC-1. Nature 2001, 413, 131–138. [Google Scholar] [CrossRef]
- Puigserver, P.; Wu, Z.; Park, C.W.; Graves, R.; Wright, M.; Spiegelman, B.M. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell 1998, 92, 829–839. [Google Scholar] [CrossRef]
- Duncan, J.G.; Fong, J.L.; Medeiros, D.M.; Finck, B.N.; Kelly, D.P. Insulin-resistant heart exhibits a mitochondrial biogenic response driven by the peroxisome proliferator-activated receptor-alpha/PGC-1alpha gene regulatory pathway. Circulation 2007, 115, 909–917. [Google Scholar] [CrossRef]
- Wu, Z.; Puigserver, P.; Andersson, U.; Zhang, C.; Adelmant, G.; Mootha, V.; Troy, A.; Cinti, S.; Lowell, B.; Scarpulla, R.C.; et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 1999, 98, 115–124. [Google Scholar] [CrossRef]
- Patti, M.E.; Butte, A.J.; Crunkhorn, S.; Cusi, K.; Berria, R.; Kashyap, S.; Miyazaki, Y.; Kohane, I.; Costello, M.; Saccone, R.; et al. Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: Potential role of PGC1 and NRF1. Proc. Natl. Acad. Sci. USA 2003, 100, 8466–8471. [Google Scholar] [CrossRef]
- St-Pierre, J.; Lin, J.; Krauss, S.; Tarr, P.T.; Yang, R.; Newgard, C.B.; Spiegelman, B.M. Bioenergetic analysis of peroxisome proliferator-activated receptor gamma coactivators 1alpha and 1beta (PGC-1alpha and PGC-1beta) in muscle cells. J. Biol. Chem. 2003, 278, 26597–26603. [Google Scholar] [CrossRef]
- Nisoli, E.; Clementi, E.; Carruba, M.O.; Moncada, S. Defective mitochondrial biogenesis: A hallmark of the high cardiovascular risk in the metabolic syndrome? Circ. Res. 2007, 100, 795–806. [Google Scholar] [CrossRef]
- Nisoli, E.; Clementi, E.; Paolucci, C.; Cozzi, V.; Tonello, C.; Sciorati, C.; Bracale, R.; Valerio, A.; Francolini, M.; Moncada, S.; et al. Mitochondrial biogenesis in mammals: The role of endogenous nitric oxide. Science 2003, 299, 896–899. [Google Scholar] [CrossRef]
- Buler, M.; Aatsinki, S.M.; Skoumal, R.; Komka, Z.; Toth, M.; Kerkela, R.; Georgiadi, A.; Kersten, S.; Hakkola, J. Energy-sensing factors coactivator peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1alpha) and AMP-activated protein kinase control expression of inflammatory mediators in liver: Induction of interleukin 1 receptor antagonist. J. Biol. Chem. 2012, 287, 1847–1860. [Google Scholar] [CrossRef]
- Kruszynska, Y.T.; Mukherjee, R.; Jow, L.; Dana, S.; Paterniti, J.R.; Olefsky, J.M. Skeletal muscle peroxisome proliferator-activated receptor-gamma expression in obesity and non-insulin-dependent diabetes mellitus. J. Clin. Investig. 1998, 101, 543–548. [Google Scholar] [CrossRef]
- Wilkins, H.M.; Weidling, I.; Koppel, S.; Wang, X.; von Schulze, A.; Swerdlow, R.H. Mitochondrial Function and Neurodegenerative Diseases. In The Molecular and Cellular Basis of Neurodegenerative Diseases; Academic Press: Cambridge, MA, USA, 2018; pp. 369–414. [Google Scholar]
- Pagel-Langenickel, I.; Bao, J.; Joseph, J.J.; Schwartz, D.R.; Mantell, B.S.; Xu, X.; Raghavachari, N.; Sack, M.N. PGC-1α integrates insulin signaling, mitochondrial regulation, and bioenergetic function in skeletal muscle. J. Biol. Chem. 2008, 283, 22464–22472. [Google Scholar] [CrossRef]
- Phielix, E.; Schrauwen-Hinderling, V.B.; Mensink, M.; Lenaers, E.; Meex, R.; Hoeks, J.; Kooi, M.E.; Moonen-Kornips, E.; Sels, J.P.; Hesselink, M.K.; et al. Lower intrinsic ADP-stimulated mitochondrial respiration underlies in vivo mitochondrial dysfunction in muscle of male type 2 diabetic patients. Diabetes 2008, 57, 2943–2949. [Google Scholar] [CrossRef]
- Chanseaume, E.; Malpuech-Brugere, C.; Patrac, V.; Bielicki, G.; Rousset, P.; Couturier, K.; Salles, J.; Renou, J.P.; Boirie, Y.; Morio, B. Diets high in sugar, fat, and energy induce muscle type-specific adaptations in mitochondrial functions in rats. J. Nutr. 2006, 136, 2194–2200. [Google Scholar] [CrossRef]
- El-Mir, M.Y.; Nogueira, V.; Fontaine, E.; Averet, N.; Rigoulet, M.; Leverve, X. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J. Biol. Chem. 2000, 275, 223–228. [Google Scholar] [CrossRef]
- Gaster, M.; Rustan, A.C.; Aas, V.; Beck-Nielsen, H. Reduced lipid oxidation in skeletal muscle from type 2 diabetic subjects may be of genetic origin: Evidence from cultured myotubes. Diabetes 2004, 53, 542–548. [Google Scholar] [CrossRef]
- Ørtenblad, N.; Mogensen, M.; Petersen, I.; Højlund, K.; Levin, K.; Sahlin, K.; Beck-Nielsen, H.; Gaster, M. Reduced insulin-mediated citrate synthase activity in cultured skeletal muscle cells from patients with type 2 diabetes: Evidence for an intrinsic oxidative enzyme defect. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2005, 1741, 206–214. [Google Scholar] [CrossRef]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yap, K.H.; Yee, G.S.; Candasamy, M.; Tan, S.C.; Md, S.; Abdul Majeed, A.B.; Bhattamisra, S.K. Catalpol Ameliorates Insulin Sensitivity and Mitochondrial Respiration in Skeletal Muscle of Type-2 Diabetic Mice Through Insulin Signaling Pathway and AMPK/SIRT1/PGC-1α/PPAR-γ Activation. Biomolecules 2020, 10, 1360. https://doi.org/10.3390/biom10101360
Yap KH, Yee GS, Candasamy M, Tan SC, Md S, Abdul Majeed AB, Bhattamisra SK. Catalpol Ameliorates Insulin Sensitivity and Mitochondrial Respiration in Skeletal Muscle of Type-2 Diabetic Mice Through Insulin Signaling Pathway and AMPK/SIRT1/PGC-1α/PPAR-γ Activation. Biomolecules. 2020; 10(10):1360. https://doi.org/10.3390/biom10101360
Chicago/Turabian StyleYap, Kah Heng, Gan Sook Yee, Mayuren Candasamy, Swee Ching Tan, Shadab Md, Abu Bakar Abdul Majeed, and Subrat Kumar Bhattamisra. 2020. "Catalpol Ameliorates Insulin Sensitivity and Mitochondrial Respiration in Skeletal Muscle of Type-2 Diabetic Mice Through Insulin Signaling Pathway and AMPK/SIRT1/PGC-1α/PPAR-γ Activation" Biomolecules 10, no. 10: 1360. https://doi.org/10.3390/biom10101360
APA StyleYap, K. H., Yee, G. S., Candasamy, M., Tan, S. C., Md, S., Abdul Majeed, A. B., & Bhattamisra, S. K. (2020). Catalpol Ameliorates Insulin Sensitivity and Mitochondrial Respiration in Skeletal Muscle of Type-2 Diabetic Mice Through Insulin Signaling Pathway and AMPK/SIRT1/PGC-1α/PPAR-γ Activation. Biomolecules, 10(10), 1360. https://doi.org/10.3390/biom10101360