Sickle Cell Disease: A Paradigm for Venous Thrombosis Pathophysiology

Abstract
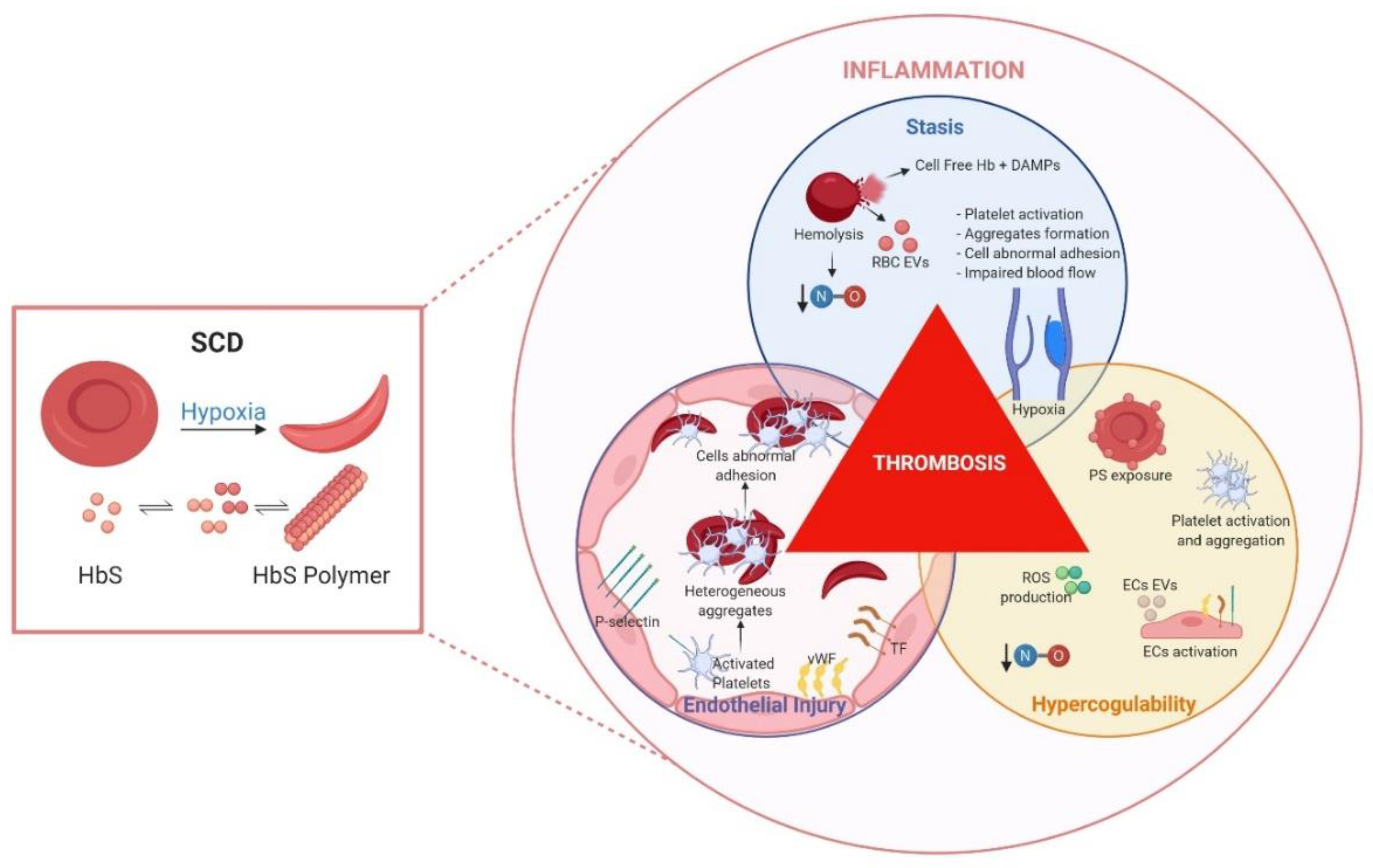
1. Venous Thromboembolism and Sickle Cell Disease
2. Animal Models of Disease Pathophysiology
2.1. Murine Models of VTE
2.2. Sickle Cell Disease Animal Models
3. Insights into VTE Pathophysiology Using SCD Mouse Models
3.1. Hypercoagulability
3.2. Inflammation and Endothelial Injury
3.3. Blood Stasis
4. Perspectives
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ACS | Acute chest syndrome |
| ADP | Adenosine diphosphate |
| BERK | Berkeley mouse |
| CLEC-2 | Platelet C–type lectin-like receptor 2 |
| CTEPH | Chronic thromboembolic pulmonary hypertension |
| DAMPs | Damage-associated molecular patterns |
| DNA | Deoxyribonucleic acid |
| DVT | Deep venous thrombosis |
| ECs | Endothelial cells |
| EPCR | Endothelial protein C receptor |
| EVs | Extracellular vesicles |
| F1.2 | Prothrombin factor 1.2 |
| Hb | Hemoglobin |
| HbSC | Heterozygote for HbS |
| HbSS | Homozygote for HbS |
| HbS | Hemoglobin S |
| HbSAntilles | Hemoglobin S Antilles mice model |
| HMGB1 | High-mobility group box 1 protein |
| HO-1 | Heme oxygenase 1 |
| I/R | Ischemia/Reperfusion |
| ICAM | Intercellular Adhesion Molecule |
| IL-X | Interleukin-X |
| IVC | Inferior vena cava |
| LCR | Locus control region |
| MCHC | Mean corpuscular hemoglobin concentration |
| NETs | Neutrophil extracellular traps |
| NLR | Nucleotide binding domain-like receptor |
| NO | Nitric oxide |
| PARs | Protease activated receptors |
| PCVs | Packed cell volumes |
| PE | Pulmonary embolism |
| PRR | Pattern recognition receptors |
| PS | Phosphatidylserine |
| RBC | Red blood cells |
| ROS | Reactive oxidative species |
| SAD | βS-AntillesD-Punjab mice model |
| SCD | Sickle cell disease |
| SS RBCs | Sickle red blood cells |
| TAT | Thrombin-antithrombin complex |
| TF | Tissue factor |
| TFPI | TF pathway inhibitor |
| TLR | Toll-like receptor |
| TM | Thrombomodulin |
| TNF-α | Tumor necrosis factor-alpha |
| VCAM | Vascular cell adhesion protein |
| VOCs | Vaso-occlusive crisis |
| VTE | Venous thromboembolism |
| vWF | von Willebrand factor |
References
- Goldhaber, S.Z.; Bounameaux, H. Pulmonary embolism and deep vein thrombosis. Lancet 2012, 379, 1835–1846. [Google Scholar] [CrossRef]
- Heit, J.A.; Spencer, F.A.; White, R.H. The epidemiology of venous thromboembolism. J. Thromb. Thrombolysis 2016, 41, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Diaz, J.A.; Obi, A.T.; Myers, D.D., Jr.; Wrobleski, S.K.; Henke, P.K.; Mackman, N.; Wakefield, T.W. Critical review of mouse models of venous thrombosis. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 556–562. [Google Scholar] [CrossRef]
- Piel, F.B.; Tatem, A.J.; Huang, Z.; Gupta, S.; Williams, T.N.; Weatherall, D.J. Global migration and the changing distribution of sickle haemoglobin: A quantitative study of temporal trends between 1960 and 2000. Lancet Glob. Health 2014, 2, e80–e89. [Google Scholar] [CrossRef]
- Ingram, V.M. A specific chemical difference between the globins of normal human and sickle-cell anaemia haemoglobin. Nature 1956, 178, 792–794. [Google Scholar] [CrossRef]
- Stuart, M.J.; Nagel, R.L. Sickle-cell disease. Lancet 2004, 364, 1343–1360. [Google Scholar] [CrossRef]
- Steinberg, M.H. Sickle cell anemia, the first molecular disease: Overview of molecular etiology, pathophysiology, and therapeutic approaches. Sci. World J. 2008, 8, 1295–1324. [Google Scholar] [CrossRef] [PubMed]
- Kato, G.J.; Piel, F.B.; Reid, C.D.; Gaston, M.H.; Ohene-Frempong, K.; Krishnamurti, L.; Smith, W.R.; Panepinto, J.A.; Weatherall, D.J.; Costa, F.F.; et al. Sickle cell disease. Nat. Rev. Dis. Primers 2018, 4, 18010. [Google Scholar] [CrossRef]
- Novelli, E.M.; Huynh, C.; Gladwin, M.T.; Moore, C.G.; Ragni, M.V. Pulmonary embolism in sickle cell disease: A case-control study. J. Thromb. Haemost. 2012, 10, 760–766. [Google Scholar] [CrossRef]
- Naik, R.P.; Streiff, M.B.; Haywood, C., Jr.; Nelson, J.A.; Lanzkron, S. Venous thromboembolism in adults with sickle cell disease: A serious and under-recognized complication. Am. J. Med. 2013, 126, 443–449. [Google Scholar] [CrossRef]
- Naik, R.P.; Streiff, M.B.; Haywood, C., Jr.; Segal, J.B.; Lanzkron, S. Venous thromboembolism incidence in the Cooperative Study of Sickle Cell Disease. J. Thromb. Haemost. 2014, 12, 2010–2016. [Google Scholar] [CrossRef] [PubMed]
- Brunson, A.; Lei, A.; Rosenberg, A.S.; White, R.H.; Keegan, T.; Wun, T. Increased incidence of VTE in sickle cell disease patients: Risk factors, recurrence and impact on mortality. Br. J. Haematol. 2017, 178, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.T.; Nelson, J.; Streiff, M.B.; Lanzkron, S.; Naik, R.P. Risk factors for venous thromboembolism in adults with hemoglobin SC or Sbeta(+) thalassemia genotypes. Thromb. Res. 2016, 141, 35–38. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Adedeji, M.O.; Cespedes, J.; Allen, K.; Subramony, C.; Hughson, M.D. Pulmonary thrombotic arteriopathy in patients with sickle cell disease. Arch. Pathol. Lab. Med. 2001, 125, 1436–1441. [Google Scholar] [PubMed]
- Faes, C.; Ilich, A.; Sotiaux, A.; Sparkenbaugh, E.M.; Henderson, M.W.; Buczek, L.; Beckman, J.D.; Ellsworth, P.; Noubouossie, D.F.; Bhoopat, L.; et al. Red blood cells modulate structure and dynamics of venous clot formation in sickle cell disease. Blood 2019, 133, 2529–2541. [Google Scholar] [CrossRef] [PubMed]
- Kassim, A.A.; Galadanci, N.A.; Pruthi, S.; DeBaun, M.R. How I treat and manage strokes in sickle cell disease. Blood 2015, 125, 3401–3410. [Google Scholar] [CrossRef] [PubMed]
- Byrnes, J.R.; Wolberg, A.S. New findings on venous thrombogenesis. Hamostaseologie 2017, 37, 25–35. [Google Scholar] [CrossRef]
- Wun, T.; Brunson, A. Sickle cell disease: An inherited thrombophilia. Hematol. Am. Soc. Hematol. Educ. Program. 2016, 2016, 640–647. [Google Scholar] [CrossRef]
- Shet, A.S.; Wun, T. How I diagnose and treat venous thromboembolism in sickle cell disease. Blood 2018, 132, 1761–1769. [Google Scholar] [CrossRef]
- Ataga, K.I.; Moore, C.G.; Hillery, C.A.; Jones, S.; Whinna, H.C.; Strayhorn, D.; Sohier, C.; Hinderliter, A.; Parise, L.V.; Orringer, E.P. Coagulation activation and inflammation in sickle cell disease-associated pulmonary hypertension. Haematologica 2008, 93, 20–26. [Google Scholar] [CrossRef]
- Sparkenbaugh, E.; Pawlinski, R. Prothrombotic aspects of sickle cell disease. J. Thromb. Haemost. 2017, 15, 1307–1316. [Google Scholar] [CrossRef] [PubMed]
- Tomer, A.; Harker, L.A.; Kasey, S.; Eckman, J.R. Thrombogenesis in sickle cell disease. J. Lab. Clin. Med. 2001, 137, 398–407. [Google Scholar] [CrossRef] [PubMed]
- Setty, B.N.; Rao, A.K.; Stuart, M.J. Thrombophilia in sickle cell disease: The red cell connection. Blood 2001, 98, 3228–3233. [Google Scholar] [CrossRef] [PubMed]
- Francis, R.B. Platelets, coagulation, and fibrinolysis in sickle cell disease: Their possible role in vascular occlusion. Blood Coagul. Fibrinolysis 1991, 2, 341–353. [Google Scholar] [CrossRef]
- Shet, A.S.; Aras, O.; Gupta, K.; Hass, M.J.; Rausch, D.J.; Saba, N.; Koopmeiners, L.; Key, N.S.; Hebbel, R.P. Sickle blood contains tissue factor-positive microparticles derived from endothelial cells and monocytes. Blood 2003, 102, 2678–2683. [Google Scholar] [CrossRef]
- Conran, N.; De Paula, E.V. Thromboinflammatory mechanisms in sickle cell disease-challenging the hemostatic balance. Haematologica 2020. [Google Scholar] [CrossRef]
- Dentali, F.; Ageno, W.; Bozzato, S.; Malato, A.; Gianni, M.; Squizzato, A.; Prisco, D. Role of factor V Leiden or G20210A prothrombin mutation in patients with symptomatic pulmonary embolism and deep vein thrombosis: A meta-analysis of the literature. J. Thromb. Haemost. 2012, 10, 732–737. [Google Scholar] [CrossRef]
- Cleuren, A.C.; van Vlijmen, B.J.; Reitsma, P.H. Transgenic mouse models of venous thrombosis: Fulfilling the expectations? Semin. Thromb. Hemost. 2007, 33, 610–616. [Google Scholar] [CrossRef]
- Diaz, J.A.; Saha, P.; Cooley, B.; Palmer, O.R.; Grover, S.P.; Mackman, N.; Wakefield, T.W.; Henke, P.K.; Smith, A.; Lal, B.K. Choosing a Mouse Model of Venous Thrombosis. Arter. Thromb. Vasc. Biol. 2019, 39, 311–318. [Google Scholar] [CrossRef]
- Fabry, M.E. Transgenic animal models of sickle cell disease. Experientia 1993, 49, 28–36. [Google Scholar] [CrossRef]
- Beuzard, Y. Mouse models of sickle cell disease. Transfus. Clin. Biol. 2008, 15, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Belcher, J.D.; Bryant, C.J.; Nguyen, J.; Bowlin, P.R.; Kielbik, M.C.; Bischof, J.C.; Hebbel, R.P.; Vercellotti, G.M. Transgenic sickle mice have vascular inflammation. Blood 2003, 101, 3953–3959. [Google Scholar] [CrossRef] [PubMed]
- Kaul, D.K.; Hebbel, R.P. Hypoxia/reoxygenation causes inflammatory response in transgenic sickle mice but not in normal mice. J. Clin. Investig. 2000, 106, 411–420. [Google Scholar] [CrossRef] [PubMed]
- Aufradet, E.; DeSouza, G.; Bourgeaux, V.; Bessaad, A.; Campion, Y.; Canet-Soulas, E.; Pialoux, V.; Chirico, E.N.; Chevrier, A.M.; Godfrin, Y.; et al. Hypoxia/reoxygenation stress increases markers of vaso-occlusive crisis in sickle SAD mice. Clin. Hemorheol. Microcirc. 2013, 54, 297–312. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, K.A., Jr.; Ou, J.; Ou, Z.; Shi, Y.; Franciosi, J.P.; Signorino, P.; Kaul, S.; Ackland-Berglund, C.; Witte, K.; Holzhauer, S.; et al. Hypoxia-induced acute lung injury in murine models of sickle cell disease. Am. J. Physiol. Lung Cell Mol. Physiol. 2004, 286, L705–L714. [Google Scholar] [CrossRef] [PubMed]
- Paszty, C.; Brion, C.M.; Manci, E.; Witkowska, H.E.; Stevens, M.E.; Mohandas, N.; Rubin, E.M. Transgenic knockout mice with exclusively human sickle hemoglobin and sickle cell disease. Science 1997, 278, 876–878. [Google Scholar] [CrossRef] [PubMed]
- Ryan, T.M.; Ciavatta, D.J.; Townes, T.M. Knockout-transgenic mouse model of sickle cell disease. Science 1997, 278, 873–876. [Google Scholar] [CrossRef]
- Gavins, F.N.; Russell, J.; Senchenkova, E.L.; De Almeida Paula, L.; Damazo, A.S.; Esmon, C.T.; Kirchhofer, D.; Hebbel, R.P.; Granger, D.N. Mechanisms of enhanced thrombus formation in cerebral microvessels of mice expressing hemoglobin-S. Blood 2011, 117, 4125–4133. [Google Scholar] [CrossRef]
- Chantrathammachart, P.; Mackman, N.; Sparkenbaugh, E.; Wang, J.G.; Parise, L.V.; Kirchhofer, D.; Key, N.S.; Pawlinski, R. Tissue factor promotes activation of coagulation and inflammation in a mouse model of sickle cell disease. Blood 2012, 120, 636–646. [Google Scholar] [CrossRef]
- Sparkenbaugh, E.M.; Chantrathammachart, P.; Wang, S.; Jonas, W.; Kirchhofer, D.; Gailani, D.; Gruber, A.; Kasthuri, R.; Key, N.S.; Mackman, N.; et al. Excess of heme induces tissue factor-dependent activation of coagulation in mice. Haematologica 2015, 100, 308–314. [Google Scholar] [CrossRef]
- Guo, Y.; Uy, T.; Wandersee, N.; Scott, P.; Weiler, H.; Holzhauer, S.; Retherford, D.; Foster, T.; Hillery, C. The protein C pathway in human and murine sicjle cell disease: Alterations in protein C, thrombomodulin (TM), and endothelial protein C receptor (EPCR) at baseline and during acuste vaso-occlusion. Blood 2016, 112, 538. [Google Scholar] [CrossRef]
- Solovey, A.; Kollander, R.; Shet, A.; Milbauer, L.C.; Choong, S.; Panoskaltsis-Mortari, A.; Blazar, B.R.; Kelm, R.J., Jr.; Hebbel, R.P. Endothelial cell expression of tissue factor in sickle mice is augmented by hypoxia/reoxygenation and inhibited by lovastatin. Blood 2004, 104, 840–846. [Google Scholar] [CrossRef] [PubMed]
- Grover, S.P.; Mackman, N. Tissue Factor: An Essential Mediator of Hemostasis and Trigger of Thrombosis. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 709–725. [Google Scholar] [CrossRef] [PubMed]
- Giesen, P.L.; Rauch, U.; Bohrmann, B.; Kling, D.; Roque, M.; Fallon, J.T.; Badimon, J.J.; Himber, J.; Riederer, M.A.; Nemerson, Y. Blood-borne tissue factor: Another view of thrombosis. Proc. Natl. Acad. Sci. USA 1999, 96, 2311–2315. [Google Scholar] [CrossRef] [PubMed]
- Furie, B.; Furie, B.C. Mechanisms of thrombus formation. N. Engl. J. Med. 2008, 359, 938–949. [Google Scholar] [CrossRef] [PubMed]
- Day, S.M.; Reeve, J.L.; Pedersen, B.; Farris, D.M.; Myers, D.D.; Im, M.; Wakefield, T.W.; Mackman, N.; Fay, W.P. Macrovascular thrombosis is driven by tissue factor derived primarily from the blood vessel wall. Blood 2005, 105, 192–198. [Google Scholar] [CrossRef]
- Yan, S.F.; Zou, Y.S.; Gao, Y.; Zhai, C.; Mackman, N.; Lee, S.L.; Milbrandt, J.; Pinsky, D.; Kisiel, W.; Stern, D. Tissue factor transcription driven by Egr-1 is a critical mechanism of murine pulmonary fibrin deposition in hypoxia. Proc. Natl. Acad. Sci. USA 1998, 95, 8298–8303. [Google Scholar] [CrossRef]
- Lawson, C.A.; Yan, S.D.; Yan, S.F.; Liao, H.; Zhou, Y.S.; Sobel, J.; Kisiel, W.; Stern, D.M.; Pinsky, D.J. Monocytes and tissue factor promote thrombosis in a murine model of oxygen deprivation. J. Clin. Investig. 1997, 99, 1729–1738. [Google Scholar] [CrossRef]
- Setty, B.N.; Betal, S.G.; Zhang, J.; Stuart, M.J. Heme induces endothelial tissue factor expression: Potential role in hemostatic activation in patients with hemolytic anemia. J. Thromb. Haemost. 2008, 6, 2202–2209. [Google Scholar] [CrossRef]
- Solovey, A.; Gui, L.; Key, N.S.; Hebbel, R.P. Tissue factor expression by endothelial cells in sickle cell anemia. J. Clin. Investig. 1998, 101, 1899–1904. [Google Scholar] [CrossRef]
- Key, N.S.; Slungaard, A.; Dandelet, L.; Nelson, S.C.; Moertel, C.; Styles, L.A.; Kuypers, F.A.; Bach, R.R. Whole blood tissue factor procoagulant activity is elevated in patients with sickle cell disease. Blood 1998, 91, 4216–4223. [Google Scholar] [CrossRef]
- Hrachovinova, I.; Cambien, B.; Hafezi-Moghadam, A.; Kappelmayer, J.; Camphausen, R.T.; Widom, A.; Xia, L.; Kazazian, H.H., Jr.; Schaub, R.G.; McEver, R.P.; et al. Interaction of P-selectin and PSGL-1 generates microparticles that correct hemostasis in a mouse model of hemophilia A. Nat. Med. 2003, 9, 1020–1025. [Google Scholar] [CrossRef] [PubMed]
- Thomas, G.M.; Brill, A.; Mezouar, S.; Crescence, L.; Gallant, M.; Dubois, C.; Wagner, D.D. Tissue factor expressed by circulating cancer cell-derived microparticles drastically increases the incidence of deep vein thrombosis in mice. J. Thromb. Haemost. 2015, 13, 1310–1319. [Google Scholar] [CrossRef] [PubMed]
- Gross, P.L.; Furie, B.C.; Merrill-Skoloff, G.; Chou, J.; Furie, B. Leukocyte-versus microparticle-mediated tissue factor transfer during arteriolar thrombus development. J. Leukoc. Biol. 2005, 78, 1318–1326. [Google Scholar] [CrossRef] [PubMed]
- Geddings, J.E.; Hisada, Y.; Boulaftali, Y.; Getz, T.M.; Whelihan, M.; Fuentes, R.; Dee, R.; Cooley, B.C.; Key, N.S.; Wolberg, A.S.; et al. Tissue factor-positive tumor microvesicles activate platelets and enhance thrombosis in mice. J. Thromb. Haemost. 2016, 14, 153–166. [Google Scholar] [CrossRef]
- Witkowski, M.; Landmesser, U.; Rauch, U. Tissue factor as a link between inflammation and coagulation. Trends Cardiovasc Med. 2016, 26, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Sparkenbaugh, E.M.; Chantrathammachart, P.; Mickelson, J.; van Ryn, J.; Hebbel, R.P.; Monroe, D.M.; Mackman, N.; Key, N.S.; Pawlinski, R. Differential contribution of FXa and thrombin to vascular inflammation in a mouse model of sickle cell disease. Blood 2014, 123, 1747–1756. [Google Scholar] [CrossRef]
- Sparkenbaugh, E.M.; Chantrathammachart, P.; Chandarajoti, K.; Mackman, N.; Key, N.S.; Pawlinski, R. Thrombin-independent contribution of tissue factor to inflammation and cardiac hypertrophy in a mouse model of sickle cell disease. Blood 2016, 127, 1371–1373. [Google Scholar] [CrossRef]
- Muller, F.; Mutch, N.J.; Schenk, W.A.; Smith, S.A.; Esterl, L.; Spronk, H.M.; Schmidbauer, S.; Gahl, W.A.; Morrissey, J.H.; Renne, T. Platelet polyphosphates are proinflammatory and procoagulant mediators in vivo. Cell 2009, 139, 1143–1156. [Google Scholar] [CrossRef]
- Sparkenbaugh, E.F.C.; Noubouossie, D.; Kirchhofer, D.K.; Gruber, A.; Key, N.; Rafal, P. FXIIa differentially regulates thrombin generation during steady state and vaso-occlusive crisis in sickle cell mice. Blood 2016, 128, 162. [Google Scholar] [CrossRef]
- Hebbel, R.P.; Belcher, J.D.; Vercellotti, G.M. The multifaceted role of ischemia/reperfusion in sickle cell anemia. J. Clin. Investig. 2020, 130, 1062–1072. [Google Scholar] [CrossRef] [PubMed]
- Pfeiler, S.; Stark, K.; Massberg, S.; Engelmann, B. Propagation of thrombosis by neutrophils and extracellular nucleosome networks. Haematologica 2017, 102, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Jansen, M.P.; Emal, D.; Teske, G.J.; Dessing, M.C.; Florquin, S.; Roelofs, J.J. Release of extracellular DNA influences renal ischemia reperfusion injury by platelet activation and formation of neutrophil extracellular traps. Kidney Int. 2017, 91, 352–364. [Google Scholar] [CrossRef]
- Noubouossie, D.F.; Whelihan, M.F.; Yu, Y.B.; Sparkenbaugh, E.; Pawlinski, R.; Monroe, D.M.; Key, N.S. In vitro activation of coagulation by human neutrophil DNA and histone proteins but not neutrophil extracellular traps. Blood 2017, 129, 1021–1029. [Google Scholar] [CrossRef] [PubMed]
- De Franceschi, L.; Baron, A.; Scarpa, A.; Adrie, C.; Janin, A.; Barbi, S.; Kister, J.; Rouyer-Fessard, P.; Corrocher, R.; Leboulch, P.; et al. Inhaled nitric oxide protects transgenic SAD mice from sickle cell disease-specific lung injury induced by hypoxia/reoxygenation. Blood 2003, 102, 1087–1096. [Google Scholar] [CrossRef] [PubMed]
- Noubouossie, D.; Key, N.S.; Ataga, K.I. Coagulation abnormalities of sickle cell disease: Relationship with clinical outcomes and the effect of disease modifying therapies. Blood Rev. 2016, 30, 245–256. [Google Scholar] [CrossRef]
- Hogan, K.A.; Weiler, H.; Lord, S.T. Mouse models in coagulation. Thromb. Haemost. 2002, 87, 563–574. [Google Scholar] [CrossRef]
- Arumugam, P.I.; Mullins, E.S.; Shanmukhappa, S.K.; Monia, B.P.; Loberg, A.; Shaw, M.A.; Rizvi, T.; Wansapura, J.; Degen, J.L.; Malik, P. Genetic diminution of circulating prothrombin ameliorates multiorgan pathologies in sickle cell disease mice. Blood 2015, 126, 1844–1855. [Google Scholar] [CrossRef]
- Sparkenbaugh, E.M.; Chen, C.; Brzoska, T.; Nguyen, J.; Wang, S.; Vercellotti, G.M.; Key, N.S.; Sundd, P.; Belcher, J.D.; Pawlinski, R. Thrombin-mediated activation of PAR-1 contributes to microvascular stasis in mouse models of sickle cell disease. Blood 2020. [Google Scholar] [CrossRef]
- Reiter, C.D.; Wang, X.; Tanus-Santos, J.E.; Hogg, N.; Cannon, R.O.; Schechter, A.N.; Gladwin, M.T. Cell-free hemoglobin limits nitric oxide bioavailability in sickle-cell disease. Nat. Med. 2002, 8, 1383–1389. [Google Scholar] [CrossRef]
- Morris, C.R.; Kato, G.J.; Poljakovic, M.; Wang, X.; Blackwelder, W.C.; Sachdev, V.; Hazen, S.L.; Vichinsky, E.P.; Morris, S.M., Jr.; Gladwin, M.T. Dysregulated arginine metabolism, hemolysis-associated pulmonary hypertension, and mortality in sickle cell disease. JAMA 2005, 294, 81–90. [Google Scholar] [CrossRef] [PubMed]
- De Franceschi, L.; Cappellini, M.D.; Olivieri, O. Thrombosis and sickle cell disease. Semin. Thromb. Hemost. 2011, 37, 226–236. [Google Scholar] [CrossRef] [PubMed]
- Hagger, D.; Wolff, S.; Owen, J.; Samson, D. Changes in coagulation and fibrinolysis in patients with sickle cell disease compared with healthy black controls. Blood Coagul. Fibrinolysis 1995, 6, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Esmon, C.T. Role of coagulation inhibitors in inflammation. Thromb. Haemost. 2001, 86, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Darbousset, R.; Schoenwaelder, S.M. Thromboinflammation: Challenges of therapeutically targeting coagulation and other host defense mechanisms. Blood 2019, 133, 906–918. [Google Scholar] [CrossRef]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [PubMed]
- Mendonca, R.; Silveira, A.A.; Conran, N. Red cell DAMPs and inflammation. Inflamm. Res. 2016, 65, 665–678. [Google Scholar] [CrossRef]
- Stark, K.; Philippi, V.; Stockhausen, S.; Busse, J.; Antonelli, A.; Miller, M.; Schubert, I.; Hoseinpour, P.; Chandraratne, S.; von Bruhl, M.L.; et al. Disulfide HMGB1 derived from platelets coordinates venous thrombosis in mice. Blood 2016, 128, 2435–2449. [Google Scholar] [CrossRef]
- Yadav, V.; Chi, L.; Zhao, R.; Tourdot, B.E.; Yalavarthi, S.; Jacobs, B.N.; Banka, A.; Liao, H.; Koonse, S.; Anyanwu, A.C.; et al. Ectonucleotidase tri(di)phosphohydrolase-1 (ENTPD-1) disrupts inflammasome/interleukin 1beta-driven venous thrombosis. J. Clin. Investig. 2019, 129, 2872–2877. [Google Scholar] [CrossRef]
- Sundd, P.; Gladwin, M.T.; Novelli, E.M. Pathophysiology of Sickle Cell Disease. Annu. Rev. Pathol. 2019, 14, 263–292. [Google Scholar] [CrossRef]
- Ansari, J.; Gavins, F.N.E. Ischemia-Reperfusion Injury in Sickle Cell Disease: From Basics to Therapeutics. Am. J. Pathol. 2019, 189, 706–718. [Google Scholar] [CrossRef] [PubMed]
- Belcher, J.D.; Chen, C.; Nguyen, J.; Milbauer, L.; Abdulla, F.; Alayash, A.I.; Smith, A.; Nath, K.A.; Hebbel, R.P.; Vercellotti, G.M. Heme triggers TLR4 signaling leading to endothelial cell activation and vaso-occlusion in murine sickle cell disease. Blood 2014, 123, 377–390. [Google Scholar] [CrossRef] [PubMed]
- Merle, N.S.; Paule, R.; Leon, J.; Daugan, M.; Robe-Rybkine, T.; Poillerat, V.; Torset, C.; Fremeaux-Bacchi, V.; Dimitrov, J.D.; Roumenina, L.T. P-selectin drives complement attack on endothelium during intravascular hemolysis in TLR-4/heme-dependent manner. Proc. Natl. Acad. Sci. USA 2019, 116, 6280–6285. [Google Scholar] [CrossRef] [PubMed]
- Silveira, A.A.A.; Mahon, O.R.; Cunningham, C.C.; Corr, E.M.; Mendonca, R.; Saad, S.T.O.; Costa, F.F.; Dunne, A.; Conran, N. S100A8 acts as an autocrine priming signal for heme-induced human Mvarphi pro-inflammatory responses in hemolytic inflammation. J. Leukoc. Biol. 2019, 106, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Jana, S.; Strader, M.B.; Meng, F.; Hicks, W.; Kassa, T.; Tarandovskiy, I.; De Paoli, S.; Simak, J.; Heaven, M.R.; Belcher, J.D.; et al. Hemoglobin oxidation-dependent reactions promote interactions with band 3 and oxidative changes in sickle cell-derived microparticles. JCI Insight 2018, 3. [Google Scholar] [CrossRef]
- Camus, S.M.; De Moraes, J.A.; Bonnin, P.; Abbyad, P.; Le Jeune, S.; Lionnet, F.; Loufrani, L.; Grimaud, L.; Lambry, J.C.; Charue, D.; et al. Circulating cell membrane microparticles transfer heme to endothelial cells and trigger vasoocclusions in sickle cell disease. Blood 2015, 125, 3805–3814. [Google Scholar] [CrossRef] [PubMed]
- Solovey, A.; Somani, A.; Belcher, J.D.; Milbauer, L.; Vincent, L.; Pawlinski, R.; Nath, K.A.; Kelm, R.J., Jr.; Mackman, N.; O’Sullivan, M.G.; et al. A monocyte-TNF-endothelial activation axis in sickle transgenic mice: Therapeutic benefit from TNF blockade. Am. J. Hematol. 2017, 92, 1119–1130. [Google Scholar] [CrossRef]
- Vats, R.; Brzoska, T.; Bennewitz, M.F.; Jimenez, M.A.; Pradhan-Sundd, T.; Tutuncuoglu, E.; Jonassaint, J.; Gutierrez, E.; Watkins, S.C.; Shiva, S.; et al. Platelet Extracellular Vesicles Drive Inflammasome-IL-1beta-Dependent Lung Injury in Sickle Cell Disease. Am. J. Respir. Crit. Care Med. 2020, 201, 33–46. [Google Scholar] [CrossRef]
- Vogel, S.; Arora, T.; Wang, X.; Mendelsohn, L.; Nichols, J.; Allen, D.; Shet, A.S.; Combs, C.A.; Quezado, Z.M.N.; Thein, S.L. The platelet NLRP3 inflammasome is upregulated in sickle cell disease via HMGB1/TLR4 and Bruton tyrosine kinase. Blood Adv. 2018, 2, 2672–2680. [Google Scholar] [CrossRef]
- Merle, N.S.; Grunenwald, A.; Rajaratnam, H.; Gnemmi, V.; Frimat, M.; Figueres, M.L.; Knockaert, S.; Bouzekri, S.; Charue, D.; Noe, R.; et al. Intravascular hemolysis activates complement via cell-free heme and heme-loaded microvesicles. JCI Insight 2018, 3. [Google Scholar] [CrossRef]
- Vercellotti, G.M.; Dalmasso, A.P.; Schaid, T.R., Jr.; Nguyen, J.; Chen, C.; Ericson, M.E.; Abdulla, F.; Killeen, T.; Lindorfer, M.A.; Taylor, R.P.; et al. Critical role of C5a in sickle cell disease. Am. J. Hematol. 2019, 94, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam, S.; Jurk, K.; Hobohm, L.; Jackel, S.; Saffarzadeh, M.; Schwierczek, K.; Wenzel, P.; Langer, F.; Reinhardt, C.; Ruf, W. Distinct contributions of complement factors to platelet activation and fibrin formation in venous thrombus development. Blood 2017, 129, 2291–2302. [Google Scholar] [CrossRef] [PubMed]
- Santiago, R.P.; Guarda, C.C.; Figueiredo, C.V.B.; Fiuza, L.M.; Aleluia, M.M.; Adanho, C.S.A.; Carvalho, M.O.S.; Pitanga, T.N.; Zanette, D.L.; Lyra, I.M.; et al. Serum haptoglobin and hemopexin levels are depleted in pediatric sickle cell disease patients. Blood Cells Mol. Dis. 2018, 72, 34–36. [Google Scholar] [CrossRef] [PubMed]
- Belcher, J.D.; Chen, C.; Nguyen, J.; Abdulla, F.; Zhang, P.; Nguyen, H.; Nguyen, P.; Killeen, T.; Miescher, S.M.; Brinkman, N.; et al. Haptoglobin and hemopexin inhibit vaso-occlusion and inflammation in murine sickle cell disease: Role of heme oxygenase-1 induction. PLoS ONE 2018, 13, e0196455. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Luo, W.; Wang, J.; Guo, C.; Wolffe, S.L.; Wang, J.; Sun, E.B.; Bradley, K.N.; Campbell, A.D.; Eitzman, D.T. Paradoxical protection from atherosclerosis and thrombosis in a mouse model of sickle cell disease. Br. J. Haematol. 2013, 162, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Lv, B.; Wang, H.; Tang, Y.; Fan, Z.; Xiao, X.; Chen, F. High-mobility group box 1 protein induces tissue factor expression in vascular endothelial cells via activation of NF-kappaB and Egr-1. Thromb. Haemost. 2009, 102, 352–359. [Google Scholar] [CrossRef]
- Xu, H.; Wandersee, N.J.; Guo, Y.; Jones, D.W.; Holzhauer, S.L.; Hanson, M.S.; Machogu, E.; Brousseau, D.C.; Hogg, N.; Densmore, J.C.; et al. Sickle cell disease increases high mobility group box 1: A novel mechanism of inflammation. Blood 2014, 124, 3978–3981. [Google Scholar] [CrossRef]
- Chen, G.; Zhang, D.; Fuchs, T.A.; Manwani, D.; Wagner, D.D.; Frenette, P.S. Heme-induced neutrophil extracellular traps contribute to the pathogenesis of sickle cell disease. Blood 2014, 123, 3818–3827. [Google Scholar] [CrossRef]
- Thalin, C.; Hisada, Y.; Lundstrom, S.; Mackman, N.; Wallen, H. Neutrophil Extracellular Traps: Villains and Targets in Arterial, Venous, and Cancer-Associated Thrombosis. Arter. Thromb. Vasc. Biol. 2019, 39, 1724–1738. [Google Scholar] [CrossRef]
- Von Bruhl, M.L.; Stark, K.; Steinhart, A.; Chandraratne, S.; Konrad, I.; Lorenz, M.; Khandoga, A.; Tirniceriu, A.; Coletti, R.; Kollnberger, M.; et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J. Exp. Med. 2012, 209, 819–835. [Google Scholar] [CrossRef]
- Fuchs, T.A.; Brill, A.; Duerschmied, D.; Schatzberg, D.; Monestier, M.; Myers, D.D., Jr.; Wrobleski, S.K.; Wakefield, T.W.; Hartwig, J.H.; Wagner, D.D. Extracellular DNA traps promote thrombosis. Proc. Natl. Acad. Sci. USA 2010, 107, 15880–15885. [Google Scholar] [CrossRef]
- Nasimuzzaman, M.; Malik, P. Role of the coagulation system in the pathogenesis of sickle cell disease. Blood Adv. 2019, 3, 3170–3180. [Google Scholar] [CrossRef] [PubMed]
- Wun, T.; Paglieroni, T.; Tablin, F.; Welborn, J.; Nelson, K.; Cheung, A. Platelet activation and platelet-erythrocyte aggregates in patients with sickle cell anemia. J. Lab. Clin. Med. 1997, 129, 507–516. [Google Scholar] [CrossRef]
- Wun, T.; Paglieroni, T.; Rangaswami, A.; Franklin, P.H.; Welborn, J.; Cheung, A.; Tablin, F. Platelet activation in patients with sickle cell disease. Br. J. Haematol. 1998, 100, 741–749. [Google Scholar] [CrossRef] [PubMed]
- Wun, T.; Cordoba, M.; Rangaswami, A.; Cheung, A.W.; Paglieroni, T. Activated monocytes and platelet-monocyte aggregates in patients with sickle cell disease. Clin. Lab. Haematol. 2002, 24, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Polanowska-Grabowska, R.; Wallace, K.; Field, J.J.; Chen, L.; Marshall, M.A.; Figler, R.; Gear, A.R.; Linden, J. P-selectin-mediated platelet-neutrophil aggregate formation activates neutrophils in mouse and human sickle cell disease. Arter. Thromb. Vasc. Biol. 2010, 30, 2392–2399. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo, A.; Chang, J.; Jang, J.-E.; Peired, A.J.; Chiang, E.Y.; Frenette, P.S. Heterotypic interactions enabled by polarized neutrophil microdomains mediate thromboinflammatory injury. Nat. Med. 2009, 15, 384–391. [Google Scholar] [CrossRef]
- Freedman, M.L.; Karpatkin, S. Elevated platelet count and megathrombocyte number in sickle cell anemia. Blood 1975, 46, 579–582. [Google Scholar] [CrossRef]
- Shet, A.S.; Hoffmann, T.J.; Jirouskova, M.; Janczak, C.A.; Stevens, J.R.; Adamson, A.; Mohandas, N.; Manci, E.A.; Cynober, T.; Coller, B.S. Morphological and functional platelet abnormalities in Berkeley sickle cell mice. Blood Cells Mol. Dis. 2008, 41, 109–118. [Google Scholar] [CrossRef]
- Annarapu, G.K.; Singhal, R.; Gupta, A.; Chawla, S.; Batra, H.; Seth, T.; Guchhait, P. HbS Binding to GP1balpha Activates Platelets in Sickle Cell Disease. PLoS ONE 2016, 11, e0167899. [Google Scholar] [CrossRef]
- Semeraro, F.; Ammollo, C.T.; Morrissey, J.H.; Dale, G.L.; Friese, P.; Esmon, N.L.; Esmon, C.T. Extracellular histones promote thrombin generation through platelet-dependent mechanisms: Involvement of platelet TLR2 and TLR4. Blood 2011, 118, 1952–1961. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, J.; Duncan, H.; Kirsh, G.; Allen, V.; King, P.; Hargraves, R.; Mendes, L.; Perera, T.; Catto, P.; Schofield, S. Prevention of pulmonary embolism and deep vein thrombosis with low dose aspirin: Pulmonary Embolism Prevention (PEP) trial. Lancet 2000, 355, 1295–1302. [Google Scholar]
- Anea, C.B.; Lyon, M.; Lee, I.A.; Gonzales, J.N.; Adeyemi, A.; Falls, G.; Kutlar, A.; Brittain, J.E. Pulmonary platelet thrombi and vascular pathology in acute chest syndrome in patients with sickle cell disease. Am. J. Hematol. 2016, 91, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Bennewitz, M.F.; Jimenez, M.A.; Vats, R.; Tutuncuoglu, E.; Jonassaint, J.; Kato, G.J.; Gladwin, M.T.; Sundd, P. Lung vaso-occlusion in sickle cell disease mediated by arteriolar neutrophil-platelet microemboli. JCI Insight 2017, 2, e89761. [Google Scholar] [CrossRef]
- Cines, D.B.; Pollak, E.S.; Buck, C.A.; Loscalzo, J.; Zimmerman, G.A.; McEver, R.P.; Pober, J.S.; Wick, T.M.; Konkle, B.A.; Schwartz, B.S.; et al. Endothelial cells in physiology and in the pathophysiology of vascular disorders. Blood 1998, 91, 3527–3561. [Google Scholar]
- Folsom, A.R.; Tang, W.; Roetker, N.S.; Kshirsagar, A.V.; Derebail, V.K.; Lutsey, P.L.; Naik, R.; Pankow, J.S.; Grove, M.L.; Basu, S.; et al. Prospective study of sickle cell trait and venous thromboembolism incidence. J. Thromb. Haemost. 2015, 13, 2–9. [Google Scholar] [CrossRef]
- Payne, H.; Ponomaryov, T.; Watson, S.P.; Brill, A. Mice with a deficiency in CLEC-2 are protected against deep vein thrombosis. Blood 2017, 129, 2013–2020. [Google Scholar] [CrossRef]
- Kaul, D.K.; Fabry, M.E.; Costantini, F.; Rubin, E.M.; Nagel, R.L. In vivo demonstration of red cell-endothelial interaction, sickling and altered microvascular response to oxygen in the sickle transgenic mouse. J. Clin. Investig. 1995, 96, 2845–2853. [Google Scholar] [CrossRef]
- Solovey, A.; Lin, Y.; Browne, P.; Choong, S.; Wayner, E.; Hebbel, R.P. Circulating activated endothelial cells in sickle cell anemia. N. Engl. J. Med. 1997, 337, 1584–1590. [Google Scholar] [CrossRef]
- Bennewitz, M.F.; Tutuncuoglu, E.; Gudapati, S.; Brzoska, T.; Watkins, S.C.; Monga, S.P.; Pradhan-Sundd, T.; Sundd, P. P-selectin-deficient mice to study pathophysiology of sickle cell disease. Blood Adv. 2020, 4, 266–273. [Google Scholar] [CrossRef]
- Wood, K.C.; Hebbel, R.P.; Granger, D.N. Endothelial Cell P-selectin Mediates a Proinflammatory and Prothrombogenic Phenotype in Cerebral Venules of Sickle Cell Transgenic Mice. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, H1608–H1614. [Google Scholar] [CrossRef] [PubMed]
- Embury, S.H.; Matsui, N.M.; Ramanujam, S.; Mayadas, T.N.; Noguchi, C.T.; Diwan, B.A.; Mohandas, N.; Cheung, A.T. The contribution of endothelial cell P-selectin to the microvascular flow of mouse sickle erythrocytes in vivo. Blood 2004, 104, 3378–3385. [Google Scholar] [CrossRef] [PubMed]
- Myers, D., Jr.; Farris, D.; Hawley, A.; Wrobleski, S.; Chapman, A.; Stoolman, L.; Knibbs, R.; Strieter, R.; Wakefield, T. Selectins influence thrombosis in a mouse model of experimental deep venous thrombosis. J. Surg. Res. 2002, 108, 212–221. [Google Scholar] [CrossRef]
- Ataga, K.I.; Kutlar, A.; Kanter, J.; Liles, D.; Cancado, R.; Friedrisch, J.; Guthrie, T.H.; Knight-Madden, J.; Alvarez, O.A.; Gordeuk, V.R.; et al. Crizanlizumab for the Prevention of Pain Crises in Sickle Cell Disease. N. Engl. J. Med. 2017, 376, 429–439. [Google Scholar] [CrossRef] [PubMed]
- Telen, M.J.; Malik, P.; Vercellotti, G.M. Therapeutic strategies for sickle cell disease: Towards a multi-agent approach. Nat. Rev. Drug Discov. 2019, 18, 139–158. [Google Scholar] [CrossRef] [PubMed]
- Brooks, E.G.; Trotman, W.; Wadsworth, M.P.; Taatjes, D.J.; Evans, M.F.; Ittleman, F.P.; Callas, P.W.; Esmon, C.T.; Bovill, E.G. Valves of the deep venous system: An overlooked risk factor. Blood 2009, 114, 1276–1279. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Byrnes, J.R.; Wolberg, A.S. Red blood cells in thrombosis. Blood 2017, 130, 1795–1799. [Google Scholar] [CrossRef]
- Klatt, C.; Kruger, I.; Zey, S.; Krott, K.J.; Spelleken, M.; Gowert, N.S.; Oberhuber, A.; Pfaff, L.; Luckstadt, W.; Jurk, K.; et al. Platelet-RBC interaction mediated by FasL/FasR induces procoagulant activity important for thrombosis. J. Clin. Investig. 2018, 128, 3906–3925. [Google Scholar] [CrossRef]
- Ogunsile, F.J.; Naik, R.; Lanzkron, S. Overcoming challenges of venous thromboembolism in sickle cell disease treatment. Expert Rev. Hematol. 2019, 12, 173–182. [Google Scholar] [CrossRef]
- Guy, A.; Gourdou-Latyszenok, V.; Le Lay, N.; Peghaire, C.; Kilani, B.; Dias, J.V.; Duplaa, C.; Renault, M.A.; Denis, C.; Villeval, J.L.; et al. Vascular endothelial cell expression of JAK2(V617F) is sufficient to promote a pro-thrombotic state due to increased P-selectin expression. Haematologica 2019, 104, 70–81. [Google Scholar] [CrossRef]
- Perner, F.; Perner, C.; Ernst, T.; Heidel, F.H. Roles of JAK2 in Aging, Inflammation, Hematopoiesis and Malignant Transformation. Cells 2019, 8, 854. [Google Scholar] [CrossRef] [PubMed]
- Connors, J.M.; Levy, J.H. COVID-19 and its implications for thrombosis and anticoagulation. Blood 2020. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Venous Thrombosis Murine Model | Characteristics | Disadvantages | Mechanisms of Thrombus Formation |
|---|---|---|---|
| Venous stasis but ligation induced injury | Complete and permanent occlusion of the inferior vena cava (IVC) and the venous flow. | The absence of blood flow, which not reproduce the clinical scenario where a thrombus is non-occlusive. | The combination of venous stasis and endothelial injury with upregulated expression of endothelial adhesion/procoagulant molecules imitates thrombosis. |
| Venous stenosis with no injury | Preservation but markedly reduced venous blood flow and minimal endothelial injury. | Occasional failure to induce persistent thrombosis and variability in thrombus size. | Endothelial activation, recruitment of immune cells and platelets, initiate thrombosis which is augmented by stasis. |
| Ferric chloride induced injury | Surgical exposure of IVC followed by topical application of ferric chloride. | Endothelial injury induced by the chemical irritant stimulus. Exposure time and concentration influence size and thrombus growth dynamics. | Oxidative damage to vascular endothelial cells. |
| Rose Bengal induced injury | This model is mainly used to induce acute thrombosis. | Endothelial injury by oxygen free radical induced oxidative stress. | Endothelial activation/injury with subsequent induce thrombus formation. |
| Electrolytic vein injury with local hypercoagulability | Non-occlusive thrombosis model that enables to study the acute and chronic deep vein thrombosis (DVT). | Substantial endothelial and vessel wall damage due to the needle access and lengthy procedural time. | Thrombus formation takes place after endothelial cell activation/injury, but blood flow is unaltered. |
| Mice Model | Type of Transgene | Phenotype | Limitations |
|---|---|---|---|
| βSAD NY1DD | Human α2-globin linked to a human β-globin LCR; human βSAD-globin gene carrying Antilles and Hb D-Punjab (β121Gln) variants linked to a human β-globin LCR | Mild phenotype Increased red cell density Low oxygen affinity and an enhanced polymerization potential Under hypoxia conditions, these mice express a more severe pathology Mice develop priapism, kidney defects, and shortened survival | Not anemic Mouse hemoglobin expression Genetic thalassemia background |
| AβS Antilles S+SAntilles | Human α2-globin and βS Antilles-globin variant (β23Ile), each linked to individual LCR HSII fragments | Moderate phenotype Anemic mice with low solubility and low oxygen affinity Slightly reduced hematocrit and haptoglobin levels Exhibit symptoms of VOC Increased reticulocyte count and plasma hemoglobin | Mouse hemoglobin expression Genetic thalassemia background |
| Berkeley model SS-BERK | Mini-LCR expressing human α1, Gγ, Aγ, δ, βS globins on a murine α- and β-globin-deficient background | Severe phenotype Express almost exclusively human sickle hemoglobin Sickle red blood cells (RBCs), intravascular hemolysis, reticulocytosis, severe anemia, leukocytosis, elevation of inflammatory cytokines, multiorgan infarcts, and pulmonary congestion Exhibit VOCs, I/R pathophysiology and increased inflammatory response Hyperalgesia | Low mean corpuscular hemoglobin concentrations (MCHC) Enlarged spleen with compensatory extramedullary hematopoiesis |
| Townes model | Human mini-LCR expressing human α1, Aγ, βS globins on a murine α- and β-globin-deficient background | Severe phenotype Expansion of red pulp, pooling of sinusoidal RBCs, vaso-occlusion, and loss of lymphoid follicular structure Marked reduction in RBC counts, Hb concentrations, PCVs, and a significantly increased reticulocyte count Hyperalgesia | Enlarged spleen with compensatory extramedullary hematopoiesis |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lizarralde-Iragorri, M.A.; Shet, A.S. Sickle Cell Disease: A Paradigm for Venous Thrombosis Pathophysiology. Int. J. Mol. Sci. 2020, 21, 5279. https://doi.org/10.3390/ijms21155279
Lizarralde-Iragorri MA, Shet AS. Sickle Cell Disease: A Paradigm for Venous Thrombosis Pathophysiology. International Journal of Molecular Sciences. 2020; 21(15):5279. https://doi.org/10.3390/ijms21155279
Chicago/Turabian StyleLizarralde-Iragorri, Maria A., and Arun S. Shet. 2020. "Sickle Cell Disease: A Paradigm for Venous Thrombosis Pathophysiology" International Journal of Molecular Sciences 21, no. 15: 5279. https://doi.org/10.3390/ijms21155279
APA StyleLizarralde-Iragorri, M. A., & Shet, A. S. (2020). Sickle Cell Disease: A Paradigm for Venous Thrombosis Pathophysiology. International Journal of Molecular Sciences, 21(15), 5279. https://doi.org/10.3390/ijms21155279