Progesterone Attenuates Stress-Induced NLRP3 Inflammasome Activation and Enhances Autophagy Following Ischemic Brain Injury

 ,
,
Abstract
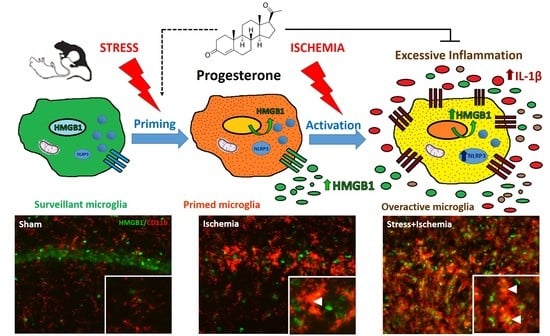
:
1. Introduction
2. Results
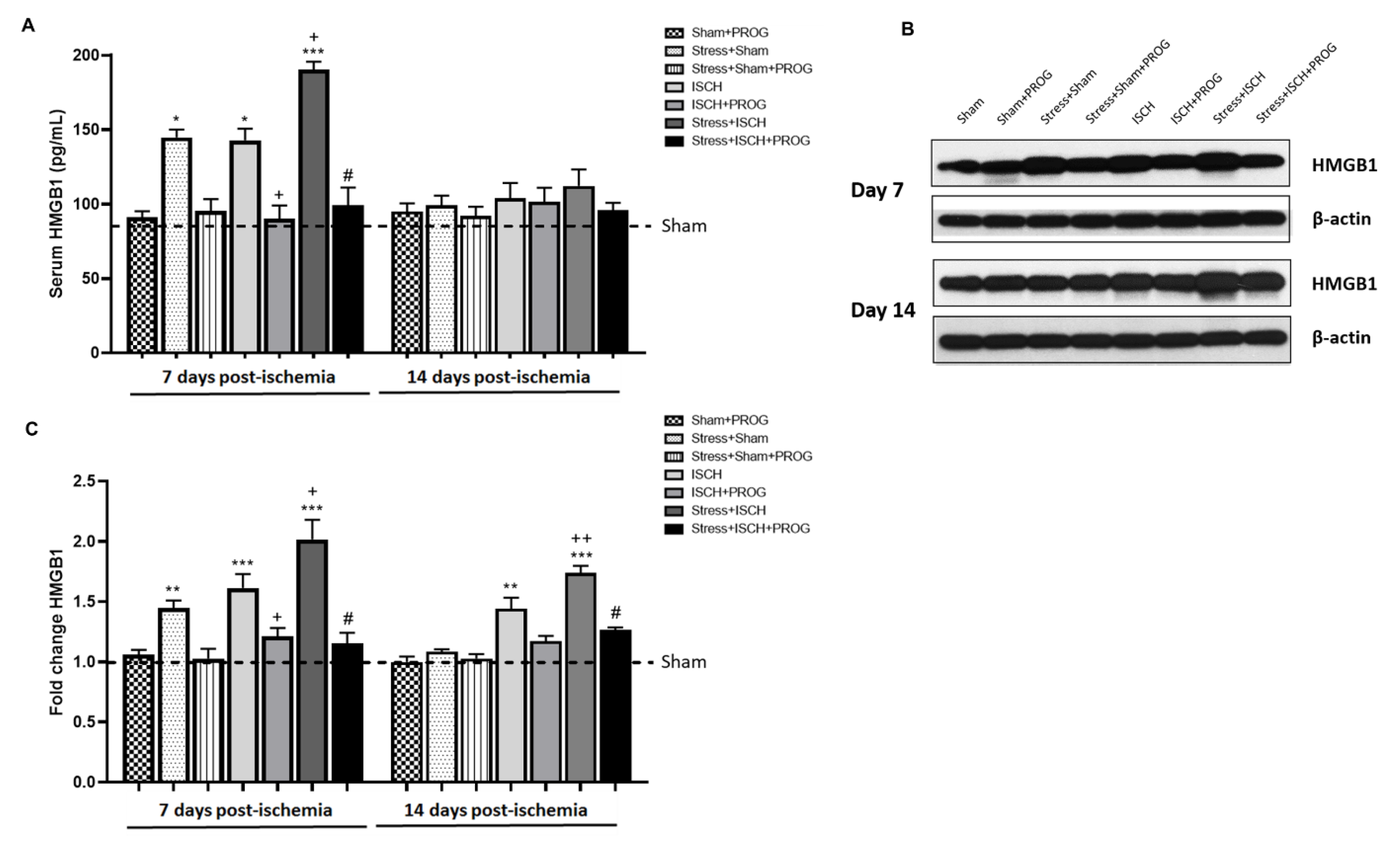
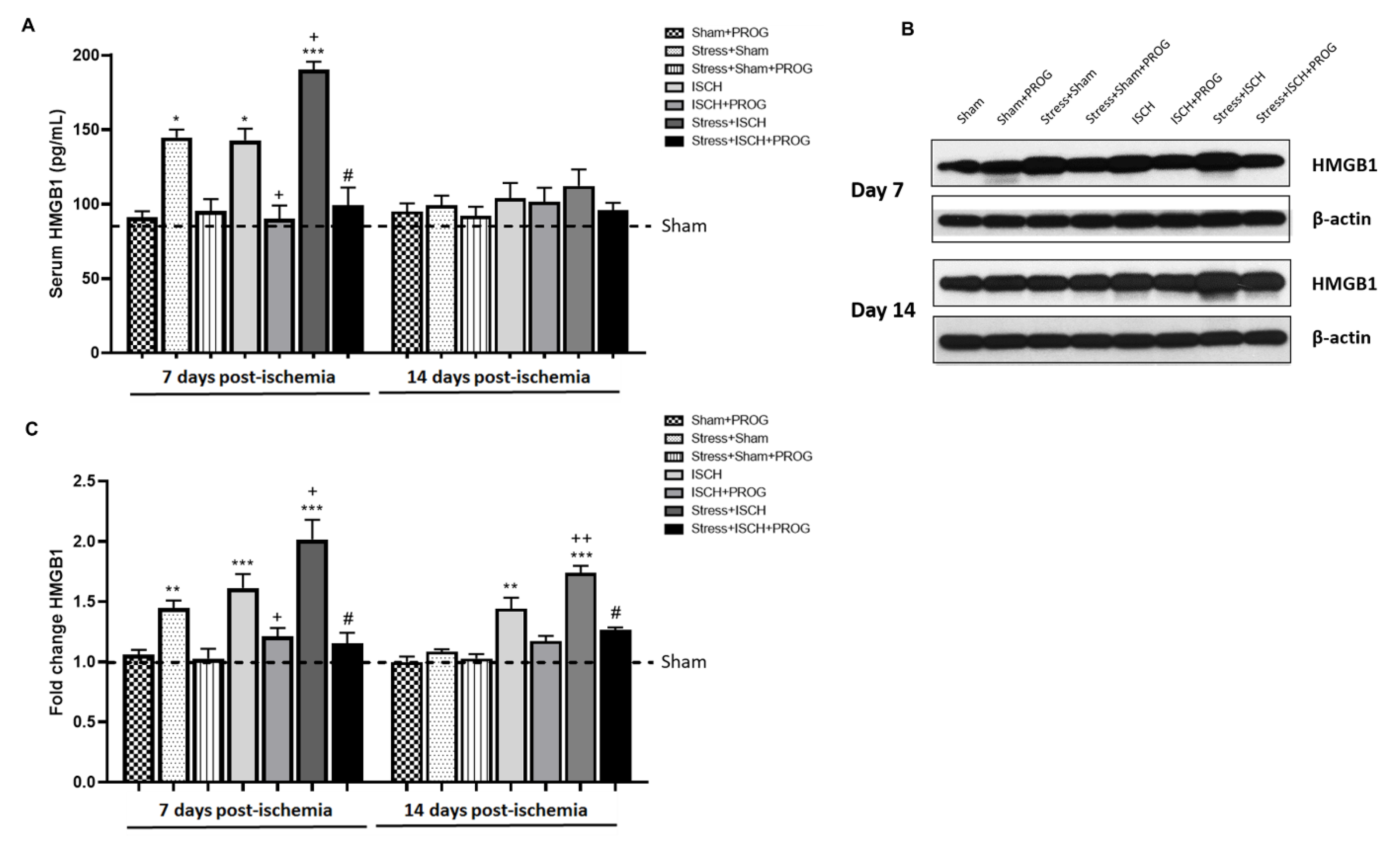
2.1. Stress Worsens HMGB1 Release Induced by Global Ischemia
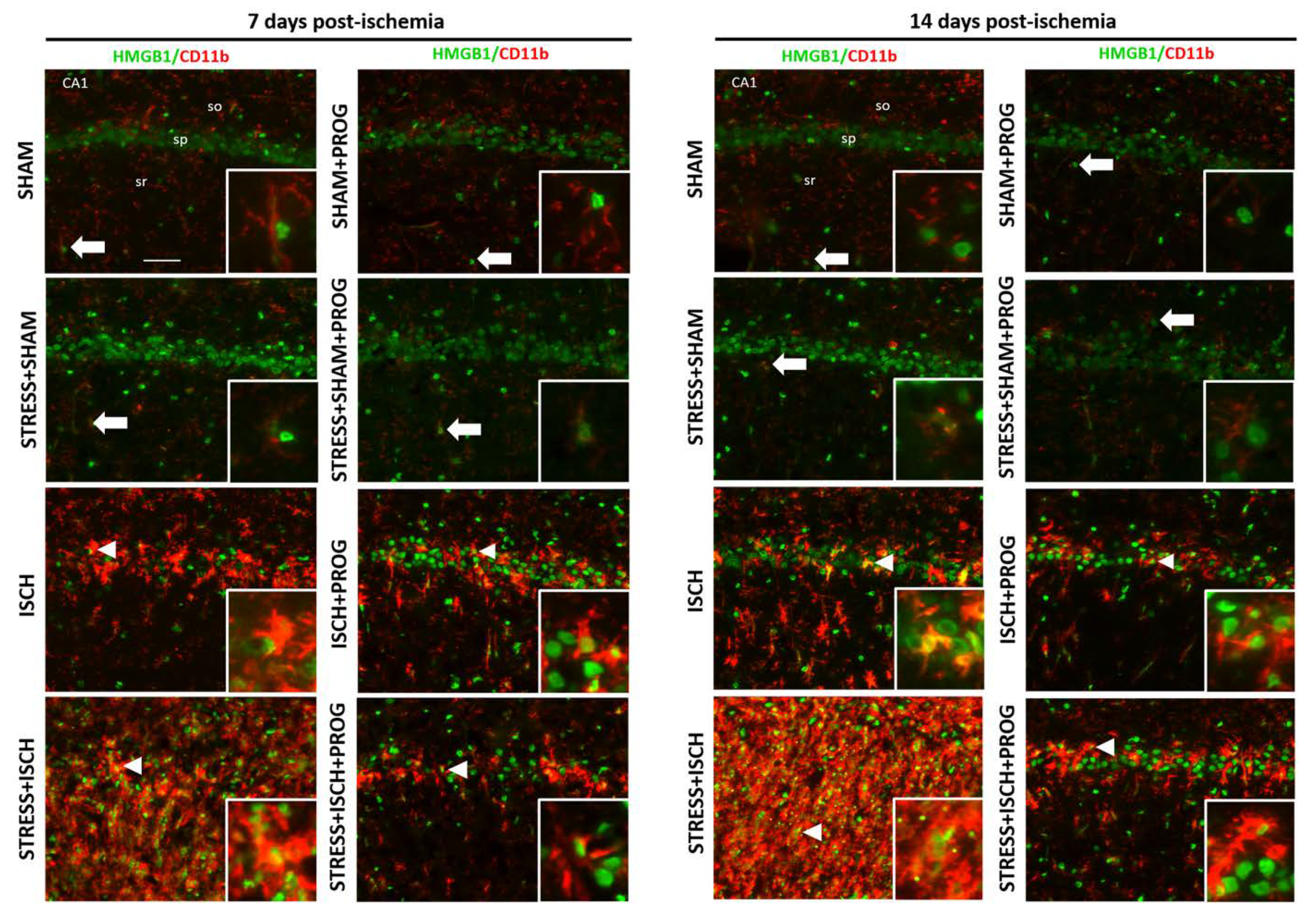
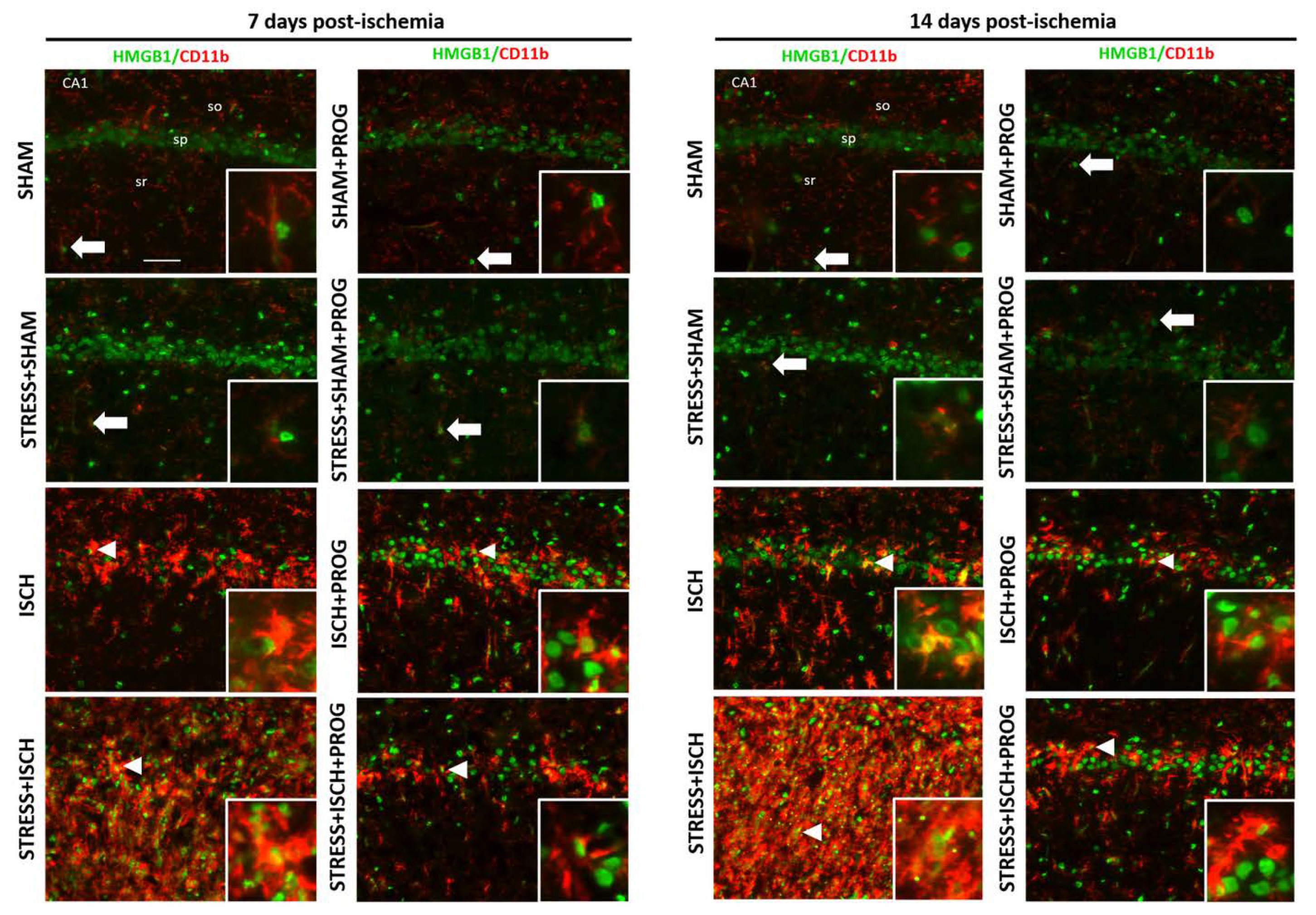
2.2. Activated Microglia Are the Main Cellular Source of HMGB1 Release
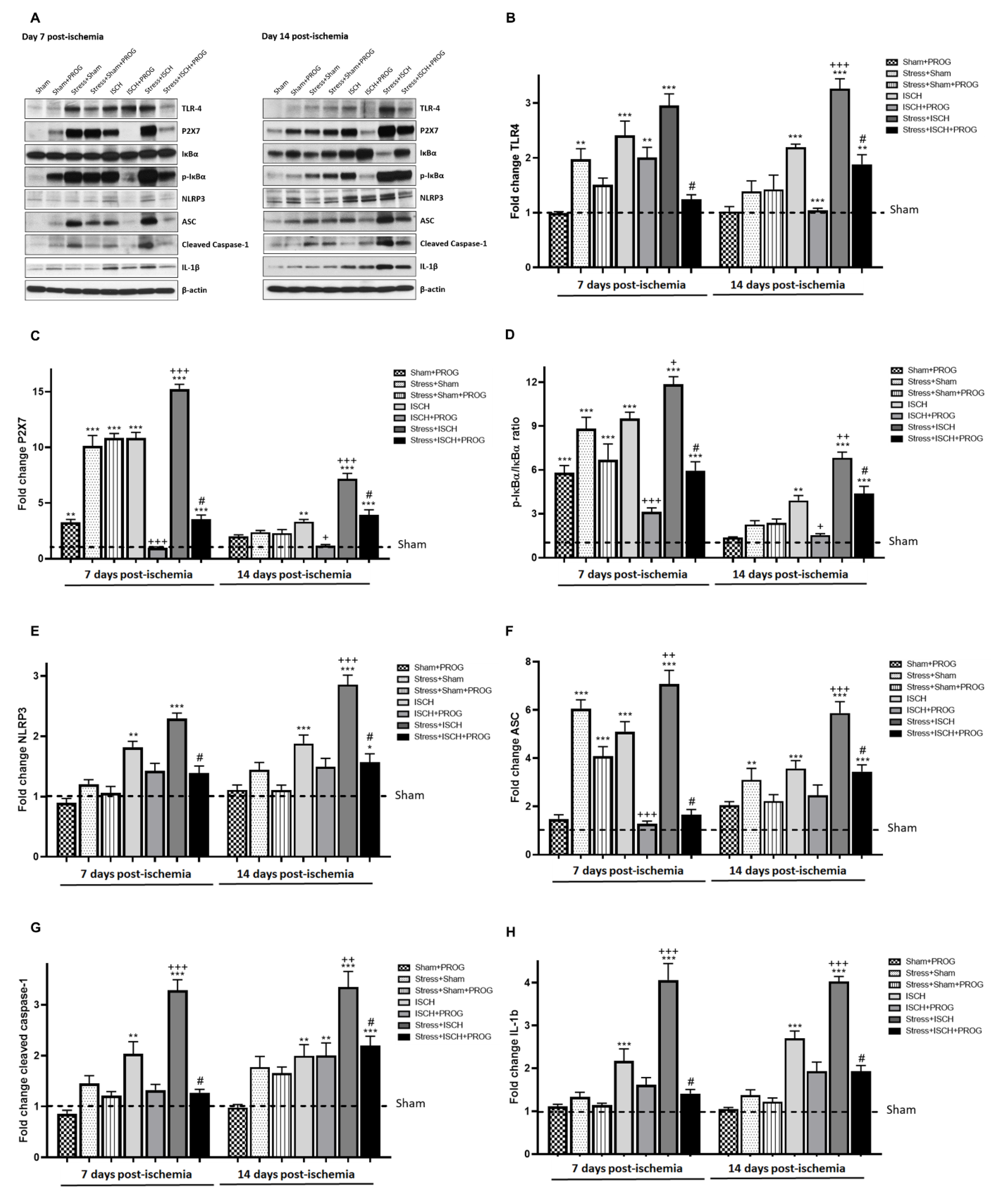
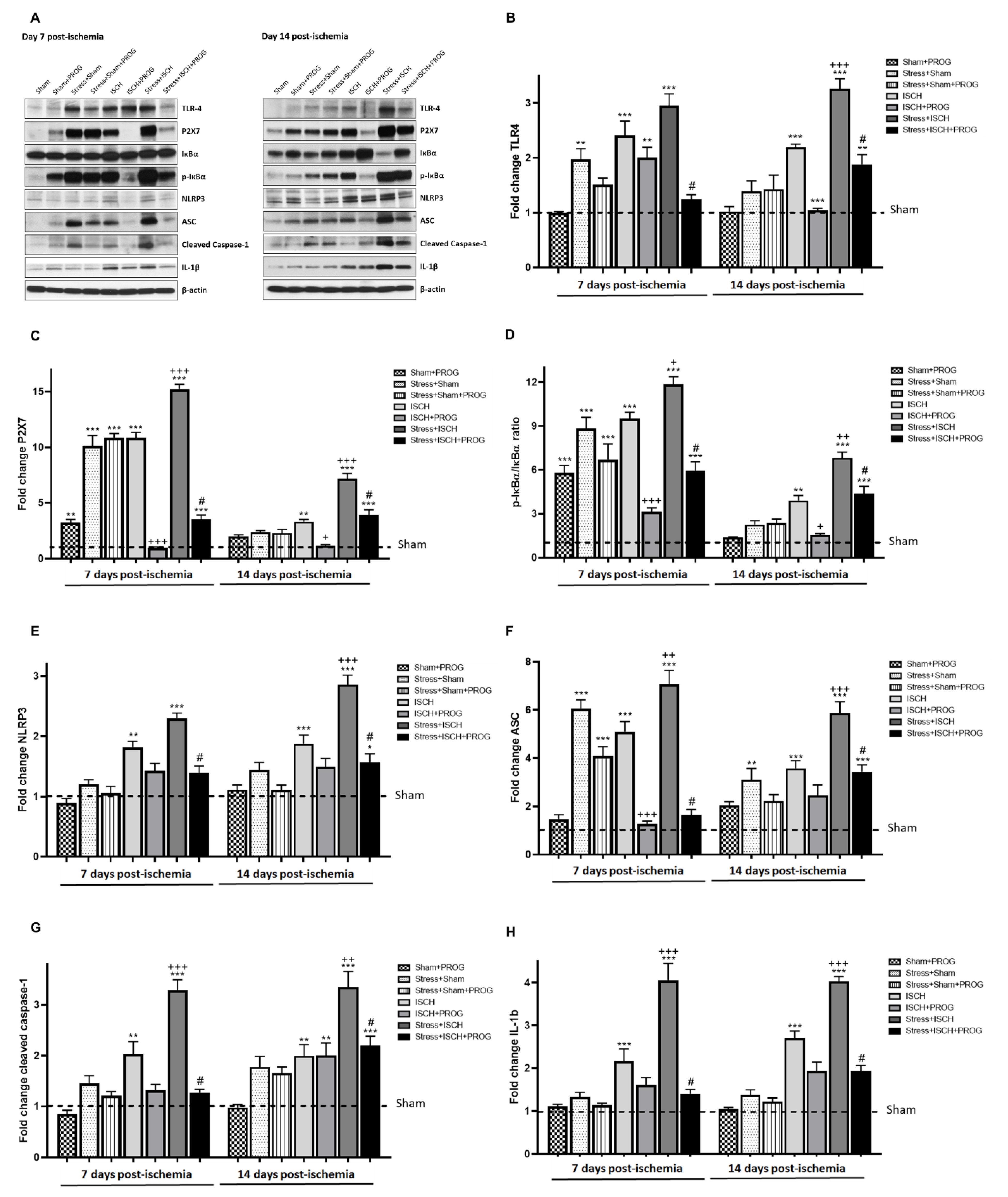
2.3. Stress Exacerbates NLRP3 Inflammasome Activation in the Ischemic Hippocampus
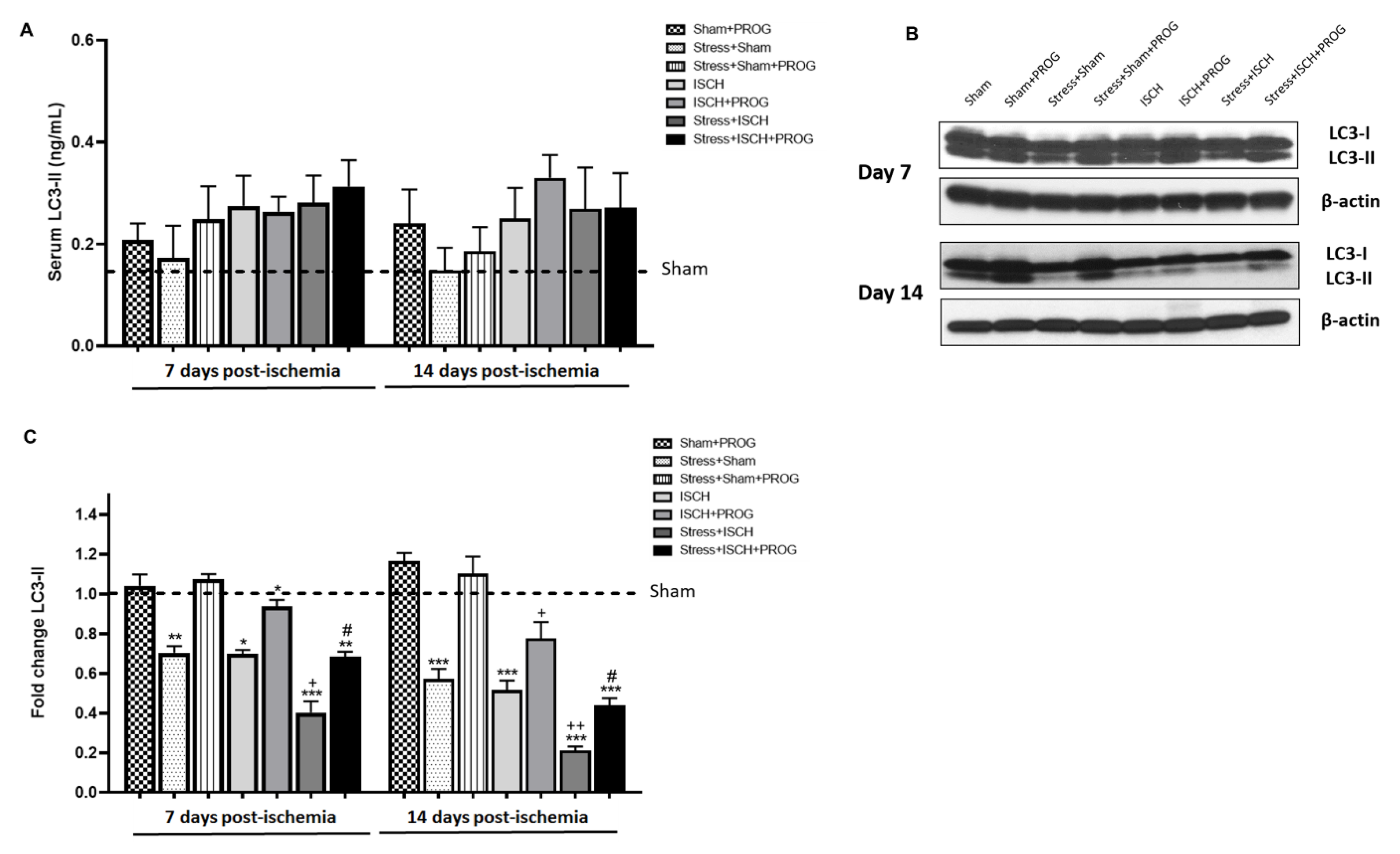
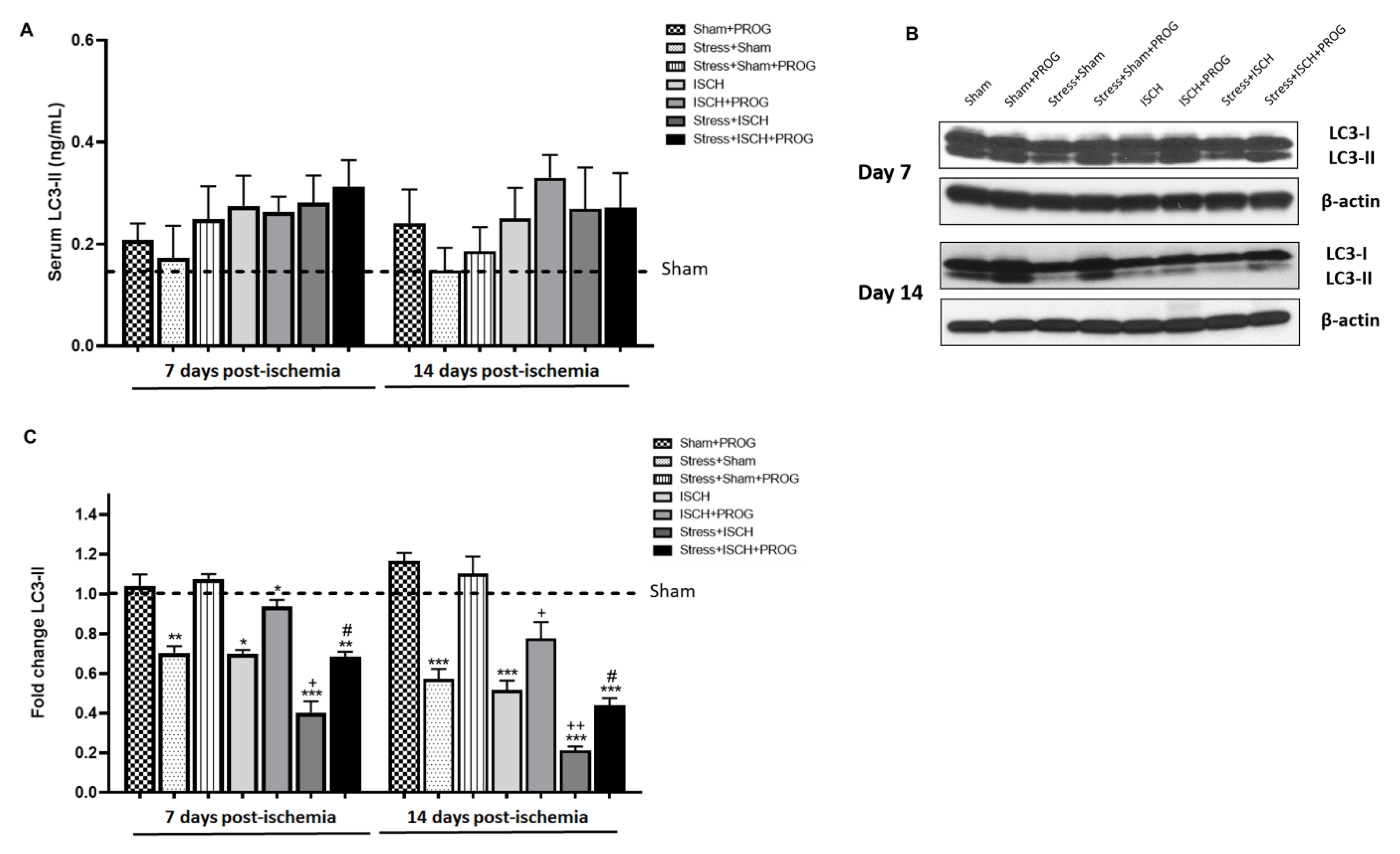
2.4. Stress Impairs Autophagy Following Global Ischemia
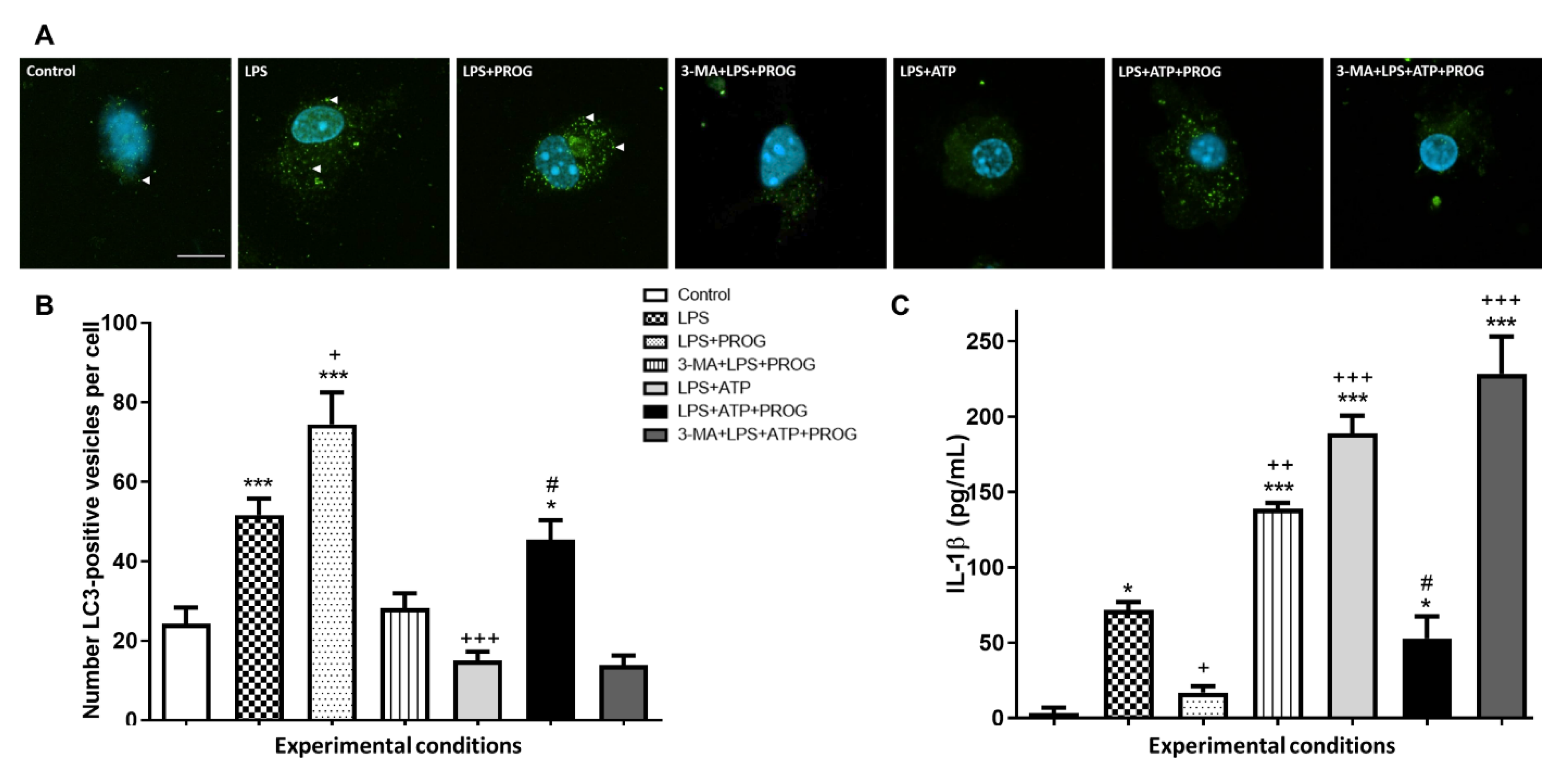
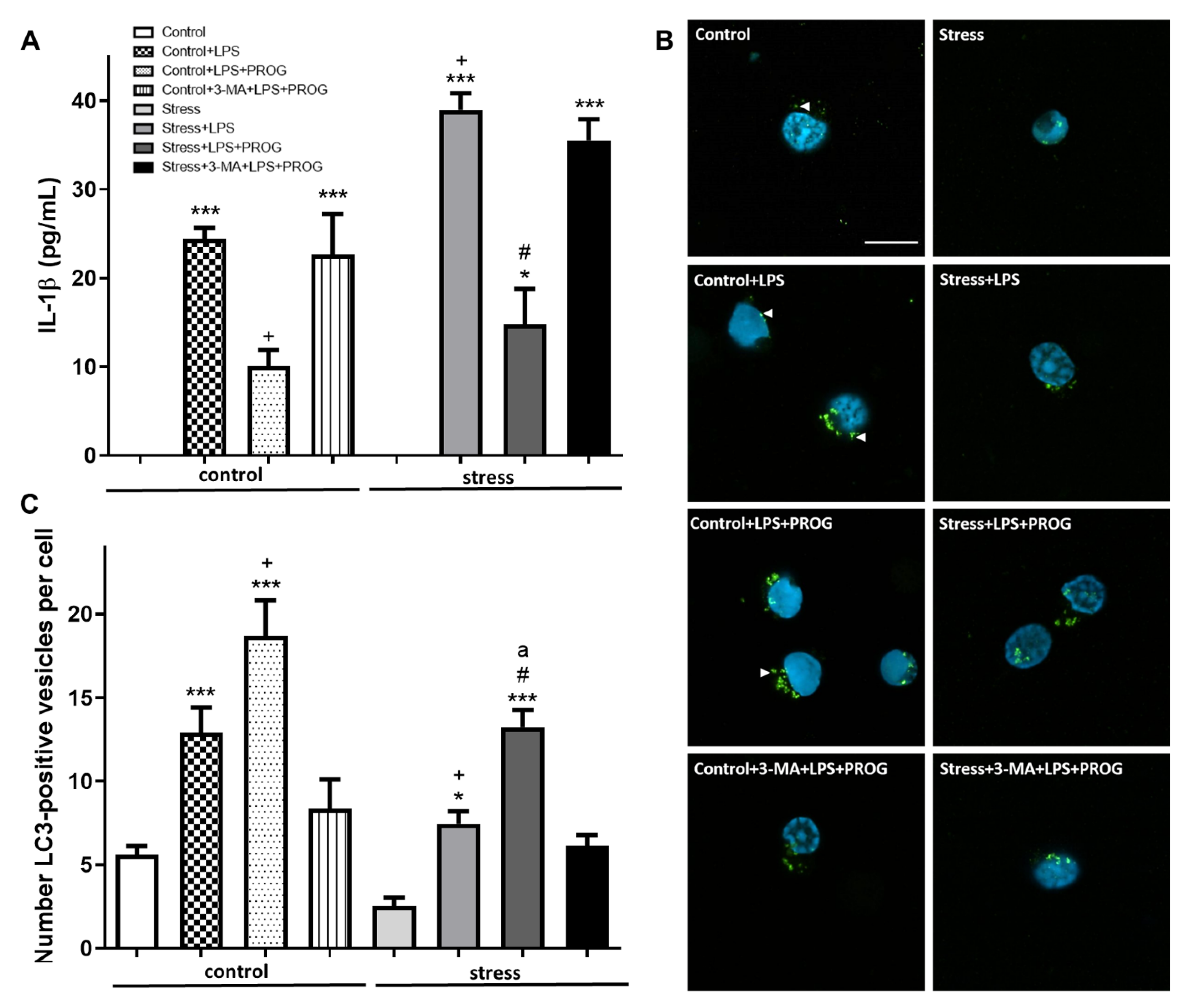
2.5. NLRP3 Inflammasome Activation In Vitro Compromises Autophagy in Microglia
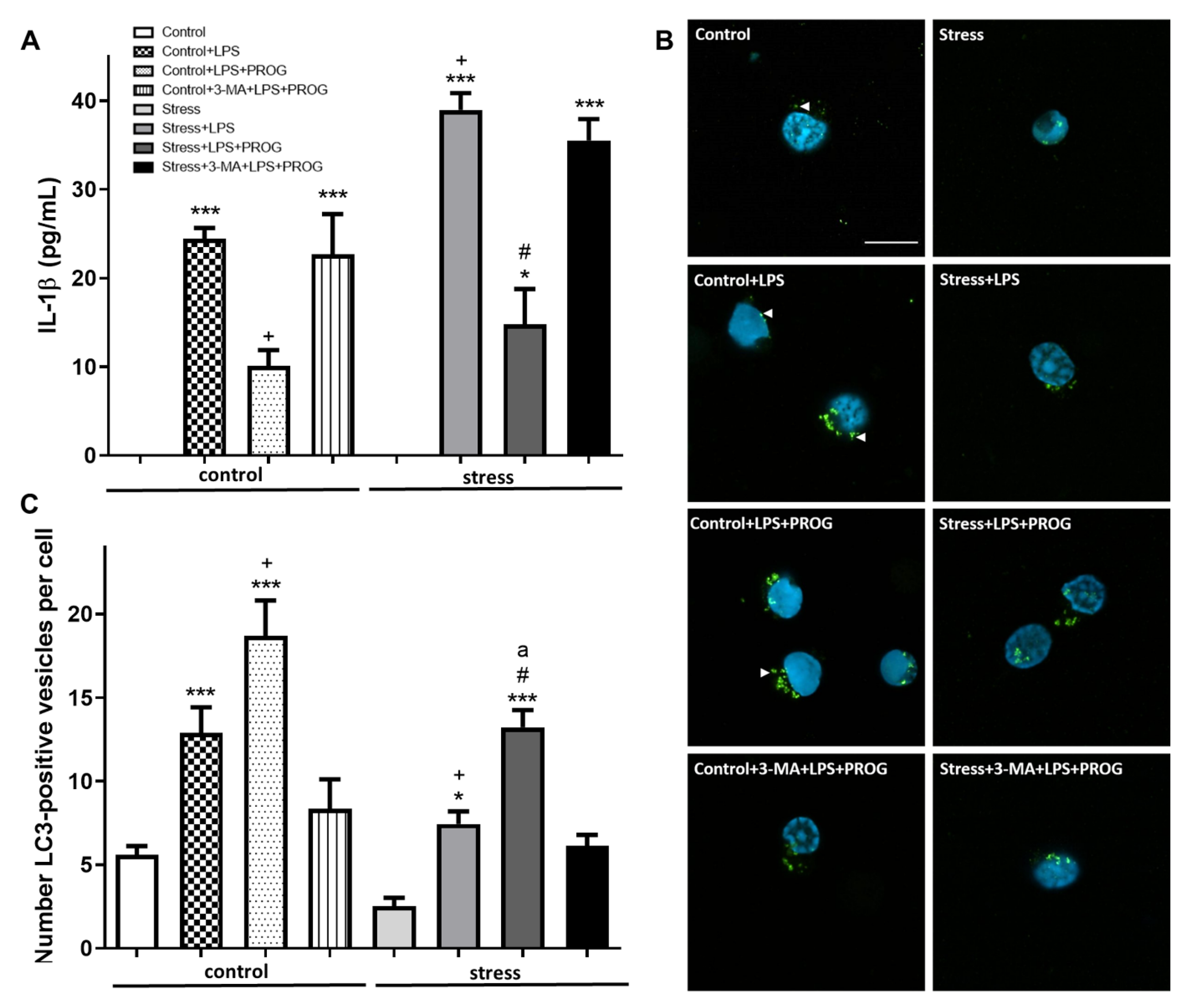
2.6. Stress-Primed Microglia Ex Vivo Show Elevated IL-1β Production and Impaired Autophagy
3. Discussion
3.1. Effects of Stress Prior to Ischemia on HMGB1 Release
3.2. Effects of Stress Exposure Prior to Ischemia on NLRP3 Inflammasome Activation
3.3. Effects of Stress Exposure Prior to Ischemia on Autophagy
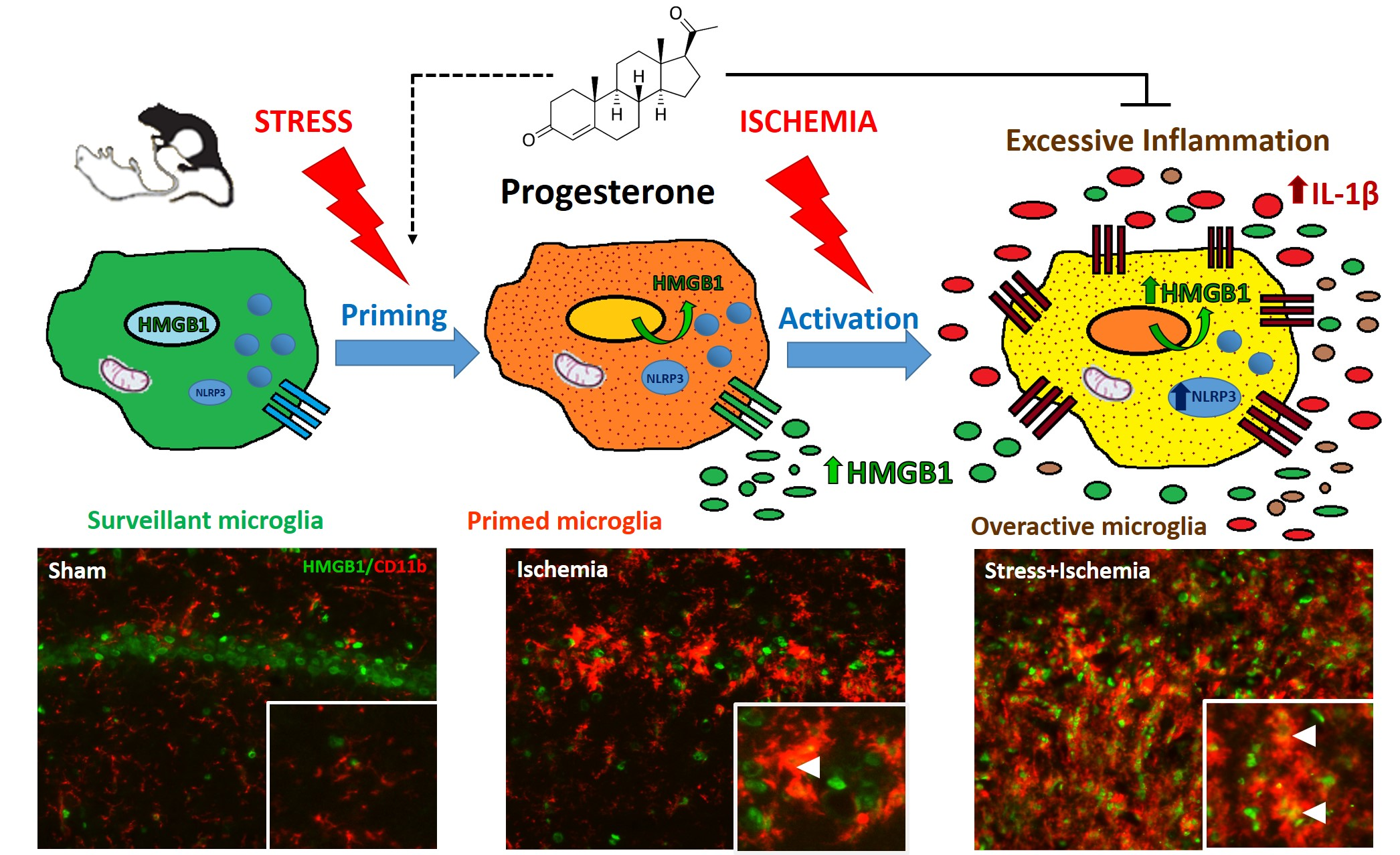
3.4. PROG Treatment of Stress-Induced Microglial Priming
4. Materials and Methods
4.1. Animals
4.2. Experimental Groups and In Vivo Treatment
4.3. Social Defeat Stress
4.4. Global Cerebral Ischemia
4.5. Tissue Collection
4.6. Serum Measurements
4.7. Immunohistochemistry
4.8. Western Blotting
4.9. In Vitro Inflammasome Assay
4.10. Neuroinflammatory Priming to Stress
4.11. Immunocytochemistry
4.12. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ATP | Adenosine triphosphate |
| ASC | Apoptosis-associated speck-like protein containing a caspase recruitment domain |
| BSA | Bovine serum albumin |
| DMSO | Dimethyl sulfoxide |
| HMGB1 | High-mobility group box-1 |
| IκBα | Nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor, alpha |
| IL | Interleukin |
| LC3 | Microtubule-associated protein light chain 3 |
| LPS | Lipopolysaccharide |
| mTOR | Mammalian target of rapamycin |
| 3-MA | 3-methyladenine |
| NF-κB | Nuclear factor of kappa light polypeptide gene enhancer in B-cells |
| NLRP3 | NOD-like receptor pyrin domain containing 3 |
| P2X7R | Purinergic receptor P2X, ligand gated ion channel, 7 |
| PROG | Progesterone |
| PGRMC1 | Progesterone-associated membrane component 1 |
| TLR4 | Toll-like receptor-4 |
References
- Everson-Rose, S.A.; Roetker, N.S.; Lutsey, P.L.; Kershaw, K.N.; Longstreth, W.T., Jr.; Sacco, R.L.; Diez Roux, A.V.; Alonso, A. Chronic stress, depressive symptoms, anger, hostility, and risk of stroke and transient ischemic attack in the multi-ethnic study of atherosclerosis. Stroke 2014, 45, 2318–2323. [Google Scholar] [CrossRef] [PubMed]
- Sumner, J.A.; Khodneva, Y.; Muntner, P.; Redmond, N.; Lewis, M.W.; Davidson, K.W.; Edmondson, D.; Richman, J.; Safford, M.M. Effects of Concurrent Depressive Symptoms and Perceived Stress on Cardiovascular Risk in Low- and High-Income Participants: Findings From the Reasons for Geographical and Racial Differences in Stroke (REGARDS) Study. J. Am. Heart Assoc. 2016, 5, e003930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, H.; Fang, F.; Arnberg, F.K.; Mataix-Cols, D.; Fernández de la Cruz, L.; Almqvist, C.; Fall, K.; Lichtenstein, P.; Thorgeirsson, G.; Valdimarsdóttir, U. Stress related disorders and risk of cardiovascular disease: Population based, sibling controlled cohort study. BMJ 2019, 10, l1255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norden, D.M.; Muccigrosso, M.M.; Godbout, J.P. Microglial priming an1d enhanced reactivity to secondary insult in aging, and traumatic CNS injury, and neurodegenerative disease. Neuropharmacology 2015, 96, 29–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haley, M.J.; Brough, D.; Quintin, J.; Allan, S.M. Microglial Priming as Trained Immunity in the Brain. Neuroscience 2019, 405, 47–54. [Google Scholar] [CrossRef]
- Weil, Z.M.; Norman, G.J.; Barker, J.M.; Su, A.J.; Nelson, R.J.; Devries, A.C. Social isolation potentiates cell death and inflammatory responses after global ischemia. Mol. Psychiatry 2008, 13, 913–915. [Google Scholar] [CrossRef]
- Neigh, G.N.; Karelina, K.; Glasper, E.R.; Bowers, S.L.; Zhang, N.; Popovich, P.G.; DeVries, A.C. Anxiety after cardiac arrest/cardiopulmonary resuscitation: Exacerbated by stress and prevented by minocycline. Stroke 2009, 40, 3601–3607. [Google Scholar] [CrossRef]
- Norman, G.J.; Zhang, N.; Morris, J.S.; Karelina, K.; Berntson, G.G.; DeVries, A.C. Social interaction modulates autonomic, inflammatory, and depressive-like responses to cardiac arrest and cardiopulmonary resuscitation. Proc. Natl. Acad. Sci. USA 2010, 107, 16342–16347. [Google Scholar] [CrossRef] [Green Version]
- Espinosa-Garcia, C.; Sayeed, I.; Yousuf, S.; Atif, F.; Sergeeva, E.G.; Neigh, G.N.; Stein, D.G. Stress primes microglial polarization after global ischemia: Therapeutic potential of progesterone. Brain Behav. Immun. 2017, 177–192. [Google Scholar] [CrossRef]
- Gaudier-Diaz, M.M.; Zhang, N.; Haines, A.H.; Surbhi; Zhou, M.; DeVries, A.C. Social interaction modulates the neuroinflammatory response to global cerebral ischemia in male mice. Brain Res. 2017, 1673, 86–94. [Google Scholar] [CrossRef]
- Gaudier-Diaz, M.M.; Haines, A.H.; Zhang, N.; DeVries, A.C. Social influences on microglial reactivity and neuronal damage after cardiac arrest/cardiopulmonary resuscitation. Physiol. Behav. 2018, 194, 437–449. [Google Scholar] [CrossRef] [PubMed]
- Frank, M.G.; Weber, M.D.; Watkins, L.R.; Maier, S.F. Stress-induced neuroinflammatory priming: A liability factor in the etiology of psychiatric disorders. Neurobiol. Stress 2015, 4, 62–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, M.D.; Frank, M.G.; Tracey, K.J.; Watkins, L.R.; Maier, S.F. Stress induces the danger-associated molecular pattern HMGB-1 in the hippocampus of male Sprague Dawley rats: A priming stimulus of microglia and the NLRP3 inflammasome. J. Neurosci. 2015, 35, 316–324. [Google Scholar] [CrossRef] [PubMed]
- Franklin, T.C.; Wohleb, E.S.; Zhang, Y.; Fogaça, M.; Hare, B.; Duman, R.S. Persistent increase in microglial RAGE contributes to chronic stress Induced priming of depressive-like behavior. Biol. Psychiatry 2018, 83, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.B.; Choi, J.S.; Yu, Y.M.; Nam, K.; Piao, C.S.; Kim, S.W.; Lee, M.H.; Han, P.L.; Park, J.S.; Lee, J.K. HMGB1, a novel cytokine-like mediator linking acute neuronal death and delayed neuroinflammation in the postischemic brain. J. Neurosci. 2006, 26, 6413–6421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.B.; Lim, C.M.; Yu, Y.M.; Lee, J.K. Induction and subcellular localization of high-mobility group box-1 (HMGB1) in the postischemic rat brain. J. Neurosci. Res. 2008, 86, 1125–1131. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.D.; Lee, H.; Kim, S.W.; Lee, H.K.; Choi, J.; Han, P.L.; Lee, J.K. Alarmin HMGB1 induces systemic and brain inflammatory exacerbation in post-stroke infection rat model. Cell Death Dis. 2018, 9, 426. [Google Scholar] [CrossRef]
- Roth, S.; Singh, V.; Tiedt, S.; Schindler, L.; Huber, G.; Geerlof, A.; Antoine, D.J.; Anfray, A.; Orset, C.; Gauberti, M.; et al. Brain-released alarmins and stress response synergize in accelerating atherosclerosis progression after stroke. Sci. Transl. Med. 2018, 10, eaao1313. [Google Scholar] [CrossRef] [Green Version]
- Oda, Y.; Tsuruta, R.; Fujita, M.; Kaneda, K.; Kawamura, Y.; Izumi, T.; Kasaoka, S.; Maruyama, I.; Maekawa, T. Prediction of the neurological outcome with intrathecal high mobility group box 1 and S100B in cardiac arrest victims: A pilot study. Resuscitation 2012, 83, 1006–1012. [Google Scholar] [CrossRef]
- Shi, X.; Li, M.; Huang, K.; Zhou, S.; Hu, Y.; Pan, S.; Gu, Y. HMGB1 binding heptamer peptide improves survival and ameliorates brain injury in rats after cardiac arrest and cardiopulmonary resuscitation. Neuroscience 2017, 360, 128–138. [Google Scholar] [CrossRef]
- Trendelenburg, G. Molecular regulation of cell fate in cerebral ischemia: Role of the inflammasome and connected pathways. J. Cereb. Blood Flow Metab. 2014, 34, 1857–1867. [Google Scholar] [CrossRef] [PubMed]
- Latz, E.; Xiao, T.S.; Stutz, A. Activation and regulation of the inflammasomes. Nat. Rev. Immunol. 2013, 13, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wang, H.; Kouadir, M.; Song, H.; Shi, F. Recent advances in the mechanisms of NLRP3 inflammasome activation and its inhibitors. Cell Death Dis. 2019, 10, 128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Liu, L.; Liu, Y.Z.; Shen, X.L.; Wu, T.Y.; Zhang, T.; Wang, W.; Wang, Y.X.; Jiang, C.L. NLRP3 Inflammasome Mediates Chronic Mild Stress-Induced Depression in Mice via Neuroinflammation. Int. J. Neuropsychopharmacol. 2015, 18, pyv006. [Google Scholar] [CrossRef]
- Thakkar, R.; Wang, R.; Sareddy, G.; Wang, J.; Thiruvaiyaru, D.; Vadlamudi, R.; Zhang, Q.; Brann, D. NLRP3 Inflammasome Activation in the Brain after Global Cerebral Ischemia and Regulation by 17β-Estradiol. Oxid. Med. Cell. Longev. 2016, 2016, 8309031. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Wang, Z.; Jia, X. Neuroprotection of Glibenclamide against Brain Injury after Cardiac Arrest via Modulation of NLRP3 Inflammasome. Conf. Proc. IEEE Eng. Med. Biol. Soc. 2019, 2019, 4209–4212. [Google Scholar] [CrossRef]
- Qian, A.; Xu, J.; Wu, C.; Liu, S.; Zhang, M. Hypothermia Inhibits Cerebral Necroptosis and NOD-Like Receptor Pyrin Domain Containing 3 Pathway in a Swine Model of Cardiac Arrest. J. Surg. Res. 2019, 244, 468–476. [Google Scholar] [CrossRef]
- Sun, Q.; Fan, J.; Billiar, T.R.; Scott, M.J. Inflammasome and autophagy regulation—A two-way street. Mol. Med. 2017, 23, 188–195. [Google Scholar] [CrossRef]
- Houtman, J.; Freitag, K.; Gimber, N.; Schmoranzer, J.; Heppner, F.L.; Jendrach, M. Beclin1-driven autophagy modulates the inflammatory response of microglia via NLRP3. Embo J. 2019, 38, e99430. [Google Scholar] [CrossRef]
- Bussi, C.; Peralta Ramos, J.M.; Arroyo, D.S.; Gaviglio, E.A.; Gallea, J.I.; Wang, J.M.; Celej, M.S.; Iribarren, P. Autophagy down regulates pro-inflammatory mediators in BV2 microglial cells and rescues both LPS and alpha-synuclein induced neuronal cell death. Sci. Rep. 2017, 7, 43153. [Google Scholar] [CrossRef] [Green Version]
- Jin, M.M.; Wang, F.; Qi, D.; Liu, W.W.; Gu, C.; Mao, C.J.; Yang, Y.P.; Zhao, Z.; Hu, L.F.; Liu, C.F. A Critical Role of Autophagy in Regulating Microglia Polarization in Neurodegeneration. Front. Aging Neurosci. 2018, 10, 378. [Google Scholar] [CrossRef] [PubMed]
- Kotoda, M.; Furukawa, H.; Miyamoto, T.; Korai, M.; Shikata, F.; Kuwabara, A.; Xiong, X.; Rutledge, C.; Giffard, R.G.; Hashimoto, T. Role of Myeloid Lineage Cell Autophagy in Ischemic Brain Injury. Stroke 2018, 49, 1488–1495. [Google Scholar] [CrossRef]
- Juan, Z.; Kewei, L.; Kaibin, H.; Yong, G.; Yafang, H.; Suyue, P.; Zhong, J. Metformin Improves Neurologic Outcome Via AMP-Activated Protein Kinase–Mediated Autophagy Activation in a Rat Model of Cardiac Arrest and Resuscitation. J. Am. Heart Assoc. 2018, 7, e008389. [Google Scholar] [CrossRef] [Green Version]
- Wei, H.; Yin, M.; Lu, Y.; Yang, Y.; Li, B.; Liao, X.X.; Dai, G.; Jing, X.; Xiong, Y.; Hu, C. Mild hypothermia improves neurological outcome in mice after cardiopulmonary resuscitation through Silent Information Regulator 1-actviated autophagy. Cell Death Discov. 2019, 5, 129. [Google Scholar] [CrossRef] [Green Version]
- Lammerding, L.; Slowik, A.; Johann, S.; Beyer, C.; Zendedel, A. Poststroke Inflammasome Expression and Regulation in the Peri-Infarct Area by Gonadal Steroids after Transient Focal Ischemia in the Rat Brain. Neuroendocrinology 2016, 103, 460–475. [Google Scholar] [CrossRef] [PubMed]
- Gibson, C.L.; Constantin, D.; Prior, M.J.; Bath, P.M.; Murphy, S.P. Progesterone suppresses the inflammatory response and nitric oxide synthase-2 expression following cerebral ischemia. Exp. Neurol. 2005, 193, 522–530. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Wang, J.; Li, X.; Liu, C.; Chen, N.; Hao, Y. Progesterone exerts neuroprotective effects by inhibiting inflammatory response after stroke. Inflamm. Res. 2009, 58, 619–624. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Cui, K.; Wang, J.; He, Y. Microglia and cyclooxygenase-2: Possible therapeutic targets of progesterone for stroke. Int. Immunopharmacol. 2011, 11, 1925–1931. [Google Scholar] [CrossRef]
- Ishrat, T.; Sayeed, I.; Atif, F.; Hua, F.; Stein, D.G. Progesterone and allopregnanolone attenuate blood-brain barrier dysfunction following permanent focal ischemia by regulating the expression of matrix metalloproteinases. Exp. Neurol. 2010, 226, 183–190. [Google Scholar] [CrossRef] [Green Version]
- Yousuf, S.; Atif, F.; Sayeed, I.; Wang, J.; Stein, D.G. Post-stroke infections exacerbate ischemic brain injury in middle-aged rats: Immunomodulation and neuroprotection by progesterone. Neuroscience 2013, 239, 92–102. [Google Scholar] [CrossRef] [Green Version]
- Habib, P.; Slowik, A.; Zendedel, A.; Johann, S.; Dang, J.; Beyer, C. Regulation of hypoxia-induced inflammatory responses and M1-M2 phenotype switch of primary rat microglia by sex steroids. J. Mol. Neurosci. 2014, 52, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Won, S.; Lee, J.K.; Stein, D.G. Recombinant tissue plasminogen activator promotes, and progesterone attenuates, microglia/macrophage M1 polarization and recruitment of microglia after MCAO stroke in rats. Brain Behav. Immun. 2015, 49, 267–279. [Google Scholar] [CrossRef] [PubMed]
- Allen, R.S.; Sayeed, I.; Oumarbaeva, Y.; Morrison, K.C.; Choi, P.H.; Pardue, M.T.; Stein, D.G. Progesterone treatment shows greater protection in brain vs. retina in a rat model of middle cerebral artery occlusion: Progesterone receptor levels may play an important role. Restor. Neurol. Neurosci. 2016, 34, 947–963. [Google Scholar] [CrossRef] [PubMed]
- Herzog, R.; Zendedel, A.; Lammerding, L.; Beyer, C.; Slowik, A. Impact of 17beta-estradiol and progesterone on inflammatory and apoptotic microRNA expression after ischemia in a rat model. J. Steroid Biochem. Mol. Biol. 2017, 167, 126–134. [Google Scholar] [CrossRef]
- Slowik, A.; Lammerding, L.; Zendedel, A.; Habib, P.; Beyer, C. Impact of steroid hormones E2 and P on the NLRP3/ASC/Casp1 axis in primary mouse astroglia and BV-2 cells after in vitro hypoxia. J. Steroid Biochem. Mol. Biol. 2018, 183, 18–26. [Google Scholar] [CrossRef]
- Zhu, X.; Fréchou, M.; Schumacher, M.; Guennoun, R. Cerebroprotection by progesterone following ischemic stroke: Multiple effects and role of the neural progesterone receptors. J. Steroid Biochem. Mol. Biol. 2019, 185, 90–102. [Google Scholar] [CrossRef]
- Yoshii, S.R.; Mizushima, N. Monitoring and Measuring Autophagy. Int. J. Mol. Sci. 2017, 18, 1865. [Google Scholar] [CrossRef]
- Chae, U.; Kim, H.S.; Lee, H.S.; Lee, S.R.; Lee, D.S. Drp1-dependent mitochondrial fission regulates p62-mediated autophagy in LPS-induced activated microglial cells. Biosci. Biotechnol. Biochem. 2019, 83, 409–416. [Google Scholar] [CrossRef]
- Frank, M.G.; Baratta, M.V.; Sprunger, D.B.; Watkins, L.R.; Maier, S.F. Microglia serve as a neuroimmune substrate for stress-induced potentiation of CNS pro-inflammatory cytokine responses. Brain Behav. Immun. 2007, 21, 47–59. [Google Scholar] [CrossRef]
- Faraco, G.; Fossati, S.; Bianchi, M.E.; Patrone, M.; Pedrazzi, M.; Sparatore, B.; Moroni, F.; Chiarugi, A. High mobility group box 1 protein is released by neural cells upon different stresses and worsens ischemic neurodegeneration in vitro and in vivo. J. Neurochem. 2007, 103, 590–603. [Google Scholar] [CrossRef]
- Qiu, J.; Nishimura, M.; Wang, Y.; Sims, J.R.; Qiu, S.; Savitz, S.I.; Salomone, S.; Moskowitz, M.A. Early release of HMGB-1 from neurons after the onset of brain ischemia. J. Cereb. Blood Flow Metab. 2008, 28, 927–938. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Yoon, E.J.; Seo, J.; Kavoussi, A.; Chung, Y.E.; Chung, S.P.; Park, I.; Kim, C.H.; You, J.S. Hypothermia inhibits the propagation of acute ischemic injury by inhibiting HMGB1. Mol. Brain 2016, 9, 81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, X.X.; Gu, L.J.; Shen, J.; Kang, X.H.; Zheng, Y.Y.; Yue, S.B.; Zhu, S.M. Probenecid protects against transient focal cerebral ischemic injury by inhibiting HMGB1 release and attenuating AQP4 expression in mice. Neurochem. Res. 2014, 39, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Halder, S.K.; Ueda, H. Amlexanox Inhibits Cerebral Ischemia-Induced Delayed Astrocytic High-Mobility Group Box 1 Release and Subsequent Brain Damage. J. Pharm. Exp. Ther. 2018, 365, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Iwata, N.; Okazaki, M.; Kamiuchi, S.; Xuan, M.; Matsuzaki, H.; Sakamoto, T.; Hibino, Y. Early Release of HMGB1 may Aggravate Neuronal Damage after Transient Focal Ischemia in Diabetic Rat Brain. Int. J. Diabetes Clin. Res. 2015, 2, 21. [Google Scholar] [CrossRef]
- Hu, J.; Liu, B.; Zhao, Q.; Jin, P.; Hua, F.; Zhang, Z.; Liu, Y.; Zan, K.; Cui, G.; Ye, X. Bone marrow stromal cells inhibits HMGB1-mediated inflammation after stroke in type 2 diabetic rats. Neuroscience 2016, 324, 11–19. [Google Scholar] [CrossRef]
- Neumann, J.T.; Cohan, C.H.; Dave, K.R.; Wright, C.B.; Perez-Pinzon, M.A. Global cerebral ischemia: Synaptic and cognitive dysfunction. Curr. Drug Targets 2013, 14, 20–35. [Google Scholar] [CrossRef]
- Feng, X.; Zhao, Y.; Yang, T.; Song, M.; Wang, C.; Yao, Y.; Fan, H. Glucocorticoid-driven NLRP3 inflammasome activation in hippocampal microglia mediates chronic stress-induced depressive-like behaviors. Front. Mol. Neurosci. 2019, 12, 210. [Google Scholar] [CrossRef]
- Hong, P.; Li, F.X.; Gu, R.N.; Fang, Y.Y.; Lai, L.Y.; Wang, Y.W.; Tao, T.; Xu, S.Y.; You, Z.J.; Zhang, H.F. Inhibition of NLRP3 Inflammasome Ameliorates Cerebral Ischemia-Reperfusion Injury in Diabetic Mice. Neural Plast. 2018, 2018, 9163521. [Google Scholar] [CrossRef] [Green Version]
- Tian, F.; Deguchi, K.; Yamashita, T.; Ohta, Y.; Morimoto, N.; Shang, J.; Zhang, X.; Liu, N.; Ikeda, Y.; Matsuura, T.; et al. In vivo imaging of autophagy in a mouse stroke model. Autophagy 2010, 6, 1107–1114. [Google Scholar] [CrossRef] [Green Version]
- Hwang, J.Y.; Gertner, M.; Pontarelli, F.; Court-Vazquez, B.; Bennett, M.V.; Ofengeim, D.; Zukin, R.S. Global ischemia induces lysosomal-mediated degradation of mTOR and activation of autophagy in hippocampal neurons destined to die. Cell Death Differ. 2017, 24, 317–329. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Liu, Z.; Wang, L. Effects of Ischemic Post-Conditioning on the Expressions of LC3-II and Beclin-1 in the Hippocampus of Rats after Cerebral Ischemia and Reperfusion. Open Life Sci. 2019, 14, 1. [Google Scholar] [CrossRef]
- Wang, X.; Sun, D.; Hu, Y.; Xu, X.; Jiang, W.; Shang, H.; Cui, D. The roles of oxidative stress and Beclin-1 in the autophagosome clearance impairment triggered by cardiac arrest. Free Radic. Biol. Med. 2019, 136, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Zhang, H.; Kai, J.; Zhu, F.; Dong, J.; Xu, Z.; Wong, M.; Zeng, L.H. Rapamycin prevents cerebral stroke by modulating apoptosis and autophagy in penumbra in rats. Ann. Clin. Transl. Neurol. 2017, 5, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, D.; Bai, Y.; Xiao, J.; Jiao, H.; He, R. Ginaton improves neurological function in ischemic stroke rats via inducing autophagy and maintaining mitochondrial homeostasis. Neuropsychiatr. Dis. Treat. 2019, 15, 1813–1822. [Google Scholar] [CrossRef] [Green Version]
- Nabavi, S.F.; Sureda, A.; Sanches-Silva, A.; Pandima Devi, K.; Ahmed, T.; Shahid, M.; Sobarzo-Sánchez, E.; Dacrema, M.; Daglia, M.; Braidy, N.; et al. Novel therapeutic strategies for stroke: The role of autophagy. Crit. Rev. Clin. Lab. Sci. 2019, 56, 182–199. [Google Scholar] [CrossRef]
- Smith, C.M.; Chen, Y.; Sullivan, M.L.; Kochanek, P.M.; Clark, R.S. Autophagy in acute brain injury: Feast, famine, or folly? Neurobiol. Dis. 2011, 43, 52–59. [Google Scholar] [CrossRef] [Green Version]
- Xiao, X.; Shang, X.; Zhai, B.; Zhang, H.; Zhang, T. Nicotine alleviates chronic stress-induced anxiety and depressive-like behavior and hippocampal neuropathology via regulating autophagy signaling. Neurochem. Int. 2018, 114, 58–70. [Google Scholar] [CrossRef]
- Zhang, X.; Bu, H.; Jiang, Y.; Sun, G.; Jiang, R.; Huang, X.; Duan, H.; Huang, Z.; Wu, Q. The antidepressant effects of apigenin are associated with the promotion of autophagy via the mTOR/AMPK/ULK1 pathway. Mol. Med. Rep. 2019, 20, 2867–2874. [Google Scholar] [CrossRef]
- Wang, B.; Wu, Q.; Lei, L.; Sun, H.; Michael, N.; Zhang, X.; Wang, Y.; Zhang, Y.; Ge, B.; Wu, X.; et al. Long-term social isolation inhibits autophagy activation, induces postsynaptic dysfunctions and impairs spatial memory. Exp. Neurol. 2019, 311, 213–224. [Google Scholar] [CrossRef]
- Segerstrom, S.C.; Miller, G.E. Psychological stress and the human immune system: A meta-analytic study of 30 years of inquiry. Psychol. Bull. 2004, 130, 601–630. [Google Scholar] [CrossRef] [Green Version]
- Facci, L.; Barbierato, M.; Marinelli, C.; Argentini, C.; Skaper, S.D.; Giusti, P. Toll-like receptors 2, -3 and -4 prime microglia but not astrocytes across central nervous system regions for ATP-dependent interleukin-1β release. Sci. Rep. 2014, 4, 6824. [Google Scholar] [CrossRef] [Green Version]
- Sanz, J.M.; Di Virgilio, F. Kinetics and mechanism of ATP-dependent IL-1 beta release from microglial cells. J. Immunol. 2000, 164, 4893–4898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gustin, A.; Kirchmeyer, M.; Koncina, E.; Felten, P.; Losciuto, S.; Heurtaux, T.; Tardivel, A.; Heuschling, P.; Dostert, C. NLRP3 Inflammasome Is Expressed and Functional in Mouse Brain Microglia but Not in Astrocytes. PLoS ONE 2015, 10, e0130624. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Zhang, X.; Chen, M.; Chen, J.; Gao, T.; Yao, S. Dexmedetomidine inhibits inflammation in microglia cells under stimulation of LPS and ATP by c-Fos/NLRP3/caspase-1 cascades. EXCLI J. 2018, 17, 302–311. [Google Scholar] [CrossRef]
- Frank, M.G.; Weber, M.D.; Fonken, L.K.; Hershman, S.A.; Watkins, L.R.; Maier, S.F. The redox state of the alarmin HMGB1 is a pivotal factor in neuroinflammatory and microglial priming: A role for the NLRP3 inflammasome. Brain Behav. Immun. 2016, 55, 215–224. [Google Scholar] [CrossRef] [Green Version]
- Fonken, L.K.; Frank, M.G.; Gaudet, A.D.; D’Angelo, H.M.; Daut, R.A.; Hampson, E.C.; Ayala, M.T.; Watkins, L.R.; Maier, S.F. Neuroinflammatory priming to stress is differentially regulated in male and female rats. Brain Behav. Immun. 2018, 70, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Gibson, C.L.; Bath, P.M. Feasibility of progesterone treatment for ischaemic stroke. J. Cereb. Blood Flow Metab. 2016, 36, 487–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stein, D.G. Effects of sex steroids on damaged neural systems. In Hormones, Brain and Behavior, 3rd ed.; Pfaff, D.W., Arnold, A.P., Etgen, A.M., Fahrbach, S.E., Rubin, R.T., Eds.; Elsevier: Oxford, UK, 2017; pp. 412–441. [Google Scholar]
- Guennoun, R.; Zhu, X.; Fréchou, M.; Gaignard, P.; Slama, A.; Liere, P.; Schumacher, M. Steroids in Stroke with Special Reference to Progesterone. Cell Mol. Neurobiol. 2019, 39, 551–568. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhao, Y.; Liu, C.; Jiang, C.; Zhao, C.; Zhu, Z. Progesterone inhibits inflammatory response pathways after permanent middle cerebral artery occlusion in rats. Mol. Med. Rep. 2011, 4, 319–324. [Google Scholar] [CrossRef] [Green Version]
- Zicari, A.; Centonze, C.; Realacci, M.; Buchetti, B.; Pietropolli, A.; Ticconi, C. Estradiol 17-beta and progesterone modulate inducible nitric oxide synthase and high mobility group box 1 expression in human endometrium. Reprod. Sci. 2008, 15, 559–566. [Google Scholar] [CrossRef] [PubMed]
- Lei, B.; Mace, B.; Dawson, H.N.; Warner, D.S.; Laskowitz, D.T.; James, M.L. Anti-inflammatory effects of progesterone in lipopolysaccharide-stimulated BV-2 microglia. PLoS ONE 2014, 9, e103969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aryanpour, R.; Pasbakhsh, P.; Zibara, K.; Namjoo, Z.; Beigi Boroujeni, F.; Shahbeigi, S.; Kashani, I.R.; Beyer, C.; Zendehdel, A. Progesterone therapy induces an M1 to M2 switch in microglia phenotype and suppresses NLRP3 inflammasome in a cuprizone-induced demyelination mouse model. Int. Immunopharmacol. 2017, 51, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.N.; Lee, S.J.; Koh, J.Y. The neurosteroids, allopregnanolone and progesterone, induce autophagy in cultured astrocytes. Neurochem. Int. 2012, 60, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kim, T.Y.; Cho, K.S.; Kim, H.N.; Koh, J.Y. Autophagy activation and neuroprotection by progesterone in the G93A-SOD1 transgenic mouse model of amyotrophic lateral sclerosis. Neurobiol. Dis. 2013, 59, 80–85. [Google Scholar] [CrossRef]
- Hong, Y.; Liu, Y.; Yu, D.; Wang, M.; Hou, Y. The neuroprotection of progesterone against Aβ-induced NLRP3-Caspase-1 inflammasome activation via enhancing autophagy in astrocytes. Int. Immunopharmacol. 2019, 74, 105669. [Google Scholar] [CrossRef]
- Mir, S.U.; Schwarze, S.R.; Jin, L.; Zhang, J.; Friend, W.; Miriyala, S.; St Clair, D.; Craven, R.J. Progesterone receptor membrane component 1/Sigma-2 receptor associates with MAP1LC3B and promotes autophagy. Autophagy 2013, 9, 1566–1578. [Google Scholar] [CrossRef] [Green Version]
- Garling, R.J.; Watts, L.T.; Sprague, S.; Digicaylioglu, M. Progesterone modulates mTOR in the hippocampus of mice after traumatic brain injury. Neural Regen. Res. 2018, 13, 434–439. [Google Scholar] [CrossRef]
- Atif, F.; Yousuf, S.; Stein, D.G. Anti-tumor effects of progesterone in human glioblastoma multiforme: Role of PI3K/Akt/mTOR signaling. J. Steroid Biochem. Mol. Biol. 2015, 146, 62–73. [Google Scholar] [CrossRef]
- Atif, F.; Yousuf, S.; Espinosa-Garcia, C.; Sergeeva, E.; Stein, D.G. Progesterone Treatment Attenuates Glycolytic Metabolism and Induces Senescence in Glioblastoma. Sci. Rep. 2019, 9, 988. [Google Scholar] [CrossRef]
- Yousuf, S.; Atif, F.; Sayeed, I.; Tang, H.; Stein, D.G. Progesterone in transient ischemic stroke: A dose-response study. Psychopharmacology 2014, 231, 3313–3323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wali, B.; Ishrat, T.; Won, S.; Stein, D.G.; Sayeed, I. Progesterone in experimental permanent stroke: A dose-response and therapeutic time-window study. Brain 2014, 137, 486–502. [Google Scholar] [CrossRef]
- Koolhaas, J.M.; de Boer, S.F.; Buwalda, B.; Meerlo, P. Social stress models in rodents: Towards enhanced validity. Neurobiol. Stress 2017, 6, 104–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stein, D.J.; Vasconcelos, M.F.; Albrechet-Souza, L.; Ceresér, K.M.M.; de Almeida, R.M.M. Microglial Over-Activation by Social Defeat Stress Contributes to Anxiety- and Depressive-Like Behaviors. Front. Behav. Neurosci. 2017, 11, 207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Björkqvist, K. Social defeat as a stressor in humans. Physiol. Behav. 2001, 73, 435–442. [Google Scholar] [CrossRef]
- Hollis, F.; Kabbaj, M. Social defeat as an animal model for depression. ILAR J. 2014, 55, 221–232. [Google Scholar] [CrossRef] [Green Version]
- Pulsinelli, W.A.; Brierley, J.B. A new model of bilateral hemispheric ischemia in the unanesthetized rat. Stroke 1979, 10, 267–272. [Google Scholar] [CrossRef] [Green Version]
- Pulsinelli, W.A.; Buchan, A.M. The four-vessel occlusion rat model: Method for complete occlusion of vertebral arteries and control of collateral circulation. Stroke 1988, 19, 913–914. [Google Scholar] [CrossRef] [Green Version]
- Baron, J.C.; Yamauchi, H.; Fujioka, M.; Endres, M. Selective neuronal loss in ischemic stroke and cerebrovascular disease. J. Cereb. Blood Flow Metab. 2014, 34, 2–18. [Google Scholar] [CrossRef] [Green Version]
- Bartsch, T.; Döhring, J.; Reuter, S.; Finke, C.; Rohr, A.; Brauer, H.; Deuschl, G.; Jansen, O. Selective neuronal vulnerability of human hippocampal CA1 neurons: Lesion evolution, temporal course, and pattern of hippocampal damage in diffusion-weighted MR imaging. J. Cereb. Blood Flow Metab. 2015, 35, 1836–1845. [Google Scholar] [CrossRef]
- Bronstein, R.; Torres, L.; Nissen, J.C.; Tsirka, S.E. Culturing microglia from the neonatal and adult central nervous system. J. Vis. Exp. 2013, 50647. [Google Scholar] [CrossRef] [PubMed]
- Saura, J.; Tusell, J.M.; Serratosa, J. High-yield isolation of murine microglia by mild trypsinization. Glia 2003, 44, 183–189. [Google Scholar] [CrossRef]
- Frank, M.G.; Wieseler-Frank, J.L.; Watkins, L.R.; Maier, S.F. Rapid isolation of highly enriched and quiescent microglia from adult rat hippocampus: Immunophenotypic and functional characteristics. J. Neurosci. Methods 2006, 151, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Wasserstein, R.L.; Lazar, N.A. The ASA Statement on p-Values: Context, Process, and Purpose. Am. Stat. 2016, 70, 129–133. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | Host | Dilution | Catalog Number | Vendor |
|---|---|---|---|---|
| (A) Primary antibodies | ||||
| HMGB1 | rabbit | 1:100/1:1000 | ab18256 | Abcam |
| NeuN | mouse | 1:1000 | MAB377 | Chemicon |
| GFAP | rat | 1:1000 | 13-0300 | Invitrogen |
| CD11b | mouse | 1:100 | MCA275R | AbD serotec |
| TLR4 | mouse | 1:500 | 76B357.1 | ThermoFisher |
| P2X7 | mouse | 1:100 | sc-514962 | Santa Cruz Biotechnology |
| NLRP3 | rabbit | 1:1000 | ab214185 | Abcam |
| IκBα | rabbit | 1:1000 | 9242 | Cell Signaling |
| Phospho-IκBα | mouse | 1:1000 | 9246 | Cell Signaling |
| ASC | mouse | 1:100 | sc-514414 | Santa Cruz Biotechnology |
| Caspase-1 | mouse | 1:100 | sc-398715 | Santa Cruz Biotechnology |
| IL-1β | rabbit | 1:500 | ab9787 | Abcam |
| LC3 | rabbit | 1:100/1:1000 | 12741 | Cell Signaling |
| β-actin | mouse | 1:10,000 | A5316 | Sigma |
| (B) Secondary antibodies | ||||
| Alexa Fluor 488 anti-rabbit | donkey | 1:200 | A21206 | Invitrogen |
| Alexa Fluor 568 anti-mouse | goat | 1:200 | A11004 | Invitrogen |
| Alexa Fluor 594 anti-rat | goat | 1:200 | A11007 | Invitrogen |
| Anti-mouse | goat | 1:2000 | 074-1806 | KPL |
| Anti-rabbit | goat | 1:2000 | 074-1506 | KPL |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Espinosa-Garcia, C.; Atif, F.; Yousuf, S.; Sayeed, I.; Neigh, G.N.; Stein, D.G. Progesterone Attenuates Stress-Induced NLRP3 Inflammasome Activation and Enhances Autophagy Following Ischemic Brain Injury. Int. J. Mol. Sci. 2020, 21, 3740. https://doi.org/10.3390/ijms21113740
Espinosa-Garcia C, Atif F, Yousuf S, Sayeed I, Neigh GN, Stein DG. Progesterone Attenuates Stress-Induced NLRP3 Inflammasome Activation and Enhances Autophagy Following Ischemic Brain Injury. International Journal of Molecular Sciences. 2020; 21(11):3740. https://doi.org/10.3390/ijms21113740
Chicago/Turabian StyleEspinosa-Garcia, Claudia, Fahim Atif, Seema Yousuf, Iqbal Sayeed, Gretchen N. Neigh, and Donald G. Stein. 2020. "Progesterone Attenuates Stress-Induced NLRP3 Inflammasome Activation and Enhances Autophagy Following Ischemic Brain Injury" International Journal of Molecular Sciences 21, no. 11: 3740. https://doi.org/10.3390/ijms21113740