YB-1 Mediates TNF-Induced Pro-Survival Signaling by Regulating NF-κB Activation

 ,
,  and
and
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
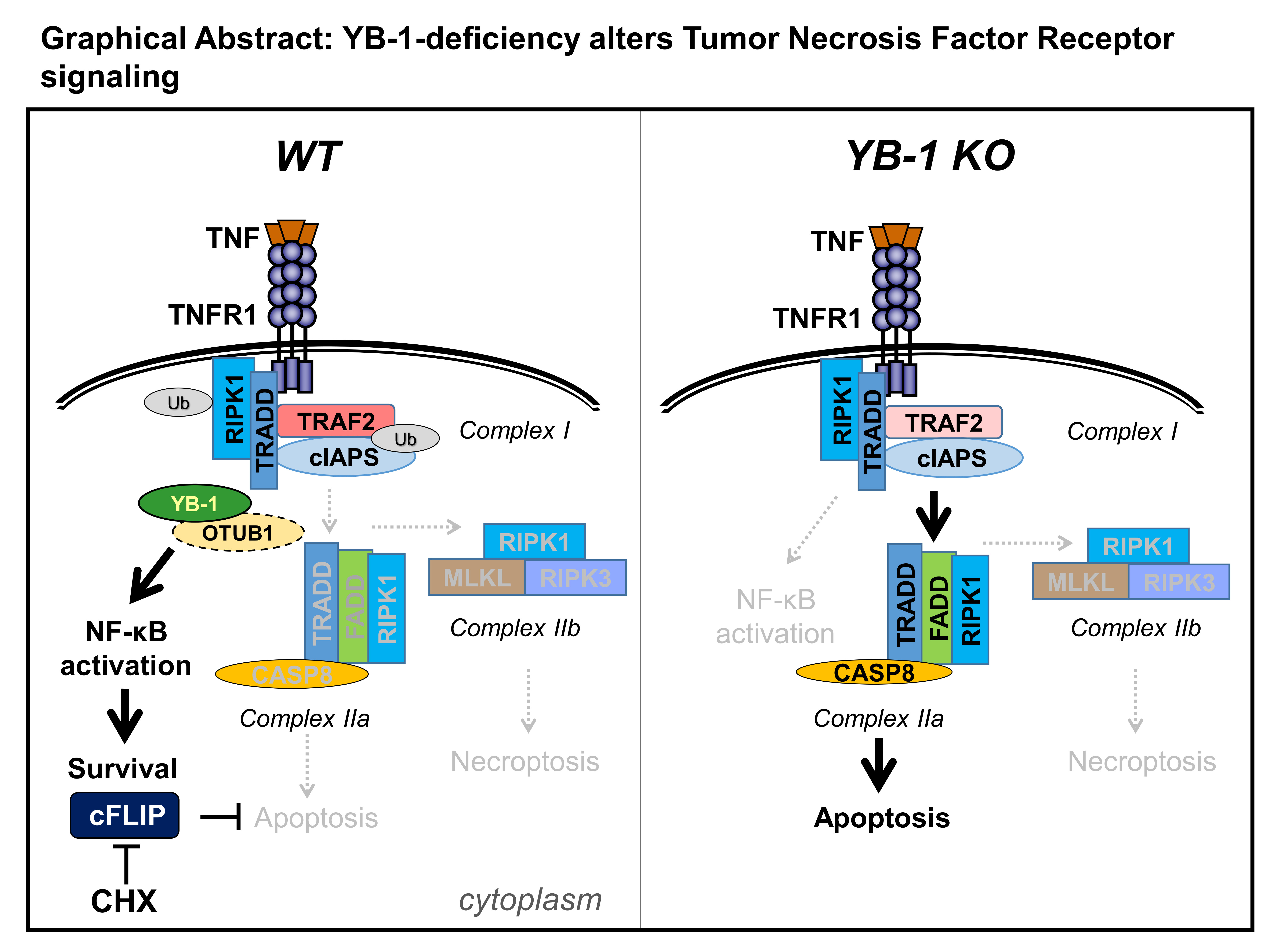
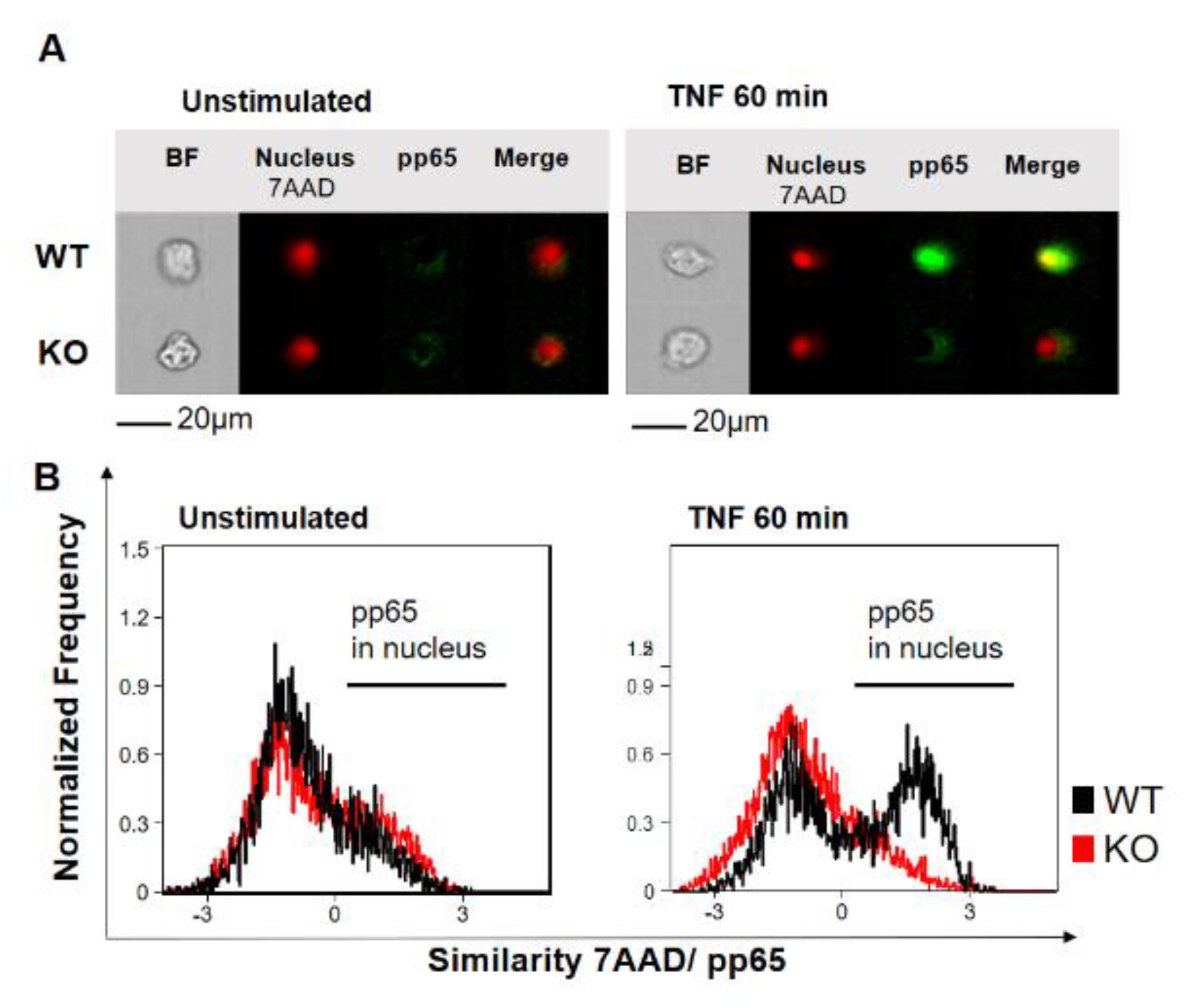
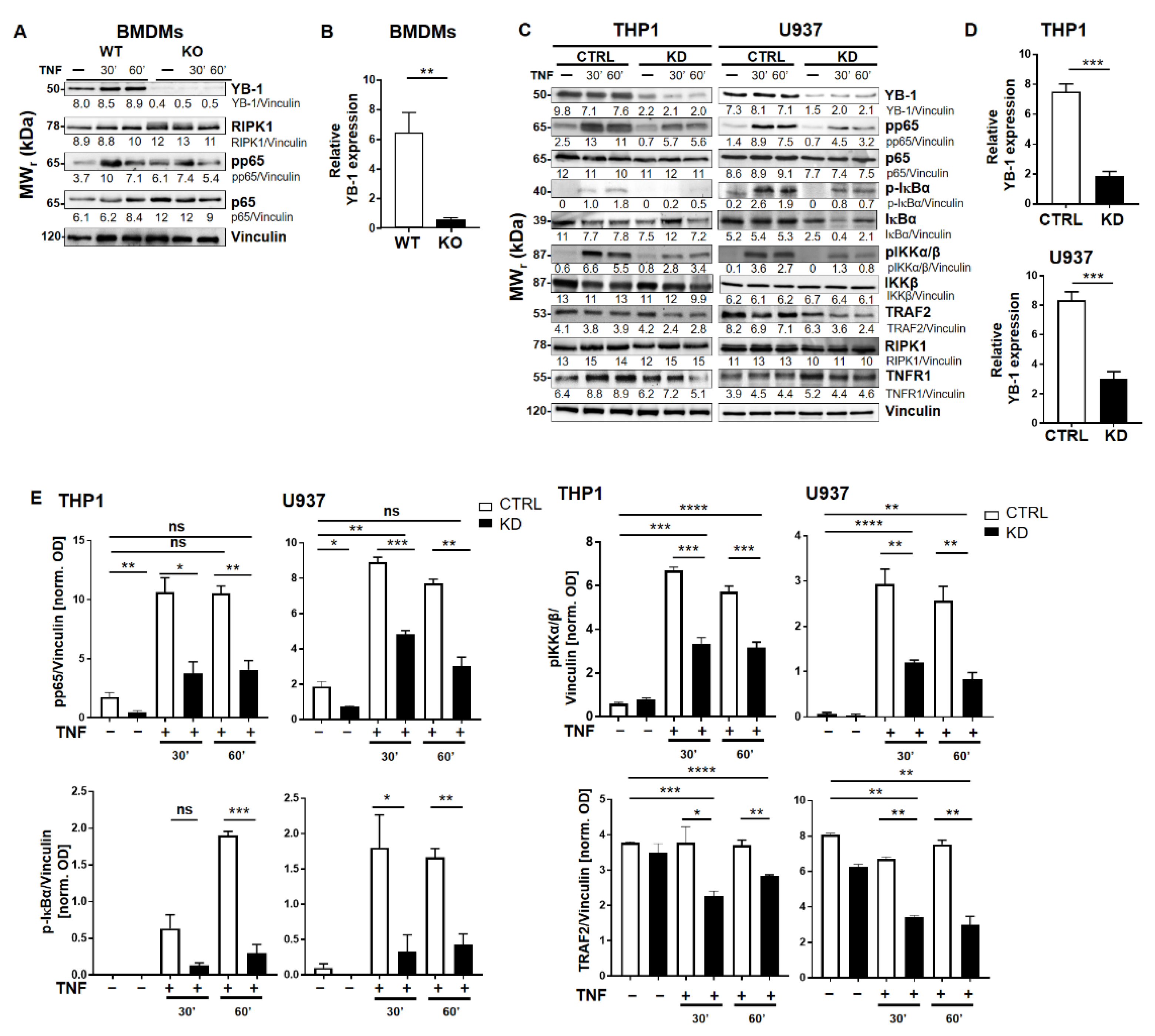
2.1. YB-1 Was Responsible for TNF-Induced NF-κB Activation
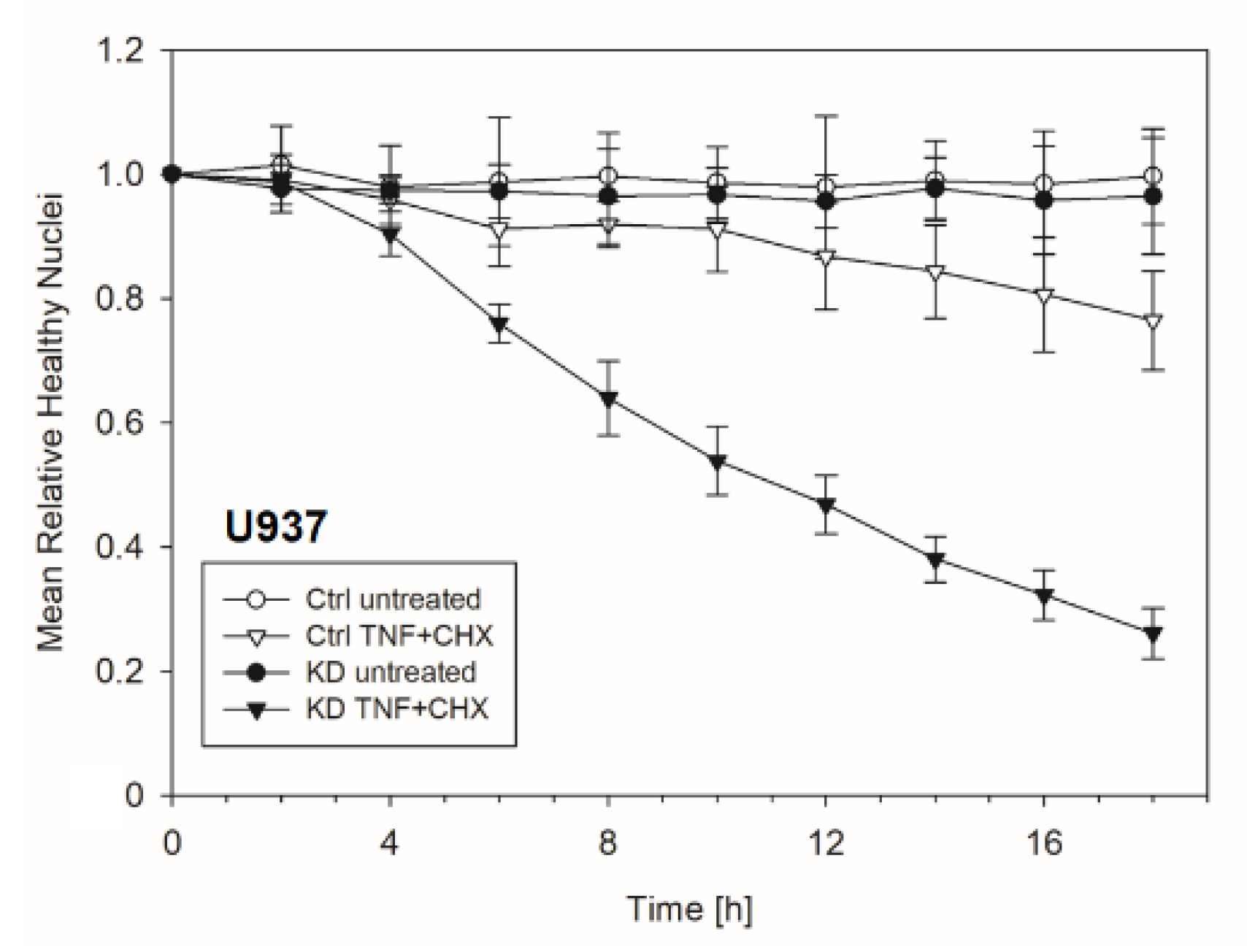
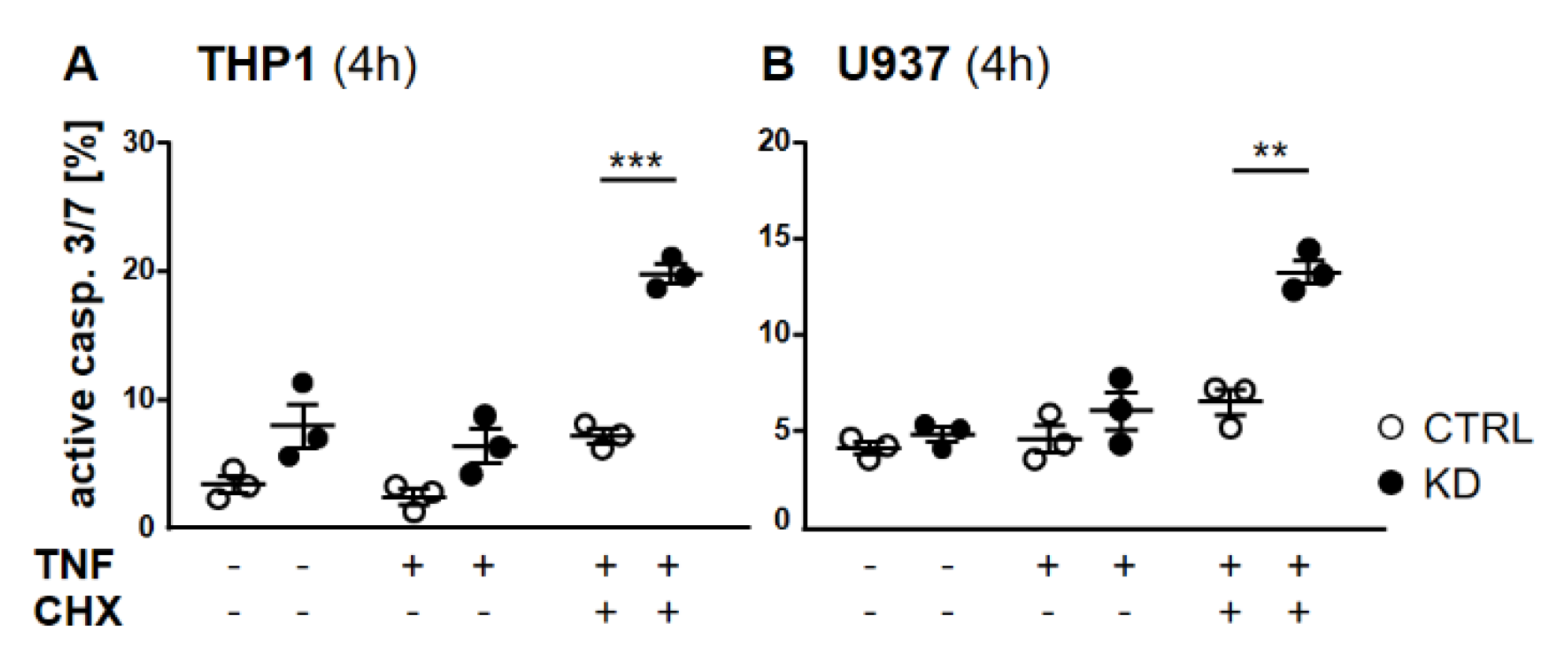
2.2. In the Absence of YB-1, TNFR Activation Induced Apoptosis
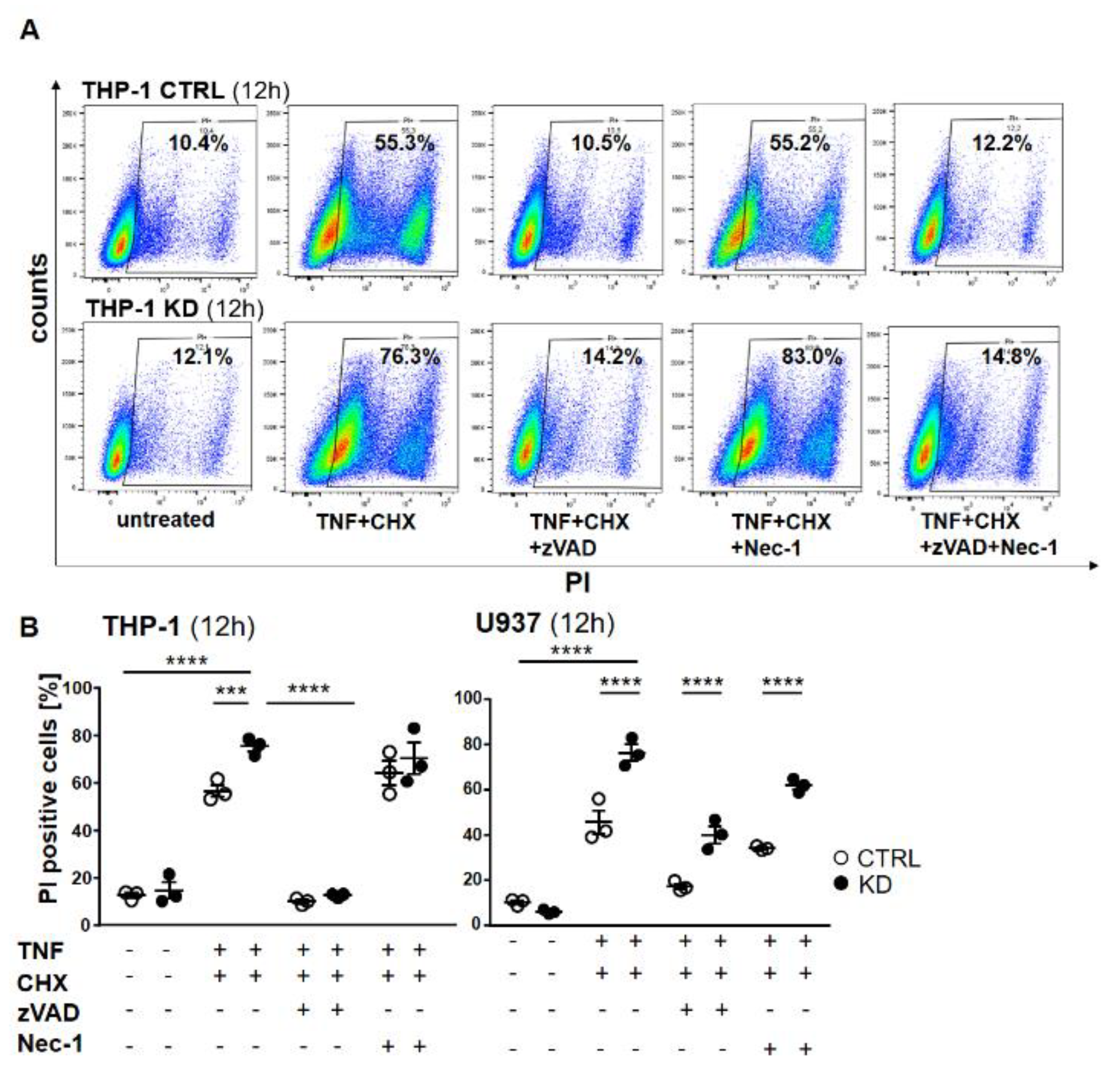
2.3. YB-1-Deficient Cells Died Largely Via Apoptosis
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Stimulation
4.2. Bone Marrow-Derived Macrophages
4.3. Lentiviral Transduction of YB-1
4.4. SDS-PAGE and Western Blotting
4.5. Imaging Flow Cytometry
4.6. Cell Death Assays
4.7. Time-Lapse Microscopy
4.8. Image Segmentation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kohno, K.; Izumi, H.; Uchiumi, T.; Ashizuka, M.; Kuwano, M. The pleiotropic functions of the Y-box-binding protein, YB-1. Bioessays 2003, 25, 691–698. [Google Scholar] [CrossRef] [PubMed]
- Lasham, A.; Print, C.G.; Woolley, A.G.; Dunn, S.E.; Braithwaite, A.W. YB-1: Oncoprotein, prognostic marker and therapeutic target? Biochem. J. 2013, 449, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Raffetseder, U.; Liehn, E.A.; Weber, C.; Mertens, P.R. Role of cold shock Y-box protein-1 in inflammation, atherosclerosis and organ transplant rejection. Eur. J. Cell. Biol. 2012, 91, 567–575. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, T.; Izumi, H.; Imamura, T.; Takano, H.; Ise, T.; Uchiumi, T.; Kuwano, M.; Kohno, K. Direct interaction of p53 with the Y-box binding protein, YB-1: A mechanism for regulation of human gene expression. Oncogene 2000, 19, 6194–6202. [Google Scholar] [CrossRef][Green Version]
- Holm, P.S.; Bergmann, S.; Jurchott, K.; Lage, H.; Brand, K.; Ladhoff, A.; Mantwill, K.; Curiel, D.T.; Dobbelstein, M.; Dietel, M.; et al. YB-1 relocates to the nucleus in adenovirus-infected cells and facilitates viral replication by inducing E2 gene expression through the E2 late promoter. J. Biol. Chem. 2002, 277, 10427–10434. [Google Scholar] [CrossRef]
- Lindquist, J.A.; Mertens, P.R. Cold shock proteins: From cellular mechanisms to pathophysiology and disease. Cell Commun. Signal. 2018, 16, 63. [Google Scholar] [CrossRef]
- Vandenabeele, P.; Galluzzi, L.; Vanden Berghe, T.; Kroemer, G. Molecular mechanisms of necroptosis: An ordered cellular explosion. Nat. Rev. Mol. Cell Biol. 2010, 11, 700–714. [Google Scholar] [CrossRef]
- Chung, J.Y.; Lu, M.; Yin, Q.; Wu, H. Structural revelations of TRAF2 function in TNF receptor signaling pathway. Adv. Exp. Med. Biol. 2007, 597, 93–113. [Google Scholar] [CrossRef]
- Hsu, H.; Xiong, J.; Goeddel, D.V. The TNF receptor 1-associated protein TRADD signals cell death and NF-kappa B activation. Cell 1995, 81, 495–504. [Google Scholar] [CrossRef]
- Emmerich, C.H.; Schmukle, A.C.; Walczak, H. The emerging role of linear ubiquitination in cell signaling. Sci. Signal. 2011, 4, re5. [Google Scholar] [CrossRef]
- Vucic, D.; Dixit, V.M.; Wertz, I.E. Ubiquitylation in apoptosis: A post-translational modification at the edge of life and death. Nat. Rev. Mol. Cell Biol. 2011, 12, 439–452. [Google Scholar] [CrossRef] [PubMed]
- Van Antwerp, D.J.; Martin, S.J.; Kafri, T.; Green, D.R.; Verma, I.M. Suppression of TNF-alpha-induced apoptosis by NF-kappaB. Science 1996, 274, 787–789. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.Y.; Mayo, M.W.; Korneluk, R.G.; Goeddel, D.V.; Baldwin, A.S., Jr. NF-kappaB antiapoptosis: Induction of TRAF1 and TRAF2 and c-IAP1 and c-IAP2 to suppress caspase-8 activation. Science 1998, 281, 1680–1683. [Google Scholar] [CrossRef] [PubMed]
- Kreuz, S.; Siegmund, D.; Scheurich, P.; Wajant, H. NF-kappaB inducers upregulate cFLIP, a cycloheximide-sensitive inhibitor of death receptor signaling. Mol. Cell. Biol. 2001, 21, 3964–3973. [Google Scholar] [CrossRef] [PubMed]
- Papa, S.; Zazzeroni, F.; Bubici, C.; Jayawardena, S.; Alvarez, K.; Matsuda, S.; Nguyen, D.U.; Pham, C.G.; Nelsbach, A.H.; Melis, T.; et al. Gadd45 beta mediates the NF-kappa B suppression of JNK signalling by targeting MKK7/JNKK2. Nat. Cell Biol. 2004, 6, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.Y.; Mayo, M.W.; Baldwin, A.S., Jr. TNF- and cancer therapy-induced apoptosis: Potentiation by inhibition of NF-kappaB. Science 1996, 274, 784–787. [Google Scholar] [CrossRef]
- Micheau, O.; Tschopp, J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 2003, 114, 181–190. [Google Scholar] [CrossRef]
- Prabhu, L.; Mundade, R.; Wang, B.; Wei, H.; Hartley, A.V.; Martin, M.; McElyea, K.; Temm, C.J.; Sandusky, G.; Liu, Y.; et al. Critical role of phosphorylation of serine 165 of YBX1 on the activation of NF-kappaB in colon cancer. Oncotarget 2015, 6, 29396–29412. [Google Scholar] [CrossRef]
- Martin, M.; Hua, L.; Wang, B.; Wei, H.; Prabhu, L.; Hartley, A.V.; Jiang, G.; Liu, Y.; Lu, T. Novel Serine 176 Phosphorylation of YBX1 Activates NF-kappaB in Colon Cancer. J. Biol. Chem. 2017, 292, 3433–3444. [Google Scholar] [CrossRef]
- Vanden Berghe, T.; Linkermann, A.; Jouan-Lanhouet, S.; Walczak, H.; Vandenabeele, P. Regulated necrosis: The expanding network of non-apoptotic cell death pathways. Nat. Rev. Mol. Cell Biol. 2014, 15, 135–147. [Google Scholar] [CrossRef]
- He, S.; Wang, L.; Miao, L.; Wang, T.; Du, F.; Zhao, L.; Wang, X. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell 2009, 137, 1100–1111. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.S.; Challa, S.; Moquin, D.; Genga, R.; Ray, T.D.; Guildford, M.; Chan, F.K. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 2009, 137, 1112–1123. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Wang, H.; Wang, Z.; He, S.; Chen, S.; Liao, D.; Wang, L.; Yan, J.; Liu, W.; Lei, X.; et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 2012, 148, 213–227. [Google Scholar] [CrossRef] [PubMed]
- Murphy, J.M.; Czabotar, P.E.; Hildebrand, J.M.; Lucet, I.S.; Zhang, J.G.; Alvarez-Diaz, S.; Lewis, R.; Lalaoui, N.; Metcalf, D.; Webb, A.I.; et al. The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity 2013, 39, 443–453. [Google Scholar] [CrossRef]
- Degterev, A.; Hitomi, J.; Germscheid, M.; Ch’en, I.L.; Korkina, O.; Teng, X.; Abbott, D.; Cuny, G.D.; Yuan, C.; Wagner, G.; et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat. Chem. Biol. 2008, 4, 313–321. [Google Scholar] [CrossRef]
- Bernhardt, A.; Fehr, A.; Brandt, S.; Jerchel, S.; Ballhause, T.M.; Philipsen, L.; Stolze, S.; Geffers, R.; Weng, H.; Fischer, K.D.; et al. Inflammatory cell infiltration and resolution of kidney inflammation is orchestrated by the cold-shock protein Y-box binding protein-1. Kidney Int. 2017, 92, 1157–1177. [Google Scholar] [CrossRef]
- Hohlfeld, R.; Brandt, S.; Bernhardt, A.; Gorny, X.; Schindele, D.; Jandrig, B.; Schostak, M.; Isermann, B.; Lindquist, J.A.; Mertens, P.R. Crosstalk between Akt signaling and cold shock proteins in mediating invasive cell phenotypes. Oncotarget 2018, 9, 19039–19049. [Google Scholar] [CrossRef]
- Salvesen, G.S.; Dixit, V.M. Caspases: Intracellular signaling by proteolysis. Cell 1997, 91, 443–446. [Google Scholar] [CrossRef]
- Thornberry, N.A.; Lazebnik, Y. Caspases: Enemies within. Science 1998, 281, 1312–1316. [Google Scholar] [CrossRef]
- Zinngrebe, J.; Montinaro, A.; Peltzer, N.; Walczak, H. Ubiquitin in the immune system. EMBO Rep. 2014, 15, 28–45. [Google Scholar] [CrossRef]
- Peltzer, N.; Darding, M.; Walczak, H. Holding RIPK1 on the Ubiquitin Leash in TNFR1 Signaling. Trends Cell Biol. 2016, 26, 445–461. [Google Scholar] [CrossRef] [PubMed]
- Wagner, S.A.; Satpathy, S.; Beli, P.; Choudhary, C. SPATA2 links CYLD to the TNF-alpha receptor signaling complex and modulates the receptor signaling outcomes. EMBO J. 2016, 35, 1868–1884. [Google Scholar] [CrossRef] [PubMed]
- Hornbeck, P.V.; Zhang, B.; Murray, B.; Kornhauser, J.M.; Latham, V.; Skrzypek, E. PhosphoSitePlus, 2014: Mutations, PTMs and recalibrations. Nucleic Acids Res. 2015, 43, D512–D520. [Google Scholar] [CrossRef] [PubMed]
- Tortola, L.; Nitsch, R.; Bertrand, M.J.M.; Kogler, M.; Redouane, Y.; Kozieradzki, I.; Uribesalgo, I.; Fennell, L.M.; Daugaard, M.; Klug, H.; et al. The Tumor Suppressor Hace1 Is a Critical Regulator of TNFR1-Mediated Cell Fate. Cell Rep. 2016, 15, 1481–1492. [Google Scholar] [CrossRef]
- Palicharla, V.R.; Maddika, S. HACE1 mediated K27 ubiquitin linkage leads to YB-1 protein secretion. Cell. Signal. 2015, 27, 2355–2362. [Google Scholar] [CrossRef]
- Dong, W.; Wang, H.; Shahzad, K.; Bock, F.; Al-Dabet, M.M.; Ranjan, S.; Wolter, J.; Kohli, S.; Hoffmann, J.; Dhople, V.M.; et al. Activated Protein C Ameliorates Renal Ischemia-Reperfusion Injury by Restricting Y-Box Binding Protein-1 Ubiquitination. J. Am. Soc. Nephrol. 2015, 26, 2789–2799. [Google Scholar] [CrossRef]
- Mellacheruvu, D.; Wright, Z.; Couzens, A.L.; Lambert, J.P.; St-Denis, N.A.; Li, T.; Miteva, Y.V.; Hauri, S.; Sardiu, M.E.; Low, T.Y.; et al. The CRAPome: A contaminant repository for affinity purification-mass spectrometry data. Nat. Methods 2013, 10, 730–736. [Google Scholar] [CrossRef]
- Weischenfeldt, J.; Porse, B. Bone Marrow-Derived Macrophages (BMM): Isolation and Applications. CSH Protoc. 2008, 2008, pdb prot5080. [Google Scholar] [CrossRef]
- Draber, P.; Kupka, S.; Reichert, M.; Draberova, H.; Lafont, E.; de Miguel, D.; Spilgies, L.; Surinova, S.; Taraborrelli, L.; Hartwig, T.; et al. LUBAC-Recruited CYLD and A20 Regulate Gene Activation and Cell Death by Exerting Opposing Effects on Linear Ubiquitin in Signaling Complexes. Cell Rep. 2015, 13, 2258–2272. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shah, A.; Plaza-Sirvent, C.; Weinert, S.; Buchbinder, J.H.; Lavrik, I.N.; Mertens, P.R.; Schmitz, I.; Lindquist, J.A. YB-1 Mediates TNF-Induced Pro-Survival Signaling by Regulating NF-κB Activation. Cancers 2020, 12, 2188. https://doi.org/10.3390/cancers12082188
Shah A, Plaza-Sirvent C, Weinert S, Buchbinder JH, Lavrik IN, Mertens PR, Schmitz I, Lindquist JA. YB-1 Mediates TNF-Induced Pro-Survival Signaling by Regulating NF-κB Activation. Cancers. 2020; 12(8):2188. https://doi.org/10.3390/cancers12082188
Chicago/Turabian StyleShah, Aneri, Carlos Plaza-Sirvent, Sönke Weinert, Jörn H. Buchbinder, Inna N. Lavrik, Peter R. Mertens, Ingo Schmitz, and Jonathan A. Lindquist. 2020. "YB-1 Mediates TNF-Induced Pro-Survival Signaling by Regulating NF-κB Activation" Cancers 12, no. 8: 2188. https://doi.org/10.3390/cancers12082188
APA StyleShah, A., Plaza-Sirvent, C., Weinert, S., Buchbinder, J. H., Lavrik, I. N., Mertens, P. R., Schmitz, I., & Lindquist, J. A. (2020). YB-1 Mediates TNF-Induced Pro-Survival Signaling by Regulating NF-κB Activation. Cancers, 12(8), 2188. https://doi.org/10.3390/cancers12082188