Interleukin Gene Variability and Periodontal Bacteria in Patients with Generalized Aggressive Form of Periodontitis

 , ,
, , 
Abstract
1. Introduction
2. Results
2.1. Genetic Analysis
2.2. Determination of the Selected Oral Bacteria
2.3. A Pilot In Vitro Study: IL-10 Levels in Unstimulated/Stimulated PBMCs
3. Discussion
3.1. Interleukin-1 Family
3.2. Interleukin-6
3.3. Interleukin-17
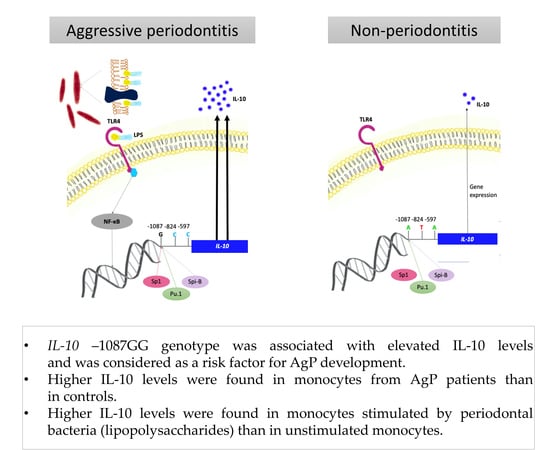
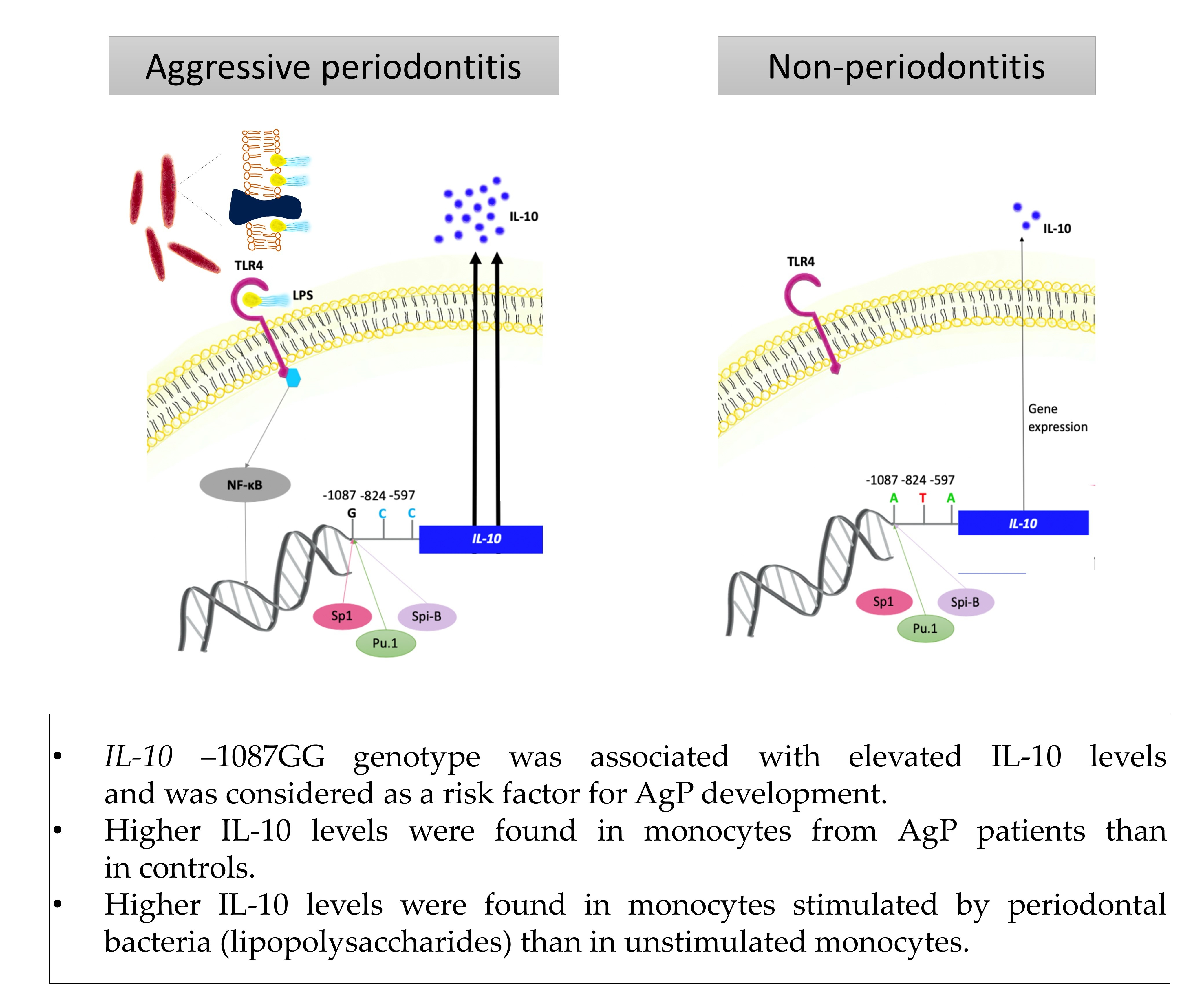
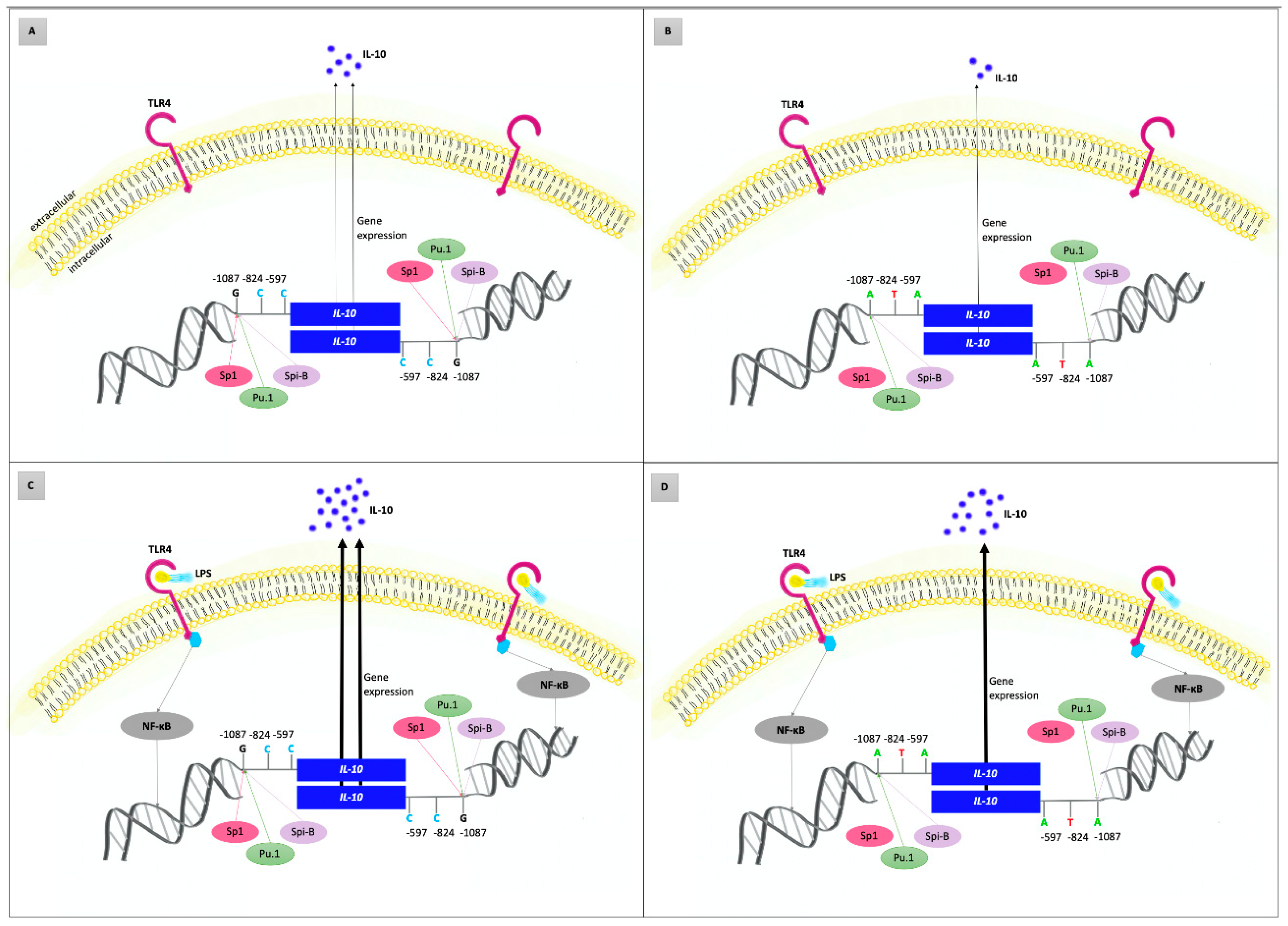
3.4. Interleukin-10
3.5. A Pilot In Vitro Study
4. Materials and Methods
4.1. Study Design, Clinical Examination and Sample Collection
4.2. Isolation of Genomic DNA and Genetic Analysis
4.3. Microbial Analysis
4.4. PBMCs Isolation, Cultivation and Stimulation by Oral Bacteria In Vitro
4.5. Determination of Interleukin-10 Levels in PBMCs
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AgP | Aggressive periodontitis |
| AS-PCR | Allele-specific polymerase chain reaction |
| cAMP | Cyclic adenosine monophosphate |
| CFU | Colony-forming units |
| CI | Confidence interval |
| CP | Chronic periodontitis |
| CPITN | Community Periodontal Index of Treatment Needs |
| dNTPs | Deoxyribonucleoside triphosphate mix |
| EDTA | Ethylenediaminetetraacetic acid |
| HWE | Hardy–Weinberg equilibrium |
| IL | Interleukin |
| IQR | Interquartile range |
| LD | Linkage disequilibrium |
| LPS | Lipopolysaccharides |
| MAF | Minor allele frequencies |
| MHC | Histocompatibility complex |
| N | Number of individuals |
| NF-1 | Nuclear factor 1 |
| NFAT | Nuclear factor activated T cells |
| NFκB | Nuclear factor kappa B |
| OR | Odds ratio |
| PAMP | Pathogen-associated molecular patterns |
| PARP-1 | Poly (ADP-ribose) polymerase-1 |
| PBMCs | Peripheral blood monocytes |
| PCR | Polymerase chain reaction |
| PWM | Pokeweed mitogen |
| RFLP-PCR | Restriction fragment length polymorphism PCR |
| SD | Standard deviation |
| SNP | Single-nucleotide polymorphism |
| Th1/2 | T helper 1/2 |
| TLR4 | Toll-like receptor 4 |
| TNF-α | Tumor necrosis factor-alpha |
| VNTR | Variable tandem repeats |
References
- Chapple, I.L.C.; Mealey, B.L.; Dyke, T.E.V.; Bartold, P.M.; Dommisch, H.; Eickholz, P.; Geisinger, M.L.; Genco, R.J.; Glogauer, M.; Goldstein, M.; et al. Periodontal Health and Gingival Diseases and Conditions on an Intact and a Reduced Periodontium: Consensus Report of Workgroup 1 of the 2017 World Workshop on the Classification of Periodontal and Peri-Implant Diseases and Conditions. J. Periodontol. 2018, 89 (Suppl. S1), S74–S84. [Google Scholar] [CrossRef] [PubMed]
- Fine, D.H.; Patil, A.G.; Loos, B.G. Classification and Diagnosis of Aggressive Periodontitis. J. Clin. Periodontol. 2018, 45 (Suppl. S20), S95–S111. [Google Scholar] [CrossRef] [PubMed]
- Papapanou, P.N.; Sanz, M.; Buduneli, N.; Dietrich, T.; Feres, M.; Fine, D.H.; Flemmig, T.F.; Garcia, R.; Giannobile, W.V.; Graziani, F.; et al. Periodontitis: Consensus Report of Workgroup 2 of the 2017 World Workshop on the Classification of Periodontal and Peri-Implant Diseases and Conditions. J. Periodontol. 2018, 89 (Suppl. S1), S173–S182. [Google Scholar] [CrossRef] [PubMed]
- Dyke, T.E.V.; Sima, C. Understanding Resolution of Inflammation in Periodontal Diseases: Is Chronic Inflammatory Periodontitis a Failure to Resolve? Periodontol. 2000 2020, 82, 205–213. [Google Scholar] [CrossRef]
- Nibali, L. Aggressive Periodontitis: Microbes and Host Response, Who to Blame? Virulence 2015, 6, 223–228. [Google Scholar] [CrossRef]
- Hajishengallis, G.; Sojar, H.; Genco, R.J.; DeNardin, E. Intracellular Signaling and Cytokine Induction upon Interactions of Porphyromonas Gingivalis Fimbriae with Pattern-Recognition Receptors. Immunol. Investig. 2004, 33, 157–172. [Google Scholar] [CrossRef] [PubMed]
- Shahabuddin, N.; Boesze-Battaglia, K.; Lally, E.T. Trends in Susceptibility to Aggressive Periodontal Disease. Int. J. Dent. Oral Health 2016, 2. [Google Scholar] [CrossRef]
- Belibasakis, G.N.; Maula, T.; Bao, K.; Lindholm, M.; Bostanci, N.; Oscarsson, J.; Ihalin, R.; Johansson, A. Virulence and Pathogenicity Properties of Aggregatibacter Actinomycetemcomitans. Pathogens 2019, 8, 222. [Google Scholar] [CrossRef]
- Sima, C.; Van Dyke, T.E. 11—Systems Medicine and Periodontal Diseases. In Translational Systems Medicine and Oral Disease; Sonis, S.T., Villa, A., Eds.; Academic Press: Cambridge, MA, USA, 2020; pp. 249–282. [Google Scholar] [CrossRef]
- Hajishengallis, G. New Developments in Neutrophil Biology and Periodontitis. Periodontol. 2000 2020, 82, 78–92. [Google Scholar] [CrossRef]
- Lamont, R.J.; Koo, H.; Hajishengallis, G. The Oral Microbiota: Dynamic Communities and Host Interactions. Nat. Rev. Microbiol. 2018, 16, 745–759. [Google Scholar] [CrossRef]
- der Velden, U.V. What Exactly Distinguishes Aggressive from Chronic Periodontitis: Is It Mainly a Difference in the Degree of Bacterial Invasiveness? Periodontol. 2000 2017, 75, 24–44. [Google Scholar] [CrossRef]
- Lang, N.P.; Lindhe, J. Clinical Periodontology and Implant. Dentistry, 6th ed.; John Wiley & Sons, Ltd.: Chichester, UK, 2015; Volume 2, pp. 557–1372. ISBN 978-0-470-67248-8. [Google Scholar]
- Meng, H.; Ren, X.; Tian, Y.; Feng, X.; Xu, L.; Zhang, L.; Lu, R.; Shi, D.; Chen, Z. Genetic Study of Families Affected with Aggressive Periodontitis. Periodontol. 2000 2011, 56, 87–101. [Google Scholar] [CrossRef]
- Nibali, L.; Donos, N.; Brett, P.M.; Parkar, M.; Ellinas, T.; Llorente, M.; Griffiths, G.S. A Familial Analysis of Aggressive Periodontitis—Clinical and Genetic Findings. J. Periodontal Res. 2008, 43, 627–634. [Google Scholar] [CrossRef] [PubMed]
- Kinane, D.F.; Hart, T.C. Genes and Gene Polymorphisms Associated with Periodontal Disease. Crit. Rev. Oral Biol. Med. 2016, 14, 430–449. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, F.M.D.; Tinoco, E.M.B.; Govil, M.; Marazita, M.L.; Vieira, A.R. Aggressive Periodontitis Is Likely Influenced by a Few Small Effect Genes. J. Clin. Periodontol. 2009, 36, 468–473. [Google Scholar] [CrossRef] [PubMed]
- Vieira, A.R.; Albandar, J.M. Role of Genetic Factors in the Pathogenesis of Aggressive Periodontitis. Periodontol. 2000 2014, 65, 92–106. [Google Scholar] [CrossRef]
- Masumoto, R.; Kitagaki, J.; Fujihara, C.; Matsumoto, M.; Miyauchi, S.; Asano, Y.; Imai, A.; Kobayashi, K.; Nakaya, A.; Yamashita, M.; et al. Identification of Genetic Risk Factors of Aggressive Periodontitis Using Genomewide Association Studies in Association with Those of Chronic Periodontitis. J. Periodontal Res. 2019, 54, 199–206. [Google Scholar] [CrossRef]
- Wang, W.-F.; Shi, J.; Chen, S.-J.; Niu, Y.-M.; Zeng, X.-T. Interleukin-1α −899 (+4845) C→T Polymorphism Is Not Associated with Aggressive Periodontitis Susceptibility: A Meta-analysis Based on 19 Case-control Studies. Biomed. Rep. 2014, 2, 378–383. [Google Scholar] [CrossRef]
- Chen, Y.-J.; Han, Y.; Mao, M.; Tan, Y.-Q.; Leng, W.-D.; Zeng, X.-T. Interleukin-1β Rs1143634 Polymorphism and Aggressive Periodontitis Susceptibility: A Meta-Analysis. Int. J. Clin. Exp. Med. 2015, 8, 2308–2316. [Google Scholar]
- Huang, W.; He, B.-Y.; Shao, J.; Jia, X.-W.; Yuan, Y.-D. Interleukin-1β Rs1143627 Polymorphism with Susceptibility to Periodontal Disease. Oncotarget 2017, 8, 31406–31414. [Google Scholar] [CrossRef]
- Ding, C.; Zhao, L.; Sun, Y.; Li, L.; Xu, Y. Interleukin-1 Receptor Antagonist Polymorphism (Rs2234663) and Periodontitis Susceptibility: A Meta-Analysis. Arch. Oral Biol. 2012, 57, 585–593. [Google Scholar] [CrossRef] [PubMed]
- Shao, M.; Huang, P.; Cheng, R.; Hu, T. Interleukin-6 Polymorphisms Modify the Risk of Periodontitis: A Systematic Review and Meta-Analysis. J. Zhejiang Univ. Sci. B 2009, 10, 920–927. [Google Scholar] [CrossRef] [PubMed]
- Albuquerque, C.M.; Cortinhas, A.J.; Morinha, F.J.; Leitão, J.C.; Viegas, C.A.; Bastos, E.M. Association of the IL-10 Polymorphisms and Periodontitis: A Meta-Analysis. Mol. Biol. Rep. 2012, 39, 9319–9329. [Google Scholar] [CrossRef] [PubMed]
- Wong, H.C.; Ooi, Y.; Pulikkotil, S.J.; Naing, C. The Role of Three Interleukin 10 Gene Polymorphisms (− 1087 A > G, − 824 C > T, − 597 A > C) in the Risk of Chronic and Aggressive Periodontitis: A Meta-Analysis and Trial Sequential Analysis. BMC Oral Health 2018, 18, 171. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Feng, G.; Deng, Y.; Song, J. Contribution of Interleukin-10-597 (-590, -597) C > A Polymorphisms to Periodontitis Susceptibility: An Updated Meta-Analysis Based on 18 Case-Control Studies. Dis. Markers 2018, 2018, 2645963. [Google Scholar] [CrossRef] [PubMed]
- da Silva, F.R.P.; dos Pessoa, L.S.; Vasconcelos, A.C.C.G.; de Aquino Lima, W.; Alves, E.H.P.; Vasconcelos, D.F.P. Polymorphisms in Interleukins 17A and 17F Genes and Periodontitis: Results from a Meta-Analysis. Mol. Biol. Rep. 2017, 44, 443–453. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.-G.; Li, J.-J.; Sun, C.-A.; Jin, Y.; Wu, W.-W. Interleukin-18 Promoter Polymorphisms and Plasma Levels Are Associated with Increased Risk of Periodontitis: A Meta-Analysis. Inflamm. Res. 2014, 63, 45–52. [Google Scholar] [CrossRef]
- da Silva, M.K.; de Carvalho, A.C.G.; Alves, E.H.P.; da Silva, F.R.P.; dos Pessoa, L.S.; Vasconcelos, D.F.P. Genetic Factors and the Risk of Periodontitis Development: Findings from a Systematic Review Composed of 13 Studies of Meta-Analysis with 71,531 Participants. Int. J. Dent. 2017, 2017, 1914073. [Google Scholar] [CrossRef]
- Maney, P.; Owens, J.L. Interleukin Polymorphisms in Aggressive Periodontitis: A Literature Review. J. Indian Soc. Periodontol. 2015, 19, 131–141. [Google Scholar] [CrossRef]
- Borilova Linhartova, P.; Vokurka, J.; Poskerova, H.; Fassmann, A.; Izakovicova Holla, L. Haplotype Analysis of Interleukin-8 Gene Polymorphisms in Chronic and Aggressive Periodontitis. Mediat. Inflamm. 2013, 2013, 342351. [Google Scholar] [CrossRef]
- Straub, R.H.; Schradin, C. Chronic Inflammatory Systemic Diseases: An Evolutionary Trade-off between Acutely Beneficial but Chronically Harmful Programs. Evol. Med. Public Health 2016, 2016, 37–51. [Google Scholar] [CrossRef] [PubMed]
- Wander, K. The Origin of Chronic Inflammatory Systemic Diseases and Their Sequelae. by Rainer H. Straub. 390 Pp. Waltham, MA: Academic Press. 2015. $140.00 (Paper or e-Book). Am. J. Hum. Biol. 2016, 28, 445–446. [Google Scholar] [CrossRef]
- Khocht, A.; Albandar, J.M. Aggressive Forms of Periodontitis Secondary to Systemic Disorders. Periodontol. 2000 2014, 65, 134–148. [Google Scholar] [CrossRef]
- Könönen, E.; Müller, H.-P. Microbiology of Aggressive Periodontitis. Periodontol. 2000 2014, 65, 46–78. [Google Scholar] [CrossRef]
- Tomita, S.; Komiya-Ito, A.; Imamura, K.; Kita, D.; Ota, K.; Takayama, S.; Makino-Oi, A.; Kinumatsu, T.; Ota, M.; Saito, A. Prevalence of Aggregatibacter Actinomycetemcomitans, Porphyromonas Gingivalis and Tannerella Forsythia in Japanese Patients with Generalized Chronic and Aggressive Periodontitis. Microb. Pathog. 2013, 61, 11–15. [Google Scholar] [CrossRef]
- Lanza, E.; Magan-Fernandez, A.; Bermejo, B.; de Rojas, J.; Marfil-Alvarez, R.; Mesa, F. Complementary Clinical Effects of Red Complex Bacteria on Generalized Periodontitis in a Caucasian Population. Oral Dis. 2016, 22, 430–437. [Google Scholar] [CrossRef] [PubMed]
- Clark, D.; Febbraio, M.; Levin, L. Aggressive Periodontitis: The Unsolved Mystery. Quintessence Int. Berl. Ger. 1985 2017, 48, 103–111. [Google Scholar] [CrossRef]
- Dinarello, C.A. Interleukin-1 and Interleukin-1 Antagonism. Blood 1991, 77, 1627–1652. [Google Scholar] [CrossRef]
- Biet, F.; Locht, C.; Kremer, L. Immunoregulatory Functions of Interleukin 18 and Its Role in Defense against Bacterial Pathogens. J. Mol. Med. 2002, 80, 147–162. [Google Scholar] [CrossRef] [PubMed]
- Steinkasserer, A.; Spurr, N.K.; Cox, S.; Jeggo, P.; Sim, R.B. The Human IL-1 Receptor Antagonist Gene (IL1RN) Maps to Chromosome 2q14–Q21, in the Region of the IL-1α and IL-1β Loci. Genomics 1992, 13, 654–657. [Google Scholar] [CrossRef]
- Dominici, R.; Cattaneo, M.; Malferrari, G.; Archi, D.; Mariani, C.; Grimaldi, L.; Biunno, I. Cloning and Functional Analysis of the Allelic Polymorphism in the Transcription Regulatory Region of Interleukin-1α. Immunogenetics 2002, 54, 82–86. [Google Scholar] [CrossRef]
- Pociot, F.; Mølvig, J.; Wogensen, L.; Worsaae, H.; Nerup, J. A TaqI Polymorphism in the Human Interleukin-1 Beta (IL-1 Beta) Gene Correlates with IL-1 Beta Secretion In Vitro. Eur. J. Clin. Investig. 1992, 22, 396–402. [Google Scholar] [CrossRef] [PubMed]
- Tarlow, J.K.; Blakemore, A.I.; Lennard, A.; Solari, R.; Hughes, H.N.; Steinkasserer, A.; Duff, G.W. Polymorphism in Human IL-1 Receptor Antagonist Gene Intron 2 Is Caused by Variable Numbers of an 86-Bp Tandem Repeat. Hum. Genet. 1993, 91, 403–404. [Google Scholar] [CrossRef] [PubMed]
- Boukortt, K.N.; Saidi-Ouahrani, N.; Boukerzaza, B.; Ouhaibi-Djellouli, H.; Hachmaoui, K.; Benaissa, F.Z.; Taleb, L.; Drabla-Ouahrani, H.; Deba, T.; Ouledhamou, S.A.; et al. Association Analysis of the IL-1 Gene Cluster Polymorphisms with Aggressive and Chronic Periodontitis in the Algerian Population. Arch. Oral Biol. 2015, 60, 1463–1470. [Google Scholar] [CrossRef]
- Puri, K.; Chhokra, M.; Dodwad, V.; Puri, N. Association of Interleukin-1 α (−889) Gene Polymorphism in Patients with Generalized Aggressive and Chronic Periodontitis. Dent. Res. J. 2015, 12, 76–82. [Google Scholar] [CrossRef]
- Ribeiro, M.S.M.; Pacheco, R.B.A.; Fischer, R.G.; Macedo, J.M.B. Interaction of IL1B and IL1RN Polymorphisms, Smoking Habit, Gender, and Ethnicity with Aggressive and Chronic Periodontitis Susceptibility. Contemp. Clin. Dent. 2016, 7, 349. [Google Scholar] [CrossRef] [PubMed]
- Ayazi, G.; Pirayesh, M.; Yari, K. Analysis of Interleukin-1β Gene Polymorphism and Its Association with Generalized Aggressive Periodontitis Disease. DNA Cell Biol. 2013, 32, 409–413. [Google Scholar] [CrossRef] [PubMed]
- Borilova Linhartova, P.; Poskerova, H.; Tomandlova, M.; Bartova, J.; Kankova, K.; Fassmann, A.; Izakovicova Holla, L. Interleukin-1 Gene Variability and Plasma Levels in Czech Patients with Chronic Periodontitis and Diabetes Mellitus. Int. J. Dent. 2019, 2019, 6802349. [Google Scholar] [CrossRef]
- Reference SNP (refSNP) Cluster Report: rs1800587. Available online: www.ncbi.nlm.nih.gov/projects/SNP/snp_ref.cgi?do_not_redirect&rs=rs1800587 (accessed on 27 April 2020).
- Reference SNP (refSNP) Cluster Report: rs1143634. Available online: www.ncbi.nlm.nih.gov/projects/SNP/snp_ref.cgi?do_not_redirect&rs=rs1143634 (accessed on 27 April 2020).
- Parkhill, J.M.; Hennig, B.J.W.; Chapple, I.L.C.; Heasman, P.A.; Taylor, J.J. Association of Interleukin-1 Gene Polymorphisms with Early-Onset Periodontitis. J. Clin. Periodontol. 2000, 27, 682–689. [Google Scholar] [CrossRef]
- Laine, M.L.; Farre, M.A.; Garcia-González, M.A.; van Dijk, L.J.; Ham, A.J.; Winkel, E.G.; Crusius, J.B.A.; Vandenbroucke, J.P.; van Winkelhoff, A.J.; Pena, A.S. Polymorphisms of the Interleukin-1 Gene Family, Oral Microbial Pathogens, and Smoking in Adult Periodontitis. J. Dent. Res. 2001, 80, 1695–1699. [Google Scholar] [CrossRef]
- Sakellari, D.; Katsares, V.; Georgiadou, M.; Kouvatsi, A.; Arsenakis, M.; Konstantinidis, A. No Correlation of Five Gene Polymorphisms with Periodontal Conditions in a Greek Population. J. Clin. Periodontol. 2006, 33, 765–770. [Google Scholar] [CrossRef] [PubMed]
- Havemose-Poulsen, A.; Sørensen, L.K.; Bendtzen, K.; Holmstrup, P. Polymorphisms Within the IL-1 Gene Cluster: Effects on Cytokine Profiles in Peripheral Blood and Whole Blood Cell Cultures of Patients With Aggressive Periodontitis, Juvenile Idiopathic Arthritis, and Rheumatoid Arthritis. J. Periodontol. 2007, 78, 475–492. [Google Scholar] [CrossRef]
- Okamura, H.; Tsutsui, H.; Komatsu, T.; Yutsudo, M.; Hakura, A.; Tanimoto, T.; Torigoe, K.; Okura, T.; Nukada, Y.; Hattori, K.; et al. Cloning of a New Cytokine That Induces IFN-γ Production by T Cells. Nature 1995, 378, 88–91. [Google Scholar] [CrossRef]
- Nolan, K.F.; Greaves, D.R.; Waldmann, H. The Human Interleukin 18 Gene IL18 Maps to 11q22.2-Q22.3, Closely Linked to the DRD2 Gene Locus and Distinct from Mapped IDDM Loci. Genomics 1998, 51, 161–163. [Google Scholar] [CrossRef] [PubMed]
- Kruse, S.; Kuehr, J.; Moseler, M.; Kopp, M.V.; Kurz, T.; Deichmann, K.A.; Foster, P.S.; Mattes, J. Polymorphisms in the IL 18 Gene Are Associated with Specific Sensitization to Common Allergens and Allergic Rhinitis. J. Allergy Clin. Immunol. 2003, 111, 117–122. [Google Scholar] [CrossRef]
- Giedraitis, V.; He, B.; Huang, W.X.; Hillert, J. Cloning and Mutation Analysis of the Human IL-18 Promoter: A Possible Role of Polymorphisms in Expression Regulation. J. Neuroimmunol. 2001, 112, 146–152. [Google Scholar] [CrossRef]
- Arimitsu, J.; Hirano, T.; Higa, S.; Kawai, M.; Naka, T.; Ogata, A.; Shima, Y.; Fujimoto, M.; Yamadori, T.; Hagiwara, K.; et al. IL-18 Gene Polymorphisms Affect IL-18 Production Capability by Monocytes. Biochem. Biophys. Res. Commun. 2006, 342, 1413–1416. [Google Scholar] [CrossRef] [PubMed]
- Noack, B.; Görgens, H.; Lorenz, K.; Ziegler, A.; Hoffmann, T.; Schackert, H.K. TLR4 and IL-18 Gene Variants in Aggressive Periodontitis. J. Clin. Periodontol. 2008, 35, 1020–1026. [Google Scholar] [CrossRef] [PubMed]
- Folwaczny, M.; Glas, J.; Török, H.-P.; Tonenchi, L.; Paschos, E.; Bauer, B.; Limbersky, O.; Folwaczny, C. Polymorphisms of the Interleukin-18 Gene in Periodontitis Patients. J. Clin. Periodontol. 2005, 32, 530–534. [Google Scholar] [CrossRef] [PubMed]
- Scapoli, C.; Mamolini, E.; Carrieri, A.; Guarnelli, M.E.; Annunziata, M.; Guida, L.; Romano, F.; Aimetti, M.; Trombelli, L. Gene–Gene Interaction among Cytokine Polymorphisms Influence Susceptibility to Aggressive Periodontitis. Genes Immun. 2011, 12, 473–480. [Google Scholar] [CrossRef]
- Martelli, F.S.; Mengoni, A.; Martelli, M.; Rosati, C.; Fanti, E. IL-18 Gene Promoter Polymorphisms Are Only Moderately Associated with Periodontal Disease in Italian Population. Clin. Cases Miner. Bone Metab. 2012, 9, 153–156. [Google Scholar]
- Reference SNP (refSNP) Cluster Report: rs1946518. Available online: www.ncbi.nlm.nih.gov/projects/SNP/snp_ref.cgi?do_not_redirect&rs=rs1946518 (accessed on 27 April 2020).
- Reference SNP (refSNP) Cluster Report: rs187238. Available online: www.ncbi.nlm.nih.gov/projects/SNP/snp_ref.cgi?do_not_redirect&rs=rs187238 (accessed on 27 April 2020).
- Reference SNP (refSNP) Cluster Report: rs4988359. Available online: www.ncbi.nlm.nih.gov/projects/SNP/snp_ref.cgi?do_not_redirect&rs=rs4988359 (accessed on 27 April 2020).
- Garbers, C.; Monhasery, N.; Aparicio-Siegmund, S.; Lokau, J.; Baran, P.; Nowell, M.A.; Jones, S.A.; Rose-John, S.; Scheller, J. The Interleukin-6 Receptor Asp358Ala Single Nucleotide Polymorphism Rs2228145 Confers Increased Proteolytic Conversion Rates by ADAM Proteases. Biochim. Biophys. Acta BBA–Mol. Basis Dis. 2014, 1842, 1485–1494. [Google Scholar] [CrossRef] [PubMed]
- Rafiq, S.; Frayling, T.M.; Murray, A.; Hurst, A.; Stevens, K.; Weedon, M.N.; Henley, W.; Ferrucci, L.; Bandinelli, S.; Corsi, A.-M.; et al. A Common Variant of the Interleukin 6 Receptor (IL-6r) Gene Increases IL-6r and IL-6 Levels, without Other Inflammatory Effects. Genes Immun. 2007, 8, 552–559. [Google Scholar] [CrossRef] [PubMed]
- Riethmueller, S.; Somasundaram, P.; Ehlers, J.C.; Hung, C.-W.; Flynn, C.M.; Lokau, J.; Agthe, M.; Düsterhöft, S.; Zhu, Y.; Grötzinger, J.; et al. Proteolytic Origin of the Soluble Human IL-6R In Vivo and a Decisive Role of N-Glycosylation. PLoS Biol. 2017, 15, e2000080. [Google Scholar] [CrossRef] [PubMed]
- Bowcock, A.M.; Kidd, J.R.; Lathrop, G.M.; Daneshvar, L.; May, L.T.; Ray, A.; Sehgal, P.B.; Kidd, K.K.; Cavalli-Sforza, L.L. The Human “Interferon-Β2/Hepatocyte Stimulating Factor/Interleukin-6” Gene: DNA Polymorphism Studies and Localization to Chromosome 7p21. Genomics 1988, 3, 8–16. [Google Scholar] [CrossRef]
- Smelaya, T.V.; Belopolskaya, O.B.; Smirnova, S.V.; Kuzovlev, A.N.; Moroz, V.V.; Golubev, A.M.; Pabalan, N.A.; Salnikova, L.E. Genetic Dissection of Host Immune Response in Pneumonia Development and Progression. Sci. Rep. 2016, 6, 1–12. [Google Scholar] [CrossRef]
- Cole, S.W.; Arevalo, J.M.G.; Takahashi, R.; Sloan, E.K.; Lutgendorf, S.K.; Sood, A.K.; Sheridan, J.F.; Seeman, T.E. Computational Identification of Gene-Social Environment Interaction at the Human IL6 Locus. Proc. Natl. Acad. Sci. USA 2010, 107, 5681–5686. [Google Scholar] [CrossRef]
- Fishman, D.; Faulds, G.; Jeffery, R.; Mohamed-Ali, V.; Yudkin, J.S.; Humphries, S.; Woo, P. The Effect of Novel Polymorphisms in the Interleukin-6 (IL-6) Gene on IL-6 Transcription and Plasma IL-6 Levels, and an Association with Systemic-Onset Juvenile Chronic Arthritis. J. Clin. Investig. 1998, 102, 1369–1376. [Google Scholar] [CrossRef]
- Nibali, L.; Pelekos, G.; D’Aiuto, F.; Chaudhary, N.; Habeeb, R.; Ready, D.; Parkar, M.; Donos, N. Influence of IL-6 Haplotypes on Clinical and Inflammatory Response in Aggressive Periodontitis. Clin. Oral Investig. 2013, 17, 1235–1242. [Google Scholar] [CrossRef]
- Pirim Gorgun, E.; Toker, H.; Korkmaz, E.M.; Poyraz, O. IL-6 and IL-10 Gene Polymorphisms in Patients with Aggressive Periodontitis: Effects on GCF, Serum and Clinic Parameters. Braz. Oral Res. 2017, 31, e12. [Google Scholar] [CrossRef][Green Version]
- Erciyas, K.; Pehlivan, S.; Sever, T.; Igci, M.; Arslan, A.; Orbak, R. Association between TNF-α, TGF-Β1, IL-10, IL-6 and IFN-γ Gene Polymorphisms and Generalized Aggressive Periodontitis. Clin. Investig. Med. 2010, 33, E85–E91. [Google Scholar] [CrossRef] [PubMed]
- Nibali, L.; Griffiths, G.S.; Donos, N.; Parkar, M.; D’Aiuto, F.; Tonetti, M.S.; Brett, P.M. Association between Interleukin-6 Promoter Haplotypes and Aggressive Periodontitis. J. Clin. Periodontol. 2008, 35, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Holla, L.I.; Fassmann, A.; Stejskalová, A.; Znojil, V.; Vanĕk, J.; Vacha, J. Analysis of the Interleukin-6 Gene Promoter Polymorphisms in Czech Patients with Chronic Periodontitis. J. Periodontol. 2004, 75, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Reference SNP (refSNP) Cluster Report: rs1800795. Available online: www.ncbi.nlm.nih.gov/projects/SNP/snp_ref.cgi?do_not_redirect&rs=rs1800795 (accessed on 27 April 2020).
- Reference SNP (refSNP) Cluster Report: rs2228145. Available online: www.ncbi.nlm.nih.gov/projects/SNP/snp_ref.cgi?do_not_redirect&rs=rs2228145 (accessed on 27 April 2020).
- Martinez, G.J.; Nurieva, R.I.; Yang, X.O.; Dong, C. Regulation and Function of Proinflammatory TH17 Cells. Ann. N. Y. Acad. Sci. 2008, 1143, 188–211. [Google Scholar] [CrossRef]
- Espinoza, J.L.; Takami, A.; Nakata, K.; Onizuka, M.; Kawase, T.; Akiyama, H.; Miyamura, K.; Morishima, Y.; Fukuda, T.; Kodera, Y.; et al. A Genetic Variant in the IL-17 Promoter Is Functionally Associated with Acute Graft-Versus-Host Disease after Unrelated Bone Marrow Transplantation. PLoS ONE 2011, 6, e26229. [Google Scholar] [CrossRef]
- Chaudhari, H.L.; Warad, S.; Ashok, N.; Baroudi, K.; Tarakji, B. Association of Interleukin-17 Polymorphism (−197G/A) in Chronic and Localized Aggressive Periodontitis. Braz. Oral Res. 2016, 30. [Google Scholar] [CrossRef]
- Saraiva, A.M.; Alves e Silva, M.R.M.; de Correia Silva, J.F.; da Costa, J.E.; Gollob, K.J.; Dutra, W.O.; Moreira, P.R. Evaluation of IL17A Expression and of IL17A, IL17F and IL23R Gene Polymorphisms in Brazilian Individuals with Periodontitis. Hum. Immunol. 2013, 74, 207–214. [Google Scholar] [CrossRef]
- Borilova Linhartova, P.; Kastovsky, J.; Lucanova, S.; Bartova, J.; Poskerova, H.; Vokurka, J.; Fassmann, A.; Kankova, K.; Izakovicova Holla, L. Interleukin-17A Gene Variability in Patients with Type 1 Diabetes Mellitus and Chronic Periodontitis: Its Correlation with IL-17 Levels and the Occurrence of Periodontopathic Bacteria. Mediat. Inflamm. 2016, 2016, 2979846. [Google Scholar] [CrossRef]
- Reference SNP (refSNP) Cluster Report: rs2275913. Available online: www.ncbi.nlm.nih.gov/projects/SNP/snp_ref.cgi?do_not_redirect&rs=rs2275913 (accessed on 27 April 2020).
- Trifunović, J.; Miller, L.; Debeljak, Ž.; Horvat, V. Pathologic Patterns of Interleukin 10 Expression—A Review. Biochem. Medica 2015, 25, 36–48. [Google Scholar] [CrossRef]
- Iyer, S.S.; Cheng, G. Role of Interleukin 10 Transcriptional Regulation in Inflammation and Autoimmune Disease. Crit. Rev. Immunol. 2012, 32, 23–63. [Google Scholar] [CrossRef]
- Kim, J.M.; Brannan, C.I.; Copeland, N.G.; Jenkins, N.A.; Khan, T.A.; Moore, K.W. Structure of the Mouse IL-10 Gene and Chromosomal Localization of the Mouse and Human Genes. J. Immunol. Baltim. Md. 1950 1992, 148, 3618–3623. [Google Scholar]
- Reuss, E.; Fimmers, R.; Kruger, A.; Becker, C.; Rittner, C.; Höhler, T. Differential Regulation of Interleukin-10 Production by Genetic and Environmental Factors—A Twin Study. Genes Immun. 2002, 3, 407–413. [Google Scholar] [CrossRef] [PubMed]
- Larsson, L.; Rymo, L.; Berglundh, T. Sp1 Binds to the G Allele of The−1087 Polymorphism in the IL-10 Promoter and Promotes IL-10 MRNA Transcription and Protein Production. Genes Immun. 2010, 11, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Turner, D.M.; Williams, D.M.; Sankaran, D.; Lazarus, M.; Sinnott, P.J.; Hutchinson, I.V. An Investigation of Polymorphism in the Interleukin-10 Gene Promoter. Eur. J. Immunogenet. 1997, 24, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Chung, E.Y.; Liu, J.; Zhang, Y.; Ma, X. Differential Expression in Lupus-Associated IL-10 Promoter Single-Nucleotide Polymorphisms Is Mediated by Poly (ADP-Ribose) Polymerase-1. Genes Immun. 2007, 8, 577–589. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Hu, B.; Feng, G.; Chen, Q.; Zhu, M.; Ying, S.; Song, J. Association of Interleukin-10-1082 (-1087) A > G Polymorphisms and Periodontitis Risk: An Updated Meta-Analysis Based on 26 Case-Control Studies. Ann. Hum. Genet. 2019, 83, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Brett, P.M.; Zygogianni, P.; Griffiths, G.S.; Tomaz, M.; Parkar, M.; D’Aiuto, F.; Tonetti, M. Functional Gene Polymorphisms in Aggressive and Chronic Periodontitis. J. Dent. Res. 2005, 84, 1149–1153. [Google Scholar] [CrossRef]
- Reichert, S.; Machulla, H.K.G.; Klapproth, J.; Zimmermann, U.; Reichert, Y.; Gläser, C.H.; Schaller, H.G.; Stein, J.; Schulz, S. The Interleukin-10 Promoter Haplotype ATA Is a Putative Risk Factor for Aggressive Periodontitis. J. Periodontal Res. 2008, 43, 40–47. [Google Scholar] [CrossRef]
- Jaradat, S.M.; Ababneh, K.T.; Jaradat, S.A.; Abbadi, M.S.; Taha, A.H.; Karasneh, J.A.; Haddad, H.I. Association of Interleukin-10 Gene Promoter Polymorphisms with Chronic and Aggressive Periodontitis. Oral Dis. 2012, 18, 271–279. [Google Scholar] [CrossRef]
- Gonzales, J.R.; Michel, J.; Diete, A.; Herrmann, J.M.; Bödeker, R.H.; Meyle, J. Analysis of genetic polymorphisms at the interleukin-10 loci in aggressive and chronic periodontitis. J. Clin. Periodontol. 2002, 29, 816–822. [Google Scholar] [CrossRef]
- Reference SNP (refSNP) Cluster Report: rs1800896. Available online: www.ncbi.nlm.nih.gov/projects/SNP/snp_ref.cgi?do_not_redirect&rs=rs1800896 (accessed on 27 April 2020).
- Reference SNP (refSNP) Cluster Report: rs1800871. Available online: www.ncbi.nlm.nih.gov/projects/SNP/snp_ref.cgi?do_not_redirect&rs=rs1800871 (accessed on 27 April 2020).
- Reference SNP (refSNP) Cluster Report: rs1800872. Available online: www.ncbi.nlm.nih.gov/projects/SNP/snp_ref.cgi?do_not_redirect&rs=rs1800872 (accessed on 27 April 2020).
- de Waal Malefyt, R.; Abrams, J.; Bennett, B.; Figdor, C.G.; de Vries, J.E. Interleukin 10(IL-10) Inhibits Cytokine Synthesis by Human Monocytes: An Autoregulatory Role of IL-10 Produced by Monocytes. J. Exp. Med. 1991, 174, 1209–1220. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.W.; de Waal Malefyt, R.; Coffman, R.L.; O’Garra, A. Interleukin-10 and the Interleukin-10 Receptor. Annu. Rev. Immunol. 2001, 19, 683–765. [Google Scholar] [CrossRef]
- Fiorentino, D.F.; Zlotnik, A.; Mosmann, T.R.; Howard, M.; O’Garra, A. IL-10 Inhibits Cytokine Production by Activated Macrophages. J. Immunol. 1991, 147, 3815–3822. [Google Scholar]
- Sultani, M.; Stringer, A.M.; Bowen, J.M.; Gibson, R.J. Anti-Inflammatory Cytokines: Important Immunoregulatory Factors Contributing to Chemotherapy-Induced Gastrointestinal Mucositis. Chemother. Res. Pract. 2012, 2012, 490804. [Google Scholar] [CrossRef] [PubMed]
- Havemose-Poulsen, A.; Sørensen, L.K.; Stoltze, K.; Bendtzen, K.; Holmstrup, P. Cytokine Profiles in Peripheral Blood and Whole Blood Cell Cultures Associated With Aggressive Periodontitis, Juvenile Idiopathic Arthritis, and Rheumatoid Arthritis. J. Periodontol. 2005, 76, 2276–2285. [Google Scholar] [CrossRef] [PubMed]
- Gemmell, E.; Seymour, G.J. Immunoregulatory Control of Th1/Th2 Cytokine Profiles in Periodontal Disease. Periodontol. 2000 2004, 35, 21–41. [Google Scholar] [CrossRef] [PubMed]
- Seymour, G.J.; Taylor, J.J. Shouts and Whispers: An Introduction to Immunoregulation in Periodontal Disease. Periodontol. 2000 2004, 35, 9–13. [Google Scholar] [CrossRef]
- Nibali, L.; Darbar, U.; Rakmanee, T.; Donos, N. Anemia of Inflammation Associated with Periodontitis: Analysis of Two Clinical Studies. J. Periodontol. 2019, 90, 1252–1259. [Google Scholar] [CrossRef]
- Armitage, G.C. Development of a Classification System for Periodontal Diseases and Conditions. Ann. Periodontol. 1999, 4, 1–6. [Google Scholar] [CrossRef]
- World Health Organization. Oral Health Surveys: Basic Methods, 4th ed.; World Health Organization: Geneva, Switzerland, 1997; Available online: apps.who.int/iris/handle/10665/41905 (accessed on 1 March 2020).
- Izakovicova Holla, L.; Borilova Linhartova, P.; Hrdlickova, B.; Marek, F.; Dolina, J.; Rihak, V.; Kala, Z. Haplotypes of the IL-1 gene cluster are associated with gastroesophageal reflux disease and Barrett’s esophagus. Hum. Immunol. 2013, 74, 1161–1169. [Google Scholar] [CrossRef]
- Linhartova, P.B.; Janos, J.; Slezakova, S.; Bartova, J.; Petanova, J.; Kuklinek, P.; Fassmann, A.; Dusek, L.; Holla, L.I. Recurrent Aphthous Stomatitis and Gene Variability in Selected Interleukins: A Case–Control Study. Eur. J. Oral Sci. 2018, 126, 485–492. [Google Scholar] [CrossRef] [PubMed]
- Borilova Linhartova, P.; Cernochova, P.; Kastovsky, J.; Vrankova, Z.; Sirotkova, M.; Izakovicova Holla, L. Genetic determinants and postorthodontic external apical root resorption in Czech children. Oral Dis. 2017, 23, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Galicia, J.C.; Tai, H.; Komatsu, Y.; Shimada, Y.; Akazawa, K.; Yoshie, H. Polymorphisms in the IL-6 Receptor (IL-6R) Gene: Strong Evidence That Serum Levels of Soluble IL-6R Are Genetically Influenced. Genes Immun. 2004, 5, 513–516. [Google Scholar] [CrossRef] [PubMed]
- Bartova, J.; Borilova Linhartova, P.; Podzimek, S.; Janatova, T.; Svobodova, K.; Fassmann, A.; Duskova, J.; Belacek, J.; Izakovicova Holla, L. The Effect of IL-4 Gene Polymorphisms on Cytokine Production in Patients with Chronic Periodontitis and in Healthy Controls. Mediators Inflamm. 2014, 2014, 185757. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymorphism | Genotype MAF | Patients with AgP N = 91 (%) | Controls N = 210 (%) | p | OR 95% CI |
|---|---|---|---|---|---|
| IL-1A −889 C/T (rs1800587) | CC | 46 (50.5) | 95 (45.2) | 1.00 | |
| CT | 32 (35.2) | 97 (46.2) | 0.68 (0.40–1.16) | ||
| TT | 13 (14.3) | 18 (8.6) | 0.12 | 1.49 (0.67–3.30) | |
| T | (31.9) | (31.7) | 0.52 | 1.01 (0.69–1.47) | |
| IL-1B +3953 C/T (rs1143634) | CC | 51 (56.0) | 111 (52.9) | 1.00 | |
| CT | 33 (36.3) | 83 (39.5) | 0.87 (0.51–1.46) | ||
| TT | 7 (7.7) | 16 (7.6) | 0.86 | 1.05 (0.41–2.71) | |
| T | (25.8) | (27.4) | 0.39 | 0.92 (0.62–1.37) | |
| IL-1 VNTR§ intron 2, 86 bp VNTR (rs2234663) | LL | 51 (56.0) | 114 (54.3) | 1.00 | |
| LS | 33 (36.3) | 79 (37.6) | 0.93 (0.55–1.58) | ||
| SS | 7 (7.7) | 17 (8.1) | 0.96 | 0.92 (0.36–2.36) | |
| S | (25.8) | (26.9) | 0.43 | 0.95 (0.64–1.41) | |
| IL-6 −174 C/G (rs1800795) | GG | 27 (29.6) | 69 (32.9) | 1.00 | |
| GC | 41 (45.1) | 101 (48.1) | 0.96 (0.54–1.71) | ||
| CC | 23 (25.3) | 40 (19.0) | 0.47 | 1.47 (0.75–2.90) | |
| C | (47.8) | (43.1) | 0.16 | 0.83 (0.58–1.17) | |
| IL-6R +48992 A/C (rs2228145) | AA | 32 (35.2) | 76 (36.2) | 1.00 | |
| AC | 46 (50.5) | 101 (48.1) | 1.08 (0.63–1.86) | ||
| CC | 13 (14.3) | 33 (15.7) | 0.91 | 0.94 (0.44–2.01) | |
| C | (39.6) | (39.8) | 0.52 | 0.99 (0.69–1.42) | |
| IL-10 −1087 A/G (rs1800896) | AA | 23 (25.3) | 54 (25.7) | 1.00 | |
| AG | 44 (48.4) | 124 (59.1) | 0.83 (0.46–1.51) | ||
| GG | 24 (26.4) | 32 (15.2) | 0.06 | 1.76 (0.86–3.62) | |
| AG + AA | 67 (73.6) | 178 (84.8) | 0.02 * | 0.50 (0.28–0.91) | |
| G | (50.5) | (44.8) | 0.12 | 1.26 (0.89–1.79) | |
| IL-10 −824 C/T (rs1800871) | CC | 49 (53.8) | 118 (56.2) | 1.00 | |
| CT | 39 (42.9) | 82 (39.0) | 1.15 (0.69–1.90) | ||
| TT | 3 (3.3) | 10 (4.8) | 0.74 | 0.72 (0.19–2.74) | |
| T | (24.7) | (24.3) | 0.49 | 1.02 (0.68–1.53) | |
| IL-10 −597 C/A (rs1800872) | CC | 50 (54.9) | 122 (58.1) | 1.00 | |
| CA | 38 (41.8) | 78 (37.1) | 1.19 (0.71–1.98) | ||
| AA | 3 (3.3) | 10 (4.8) | 0.68 | 0.73 (0.19–2.77) | |
| A | (24.2) | (23.3) | 0.45 | 1.05 (0.70–1.58) | |
| IL-17A −197 A/G (rs2275913) | GG | 46 (50.5) | 92 (43.8) | 1.00 | |
| GA | 34 (37.4) | 92 (43.8) | 0.74 (0.44–1.25) | ||
| AA | 11 (12.1) | 26 (12.4) | 0.53 | 0.85 (0.38–1.86) | |
| A | (30.8) | (34.4) | 0.23 | 1.17 (0.81–1.71) | |
| IL-18 −607 A/C (rs1946518) | CC | 25 (27.5) | 71 (34.0) | 1.00 | |
| AC | 54 (59.3) | 109 (52.2) | 1.41 (0.80–2.46) | ||
| AA | 12 (13.2) | 29 (13.8) | 0.48 | 0.85 (0.38–1.92) | |
| A | (42.9) | (39.8) | 0.28 | 1.13 (0.79–1.60) | |
| IL-18 −137 C/G (rs187238) | GG | 45 (49.4) | 97 (46.4) | 1.00 | |
| GC | 42 (46.2) | 93 (44.5) | 1.03 (0.62–1.71) | ||
| CC | 4 (4.4) | 19 (9.1) | 0.45 (0.15–1.41) | ||
| C | (27.5) | (31.7) | 0.20 | 1.21 (0.82–1.77) | |
| IL-18 −140 C/G (rs4988359) | CC | 39(42.9) | 85 (40.7) # | 1.00 | |
| GC | 45 (49.4) | 97 (46.4) | 1.01 (0.60–1.70) | ||
| GG | 7 (7.7) | 25 (12.0) | 0.52 | 0.61 (0.24–1.53) | |
| G | (32.4) | (36.4) | 0.26 | 1.15 (0.79–1.66) |
| Gene | Haplotype ‡ | Patients with AgP N = 91 (%) | Controls N = 210 (%) | p | OR 95% CI | ||
|---|---|---|---|---|---|---|---|
| IL-1 | C | C | A | (45.2) | (42.8) | 0.54 | 1.12 (0.79–1.59) |
| T | T | A | (20.5) | (21.9) | 0.65 | 0.91 (0.60–1.38) | |
| C | C | B | (20.0) | (21.6) | 0.58 | 0.89 (0.59–1.35) | |
| T | C | A | (7.0) | (5.5) | 0.58 | 1.21 (0.62–2.37) | |
| C | T | A | (1.4) | (2.9) | 0.36 | 0.57 (0.16–2.04) | |
| T | C | B | (2.0) | (2.7) | 0.74 | 0.77 (0.15–3.84) | |
| T | T | B | (2.4) | (1.6) | 0.31 | 2.33 (0.47–11.65) | |
| C | T | B | (1.5) | (1.0) | 0.64 | 1.54 (0.26–9.32) | |
| IL-10 | G | C | C | (50.5) | (44.4) | 0.17 | 1.27 (0.90–1.81) |
| A | C | C | (24.2) | (31.1) | 0.09 | 0.71 (0.48–1.06) | |
| A | T | A | (23.6) | (22.7) | 0.84 | 0.69 (0.57–1.58) | |
| A | T | C | (1.1) | (1.2) | 0.92 | 0.92 (0.18–4.80) | |
| G | T | A | (0.0) | (0.4) | 1.00 | 0.00 (0.00–0.00) | |
| A | C | A | (0.6) | (0.2) | 0.56 | 2.32 (0.14–37.22) | |
| IL-18# | C | G | C | (52.5) | (53.3) | 0.81 | 0.96 (0.68–1.36) |
| A | C | G | (25.7) | (27.2) | 0.67 | 0.92 (0.62–1.36) | |
| A | G | C | (13.3) | (8.7) | 0.08 | 1.66 (0.96–2.89) | |
| C | G | G | (4.1) | (5.7) | 0.45 | 0.72 (0.30–1.17) | |
| A | C | C | (1.2) | (2.1) | 0.46 | 0.57 (0.12–2.71) | |
| A | G | G | (2.6) | (1.5) | 0.40 | 1.66 (0.52–5.30) | |
| C | C | G | (0.0) | (1.1) | 1.00 | 0.00 (0.00–0.00) | |
| C | C | C | (0.6) | (0.4) | 0.91 | 1.15 (0.10–12.75) | |
| Bacteria | Presence | Patients with AgP N = 25 (%) | Controls N = 51 (%) | p | OR 95% CI | Non-Smokers with AgP | Non-Smokers Controls | p | OR 95%CI |
| A. actinomycetemcomitans | negative | 16 (64.0) | 37 (72.5) | 10 (71.4) | 33 (73.3) | ||||
| positive | 9 (36.0) | 14 (27.5) | 0.31 | 1.49 (0.53–4.13) | 4 (28.6) | 12 (26.7) | 0.57 | 1.10 (0.29–4.18) | |
| T. forsythia | negative | 1 (4.0) | 23 (45.1) | 1 (7.1) | 21 (46.7) | ||||
| positive | 24 (96.0) | 28 (54.9) | 0.000137 * | 19.71 (2.48–157.03) | 13 (92.9) | 24 (53.3) | 0.0064 | 11.38 (1.37–94.45) | |
| P. gingivalis | negative | 4 (16.0) | 42 (82.4) | 1 (7.1) | 37 (82.2) | ||||
| positive | 21 (84.0) | 9 (17.6) | <0.000001 * | 24.50 (6.75–88.92) | 13 (92.9) | 8 (17.8) | 0.000001 | 60.13 (6.85–528.08) | |
| T. denticola | negative | 3 (12.0) | 28 (54.9) | 1 (7.1) | 25 (55.6) | ||||
| positive | 22 (88.0) | 23 (45.1) | 0.000262 * | 8.93 (2.37–33.63) | 13 (92.9) | 20 (44.4) | 0.0012 | 16.25 (1.96–135.01) | |
| P. micra | negative | 3 (12.0) | 22 (43.1) | 3 (21.4) | 20 (44.4) | ||||
| positive | 22 (88.0) | 29 (56.9) | 0.005410 * | 5.56 (1.48–20.98) | 11 (78.6) | 25 (55.6) | 0.11 | 2.93 (0.72–11.96) | |
| P. intermedia | negative | 9 (36.0) | 28 (54.9) | 4 (28.6) | 26 (57.8) | ||||
| positive | 16 (64.0) | 23 (45.1) | 0.10 | 2.16 (0.81–5.80) | 10 (71.4) | 19 (42.2) | 0.054 | 3.42 (0.93–12.57) | |
| F. nucleatum | negative | 0 (0.0) | 3 (5.9) | 5 (35.7) | 3 (6.67) | ||||
| positive | 25 (100.0) | 48 (94.1) | 0.30 | 1.56 (0.15–15.81) | 9 (64.3) | 42 (93.3) | 0.014 | 0.13 (0.03–0.64) |
| PBMCs | IL-10 Levels (pg/mL) | p | ||
|---|---|---|---|---|
| Median | IQR | Minimum–Maximum | ||
| Unstimulated (negative control) | 1.8 | 0.4–6.2 | 0–13.9 | |
| With PWM (positive control) | 85.7 | 61.7–302.0 | 14.4–3447.0 | 0.00013 |
| Stimulated by bacteria | ||||
| A. actinomycetemcomitans | 30.1 | 15.9–136.9 | 4.4–663.1 | 0.00009 |
| T. forsythia | 75.9 | 29.2–173.6 | 0.6–320.1 | 0.00035 |
| P. gingivalis | 0.7 | 0.2–5.0 | 0.0–45.7 | 0.287 |
| P. intermedia | 5.3 | 2.3–13.0 | 0.0–173.6 | 0.034 (Pcorr > 0.05) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borilova Linhartova, P.; Danek, Z.; Deissova, T.; Hromcik, F.; Lipovy, B.; Szaraz, D.; Janos, J.; Fassmann, A.; Bartova, J.; Drizhal, I.; et al. Interleukin Gene Variability and Periodontal Bacteria in Patients with Generalized Aggressive Form of Periodontitis. Int. J. Mol. Sci. 2020, 21, 4728. https://doi.org/10.3390/ijms21134728
Borilova Linhartova P, Danek Z, Deissova T, Hromcik F, Lipovy B, Szaraz D, Janos J, Fassmann A, Bartova J, Drizhal I, et al. Interleukin Gene Variability and Periodontal Bacteria in Patients with Generalized Aggressive Form of Periodontitis. International Journal of Molecular Sciences. 2020; 21(13):4728. https://doi.org/10.3390/ijms21134728
Chicago/Turabian StyleBorilova Linhartova, Petra, Zdenek Danek, Tereza Deissova, Filip Hromcik, Bretislav Lipovy, David Szaraz, Julius Janos, Antonin Fassmann, Jirina Bartova, Ivo Drizhal, and et al. 2020. "Interleukin Gene Variability and Periodontal Bacteria in Patients with Generalized Aggressive Form of Periodontitis" International Journal of Molecular Sciences 21, no. 13: 4728. https://doi.org/10.3390/ijms21134728
APA StyleBorilova Linhartova, P., Danek, Z., Deissova, T., Hromcik, F., Lipovy, B., Szaraz, D., Janos, J., Fassmann, A., Bartova, J., Drizhal, I., & Izakovicova Holla, L. (2020). Interleukin Gene Variability and Periodontal Bacteria in Patients with Generalized Aggressive Form of Periodontitis. International Journal of Molecular Sciences, 21(13), 4728. https://doi.org/10.3390/ijms21134728