Dispersive Solid-Phase Extraction using Magnetic Carbon Nanotube Composite for the Determination of Emergent Mycotoxins in Urine Samples


Abstract

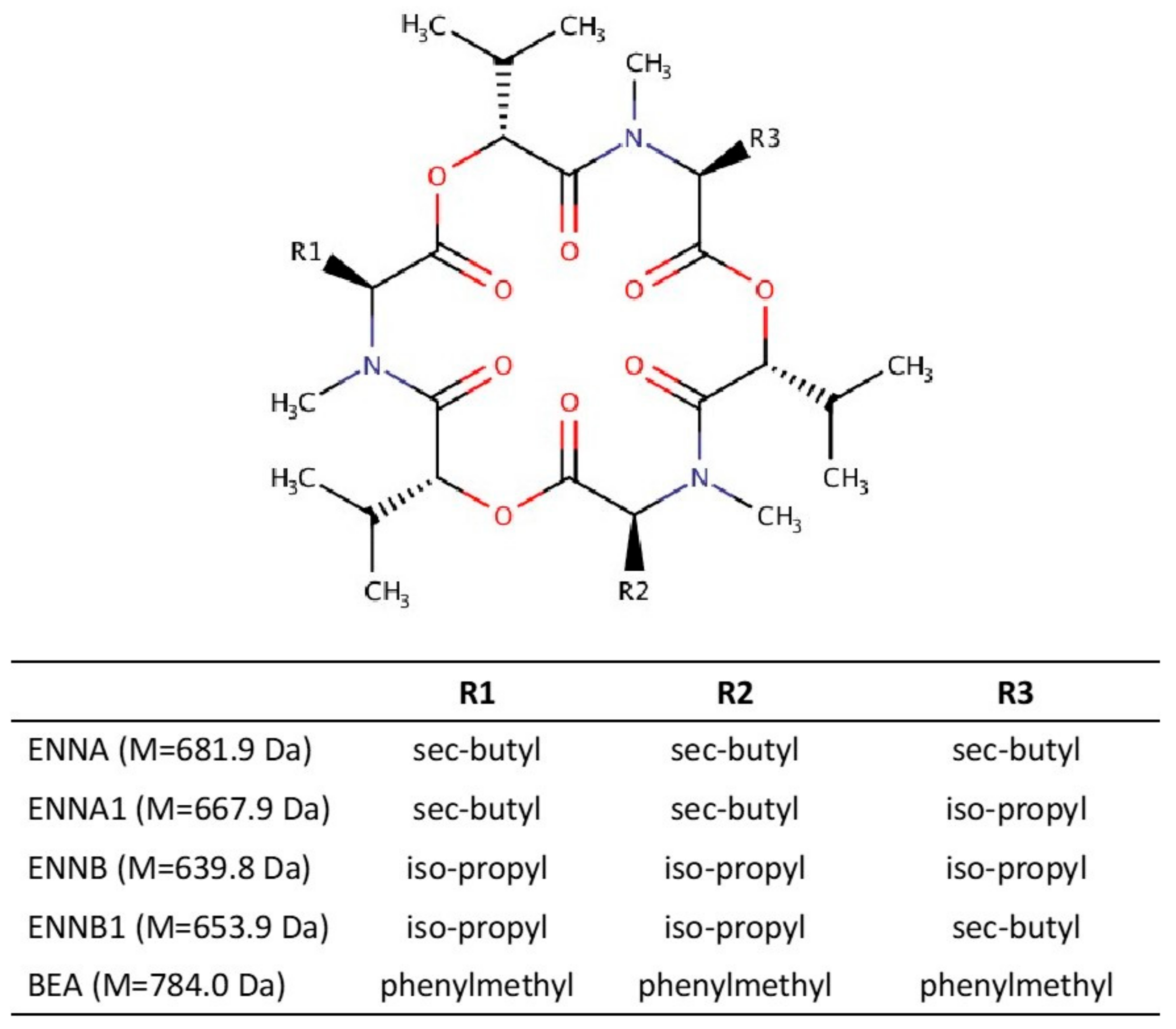
1. Introduction
2. Results and Discussion
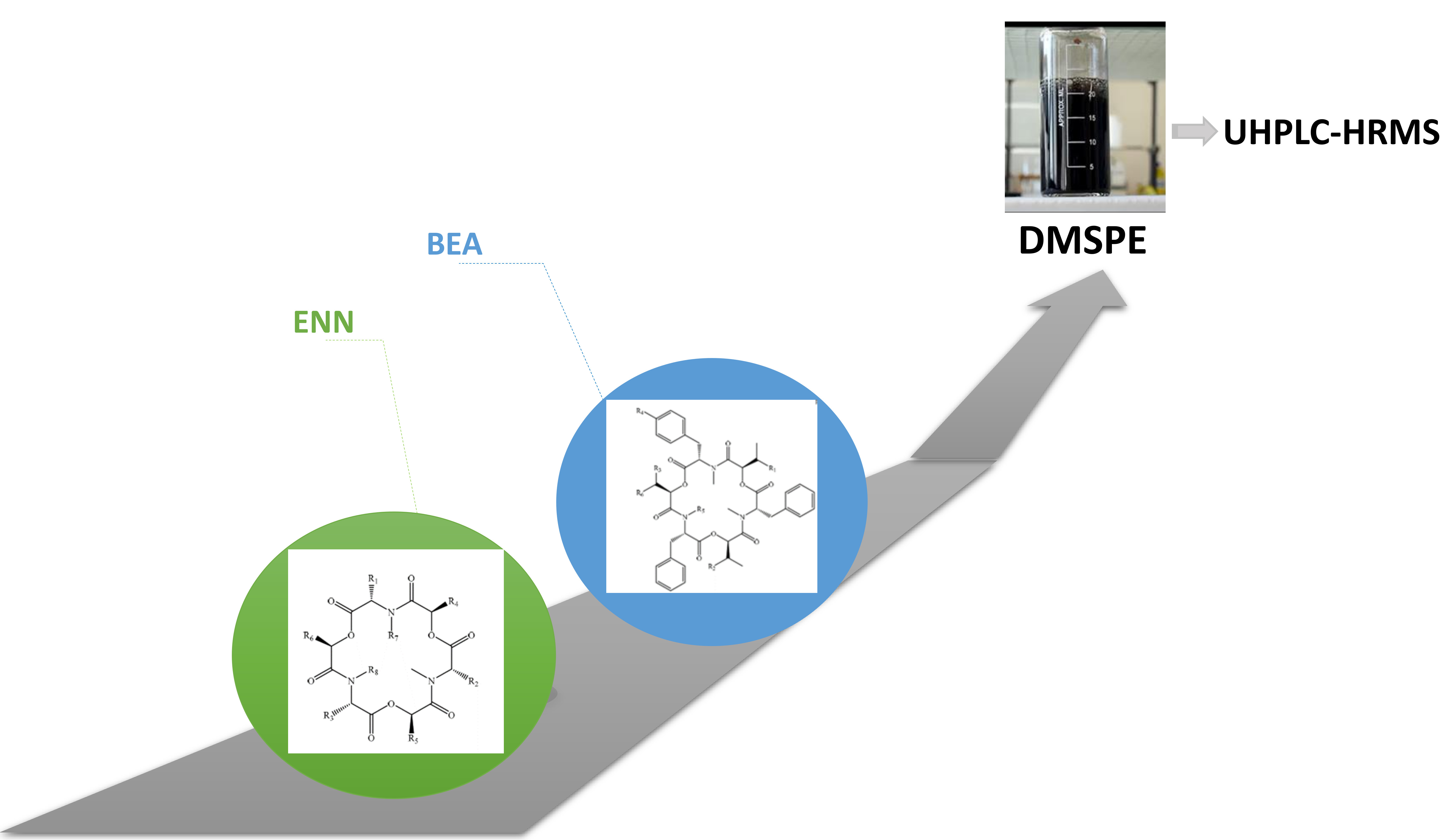
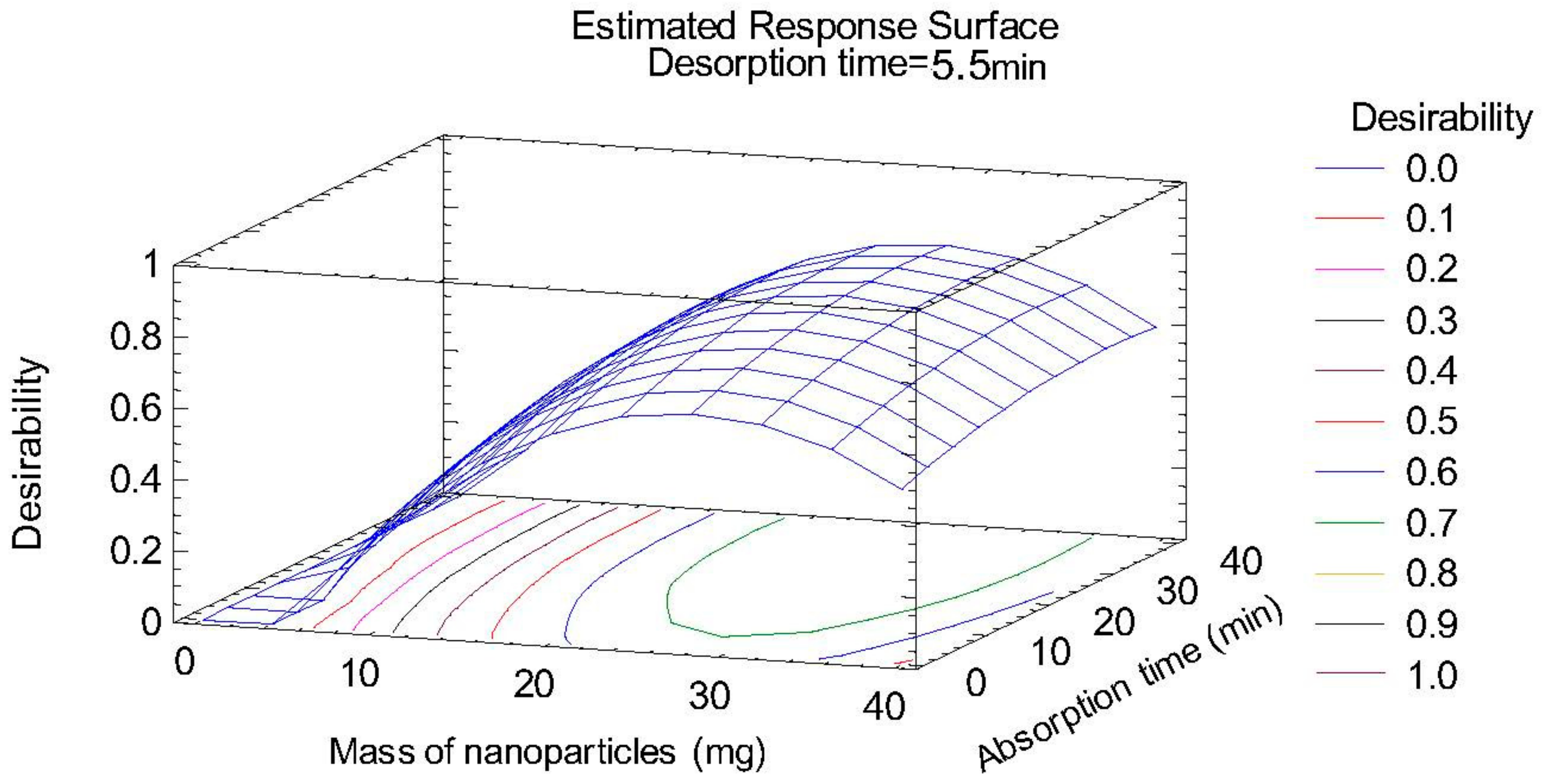
2.1. Optimization of Sample Treatment
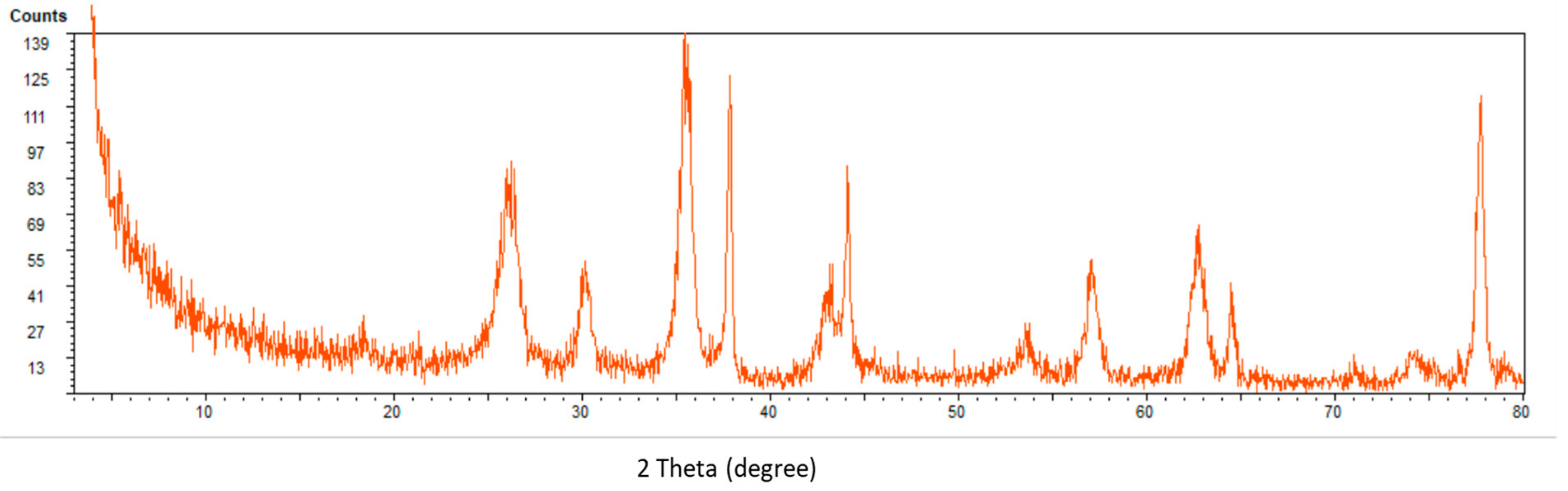
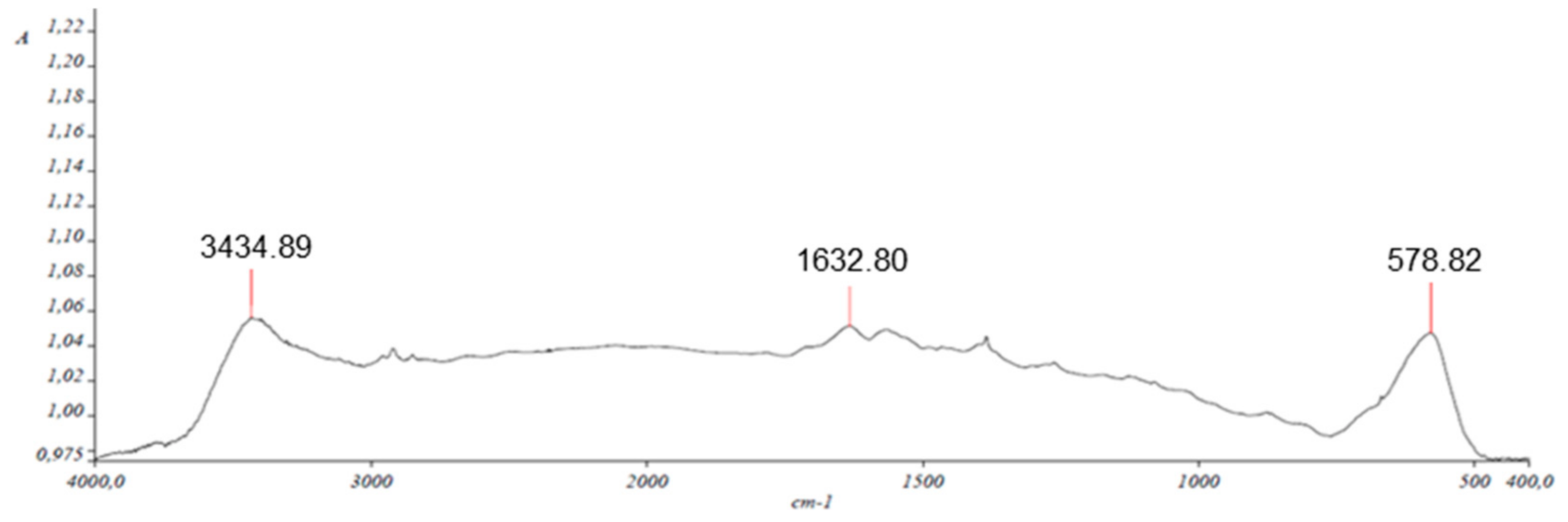
2.2. Characterization of Nanocomposite
2.3. Method Validation
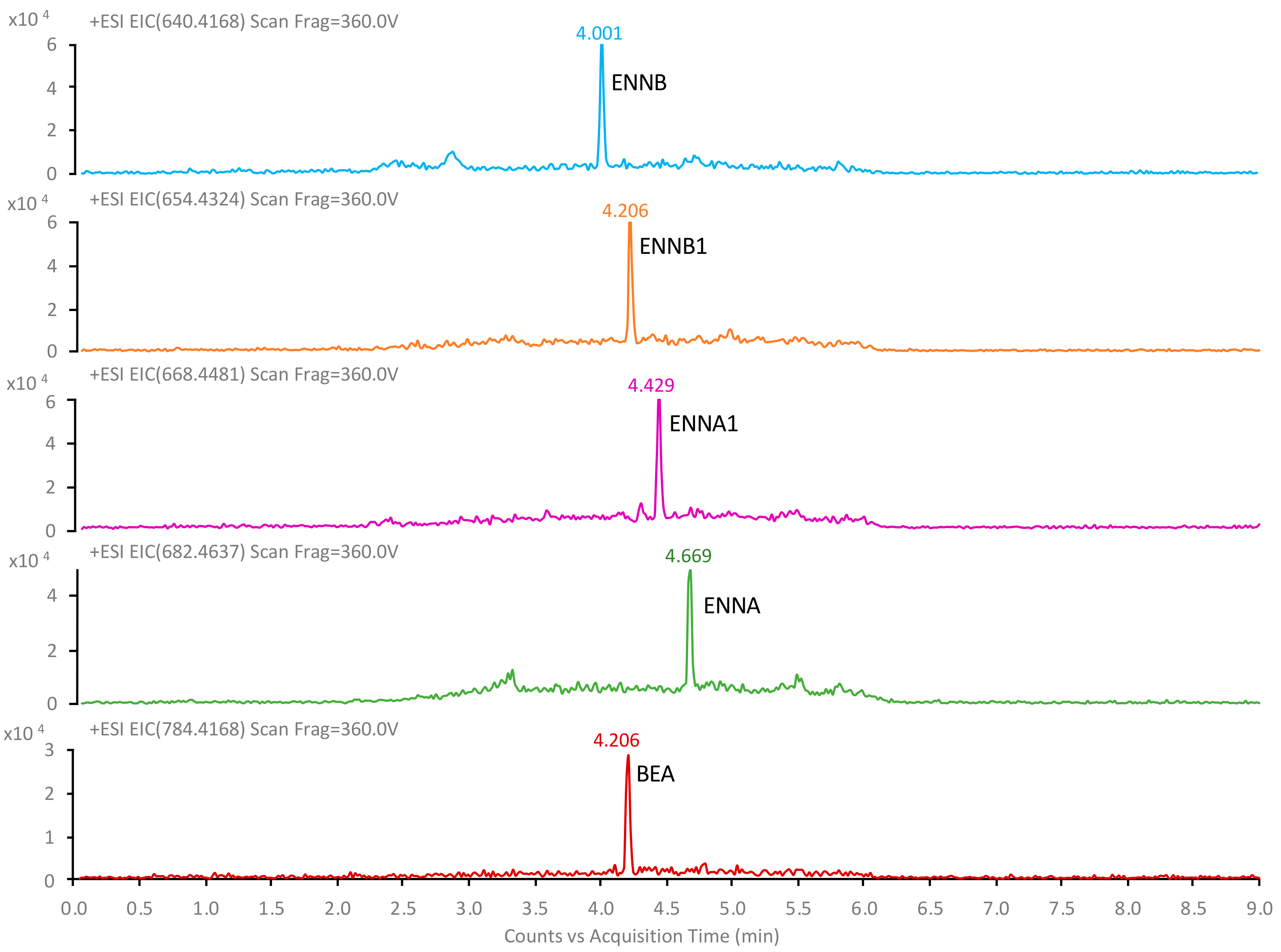
2.4. Application to Urine Samples
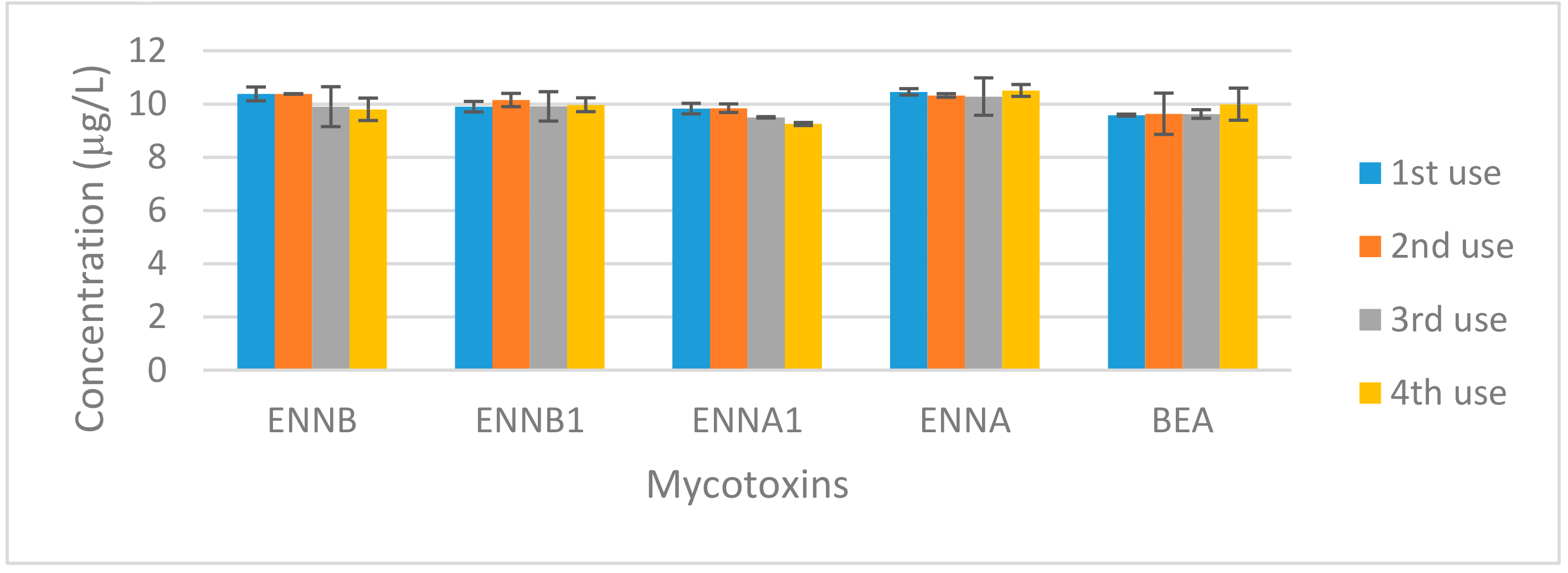
2.5. Nanomaterial Reuse
3. Conclusions
4. Materials and Methods
4.1. Chemicals, Reagents, and Standards
4.2. Instrumentation and Software
4.3. Synthesis of Fe3O4@MWCNTs Composite
4.4. Sample Preparation
4.5. UHPLC-HRMS Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Commission of the European Communities. Regulation (EC) No. 1881/2006 setting maximum levels for certain contaminants in foodstuffs. Off. J. Eur. Commun. 2006, L364, 5–24. [Google Scholar]
- European Commission. Commission recommendation No. 2012/154/UE on the monitoring of the presence of ergot alkaloids in feed and food. Off. J. Eur. Commun. 2012, L77, 20–21. [Google Scholar]
- Commission of the European Communities. Commission recommendation No. 2013/165/EU on the presence of T-2 and HT-2 toxin in cereals and cereal products. Off. J. Eur. Commun. 2013, L91, 12–15. [Google Scholar]
- Mamur, S.; Yuzbasioglu, D.; Yılmaz, S.; Erikel, E.; Unal, F. Assessment of cytotoxic and genotoxic effects of enniatin-A in vitro. Food Addit. Contam. Part A 2018, 35, 1633–1644. [Google Scholar] [CrossRef]
- EFSA Panel on Contaminants in the Food Chain (CONTAM). Scientific opinion on the risks to human and animal health related to the presence of beauvericin and enniatins in food and feed. EFSA J. 2014, 12, 3802. [Google Scholar] [CrossRef]
- Wang, X.; Gong, X.; Li, P.; Lai, D.; Zhou, L. Structural diversity and biological activities of cyclic depsipeptides from fungi. Molecules 2018, 23, 169. [Google Scholar] [CrossRef]
- Juan, C.; Mañes, J.; Raiola, A.; Ritieni, A. Evaluation of beauvericin and enniatins in Italian cereal products and multicereal food by liquid chromatography coupled to triple quadrupole mass spectrometry. Food Chem. 2013, 140, 755–762. [Google Scholar] [CrossRef] [PubMed]
- Juan, C.; Ritieni, A.; Mañes, J. Occurrence of Fusarium mycotoxins in Italian cereal and cereal products from organic farming. Food Chem 2013, 141, 1747–1755. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.-B.; Song, N.-E.; Nam, T.G.; Lee, S.; Seo, D.; Yoo, M. Occurrence of emerging mycotoxins in cereals and cereal-based products from the Korean market using LC-MS/MS. Food Addit. Contam. Part A 2019, 36, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Xu, W.; Zhang, J.; Xu, J.; Li, F. Natural occurrence of beauvericin and enniatins in corn-and wheat-based samples harvested in 2017 collected from Shandong province, China. Toxins 2019, 11, 9. [Google Scholar] [CrossRef] [PubMed]
- Arroyo-Manzanares, N.; Rodríguez-Estévez, V.; Arenas-Fernández, P.; García-Campaña, A.M.; Gámiz-Gracia, L. Occurrence of mycotoxins in swine feeding from Spain. Toxins 2019, 11, 342. [Google Scholar] [CrossRef] [PubMed]
- Uhlig, S.; Torp, M.; Heier, B.T. Beauvericin and enniatins A, A1, B and B1 in Norwegian grain: A survey. Food Chem. 2006, 94, 193–201. [Google Scholar] [CrossRef]
- Huybrechts, B.; Martins, J.C.; Debongnie, P.; Uhlig, S.; Callebau, A. Fast and sensitive LC–MS/MS method measuring human mycotoxin exposure using biomarkers in urine. Arch. Toxicol. 2015, 89, 1993–2005. [Google Scholar] [CrossRef] [PubMed]
- Serrano, A.B.; Capriotti, A.L.; Cavaliere, C.; Piovesana, S.; Samperi, R.; Ventura, S.; Laganà, A. Development of a rapid LC-MS/MS method for the determination of emerging fusarium mycotoxins enniatins and beauvericin in human biological fluids. Toxins 2015, 7, 3554–3571. [Google Scholar] [CrossRef]
- Juan, C.; Manyes, L.; Font, G.; Juan-Garcia, A. Evaluation of immunologic effect of Enniatin A and quantitative determination in feces, urine and serum on treated Wistar rats. Toxicon 2014, 87, 45–53. [Google Scholar] [CrossRef]
- Escrivá, L.; Font, G.; Manyes, L. Quantitation of enniatins in biological samples of Wistar rats after oral administration by LC-MS/MS. Toxicol Mech. Methods 2015, 25, 552–558. [Google Scholar] [CrossRef]
- Escrivá, L.; Manyes, L.; Font, G.; Berrada, H. Mycotoxin analysis of human urine by LC-MS/MS: A comparative extraction study. Toxins 2017, 9, 330. [Google Scholar] [CrossRef]
- Lauwers, M.; De Baere, S.; Letor, B.; Rychlik, M.; Croubels, S.; Devreese, M. Multi LC-MS/MS and LC-HRMS methods for determination of 24 mycotoxins including major phase I and II biomarker metabolites in biological matrices from pigs and broiler chickens. Toxins 2019, 11, 171. [Google Scholar] [CrossRef]
- Rodríguez-Carrasco, Y.; Izzo, L.; Gaspari, A.; Graziani, G.; Mañesa, J.; Ritieni, A. Urinary levels of enniatin B and its phase I metabolites: First human pilot T biomonitoring study. Food Chem. Toxicol. 2018, 118, 454–459. [Google Scholar] [CrossRef]
- Taevernier, L.; Bracke, N.; Veryser, L.; Wynendaele, E.; Gevaert, B.; Peremans, K.; De Spiegeleer, B. Blood-brain barrier transport kinetics of the cyclic depsipeptide mycotoxins beauvericin and enniatins. Toxicol. Lett. 2016, 258, 175–184. [Google Scholar] [CrossRef]
- Fraeyman, S.; Devreese, M.; Antonissen, G.; De Baere, S.; Rychlik, M.; Croubels, S. Comparative Oral Bioavailability, Toxicokinetics, and Biotransformation of Enniatin B1 and Enniatin B in Broiler Chickens. J. Agric. Food Chem. 2016, 64, 7259–7264. [Google Scholar] [CrossRef] [PubMed]
- Devreese, M.; De Baere, S.; De Backer, P.; Croubels, S. Quantitative determination of the Fusarium mycotoxins beauvericin, enniatin A, A1, B and B1 in pig plasma using high performance liquid chromatography–tandem mass spectrometry. Talanta 2013, 106, 212–219. [Google Scholar] [CrossRef] [PubMed]
- Devreese, M.; Broekaert, N.; De Mil, T.; Fraeyman, S.; De Backer, P.; Croubels, S. Pilot toxicokinetic study and absolute oral bioavailability of the Fusarium mycotoxin enniatin B1 in pigs. Food Chem. Toxicol. 2014, 63, 161–165. [Google Scholar] [CrossRef] [PubMed]
- Tolosa, J.; Font, G.; Mañes, J.; Ferrer, E. Multimycotoxin analysis in water and fish plasma by liquid chromatography-tandem mass spectrometry. Chemosphere 2016, 145, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Manyes, L.; Escriva, L.; BelenSerrano, A.; Rodriguez-Carrasco, Y.; Tolosa, J.; Meca, G.; Font, G. A preliminary study in Wistar rats with enniatin A contaminated feed. Toxicol. Mech. Methods 2014, 24, 179–190. [Google Scholar] [CrossRef]
- Kongkapan, J.; Giorgi, M.; Poapolathep, S.; Isariyodom, S.; Poapolathep, A. Toxic kinetics and tissue distribution of nivalenol in broiler chickens. Toxicon 2016, 111, 31–36. [Google Scholar] [CrossRef]
- Tolosa, J.; Font, G.; Mañes, J.; Ferrer, E. Natural occurrence of emerging Fusarium mycotoxins in feed and fish from aquaculture. J. Agric. Food Chem. 2014, 62, 12462–12470. [Google Scholar] [CrossRef]
- Rodríguez-Carrasco, Y.; Heilos, D.; Richter, L.; Süssmuth, R.D.; Heffeter, P.; Sulyok, M.; Kenner, L.; Berger, W.; Dornetshuber-Fleiss, R. Mouse tissue distribution and persistence of the food-born fusariotoxins Enniatin B and Beauvericin. Toxicol. Lett. 2016, 247, 35–44. [Google Scholar] [CrossRef]
- Herrero-Latorre, C.; Barciela-García, J.; García-Martín, S.; Peña-Crecente, R.M.; Otárola-Jiménez, J. Magnetic solid-phase extraction using carbon nanotubes as sorbents: A review. Anal. Chim. Acta 2015, 892, 10–26. [Google Scholar] [CrossRef]
- Jing, W.; Zhou, Y.; Wang, J.; Ni, M.; Bi, W.; Chen, D.D.Y. Dispersive Magnetic Solid-Phase Extraction Coupled to Direct Analysis in Real Time Mass Spectrometry for High-Throughput analysis of trace environmental contaminants. Anal. Chem. 2019, 9, 11240–11246. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, Y.; Yang, S. Simultaneous removal of Co (II) and 1-naphthol by core-shell structured Fe3O4@cyclodextrin magnetic nanoparticles. Carbohydr. Polym. 2014, 114, 521–529. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Lee, H.K. Study and comparison of polydopamine and its derived carbon decorated nanoparticles in the magnetic solid-phase extraction of estrogens. J. Chromatogr. A 2015, 1414, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Gopal, J.; Abdelhamid, H.N.; Hua, P.Y.; Wu, H.F. Chitosan nanomagnets for effective extraction and sensitive mass spectrometric detection of pathogenic bacterial endotoxin from human urine. J. Mater. Chem. 2013, 1, 2463–2475. [Google Scholar] [CrossRef]
- Benedé, J.L.; Chisvert, A.; Giokas, D.L.; Salvador, A. Development of stir bar sorptive-dispersive microextraction mediated by magnetic nanoparticles and its analytical application to the determination of hydrophobic organic compounds in aqueous media. J. Chromatogr. A 2014, 1362, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Sun, Y.; Jiang, C.; Sun, X.; Gao, Y.; Wang, Y.; Zhang, H.; Son, D. Magnetic solid-phase extraction of five pyrethroids from environmental water samples followed by ultrafast liquid chromatography analysis. Talanta 2012, 98, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Asgharinezhad, A.A.; Ebrahimzadeh, H. Coextraction of acidic, basic and amphiprotic pollutants using multiwalled carbon nanotubes/magnetite nanoparticles@polypyrrole composite. J. Chromatogr. A 2015, 1412, 1–11. [Google Scholar] [CrossRef]
- Upadhyay, J.; Kumar, A.; Gogoi, B.; Buragohain, A.K. Antibacterial and hemolysis activity of polypyrrole nanotubes decorated with silver nanoparticles by an in-situ reduction process. Mater. Sci. Eng. C 2015, 54, 8–13. [Google Scholar] [CrossRef]
- Periyasamy, S.; Gopalakannan, V.; Viswanathan, N. Fabrication of magnetic particles imprinted cellulose based biocomposites for chromium (VI) removal. Carbohyd. Polym. 2017, 174, 352–359. [Google Scholar] [CrossRef]
- Martin, J.D. XPowder. 2006. Available online: www.xpowder.com (accessed on 6 December 2019).
- Commission of the European Communities. Regulation (EC) No. 401/2006 of laying down the methods of sampling and analysis for the official control of the levels of mycotoxins in foodstuffs. Off. J. Eur. Commun. 2006, L70, 12–34. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mycotoxin | Equation | Linear Range (μg/L) | Linearity R2 | LOD (μg/L) | LOQ (μg/L) | |
|---|---|---|---|---|---|---|
| ENNA | y = 629312 x + 871432 | 0.04–50 | 0.996 | 0.01 | 0.04 | |
| ENNA1 | y = 1755799 x + 1381024 | 0.10–50 | 0.995 | 0.03 | 0.10 | |
| ENNB | y = 833213 x + 669371 | 0.04–50 | 0.995 | 0.01 | 0.04 | |
| ENNB1 | y = 1883668 x + 3420346 | 0.04–50 | 0.992 | 0.01 | 0.04 | |
| BEA | y = 1468318 x – 1256332 | 0.04–50 | 0.993 | 0.01 | 0.04 | |
| Matrix Effect (%) | Trueness (%) | |||||
| 0.1 μg/L | 5 μg/L | 25 μg/L | 0.1 μg/L | 5 μg/L | 25 μg/L | |
| ENNA | –12.5 | –18.6 | –17.0 | 97.0 | 92.2 | 89.3 |
| ENNA1 | –33.6 | –34.3 | –22.3 | 96.9 | 92.4 | 93.9 |
| ENNB | –33.7 | –37.8 | –37.2 | 97.6 | 95.7 | 98.9 |
| ENNB1 | –5.1 | –8.7 | –12.1 | 98.6 | 98.3 | 98.0 |
| BEA | –35.5 | –20.3 | –35.1 | 96.7 | 90.5 | 94.4 |
| Repeatability, %RSD (n = 9) | Intermediate Precision, %RSD (n = 12) | |||||
| 0.1 μg/L | 5 μg/L | 25 μg/L | 0.1 μg/L | 5 μg/L | 25 μg/L | |
| ENNA | 6.0 | 6.7 | 6.9 | 10.1 | 8.2 | 7.4 |
| ENNA1 | 6.8 | 8.0 | 8.6 | 7.5 | 8.5 | 10.2 |
| ENNB | 8.9 | 9.4 | 8.7 | 10.2 | 9.3 | 9.9 |
| ENNB1 | 8.5 | 6.9 | 5.9 | 8.6 | 9.1 | 11.7 |
| BEA | 8.7 | 8.8 | 9.0 | 8.9 | 9.0 | 9.1 |
| Compound | tR (min) | Formula | m/z Theoretical | m/z Experimental | Error (ppm) | Q1, m/z | Q2, m/z |
|---|---|---|---|---|---|---|---|
| ENNB | 4.00 | C33H58N3O9+ | 640.4168 | 640.4173 | 0.8 | 196.1341 | 214.1441 |
| ENNB1 | 4.21 | C34H60N3O9+ | 654.4324 | 654.4326 | 0.3 | 196.1333 | 210.1489 |
| BEA | 4.21 | C45H58N3O9+ | 784.4168 | 784.4163 | −0.6 | 244.1334 | 262.1438 |
| ENNA1 | 4.43 | C35H62N3O9+ | 668.4481 | 668.4485 | 0.6 | 210.1491 | 228.1592 |
| ENNA | 4.67 | C36H64N3O9+ | 682.4637 | 682.4636 | −0.1 | 210.1491 | 228.1593 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arroyo-Manzanares, N.; Peñalver-Soler, R.; Campillo, N.; Viñas, P. Dispersive Solid-Phase Extraction using Magnetic Carbon Nanotube Composite for the Determination of Emergent Mycotoxins in Urine Samples. Toxins 2020, 12, 51. https://doi.org/10.3390/toxins12010051
Arroyo-Manzanares N, Peñalver-Soler R, Campillo N, Viñas P. Dispersive Solid-Phase Extraction using Magnetic Carbon Nanotube Composite for the Determination of Emergent Mycotoxins in Urine Samples. Toxins. 2020; 12(1):51. https://doi.org/10.3390/toxins12010051
Chicago/Turabian StyleArroyo-Manzanares, Natalia, Rosa Peñalver-Soler, Natalia Campillo, and Pilar Viñas. 2020. "Dispersive Solid-Phase Extraction using Magnetic Carbon Nanotube Composite for the Determination of Emergent Mycotoxins in Urine Samples" Toxins 12, no. 1: 51. https://doi.org/10.3390/toxins12010051
APA StyleArroyo-Manzanares, N., Peñalver-Soler, R., Campillo, N., & Viñas, P. (2020). Dispersive Solid-Phase Extraction using Magnetic Carbon Nanotube Composite for the Determination of Emergent Mycotoxins in Urine Samples. Toxins, 12(1), 51. https://doi.org/10.3390/toxins12010051