Synthesis, Cyclooxygenases Inhibition Activities and Interactions with BSA of N-substituted 1H-pyrrolo[3,4-c]pyridine-1,3(2H)-diones Derivatives

 , , ,
, , ,  and
and
Abstract
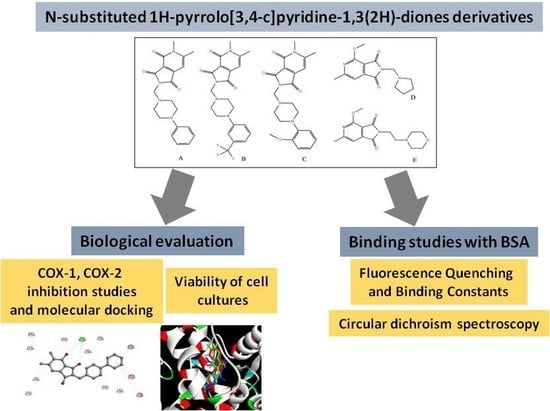
:
1. Introduction
2. Results and Discussion
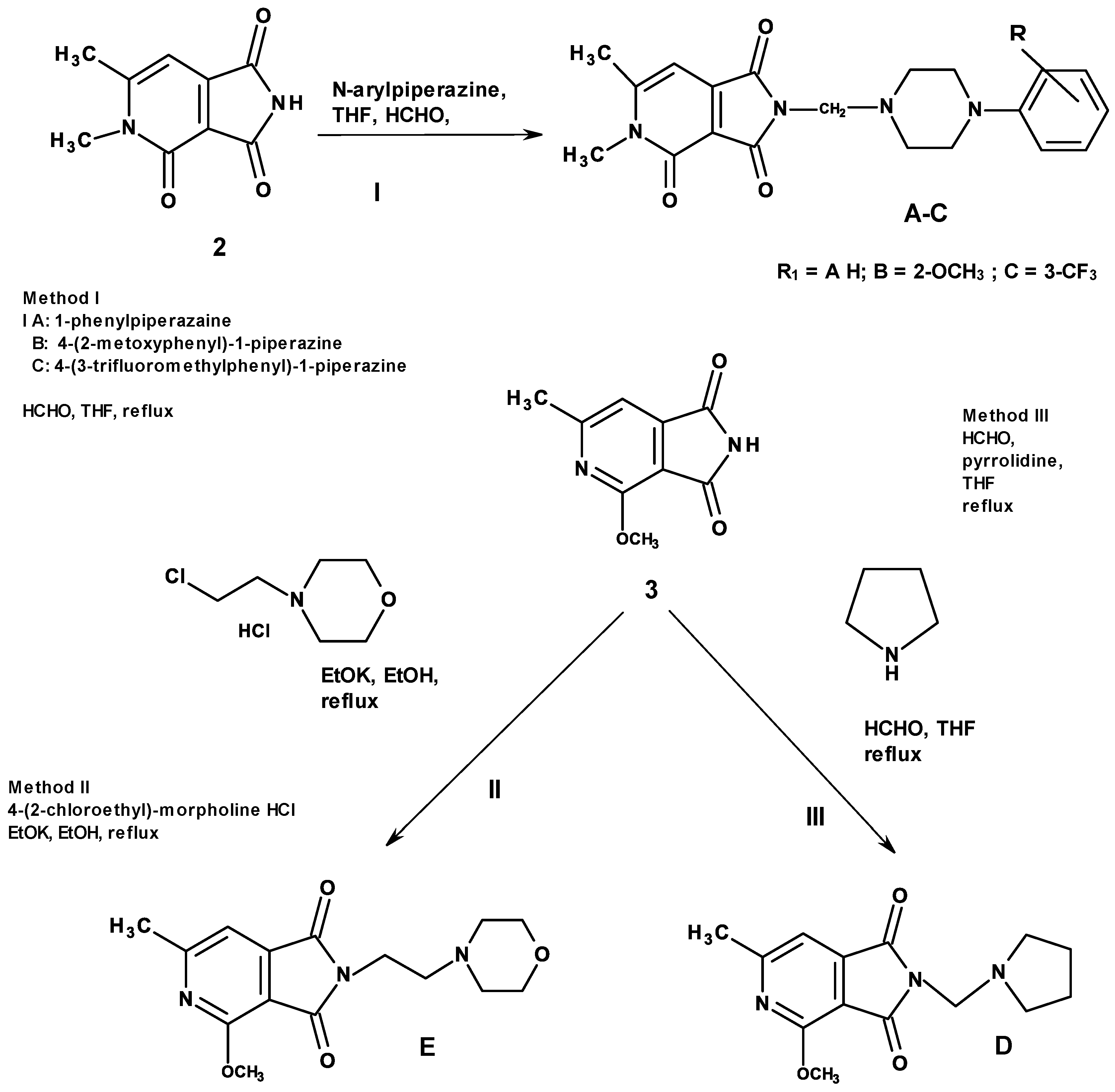
2.1. Chemistry
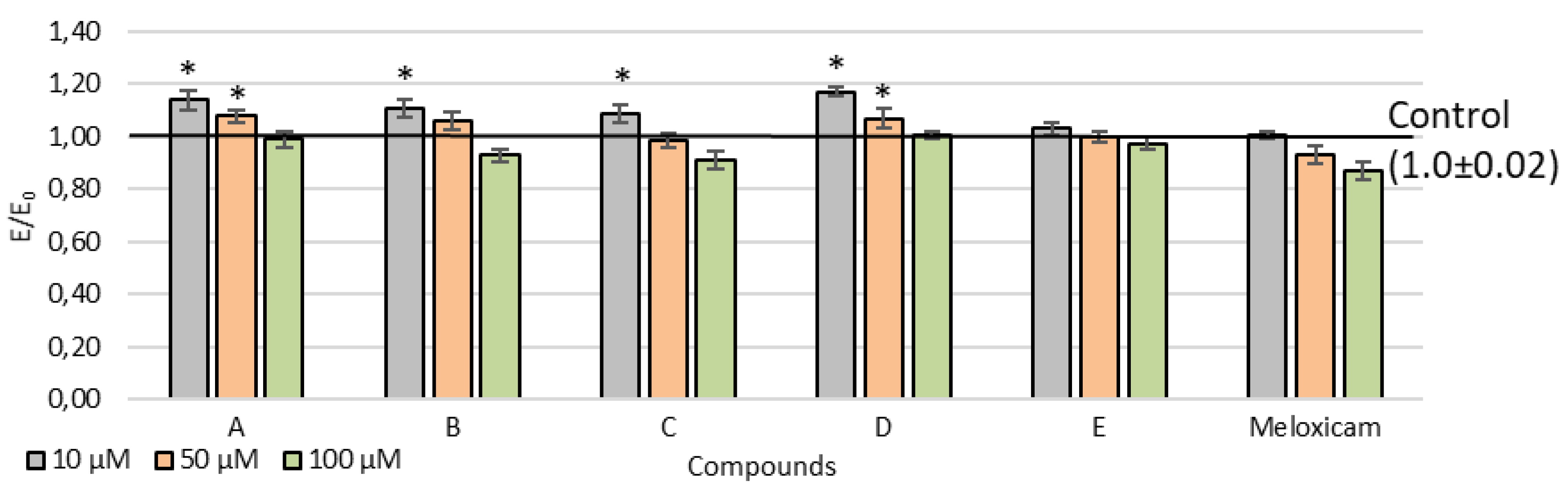
2.2. Viability of Cell Cultures
2.3. Cyclooxygenase Inhibition
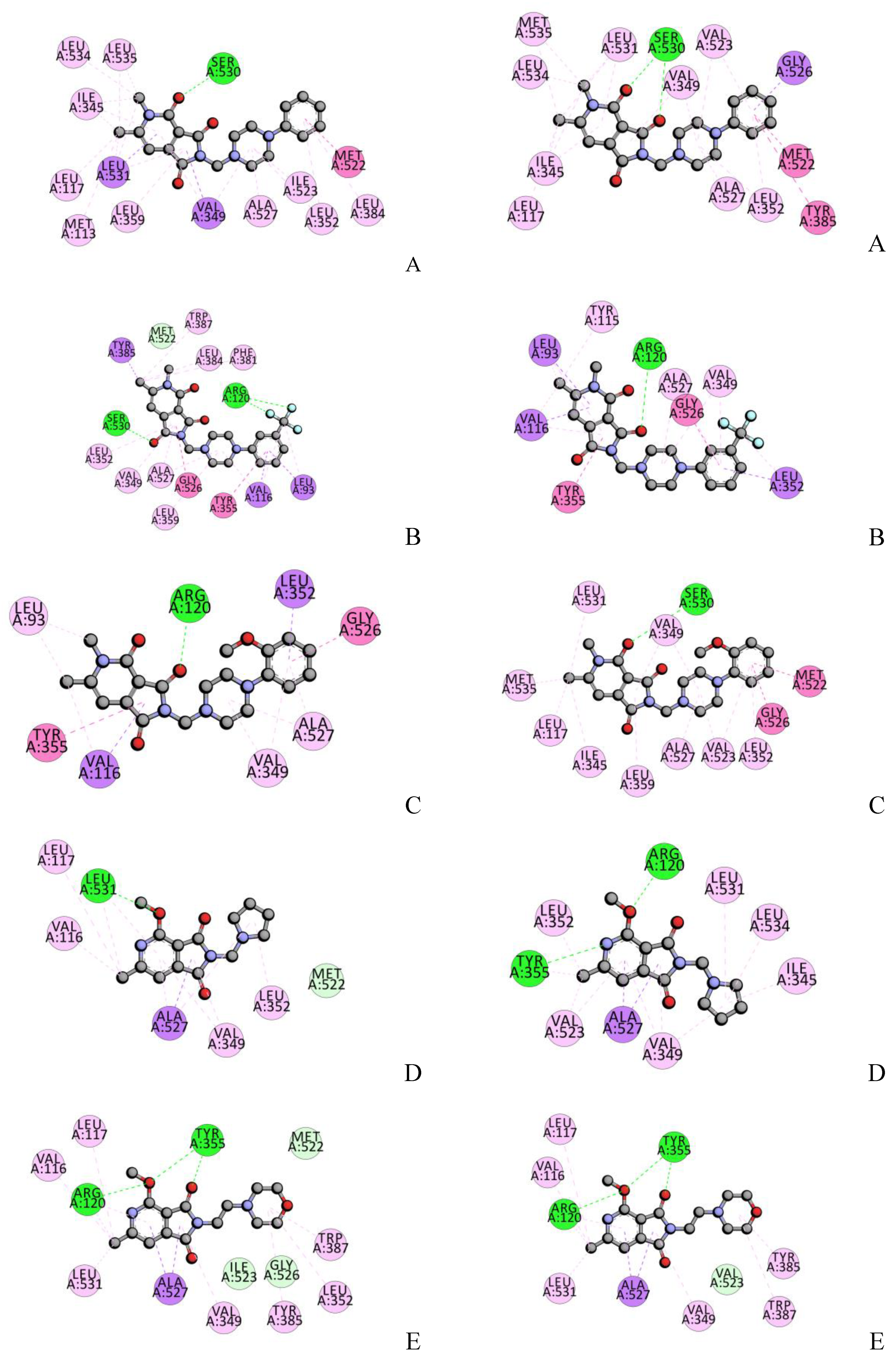
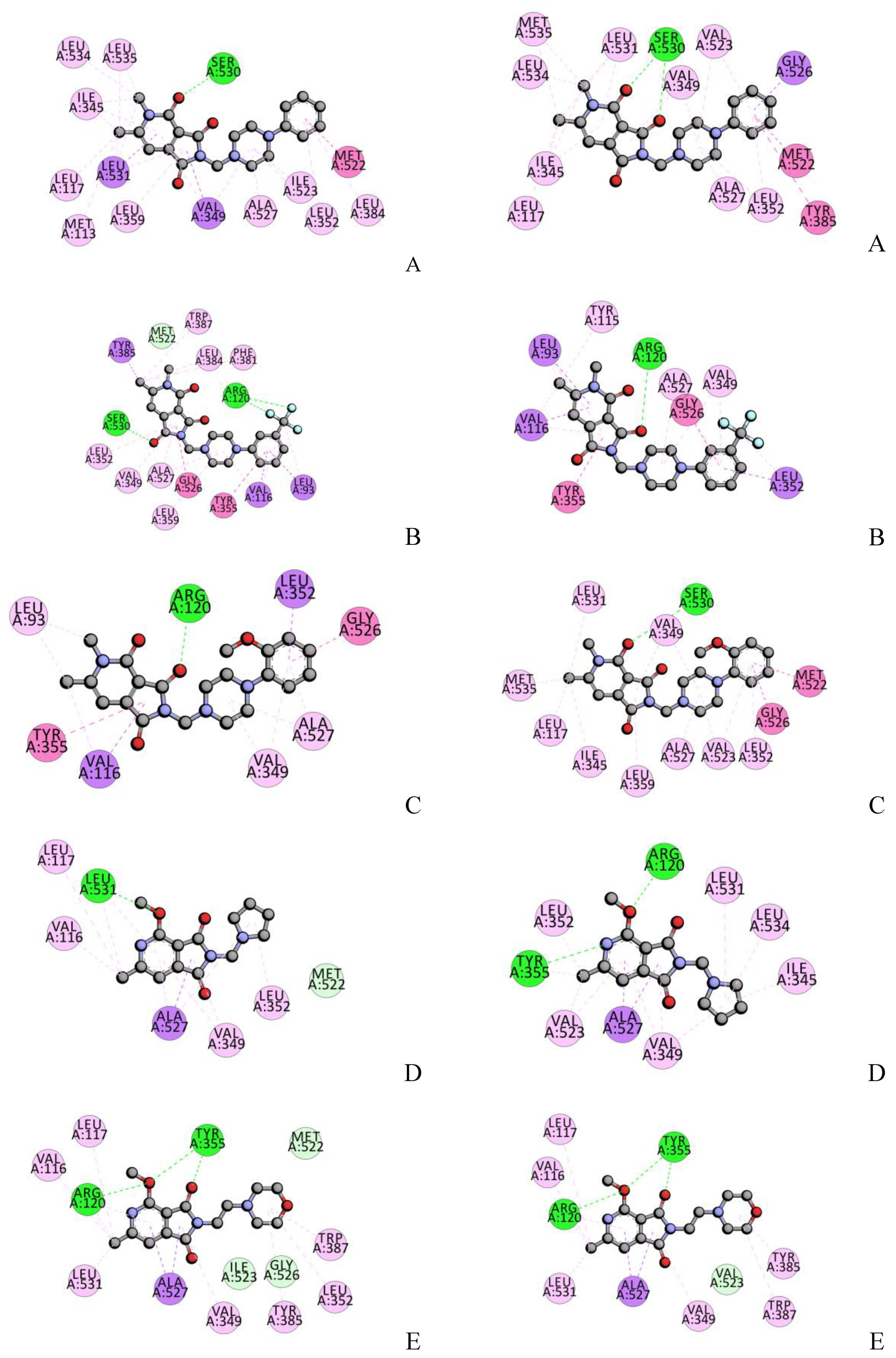
2.4. Molecular Docking Studies
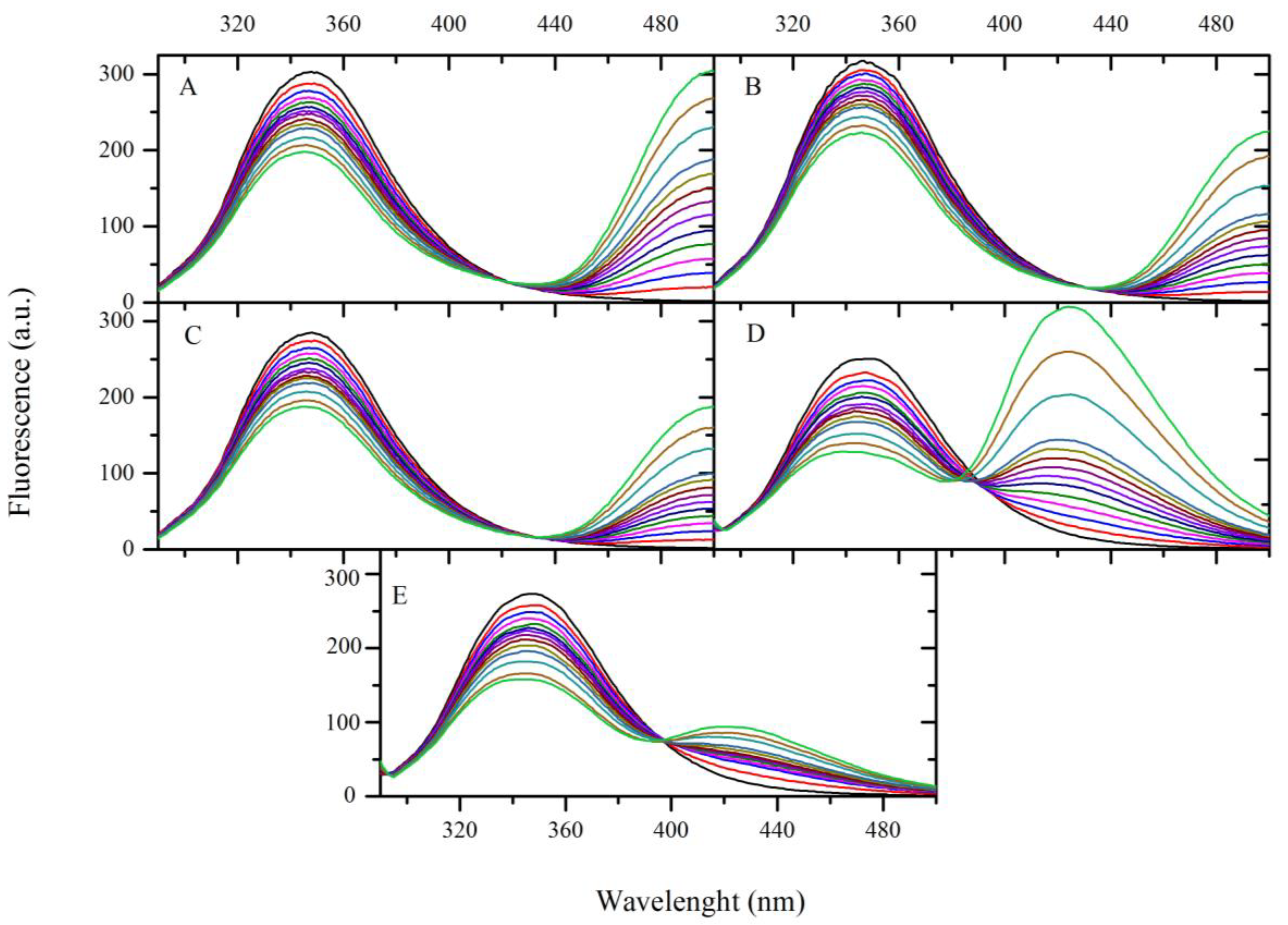
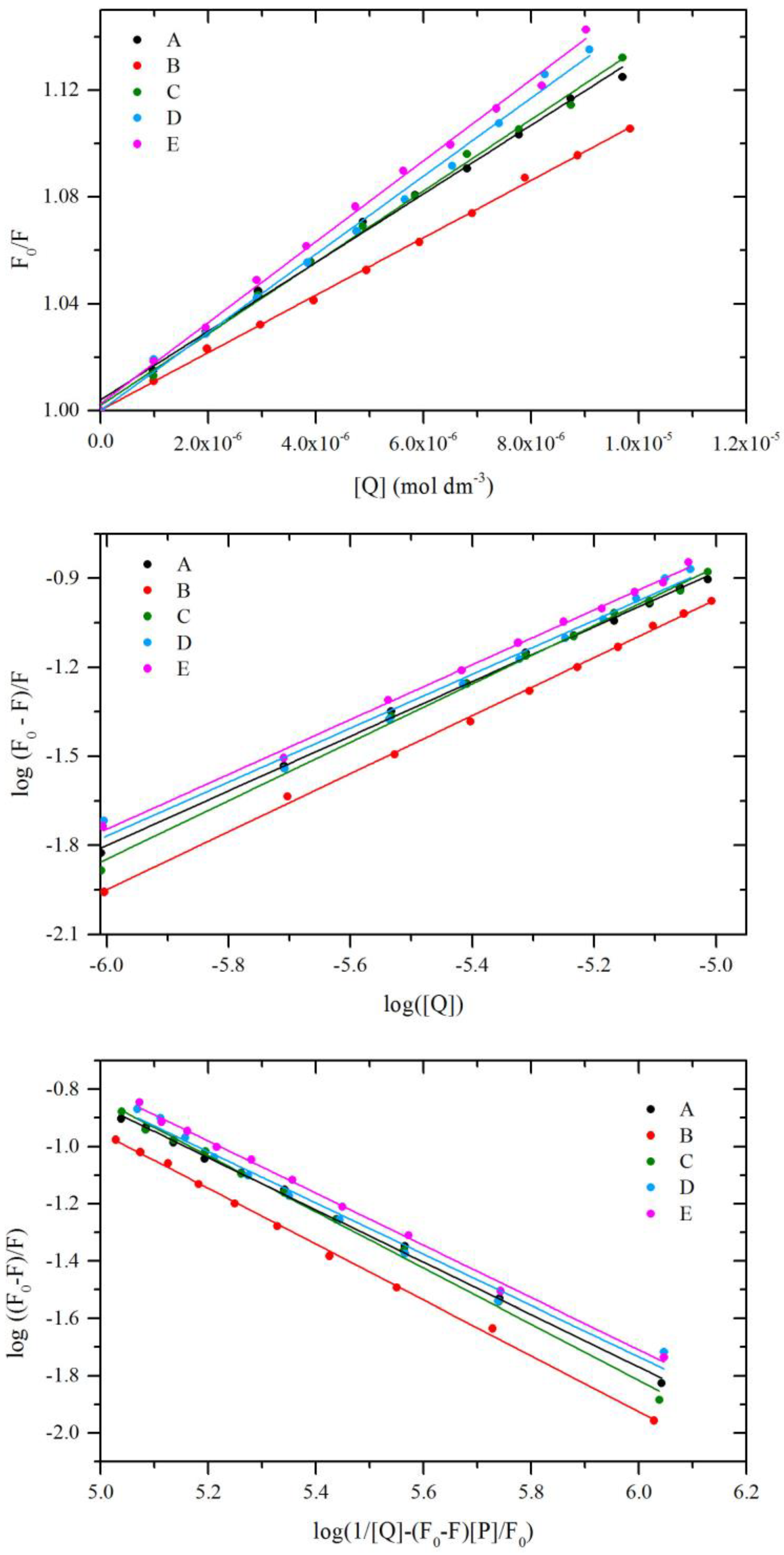
2.5. Fluorescence Quenching of BSA by Compound A–E
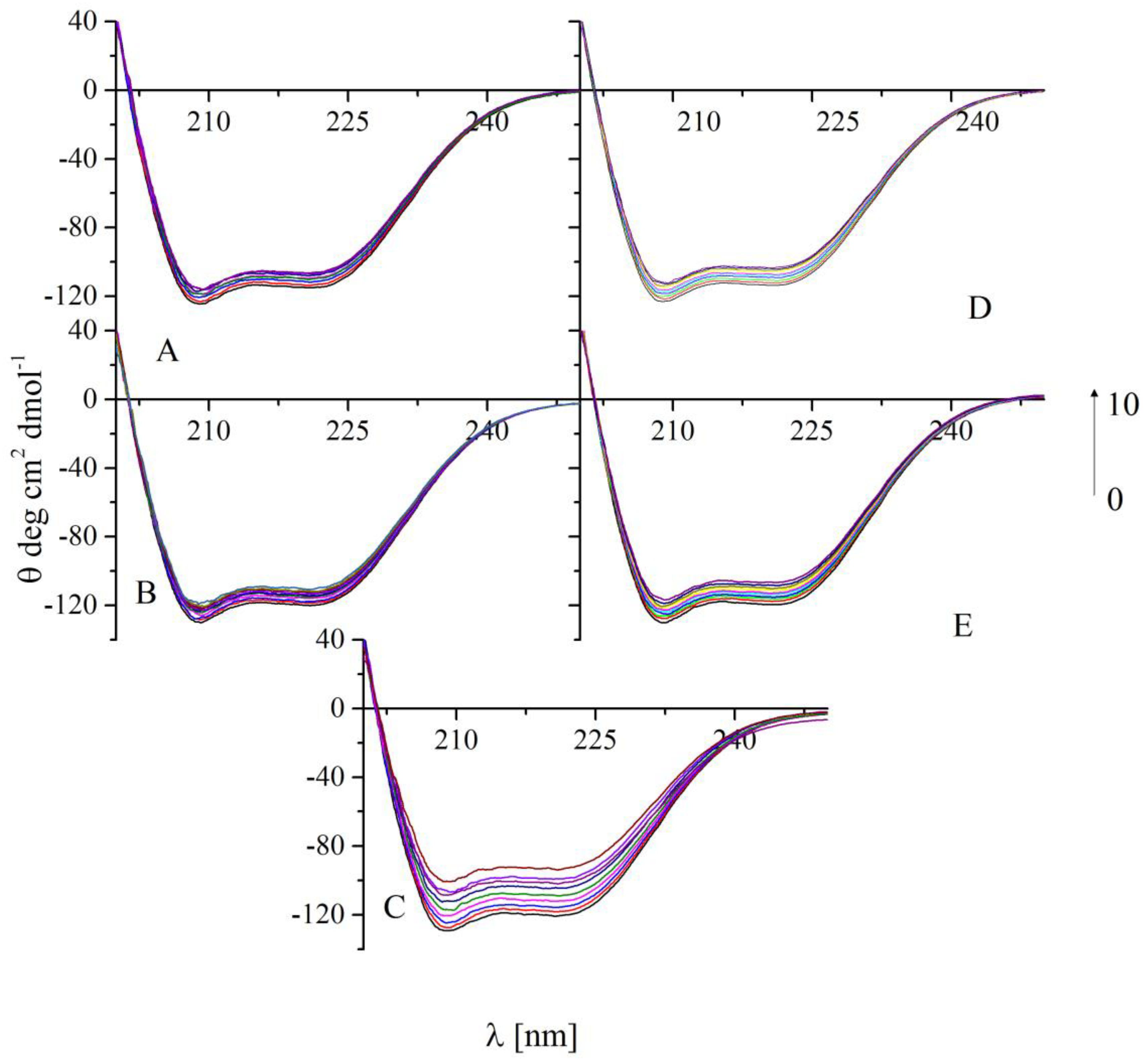
2.6. Circular Dichroism Spectra
2.7. Binding Constants
2.8. Thermodynamic studies
2.9. Site Markers Studies
2.10. Molecular Docking-Interactions with BSA
3. Materials and Methods
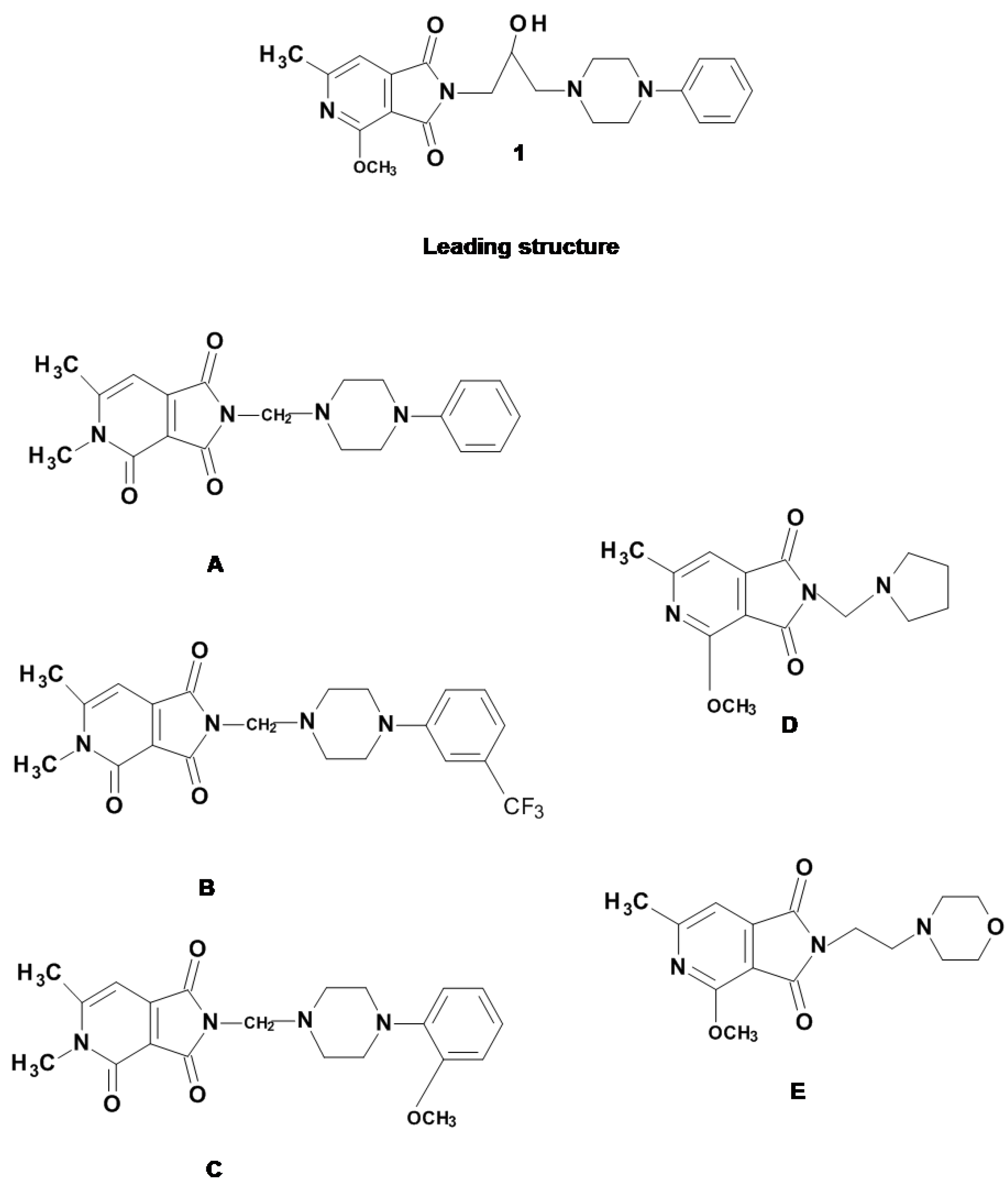
3.1. Chemistry
3.1.1. General Method for the Preparation of the Mannich Bases A–C (Scheme 1)
3.1.2. Method for the Preparation of the imides D, E (Scheme 1)
3.2. Cell Line
3.3. Tested Compounds
3.4. MTT Assay
3.5. Cyclooxygenase Inhibition Assay
3.6. Statistical Analysis
3.7. Spectroscopic Studies
3.8. Molecular Docking
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| COX | Cyclooxygenases |
| BSA | bovine serum albumin |
| HSA | human serum albumin |
| TLC | thin layer-chromatography |
| THF | Tetrahydrofuran |
| CD | circular dichroism |
| DMSO | dimethyl sulfoxide |
| NHDF | Normal Human Dermal Fibroblasts |
References
- Bailleux, V.; Vallée, L.; Nuyts, J.; Vamecq, J. Synthesis and anticonvulsant activity of two N-(2, 6-dimethylphenyl) pyridinedicarboximides. Biomed. Pharmacother. 1995, 49, 75–78. [Google Scholar] [CrossRef]
- Da Settimo, A.; Primofiore, G.; Da Settimo, F.; Simorini, F.; La Motta, C.; Martinelli, A.; Boldrini, E. Synthesis of pyrrolo[3,4-c]pyridine derivatives possessing an acid group and their in vitro and in vivo evaluation as aldose reductase inhibitors. Eur. J. Med. Chem. 1996, 31, 49–58. [Google Scholar] [CrossRef]
- Deraeve, C.; Dorobantu, I.M.; Rebbah, F.; Le Quéméner, F.; Constant, P.; Quémard, A.; Bernardes-Génisson, V.; Bernadou, J.; Pratviel, G. Chemical synthesis, biological evaluation and structure–activity relationship analysis of azaisoindolinones, a novel class of direct enoyl-ACP reductase inhibitors as potential antimycobacterial agents. Bioorg. Med. Chem. 2011, 19, 6225–6232. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.Z.; Maddali, K.; Metifiot, M.; Smith, S.J.; Vu, B.C.; Marchand, C.; Hughes, S.H.; Pommier, Y.; Burke, T.R. Bicyclic Hydroxy-1H-pyrrolopyridine-trione Containing HIV-1 Integrase Inhibitors. Chem. Biol. Drug Des. 2012, 79, 157–165. [Google Scholar] [CrossRef]
- Vane, J.R.; Botting, R.M. Anti-inflammatory drugs and their mechanism of action. Inflamm. Res. 1998, 47, 78–87. [Google Scholar] [CrossRef]
- Martel-Pelletier, J.; Lajeunesse, D.; Reboul, P.; Pelletier, J.P. Therapeutic role of dual inhibitors of 5-LOX and COX, selective and non-selective non-steroidal anti-inflammatory drugs. Ann. Rheum. Dis. 2003, 62, 501–509. [Google Scholar] [CrossRef]
- Zarghi, A.; Arfaei, S. Selective COX-2 inhibitors: A review of their structure-activity relationships. Iran. J. Pharm. Res. 2011, 10, 655–683. [Google Scholar]
- Turini, M.E.; DuBois, R.N. Cyclooxygenase-2: A therapeutic target. Annu. Rev. Med. 2002, 53, 35–57. [Google Scholar] [CrossRef]
- Śladowska, H.; Filipek, B.; Szkatuła, D.; Sabiniarz, A.; Kardasz, M.; Potoczek, J.; Sieklucka-Dziuba, M.; Rajtar, G.; Kleinrok, Z.; Lis, T. Investigations on the synthesis and pharmacological properties of 4-alkoxy-2-[2-hydroxy-3-(4-aryl-1-piperazinyl)propyl]-6-methyl-1H-pyrrolo[3,4-c]pyridine-1,3(2H)-diones. Farmaco 2003, 57, 897–908. [Google Scholar] [CrossRef]
- Sladowska, H. Investigations on the synthesis and properties of arylpiperazinylalkyl derivatives of some dihydro- and tetrahydropyrido[2,3-d]pyrimidines. Farmaco 1993, 48, 77–84. [Google Scholar] [CrossRef]
- Johnson, R.S.; Lovett, T.O.; Stevens, T.S. The alkaloids of Gelsemium sempervirens. Part IV. Derivatives of pyridine, isoquinoline, and indol-2(3H)-one as possible initial materials for synthesis of gelsemine. J. Chem. Soc. C Org. 1970, 6, 796. [Google Scholar] [CrossRef]
- Von Dobeneck, H.; Hansen, B. Aza-isoindole, I Aza-phthalimidine. Chem. Ber. 1972, 105, 3611–3621. [Google Scholar] [CrossRef]
- Sladowska, H.; Szkatuła, D.; Filipek, B.; Maciag, D.; Sapa, J.; Zygmunt, M. Synthesis and properties of 2-(4-substituted)butyl derivatives of some 2,3-dihydro-1,3-dioxo-1H-pyrrolo[3,4-c]pyridines. Pharmazie 2001, 56, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Śladowska, H.; Potoczek, J.; Sokołowska, M.; Rajtar, G.; Sieklucka-Dziuba, M.; Kocki, T.; Kleinrok, Z. Investigations on the synthesis and properties of N-phenyl derivatives of 1,4-dioxo (1,4,5-trioxo)-1,2,3,4-tetra (1,2,3,4,5,6-hexa) hydropyrido- [3,4-d] pyridazines and allied compounds. Farmaco 1998, 53, 468–474. [Google Scholar] [CrossRef]
- Śladowska, H.; Stanasiuk, J.; Sieklucka-Dziuba, M.; Saran, T.; Kleinrok, Z. Investigations on the synthesis and properties of 4-aminosubstituted 2,6,7-trimethyl-1,5-dioxo-1,2,5,6-tetrahydropyrido [3,4-d] pyridazines. Farmaco 1998, 53, 475–479. [Google Scholar] [CrossRef]
- Roman, G. Mannich bases in medicinal chemistry and drug design. Eur. J. Med. Chem. 2015, 89, 743–816. [Google Scholar] [CrossRef]
- Śladowska, H.; Filipek, B.; Szkatuła, D.; Sapa, J.; Bednarski, M.; Ciołkowska, M. Investigations on the synthesis and pharmacological properties of N-substituted derivatives of 4-alkoxy-6-methyl-1H-pyrrolo[3,4-c]pyridine-1,3(2H) -diones. Farmaco 2005, 60, 53–59. [Google Scholar] [CrossRef]
- Sladowska, H.; Sabiniarz, A.; Szkatula, D.; Filipek, B.; Sapa, J. Synthesis and properties of 4-alkoxy-2-[2-hydroxy-3-(4-o,m,p-halogenoaryl-1 -piperazinyl)propyl]-6-methyl-1H-pyrrolo-[3,4-c]pyridine-1,3(2H)-diones with analgesic and sedative activities. Acta Pol. Pharm. 2006, 63, 245–254. [Google Scholar]
- Ogino, K.; Harada, Y.; Kawamura, M.; Hatanaka, K.; Tsuda, S.; Kanai, K.; Katori, M. An inhibitory effect of meloxicam, a novel nonsteroidal anti-inflammatory drug, on COX-2. Jpn. J. Pharmacol. 1996, 71 (Suppl. 2). [Google Scholar]
- Świątek, P.; Strzelecka, M.; Urniaz, R.; Gębczak, K.; Gębarowski, T.; Gąsiorowski, K.; Malinka, W. Synthesis, COX-1/2 inhibition activities and molecular docking study of isothiazolopyridine derivatives. Bioorg. Med. Chem. 2017, 25, 316–326. [Google Scholar]
- Ali, M.S.; Al-Lohedan, H.A. Sulfadiazine binds and unfolds bovine serum albumin: An in vitro study. Mol. Biol. Rep. 2013, 40, 6081–6090. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.Z.; Huang, X.Z.; Xu, J.H.; Zheng, Z.Z.; Wang, Z.B. The Methods of Fluorescence Analysis, 2nd ed.; Science: Beijing, China, 1990. [Google Scholar]
- Lakowicz, J.R. (Ed.) Principles of Fluorescence Spectroscopy, 3rd ed.; Springer US: Boston, MA, USA, 2006; ISBN 978-0-387-31278-1. [Google Scholar]
- Lakowicz, J.R.; Weber, G. Quenching of fluorescence by oxygen. Probe for structural fluctuations in macromolecules. Biochemistry 1973, 12, 4161–4170. [Google Scholar] [CrossRef] [PubMed]
- Ware, W.R. Oxygen quenching of fluorescence in solution: An experimental study of the diffusion process. J. Phys. Chem. 1962, 66, 455–458. [Google Scholar] [CrossRef]
- Kelly, S.M.; Jess, T.J.; Price, N.C. How to study proteins by circular dichroism. Biochim. Biophys. Acta Proteins Proteom. 2005, 1751, 119–139. [Google Scholar] [CrossRef]
- Kelly, S.M.; Price, N.C. The Use of Circular Dichroism in the Investigation of Protein Structure and Function. Curr. Protein Pept. Sci. 2000, 1, 349–384. [Google Scholar] [CrossRef] [Green Version]
- Lu, Z.X.; Cui, T.; Shi, Q.L. Applications of Circular Dichroism (CD) and Optical Rotatory Dispersion (ORD) in Molecular Biology; Science Press: Beijing, China, 1987. [Google Scholar]
- Wei, X.L.; Xiao, J.B.; Wang, Y.; Bai, Y. Which model based on fluorescence quenching is suitable to study the interaction between trans-resveratrol and BSA? Spectrochim. Acta Part. A Mol. Biomol. Spectrosc. 2010, 75, 299–304. [Google Scholar] [CrossRef]
- Van De Weert, M.; Stella, L. Fluorescence quenching and ligand binding: A critical discussion of a popular methodology. J. Mol. Struct. 2011, 998, 145–150. [Google Scholar] [CrossRef]
- Bi, S.; Song, D.; Tian, Y.; Zhou, X.; Liu, Z.; Zhang, H. Molecular spectroscopic study on the interaction of tetracyclines with serum albumins. Spectrochim. Acta Part. A Mol. Biomol. Spectrosc. 2005, 61, 629–636. [Google Scholar] [CrossRef]
- Mohammadnia, F.; Fatemi, M.H.; Taghizadeh, S.M. Study on the interaction of anti-inflammatory drugs with human serum albumin using molecular docking, quantitative structure-activity relationship, and fluorescence spectroscopy. Luminescence 2020, 35, 266–273. [Google Scholar] [CrossRef]
- Dufour, C.; Dangles, O. Flavonoid-serum albumin complexation: Determination of binding constants and binding sites by fluorescence spectroscopy. Biochim. Biophys. Acta Gen. Subj. 2005, 1721, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Wani, T.A.; Bakheit, A.H.; Al-Majed, A.R.A.; Bhat, M.A.; Zargar, S. Study of the interactions of bovine serum albumin with the new anti-inflammatory agent 4-(1,3-dsioxo-1,3-dihydro-2H-isoindol-2-yl)-N-[(4-ethoxy-phenyl) methylidene]benzohydrazide using a multi-spectroscopic approach and molecular docking. Molecules 2017, 22, 1258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suryawanshi, V.D.; Walekar, L.S.; Gore, A.H.; Anbhule, P.V.; Kolekar, G.B. Spectroscopic analysis on the binding interaction of biologically active pyrimidine derivative with bovine serum albumin. J. Pharm. Anal. 2016, 6, 56–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wani, T.A.; Bakheit, A.H.; Zargar, S.; Bhat, M.A.; Al-Majed, A.A. Molecular docking and experimental investigation of new indole derivative cyclooxygenase inhibitor to probe its binding mechanism with bovine serum albumin. Bioorg. Chem. 2019, 89, 103010. [Google Scholar] [CrossRef]
- Abdelhameed, A.S.; Bakheit, A.H.; Mohamed, M.S.; Eldehna, W.M.; Abdel-Aziz, H.A.; Attia, M.I. Synthesis and biophysical insights into the binding of a potent anti-proliferative non-symmetric bis-isatin derivative with bovine serum albumin: Spectroscopic and molecular docking approaches. Appl. Sci. 2017, 7, 617. [Google Scholar] [CrossRef]
- Klotz, I.M.; Urquhart, J.M. The Binding of Organic Ions by Proteins. Effect of Temperature. J. Am. Chem. Soc. 1949, 71, 847–851. [Google Scholar] [CrossRef]
- Ross, P.D.; Subramanian, S. Thermodynamics of protein association reactions: Forces contributing to stability. Biochemistry 1981, 20, 3096–3102. [Google Scholar] [CrossRef]
- Sudlow, G.; Birkett, D.J.; Wade, D.N. The Characterization of two specific drug binding sites on human serum albumin. Mol. Pharmacol. 1975, 11, 824–832. [Google Scholar]
- Sudlow, G.; Birkett, D.J.; Wade, D.N. Further characterization of specific drug binding sites on human serum albumin. Mol. Pharmacol. 1976, 12, 1052–1061. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perdew, J.P.; Wang, Y. Accurate and simple analytic representation of the electron-gas correlation energy. Phys. Rev. B 1992, 45, 13244–13249. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian~16 {R}evision {A}.03 2016.
- Xu, S.; Hermanson, D.J.; Banerjee, S.; Ghebreselasie, K.; Clayton, G.M.; Garavito, R.M.; Marnett, L.J. Oxicams bind in a novel mode to the cyclooxygenase active site via a two-water-mediated h-bonding network. J. Biol. Chem. 2014, 289, 6799–6808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.M.; Chen, C.C. Gemdock: A generic evolutionary method for molecular docking. Proteins Struct. Funct. Genet. 2004, 55, 288–304. [Google Scholar] [CrossRef]
- Ghuman, J.; Zunszain, P.A.; Petitpas, I.; Bhattacharya, A.A.; Otagiri, M.; Curry, S. Structural basis of the drug-binding specificity of human serum albumin. J. Mol. Biol. 2005, 353, 38–52. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds A–E. are available from Dominika Szkatuła. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Cyclooxygenase Inhibition Assay IC50 [µM] | COX Selectivity Ratio IC50(COX-2)/IC50(COX-1) | |
|---|---|---|---|
| COX-1 | COX-2 | ||
| A | 81.6 | 54.9 | 0.67 |
| B | 93.2 | 57.1 | 0.61 |
| C | 95.8 | 66.2 | 0.69 |
| D | 76.6 | 56.8 | 0.74 |
| E | 99.2 | 54.2 | 0.55 |
| Meloxicam | 83.7 | 59.2 | 0.71 |
| COX-1 | COX-2 | |||||
|---|---|---|---|---|---|---|
| Total Binding Energy [kcal/mol] | Van der Waal’s Force [kcal/mol] | H Bond [kcal/mol] | Total Binding Energy [kcal/mol] | Van der Waal’s Force [kcal/mol] | H Bond [kcal/mol] | |
| A | −105.99 | −103.49 | −2.50 | −118.57 | −112.0 | −6.57 |
| B | −103.86 | −91.78 | −12.08 | −113.46 | −105.35 | −8.01 |
| C | −102.84 | −88.84 | −14.00 | −104.42 | −100.92 | −3.50 |
| D | −107.83 | −98.27 | −9.56 | −112.51 | −92.86 | −19.65 |
| E | −101.03 | −88.20 | −12.83 | −115.90 | −102.53 | −13.37 |
| Meloxicam | −102.27 | −94.98 | −7.29 | −104.52 | −92.92 | −11.60 |
| COX-1 | COX-2 | |||
|---|---|---|---|---|
| H-Bonding | Hydrophobic Interactions | H-Bonding | Hydrophobic Interactions | |
| Residue– Atom of Ligand | Residue | Residue– Atom of Ligand | Residue | |
| A | Ser530-O | Ile345, Val349, Leu352, Leu359, Phe518, Ile523, Gly526, Ala527, Ser530, Leu531 | Ser530-O Ser530-O | Ile345, Val349, Leu352, Leu359, Phe518, Met522, Val523, Gly526, Ala527, Ser530, Leu531 |
| B | Ser530-O Arg120-F Arg120-F | Leu93, Val116, Arg120, Val349, Leu352, Tyr355, Tyr385, TRp387, Phe518, Ile523, Gly526, Ala527 | Arg120-O | Leu93, Val116, Arg120, Val349, Leu352, Tyr355, Leu359, Val523, Gly526, Ala527 |
| C | Arg120-O | Leu93, Val116, Arg120, Val349, Leu352, Tyr355, Gly526, Ala527 | Ser530-O | Ile345, Val349, Leu352, Tyr387, Phe518, Gly526, Ala527, Ser530, Leu531 |
| D | Leu531-O | Arg120, Val349, Leu352, Ile523, Gly526, Ala527, Leu531 | Arg120-O Tyr355-N | Arg120, Val349, Tyr355, Ala527, Ser530, Leu531 |
| E | Arg120-O Tyr355-O Tyr355-O | Val116, Val349, Leu352, Tyr355, Tyr385, TRp387, Ile523, Gly526, Ala527 | Arg120-O Tyr355-O Tyr355-O | Arg120, Val349, Tyr355, Tyr385, Val523, Gly 526, Ala527 |
| Quenching | Double log | Modified Double log | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| T [K] | Ksv × 104 [dm3·mol−1] | kq × 1012 [dm3·mol−1·s−1] | R2 | Kb × 103 [dm3·mol−1] | n | R2 | Kb × 103 [dm3·mol−1] | n | R2 | |
| A | 298 | 1.28 | 1.28 | 0.987 | 5.37 | 0.93 | 0.998 | 5.23 | 0.92 | 0.998 |
| 303 | 1.20 | 1.20 | 0.997 | 2.14 | 0.86 | 0.997 | 2.00 | 0.85 | 0.993 | |
| 310 | 1.09 | 1.09 | 0.992 | 0.83 | 0.80 | 0.993 | 0.74 | 0.78 | 0.996 | |
| B | 298 | 1.07 | 1.07 | 0.996 | 8.32 | 0.98 | 0.998 | 8.32 | 0.97 | 0.998 |
| 303 | 1.02 | 1.02 | 0.998 | 3.98 | 0.92 | 0.998 | 3.89 | 0.91 | 0.999 | |
| 310 | 0.99 | 0.99 | 0.996 | 1.70 | 0.85 | 0.999 | 1.58 | 0.84 | 0.999 | |
| C | 298 | 1.33 | 1.33 | 0.987 | 11.51 | 0.98 | 0.997 | 11.14 | 0.98 | 0.996 |
| 303 | 1.30 | 1.30 | 0.994 | 5.25 | 0.92 | 0.998 | 5.01 | 0.92 | 0.997 | |
| 310 | 1.28 | 1.28 | 0.991 | 2.24 | 0.85 | 0.997 | 2.04 | 0.84 | 0.997 | |
| D | 298 | 1.46 | 1.46 | 0.997 | 4.82 | 0.91 | 0.996 | 4.34 | 0.90 | 0.989 |
| 303 | 1.43 | 1.43 | 0.996 | 2.34 | 0.85 | 0.997 | 2.03 | 0.83 | 0.997 | |
| 310 | 1.34 | 1.34 | 0.991 | 1.41 | 0.81 | 0.999 | 1.23 | 0.79 | 0.998 | |
| E | 298 | 1.51 | 1.51 | 0.998 | 6.24 | 0.92 | 0.997 | 5.74 | 0.91 | 0.997 |
| 303 | 1.48 | 1.48 | 0.991 | 3.55 | 0.87 | 0.992 | 3.16 | 0.86 | 0.991 | |
| 310 | 1.41 | 1.41 | 0.994 | 1.55 | 0.82 | 0.991 | 1.32 | 0.80 | 0.991 | |
| A | B | C | D | E | |
|---|---|---|---|---|---|
| BSA/compound molar ratio | α-helix(%) | ||||
| 1:0 | 58.7% | 61.8% | 61.6% | 59.0% | 61.7% |
| 1:0.5 | 57.5% | 60.7% | 60.1% | 57.8% | 60.5% |
| 1:1 | 56.6% | 60.6% | 58.1% | 57.2% | 59.9% |
| 1:2 | 55.0% | 58.9% | 56.4% | 56.0% | 58.7% |
| 1:4 | 54.4% | 58.4% | 53.9% | 54.8% | 57.5% |
| 1:6 | 54.1% | 56.7% | 48.1% | 53.0% | 56.3% |
| 1:8 | 53.1% | 53.1% | 47.4% | 52.6% | 55.4% |
| 1:10 | 52.8% | 52.8% | 44.5% | 52.3% | 53.9% |
| T [K] | logKb | n | R2 | ΔG° [kJmol−1] | ΔH° [kJmol−1] | ΔS° [Jmol−1K−1] | |
|---|---|---|---|---|---|---|---|
| A | 298 | 3.73 ± 0.07 | 0.93 ± 0.01 | 0.998 | −21.16 | −11.88 | −327.53 |
| 303 | 3.33 ± 0.13 | 0.86 ± 0.04 | 0.997 | ||||
| 310 | 2.87 ± 0.16 | 0.80 ± 0.03 | 0.993 | ||||
| B | 298 | 3.92 ± 0.07 | 0.98 ± 0.01 | 0.998 | −22.28 | −10.00 | −260.88 |
| 303 | 3.60 ± 0.06 | 0.92 ± 0.01 | 0.998 | ||||
| 310 | 3.23 ± 0.03 | 0.85 ± 0.01 | 0.999 | ||||
| C | 298 | 4.06 ± 0.09 | 0.98 ± 0.02 | 0.997 | −23.07 | −10.38 | 270.95 |
| 303 | 3.72 ± 0.08 | 0.92 ± 0.02 | 0.998 | ||||
| 310 | 3.35 ± 0.08 | 0.85 ± 0.02 | 0.997 | ||||
| D | 298 | 3.68 ± 0.10 | 0.91 ± 0.03 | 0.996 | −20.83 | −7.73 | −189.67 |
| 303 | 3.37 ± 0.07 | 0.85 ± 0.01 | 0.997 | ||||
| 310 | 3.15 ± 0.05 | 0.81 ± 0.01 | 0.999 | ||||
| E | 298 | 3.80 ± 0.08 | 0.92 ± 0.02 | 0.997 | −21.68 | −8.92 | −226.56 |
| 303 | 3.55 ± 0.12 | 0.87 ± 0.03 | 0.992 | ||||
| 310 | 3.19 ± 0.15 | 0.82 ± 0.03 | 0.991 |
| System | Kb [dm3mol−1] | R2 | Kq [dm3mol−1] | R2 |
|---|---|---|---|---|
| BSA+A | 5.37 × 103 | 0.998 | 1.28 × 1012 | 0.996 |
| BSA+A+PHB | 5.12 × 103 | 0.983 | 1.22 × 1012 | 0.982 |
| BSA+A+IBP | 1.17 × 103 | 0.982 | 5.46 × 1011 | 0.977 |
| BSA+B | 8.32 × 103 | 0.998 | 1.07 × 1012 | 0.995 |
| BSA+B+PHB | 8.16 × 103 | 0.981 | 1.01 × 1012 | 0.982 |
| BSA+B+IBP | 1.02 × 103 | 0.986 | 3.81 × 1011 | 0.991 |
| BSA+C | 1.15 × 104 | 0.997 | 1.33 × 1012 | 0.987 |
| BSA+C+PHB | 1.02 × 104 | 0.985 | 1.28 × 1012 | 0.989 |
| BSA+C+IBP | 5.82 × 103 | 0.975 | 6.97 × 1011 | 0.976 |
| BSA+D | 4.82 × 103 | 0.996 | 1.46 × 1012 | 0.997 |
| BSA+D+PHB | 1.50 × 102 | 0.987 | 5.11 × 1011 | 0.988 |
| BSA+D+IBP | 1.38 × 102 | 0.981 | 1.51 × 1011 | 0.983 |
| BSA+E | 6.24 × 103 | 0.997 | 1.51 × 1012 | 0.998 |
| BSA+E+PHB | 4.74 × 102 | 0.985 | 1.22 × 1011 | 0.985 |
| BSA+E+IBP | 1.94 × 102 | 0.969 | 1.03 × 1011 | 0992 |
| Binding Site | ΔG° [kJmol−1] | ΔE1 [kJmol−1] | ΔE2 [kJmol−1] | ΔE3 [kJmol−1] | |
|---|---|---|---|---|---|
| A | site I | −31.50 | −32.23 | −31.50 | −3.77 |
| site II | −35.56 | −39.28 | −35.06 | −4.22 | |
| B | site I | −29.29 | −34.26 | −30.63 | −3.64 |
| site II | −35.14 | −40.12 | −35.68 | −4.39 | |
| C | site I | −31.42 | −36.40 | −33.26 | −3.13 |
| site II | −36.52 | −41.54 | −37.57 | −3.93 | |
| D | site I | −26.70 | −30.41 | −26.98 | −3.55 |
| site II | −27.44 | −31.21 | −26.94 | −4.23 | |
| E | site I | −26.52 | −31.51 | −27.57 | −4.14 |
| site II | −27.48 | −32.46 | −28.53 | −3.93 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krzyżak, E.; Szkatuła, D.; Wiatrak, B.; Gębarowski, T.; Marciniak, A. Synthesis, Cyclooxygenases Inhibition Activities and Interactions with BSA of N-substituted 1H-pyrrolo[3,4-c]pyridine-1,3(2H)-diones Derivatives. Molecules 2020, 25, 2934. https://doi.org/10.3390/molecules25122934
Krzyżak E, Szkatuła D, Wiatrak B, Gębarowski T, Marciniak A. Synthesis, Cyclooxygenases Inhibition Activities and Interactions with BSA of N-substituted 1H-pyrrolo[3,4-c]pyridine-1,3(2H)-diones Derivatives. Molecules. 2020; 25(12):2934. https://doi.org/10.3390/molecules25122934
Chicago/Turabian StyleKrzyżak, Edward, Dominika Szkatuła, Benita Wiatrak, Tomasz Gębarowski, and Aleksandra Marciniak. 2020. "Synthesis, Cyclooxygenases Inhibition Activities and Interactions with BSA of N-substituted 1H-pyrrolo[3,4-c]pyridine-1,3(2H)-diones Derivatives" Molecules 25, no. 12: 2934. https://doi.org/10.3390/molecules25122934