Dual Leucine Zipper Kinase Is Constitutively Active in the Adult Mouse Brain and Has Both Stress-Induced and Homeostatic Functions

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Design
2.2. Tissue Harvesting
2.3. Antibodies and DLKi
2.4. Tissue Processing for Western Blot Analysis
2.5. Immunohistochemistry and Immunofluorescence Analysis
2.6. RNA-Seq Analysis
2.7. Pharmacokinetic Assessment of DLKi in Plasma and Brain Tissue
2.8. Statistical Analysis
3. Results
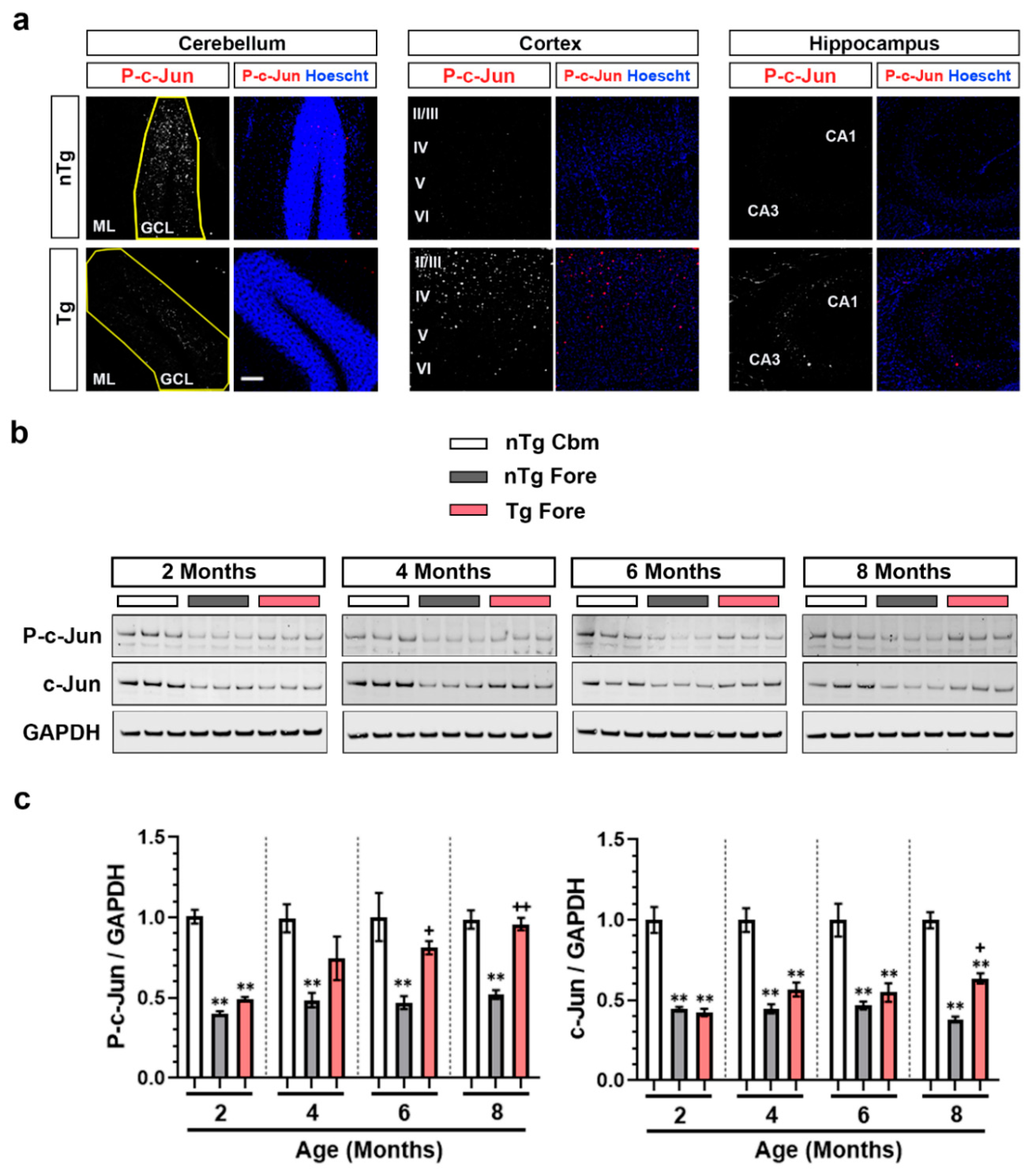
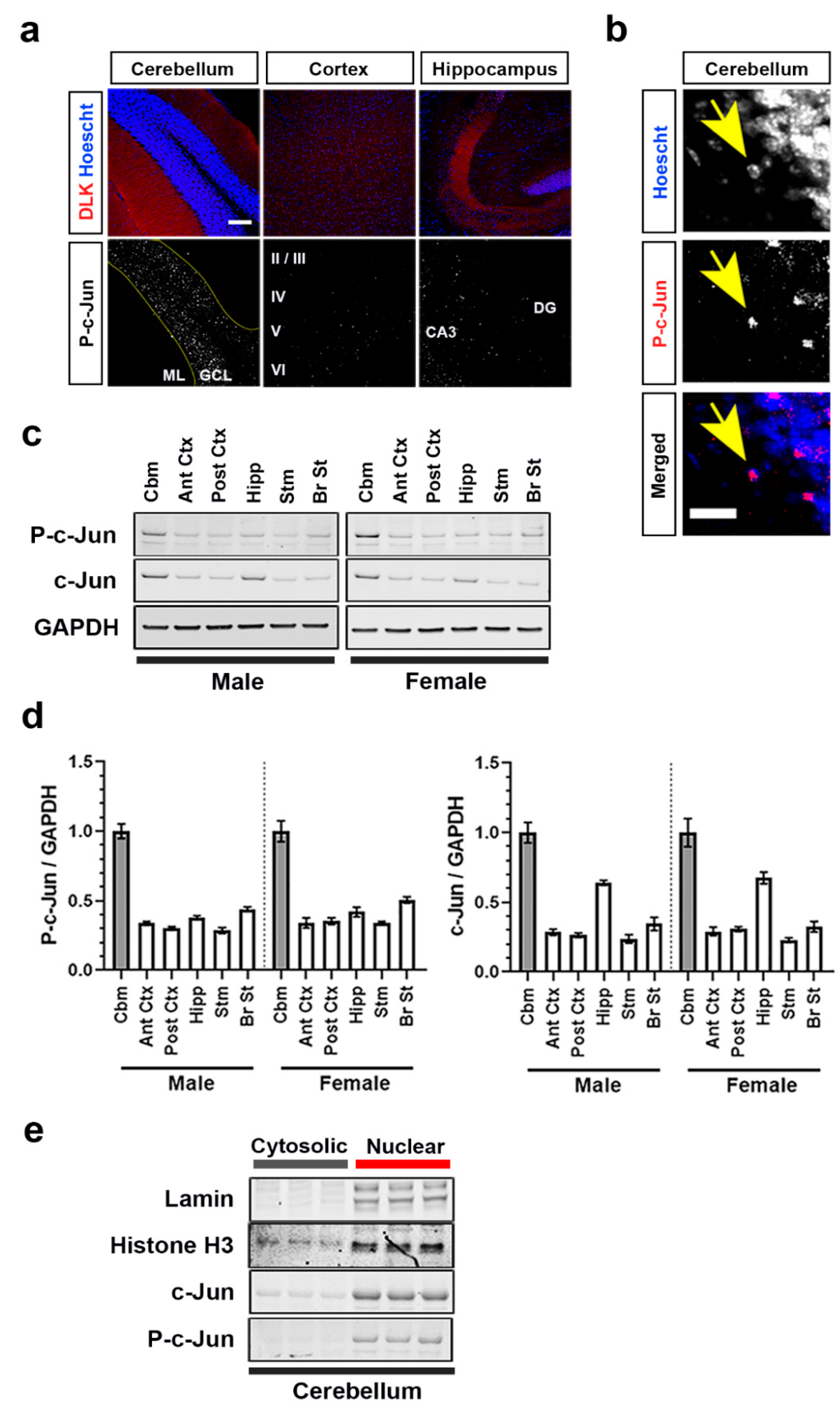
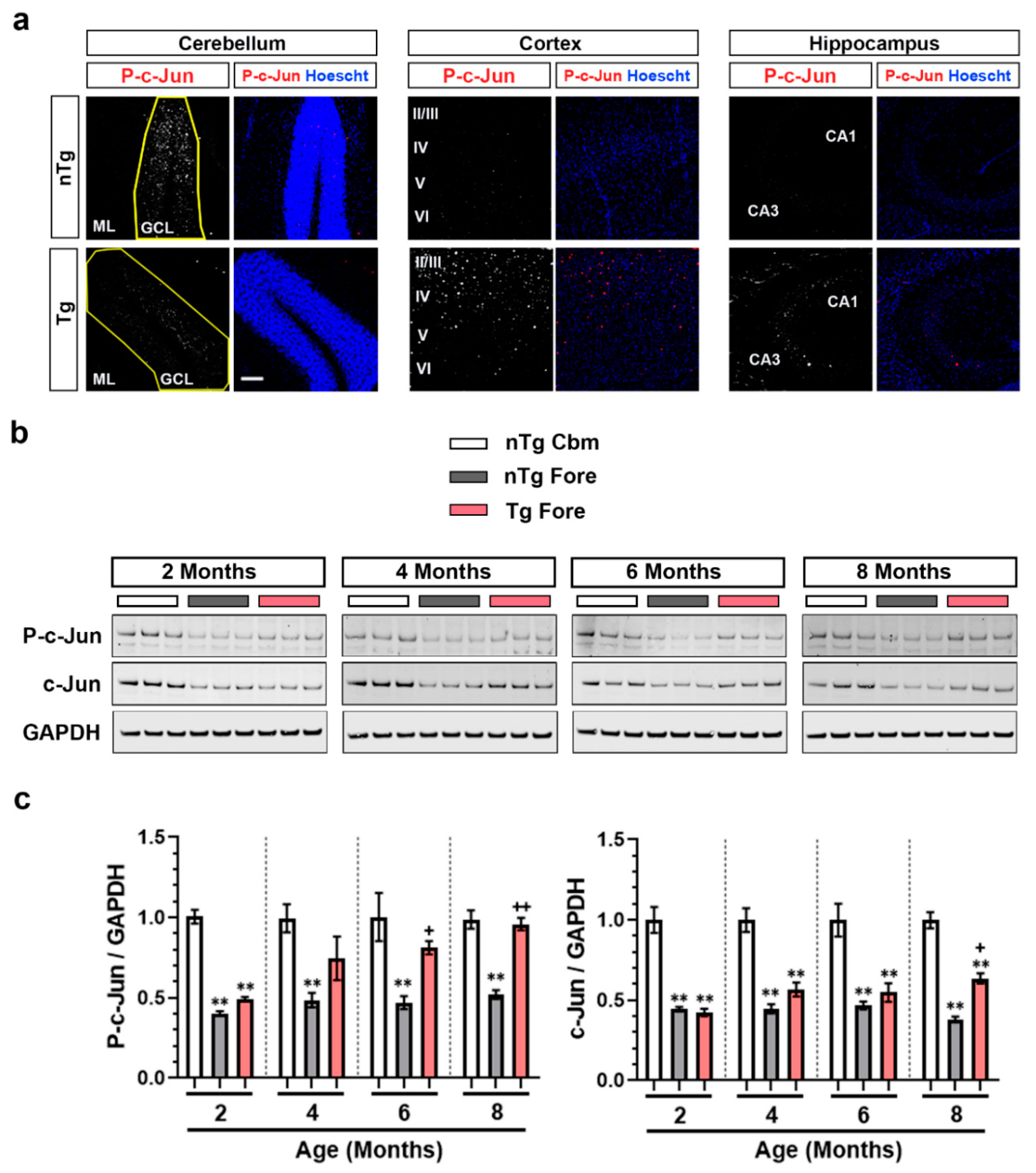
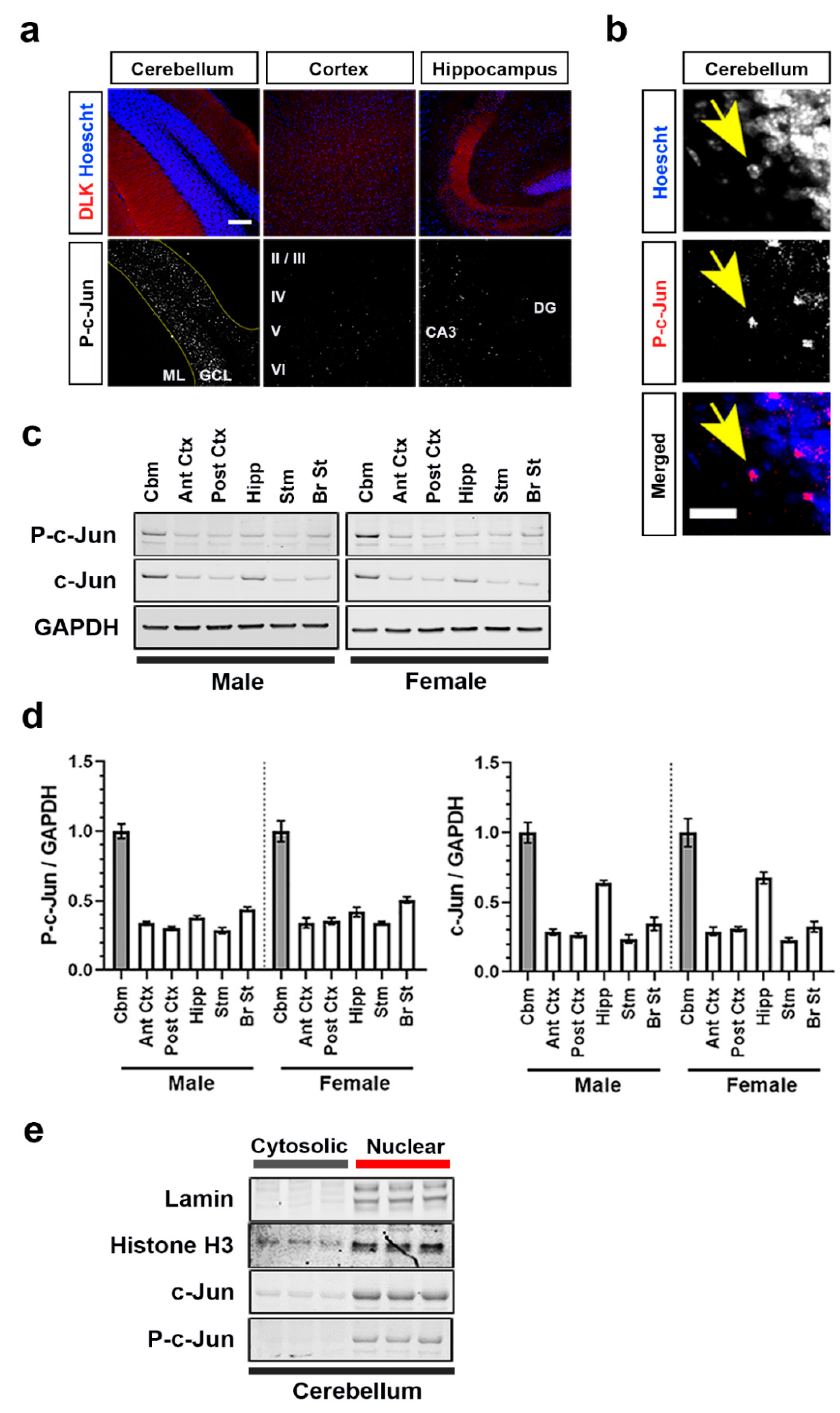
3.1. Brain Region-Dependent Constitutive or Induced Phosphorylation of c-Jun
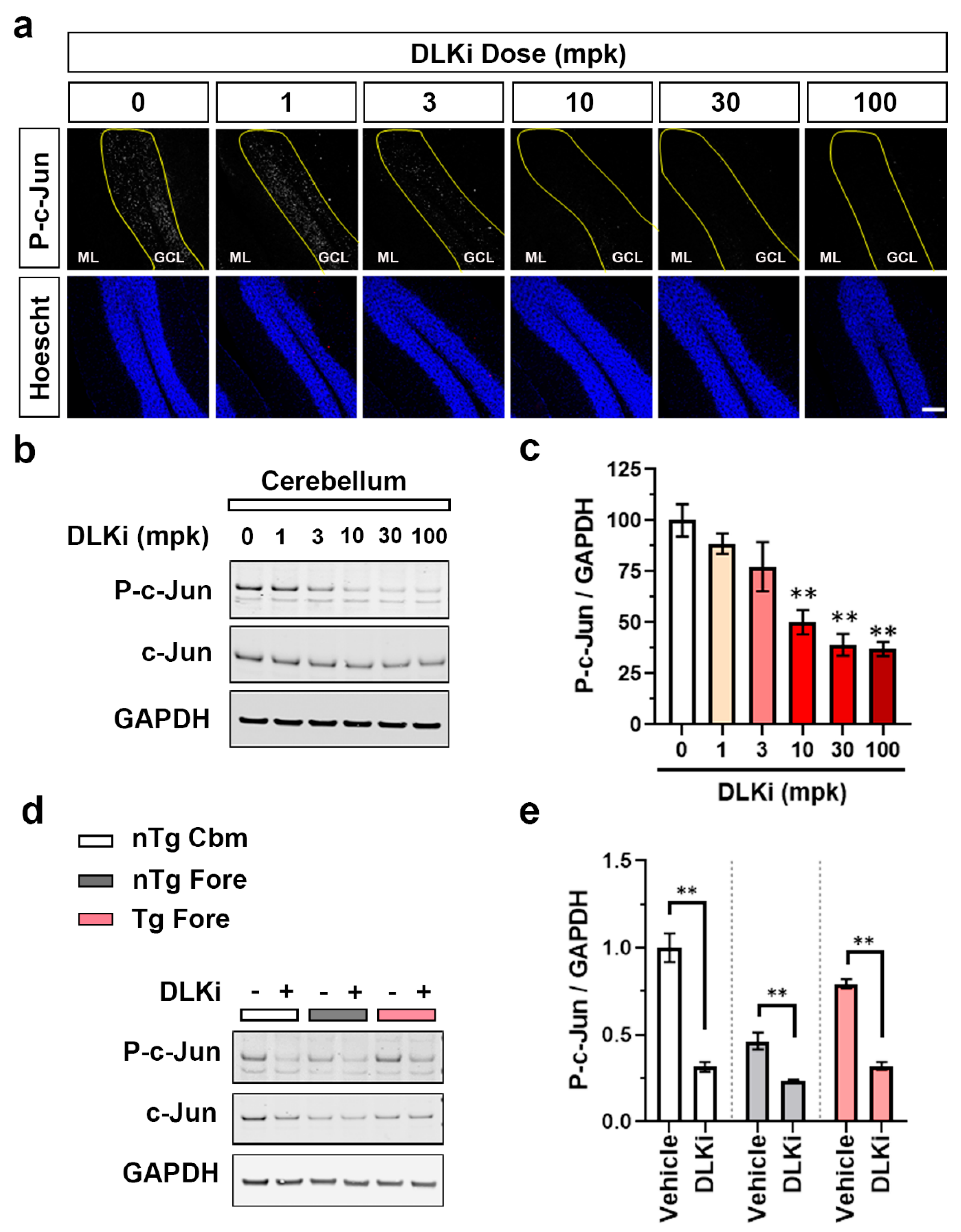
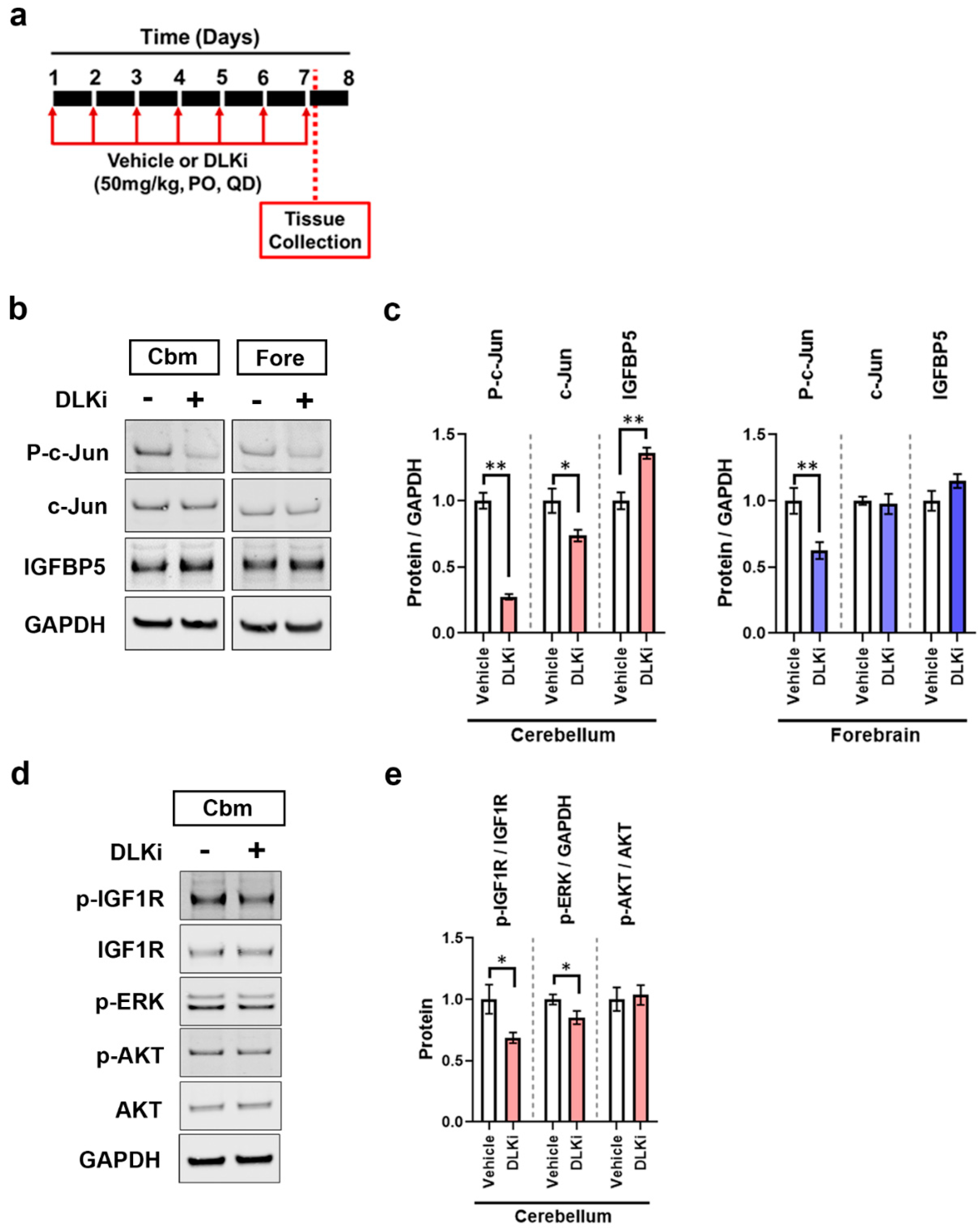
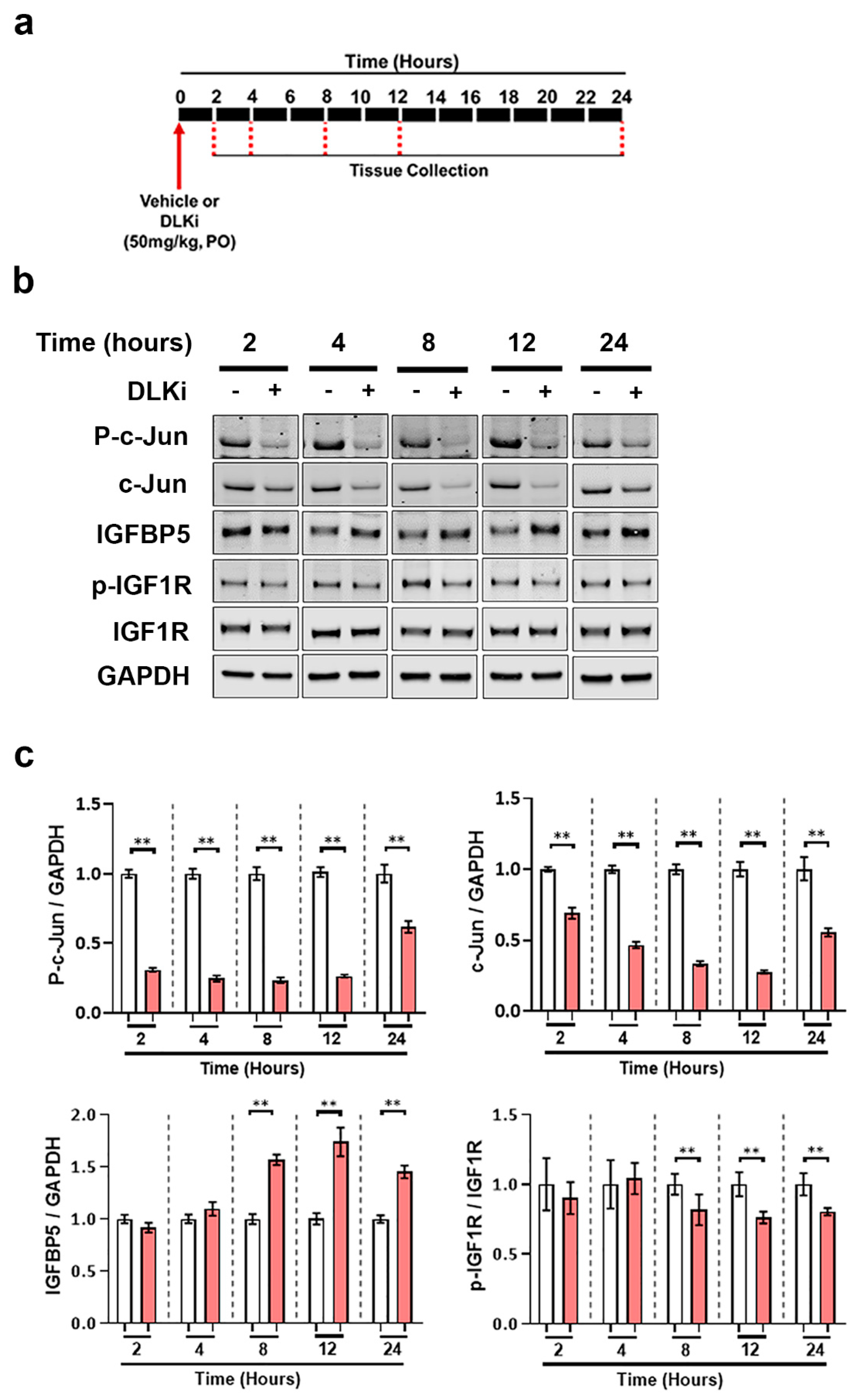
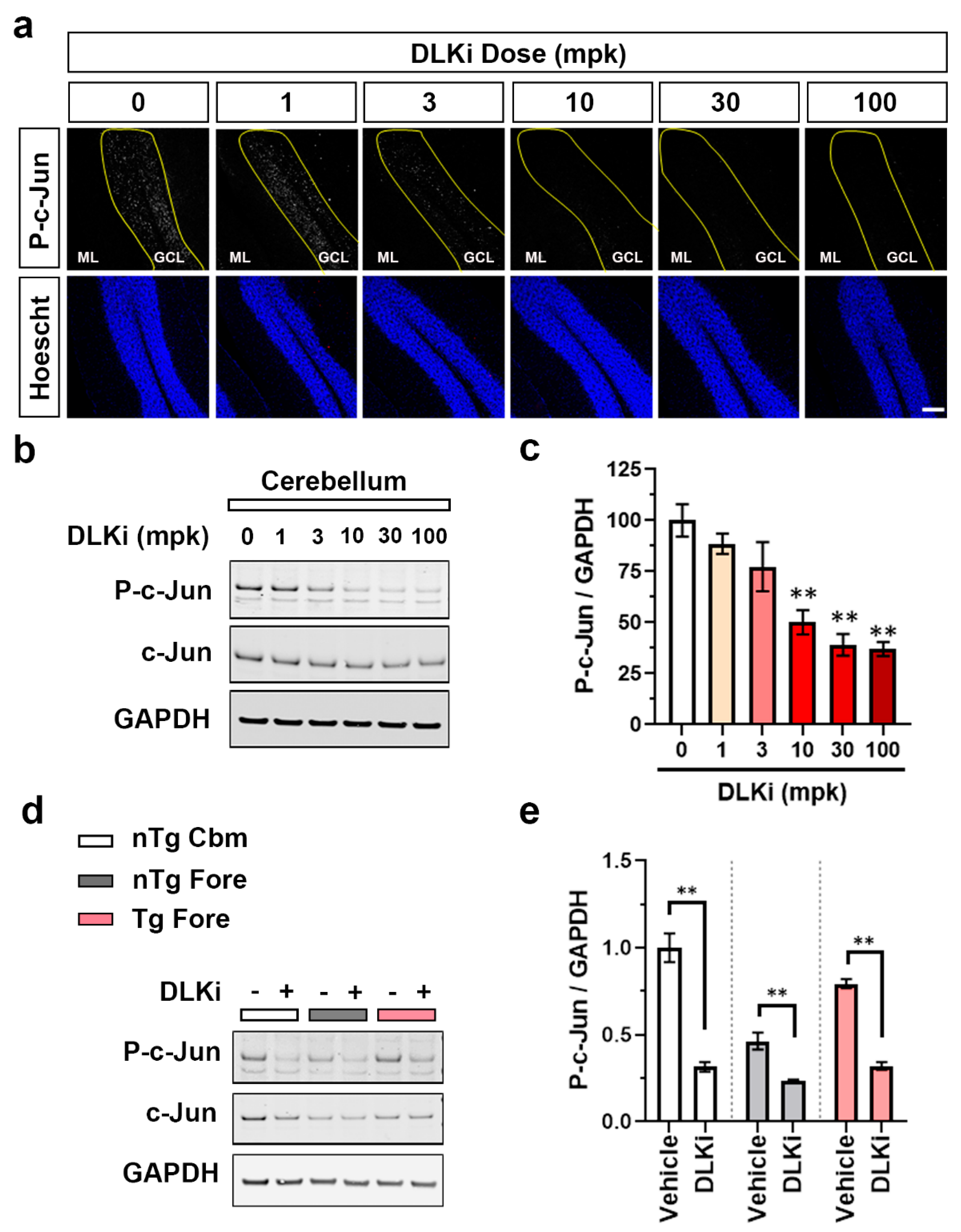
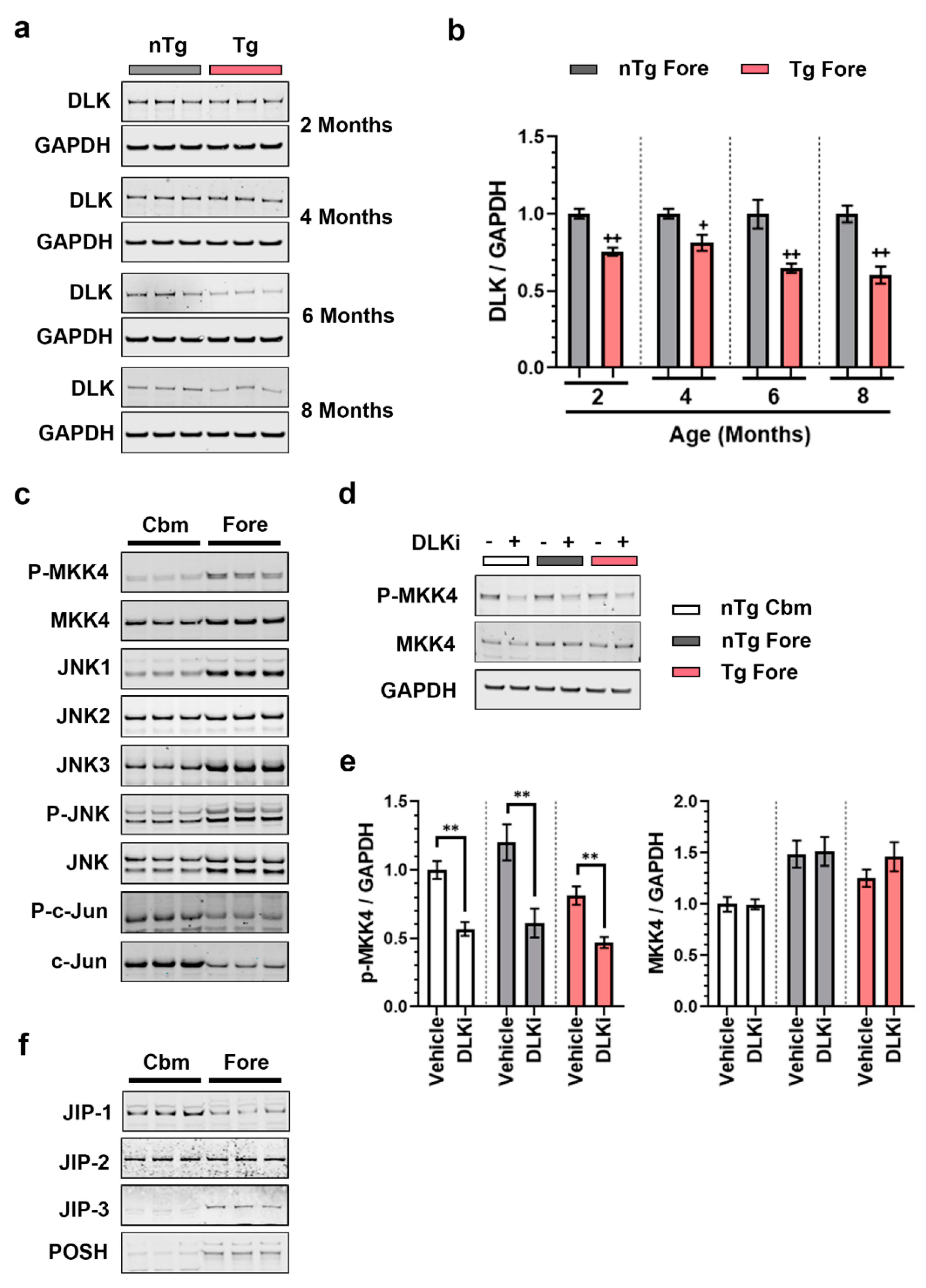
3.2. Constitutive Cerebellar p-c-Jun and tau-Induced Forebrain p-c-Jun Are Both DLK Dependent
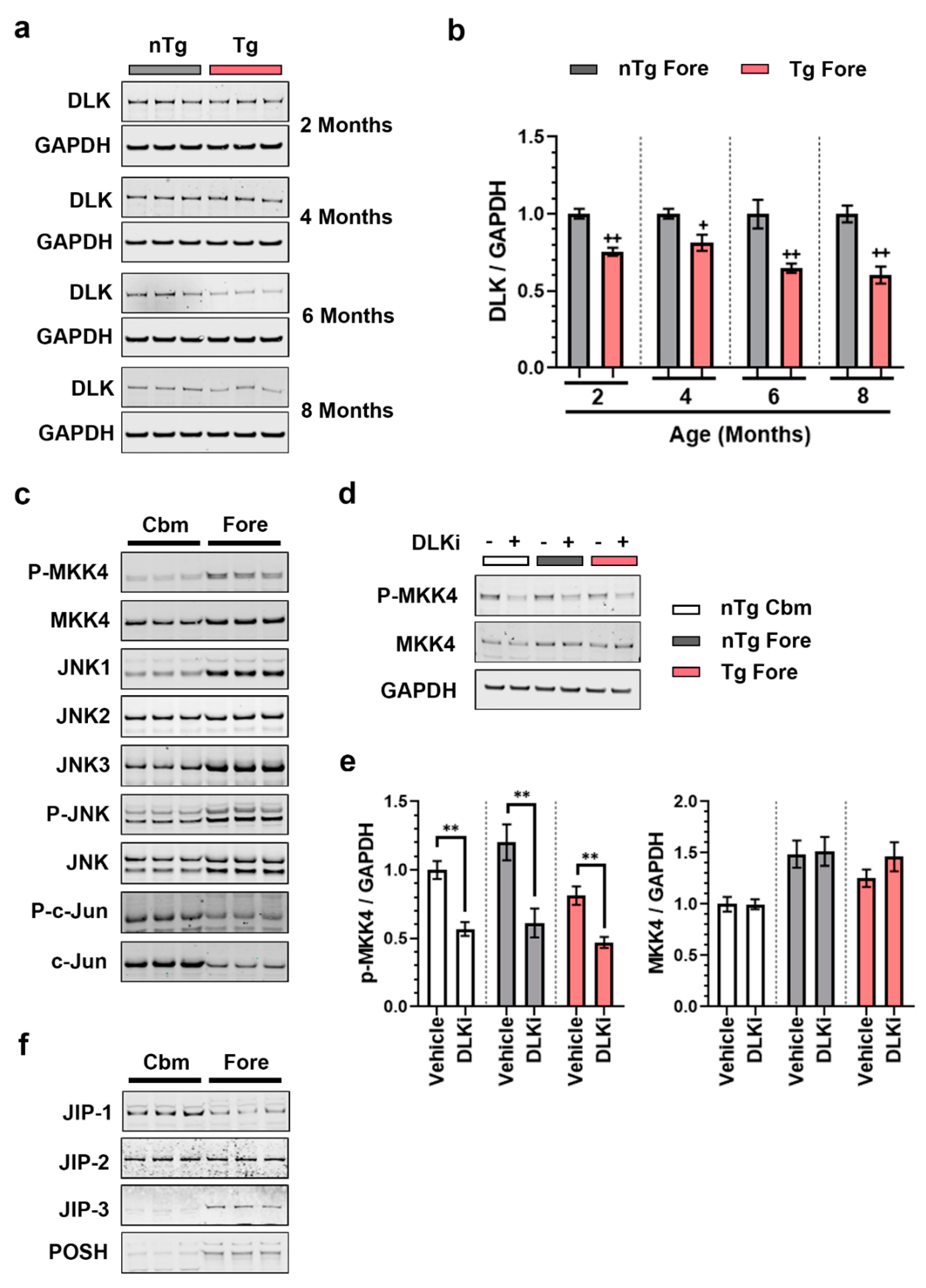
3.3. DLK Is Constitutively Active in Adult Mouse Brain
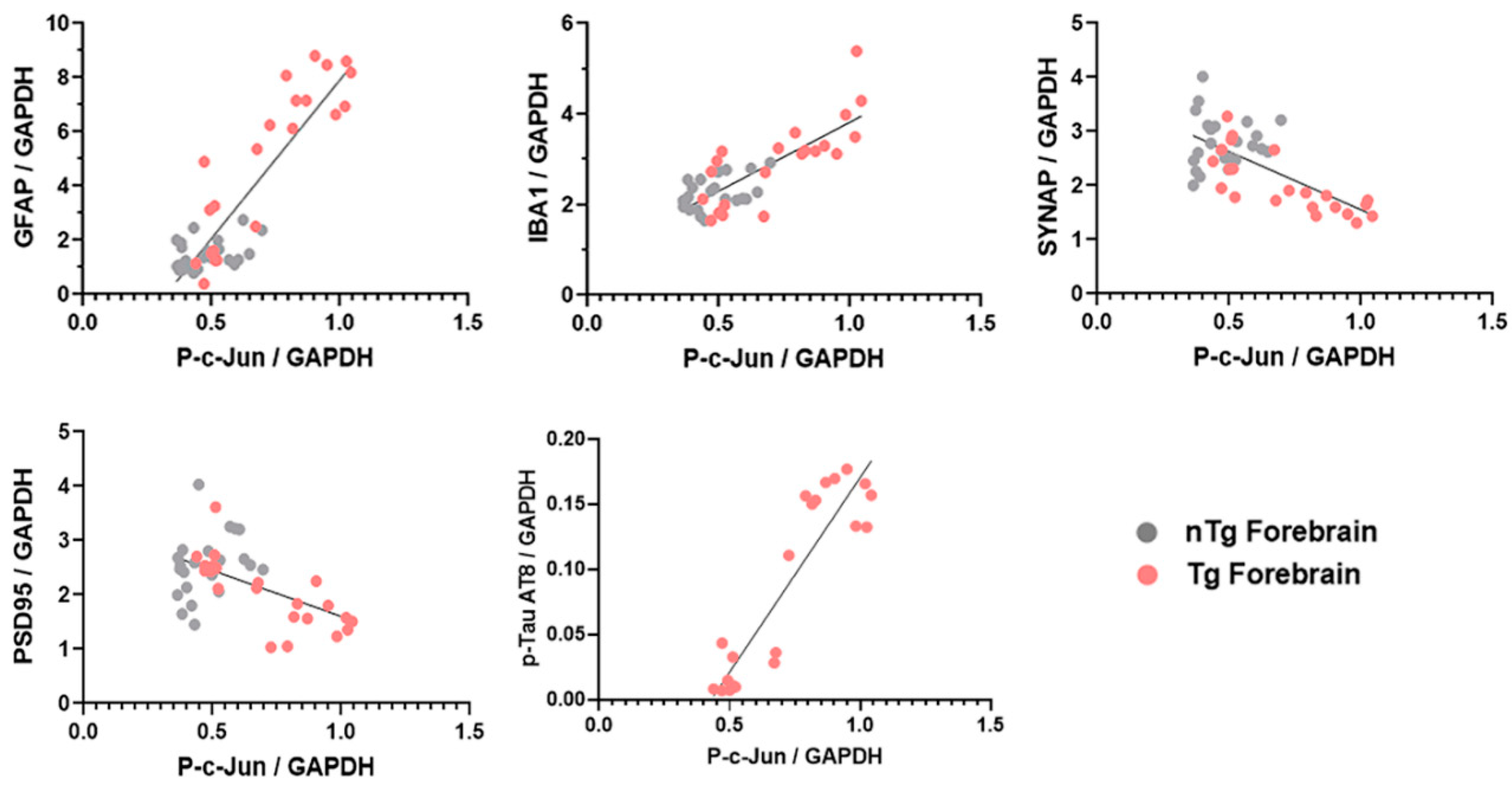
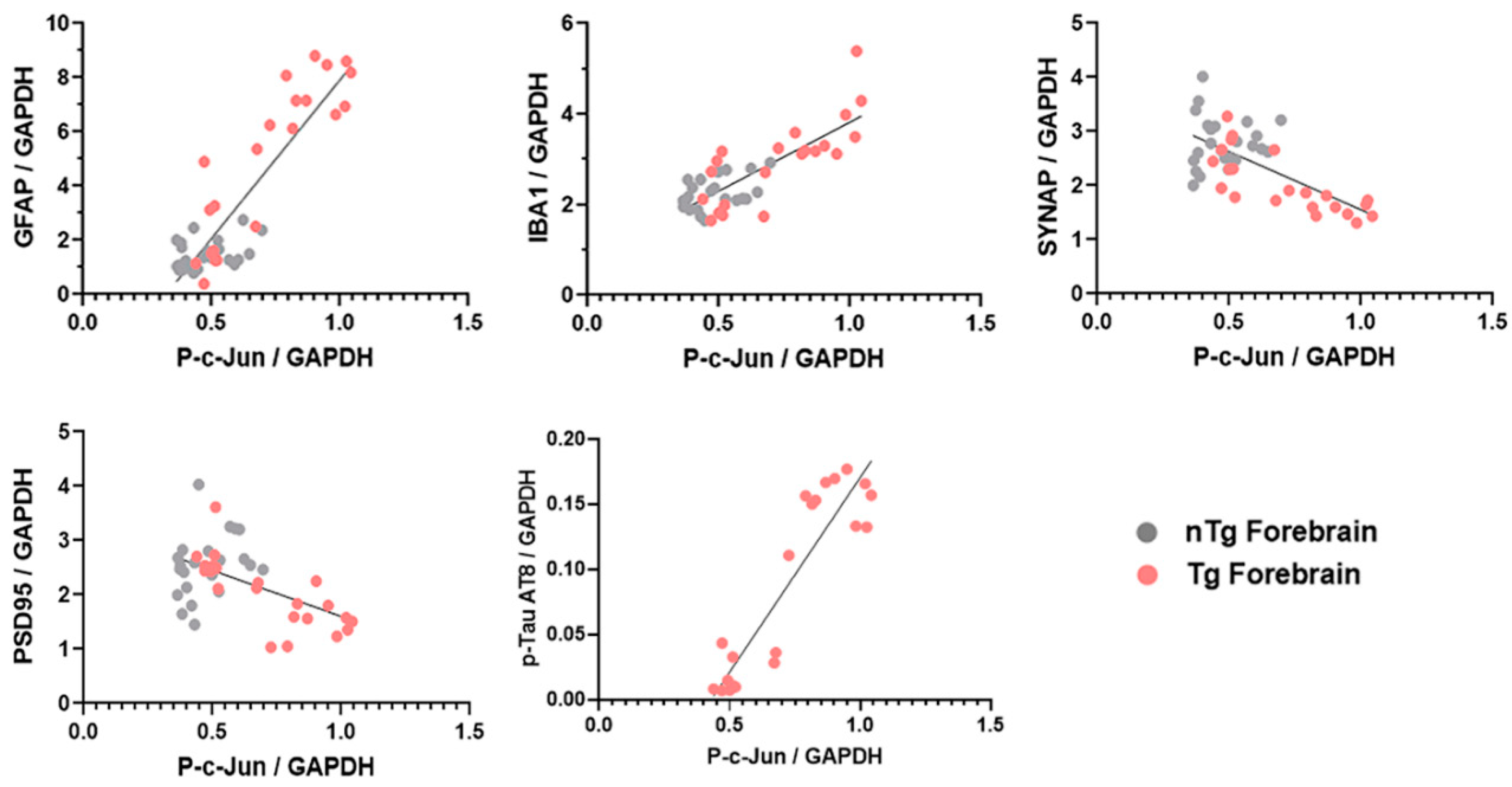
3.4. Forebrain p-c-Jun Correlates with Markers of Neuronal Injury in rTg4510 Mouse Model
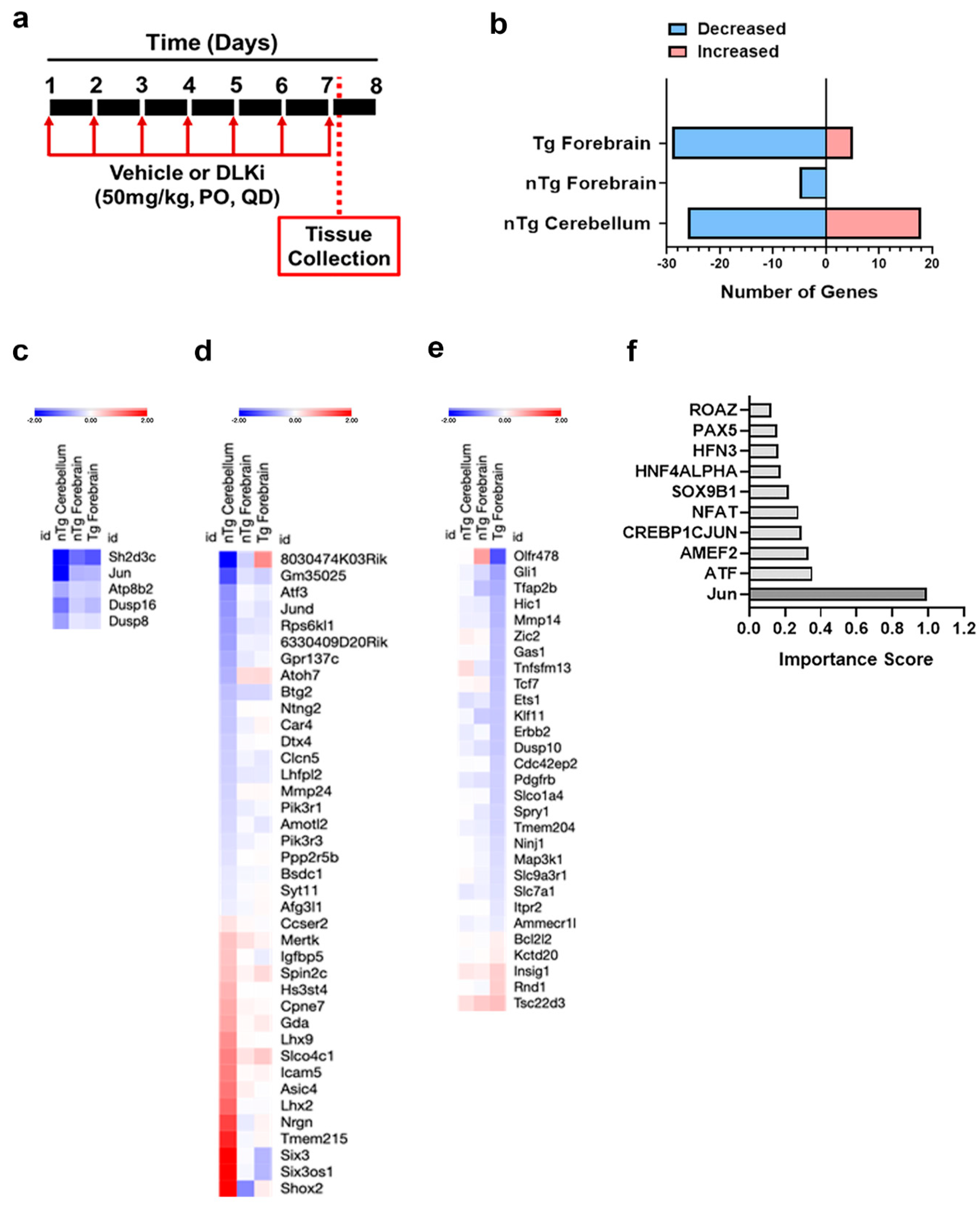
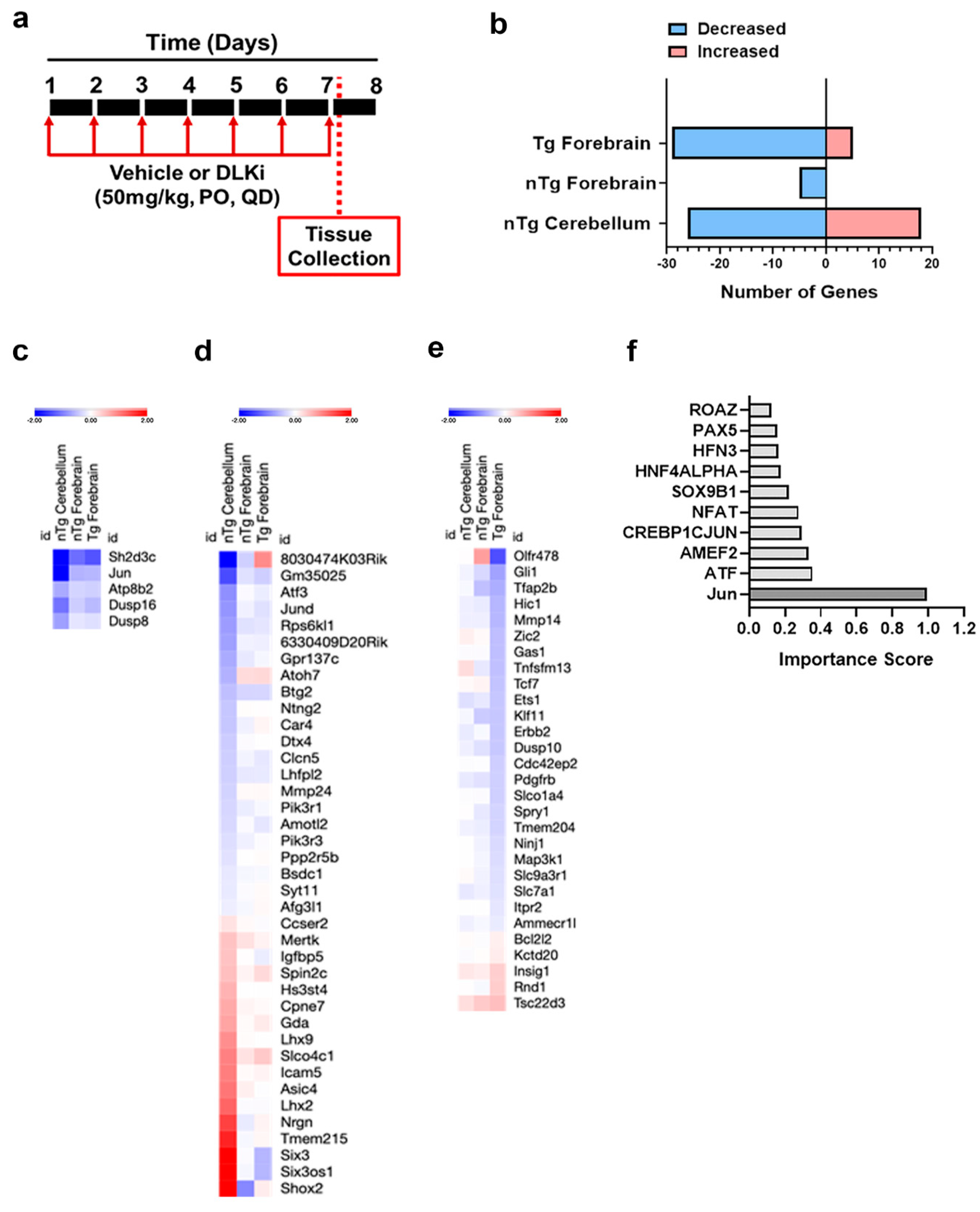
3.5. Downstream Consequences of DLK Activity Are Regionally Distinct
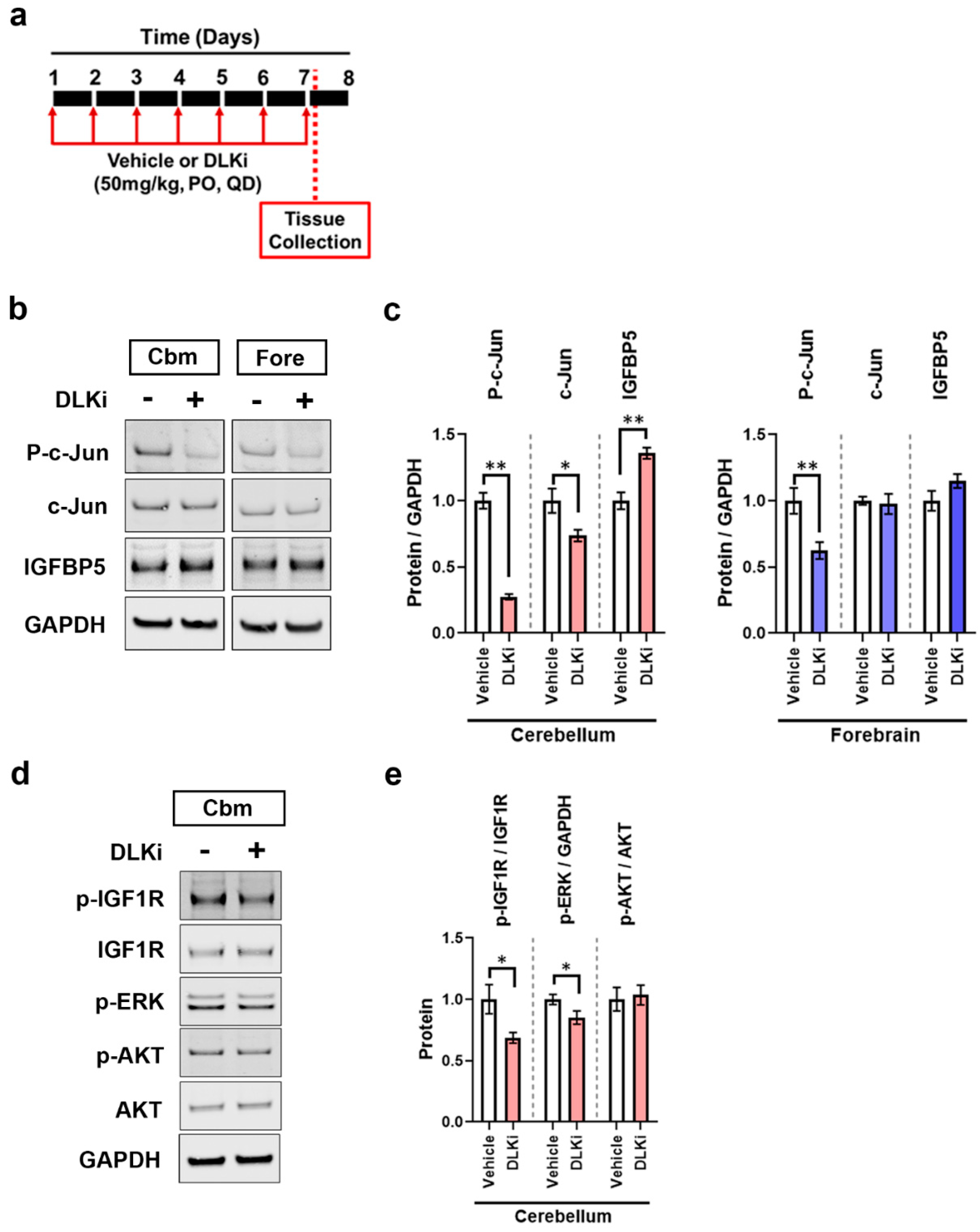
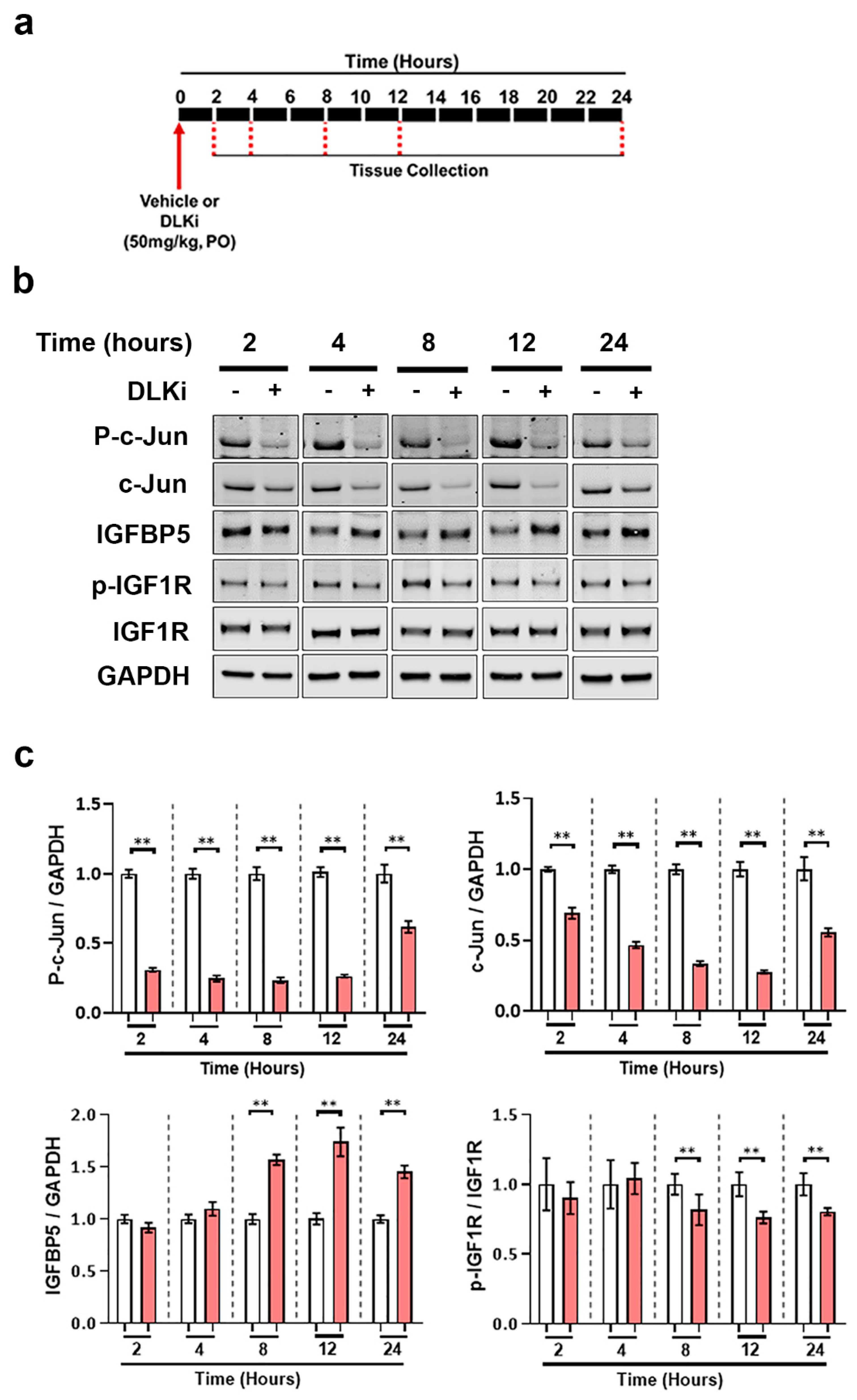
3.6. DLK Upregulates IGFBP5 Levels Specifically in the Cerebellum
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Conforti, L.; Gilley, J.; Coleman, M.P. Wallerian degeneration: An emerging axon death pathway linking injury and disease. Nat. Rev. Neurosci. 2014, 1515, 394–409. [Google Scholar] [CrossRef]
- Miller, B.R.; Press, C.; Daniels, R.W.; Sasaki, Y.; Milbrandt, J.; DiAntonio, A. A dual leucine kinase-dependent axon self-destruction program promotes Wallerian degeneration. Nat. Neurosci. 2009, 12, 387–389. [Google Scholar] [CrossRef]
- Tedeschi, A.; Bradke, F. The DLK signalling pathway--a double-edged sword in neural development and regeneration. EMBO Rep. 2013, 14, 605–614. [Google Scholar] [CrossRef] [Green Version]
- Hammarlund, M.; Nix, P.; Hauth, L.; Jorgensen, E.M.; Bastiani, M. Axon regeneration requires a conserved MAP kinase pathway. Science 2009, 323, 802–806. [Google Scholar] [CrossRef] [Green Version]
- Watkins, T.A.; Wang, B.; Huntwork-Rodriguez, S.; Yang, J.; Jiang, Z.; Eastham-Anderson, J.; Modrusan, Z.; Kaminker, J.S.; Tessier-Lavigne, M.; Lewcock, J.W. DLK initiates a transcriptional program that couples apoptotic and regenerative responses to axonal injury. Proc. Natl. Acad. Sci. USA 2013, 110, 4039–4044. [Google Scholar] [CrossRef] [Green Version]
- Welsbie, D.S.; Yang, Z.; Ge, Y.; Mitchell, K.L.; Zhou, X.; Martin, S.E.; Berlinicke, C.A.; Hackler, L., Jr.; Fuller, J.; Fu, J.; et al. Functional genomic screening identifies dual leucine zipper kinase as a key mediator of retinal ganglion cell death. Proc. Natl. Acad. Sci. USA 2013, 110, 4045–4050. [Google Scholar] [CrossRef] [Green Version]
- Xiong, X.; Collins, C.A. A conditioning lesion protects axons from degeneration via the Wallenda/DLK MAP kinase signaling cascade. J. Neurosci. 2012, 32, 610–615. [Google Scholar] [CrossRef]
- Xiong, X.; Wang, X.; Ewanek, R.; Bhat, P.; Diantonio, A.; Collins, C.A. Protein turnover of the Wallenda/DLK kinase regulates a retrograde response to axonal injury. J. Cell Biol. 2010, 191, 211–223. [Google Scholar] [CrossRef] [Green Version]
- Yan, D.; Wu, Z.; Chisholm, A.D.; Jin, Y. The DLK-1 kinase promotes mRNA stability and local translation in C. elegans synapses and axon regeneration. Cell 2009, 138, 1005–1018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirai, S.; Cui, D.F.; Miyata, T.; Ogawa, M.; Kiyonari, H.; Suda, Y.; Aizawa, S.; Banba, Y.; Ohno, S. The c-Jun N-terminal kinase activator dual leucine zipper kinase regulates axon growth and neuronal migration in the developing cerebral cortex. J. Neurosci. 2006, 26, 11992–12002. [Google Scholar] [CrossRef] [PubMed]
- Hirai, S.; Banba, Y.; Satake, T.; Ohno, S. Axon formation in neocortical neurons depends on stage-specific regulation of microtubule stability by the dual leucine zipper kinase-c-Jun N-terminal kinase pathway. J. Neurosci. 2011, 31, 6468–6480. [Google Scholar] [CrossRef] [PubMed]
- Nakata, K.; Abrams, B.; Grill, B.; Goncharov, A.; Huang, X.; Chisholm, A.D.; Jin, Y. Regulation of a DLK-1 and p38 MAP kinase pathway by the ubiquitin ligase RPM-1 is required for presynaptic development. Cell 2005, 120, 407–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itoh, A.; Horiuchi, M.; Wakayama, K.; Xu, J.; Bannerman, P.; Pleasure, D.; Itoh, T. ZPK/DLK, a mitogen-activated protein kinase kinase kinase, is a critical mediator of programmed cell death of motoneurons. J. Neurosci. 2011, 31, 7223–7228. [Google Scholar] [CrossRef]
- Klinedinst, S.; Wang, X.; Xiong, X.; Haenfler, J.M.; Collins, C.A. Independent pathways downstream of the Wnd/DLK MAPKKK regulate synaptic structure, axonal transport, and injury signaling. J. Neurosci. 2013, 33, 12764–12778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crawley, O.; Giles, A.C.; Desbois, M.; Kashyap, S.; Birnbaum, R.; Grill, B. A MIG-15/JNK-1 MAP kinase cascade opposes RPM-1 signaling in synapse formation and learning. PLoS Genet. 2017, 13, e1007095. [Google Scholar] [CrossRef]
- Pozniak, C.D.; Sengupta Ghosh, A.; Gogineni, A.; Hanson, J.E.; Lee, S.H.; Larson, J.L.; Solanoy, H.; Bustos, D.; Li, H.; Ngu, H.; et al. Dual leucine zipper kinase is required for excitotoxicity-induced neuronal degeneration. J. Exp. Med. 2013, 210, 2553–2567. [Google Scholar] [CrossRef]
- Huang, Y.A.; Zhou, B.; Wernig, M.; Sudhof, T.C. ApoE2, ApoE3, and ApoE4 Differentially Stimulate APP Transcription and Abeta Secretion. Cell 2017, 168, 427–441.e21. [Google Scholar] [CrossRef] [Green Version]
- Le Pichon, C.E.; Meilandt, W.J.; Dominguez, S.; Solanoy, H.; Lin, H.; Ngu, H.; Gogineni, A.; Sengupta Ghosh, A.; Jiang, Z.; Lee, S.H.; et al. Loss of dual leucine zipper kinase signaling is protective in animal models of neurodegenerative disease. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [Green Version]
- Lewcock, J.W.; Genoud, N.; Lettieri, K.; Pfaff, S.L. The ubiquitin ligase Phr1 regulates axon outgrowth through modulation of microtubule dynamics. Neuron 2007, 56, 604–620. [Google Scholar] [CrossRef] [Green Version]
- Yan, D.; Jin, Y. Regulation of DLK-1 kinase activity by calcium-mediated dissociation from an inhibitory isoform. Neuron 2012, 76, 534–548. [Google Scholar] [CrossRef] [Green Version]
- Holland, S.M.; Collura, K.M.; Ketschek, A.; Noma, K.; Ferguson, T.A.; Jin, Y.; Gallo, G.; Thomas, G.M. Palmitoylation controls DLK localization, interactions and activity to ensure effective axonal injury signaling. Proc. Natl. Acad. Sci. USA 2016, 113, 763–768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Zhang, Y.V.; Asghari Adib, E.; Stanchev, D.T.; Xiong, X.; Klinedinst, S.; Soppina, P.; Jahn, T.R.; Hume, R.I.; Rasse, T.M.; et al. Restraint of presynaptic protein levels by Wnd/DLK signaling mediates synaptic defects associated with the kinesin-3 motor Unc-104. Elife 2017, 6, e24271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valakh, V.; Frey, E.; Babetto, E.; Walker, L.J.; DiAntonio, A. Cytoskeletal disruption activates the DLK/JNK pathway, which promotes axonal regeneration and mimics a preconditioning injury. Neurobiol. Dis. 2015, 77, 13–25. [Google Scholar] [CrossRef] [Green Version]
- Collins, C.A.; Wairkar, Y.P.; Johnson, S.L.; DiAntonio, A. Highwire restrains synaptic growth by attenuating a MAP kinase signal. Neuron 2006, 51, 57–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huntwork-Rodriguez, S.; Wang, B.; Watkins, T.; Ghosh, A.S.; Pozniak, C.D.; Bustos, D.; Newton, K.; Kirkpatrick, D.S.; Lewcock, J.W. JNK-mediated phosphorylation of DLK suppresses its ubiquitination to promote neuronal apoptosis. J. Cell Biol. 2013, 202, 747–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nix, P.; Hisamoto, N.; Matsumoto, K.; Bastiani, M. Axon regeneration requires coordinate activation of p38 and JNK MAPK pathways. Proc. Natl. Acad. Sci. USA 2011, 108, 10738–10743. [Google Scholar] [CrossRef] [Green Version]
- Larhammar, M.; Huntwork-Rodriguez, S.; Rudhard, Y.; Sengupta-Ghosh, A.; Lewcock, J.W. The Ste20 Family Kinases MAP4K4, MINK1, and TNIK Converge to Regulate Stress-Induced JNK Signaling in Neurons. J. Neurosci. 2017, 37, 11074–11084. [Google Scholar] [CrossRef]
- Xu, Z.; Maroney, A.C.; Dobrzanski, P.; Kukekov, N.V.; Greene, L.A. The MLK family mediates c-Jun N-terminal kinase activation in neuronal apoptosis. Mol. Cell. Biol. 2001, 21, 4713–4724. [Google Scholar] [CrossRef] [Green Version]
- Mata, M.; Merritt, S.E.; Fan, G.; Yu, G.G.; Holzman, L.B. Characterization of dual leucine zipper-bearing kinase, a mixed lineage kinase present in synaptic terminals whose phosphorylation state is regulated by membrane depolarization via calcineurin. J. Biol. Chem. 1996, 271, 16888–16896. [Google Scholar] [CrossRef] [Green Version]
- Asaoka, Y.; Nishina, H. Diverse physiological functions of MKK4 and MKK7 during early embryogenesis. J. Biochem. 2010, 148, 393–401. [Google Scholar] [CrossRef]
- Bode, A.M.; Dong, Z. The functional contrariety of JNK. Mol. Carcinog. 2007, 46, 591–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.M.; Onesime, D.; Reddy, C.D.; Dhanasekaran, N.; Reddy, E.P. JLP: A scaffolding protein that tethers JNK/p38MAPK signaling modules and transcription factors. Proc. Natl. Acad. Sci. USA 2002, 99, 14189–14194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mooney, L.M.; Whitmarsh, A.J. Docking interactions in the c-Jun N-terminal kinase pathway. J. Biol. Chem. 2004, 279, 11843–11852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitmarsh, A.J. The JIP family of MAPK scaffold proteins. Biochem. Soc. Trans. 2006, 34, 828–832. [Google Scholar] [CrossRef]
- Xu, Z.; Kukekov, N.V.; Greene, L.A. POSH acts as a scaffold for a multiprotein complex that mediates JNK activation in apoptosis. EMBO J. 2003, 22, 252–261. [Google Scholar] [CrossRef] [Green Version]
- Rishal, I.; Fainzilber, M. Axon-soma communication in neuronal injury. Nat. Rev. Neurosci. 2014, 15, 32–42. [Google Scholar] [CrossRef]
- Coffey, E.T. Nuclear and cytosolic JNK signalling in neurons. Nat. Rev. Neurosci. 2014, 15, 285–299. [Google Scholar] [CrossRef]
- Herdegen, T.; Skene, P.; Bahr, M. The c-Jun transcription factor--bipotential mediator of neuronal death, survival and regeneration. Trends Neurosci. 1997, 20, 227–231. [Google Scholar] [CrossRef]
- Patel, S.; Meilandt, W.J.; Erickson, R.I.; Chen, J.; Deshmukh, G.; Estrada, A.A.; Fuji, R.N.; Gibbons, P.; Gustafson, A.; Harris, S.F.; et al. Selective Inhibitors of Dual Leucine Zipper Kinase (DLK, MAP3K12) with Activity in a Model of Alzheimer’s Disease. J. Med. Chem. 2017, 60, 8083–8102. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Genome Project Data Processing, S. LThe Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Babicki, S.; Arndt, D.; Marcu, A.; Liang, Y.; Grant, J.R.; Maciejewski, A.; Wishart, D.S. Heatmapper: Web-enabled heat mapping for all. Nucleic Acids Res. 2016, 44, W147–W153. [Google Scholar] [CrossRef] [PubMed]
- Gotea, V.; Ovcharenko, I. DiRE: Identifying distant regulatory elements of co-expressed genes. Nucleic Acids Res. 2008, 36, W133–W139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santacruz, K.; Lewis, J.; Spires, T.; Paulson, J.; Kotilinek, L.; Ingelsson, M.; Guimaraes, A.; DeTure, M.; Ramsden, M.; McGowan, E.; et al. Tau suppression in a neurodegenerative mouse model improves memory function. Science 2005, 309, 476–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suenaga, J.; Cui, D.F.; Yamamoto, I.; Ohno, S.; Hirai, S. Developmental changes in the expression pattern of the JNK activator kinase MUK/DLK/ZPK and active JNK in the mouse cerebellum. Cell Tissue Res. 2006, 325, 189–195. [Google Scholar] [CrossRef]
- Yuan, Z.; Gong, S.; Luo, J.; Zheng, Z.; Song, B.; Ma, S.; Guo, J.; Hu, C.; Thiel, G.; Vinson, C.; et al. Opposing roles for ATF2 and c-Fos in c-Jun-mediated neuronal apoptosis. Mol. Cell. Biol. 2009, 29, 2431–2442. [Google Scholar] [CrossRef] [Green Version]
- Patricelli, M.P.; Nomanbhoy, T.K.; Wu, J.; Brown, H.; Zhou, D.; Zhang, J.; Jagannathan, S.; Aban, A.; Okerberg, E.; Herring, C.; et al. In situ kinase profiling reveals functionally relevant properties of native kinases. Chem. Biol. 2011, 18, 699–710. [Google Scholar] [CrossRef] [Green Version]
- Pellet, J.B.; Haefliger, J.A.; Staple, J.K.; Widmann, C.; Welker, E.; Hirling, H.; Bonny, C.; Nicod, P.; Catsicas, S.; Waeber, G.; et al. Spatial, temporal and subcellular localization of islet-brain 1 (IB1), a homologue of JIP-1, in mouse brain. Eur. J. Neurosci. 2000, 12, 621–632. [Google Scholar] [CrossRef]
- Larhammar, M.; Huntwork-Rodriguez, S.; Jiang, Z.; Solanoy, H.; Sengupta Ghosh, A.; Wang, B.; Kaminker, J.S.; Huang, K.; Eastham-Anderson, J.; Siu, M.; et al. Dual leucine zipper kinase-dependent PERK activation contributes to neuronal degeneration following insult. Elife 2017, 6. [Google Scholar] [CrossRef]
- Chauvin, S.; Sobel, A. Neuronal stathmins: A family of phosphoproteins cooperating for neuronal development, plasticity and regeneration. Prog. Neurobiol. 2015, 126, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.S.; Wang, B.; Pozniak, C.D.; Chen, M.; Watts, R.J.; Lewcock, J.W. DLK induces developmental neuronal degeneration via selective regulation of proapoptotic JNK activity. J. Cell Biol. 2011, 194, 751–764. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, C.A.; Knudson, K.M.; Peng, B.J.; Teixeiro, E.; Daniels, M.A. The POSH/JIP-1 scaffold network regulates TCR-mediated JNK1 signals and effector function in CD8(+) T cells. Eur. J. Immunol. 2013, 43, 3361–3371. [Google Scholar] [CrossRef] [PubMed]
- Whitmarsh, A.J.; Kuan, C.Y.; Kennedy, N.J.; Kelkar, N.; Haydar, T.F.; Mordes, J.P.; Appel, M.; Rossini, A.A.; Jones, S.N.; Flavell, R.A.; et al. Requirement of the JIP1 scaffold protein for stress-induced JNK activation. Genes Dev. 2001, 15, 2421–2432. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.G.; Wang, R.M.; Yin, X.H.; Pan, J.; Xu, T.L.; Zhang, G.Y. Knock-down of POSH expression is neuroprotective through down-regulating activation of the MLK3-MKK4-JNK pathway following cerebral ischaemia in the rat hippocampal CA1 subfield. J. Neurochem. 2005, 95, 784–795. [Google Scholar] [CrossRef]
- Mardinly, A.R.; Spiegel, I.; Patrizi, A.; Centofante, E.; Bazinet, J.E.; Tzeng, C.P.; Mandel-Brehm, C.; Harmin, D.A.; Adesnik, H.; Fagiolini, M.; et al. Sensory experience regulates cortical inhibition by inducing IGF1 in VIP neurons. Nature 2016, 531, 371–375. [Google Scholar] [CrossRef] [Green Version]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goodwani, S.; Fernandez, C.; Acton, P.J.; Buggia-Prevot, V.; McReynolds, M.L.; Ma, J.; Hu, C.H.; Hamby, M.E.; Jiang, Y.; Le, K.; et al. Dual Leucine Zipper Kinase Is Constitutively Active in the Adult Mouse Brain and Has Both Stress-Induced and Homeostatic Functions. Int. J. Mol. Sci. 2020, 21, 4849. https://doi.org/10.3390/ijms21144849
Goodwani S, Fernandez C, Acton PJ, Buggia-Prevot V, McReynolds ML, Ma J, Hu CH, Hamby ME, Jiang Y, Le K, et al. Dual Leucine Zipper Kinase Is Constitutively Active in the Adult Mouse Brain and Has Both Stress-Induced and Homeostatic Functions. International Journal of Molecular Sciences. 2020; 21(14):4849. https://doi.org/10.3390/ijms21144849
Chicago/Turabian StyleGoodwani, Sunil, Celia Fernandez, Paul J. Acton, Virginie Buggia-Prevot, Morgan L. McReynolds, Jiacheng Ma, Cheng Hui Hu, Mary E. Hamby, Yongying Jiang, Kang Le, and et al. 2020. "Dual Leucine Zipper Kinase Is Constitutively Active in the Adult Mouse Brain and Has Both Stress-Induced and Homeostatic Functions" International Journal of Molecular Sciences 21, no. 14: 4849. https://doi.org/10.3390/ijms21144849