Characterization of the Gut Microbiota of Individuals at Different T2D Stages Reveals a Complex Relationship with the Host

 , ,
, , 
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Subjects
2.2. Anthropometrical Evaluation
2.3. Dietary Assessment
2.4. Types of Physical Activity
2.5. Biochemical Profile
2.6. DNA Extraction from Feces
2.7. Construction of the V3-16S rRNA Gene Library and High Throughput DNA Sequencing
2.8. Microbial Diversity Analysis
2.9. Analysis of Bacterial Enrichment among Study Groups
2.10. Predictive Functional Metagenome of the Gut Microbiota using PICRUSt
2.11. Multivariate Association of the Gut Microbiota with All Measured Variables
2.12. Statistical Analyses
3. Results
3.1. Analysis of Physical Activity, Intake of Nutrients, and Anthropometrical Profile Data Contrast the T2D Groups with the Control and Prediabetes Groups
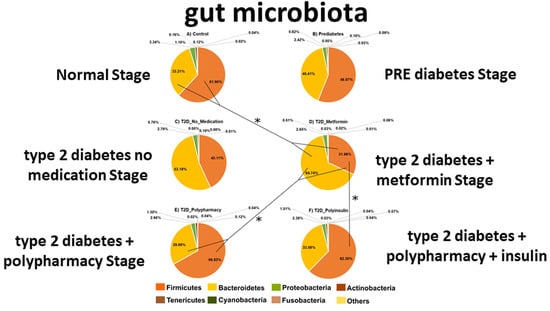
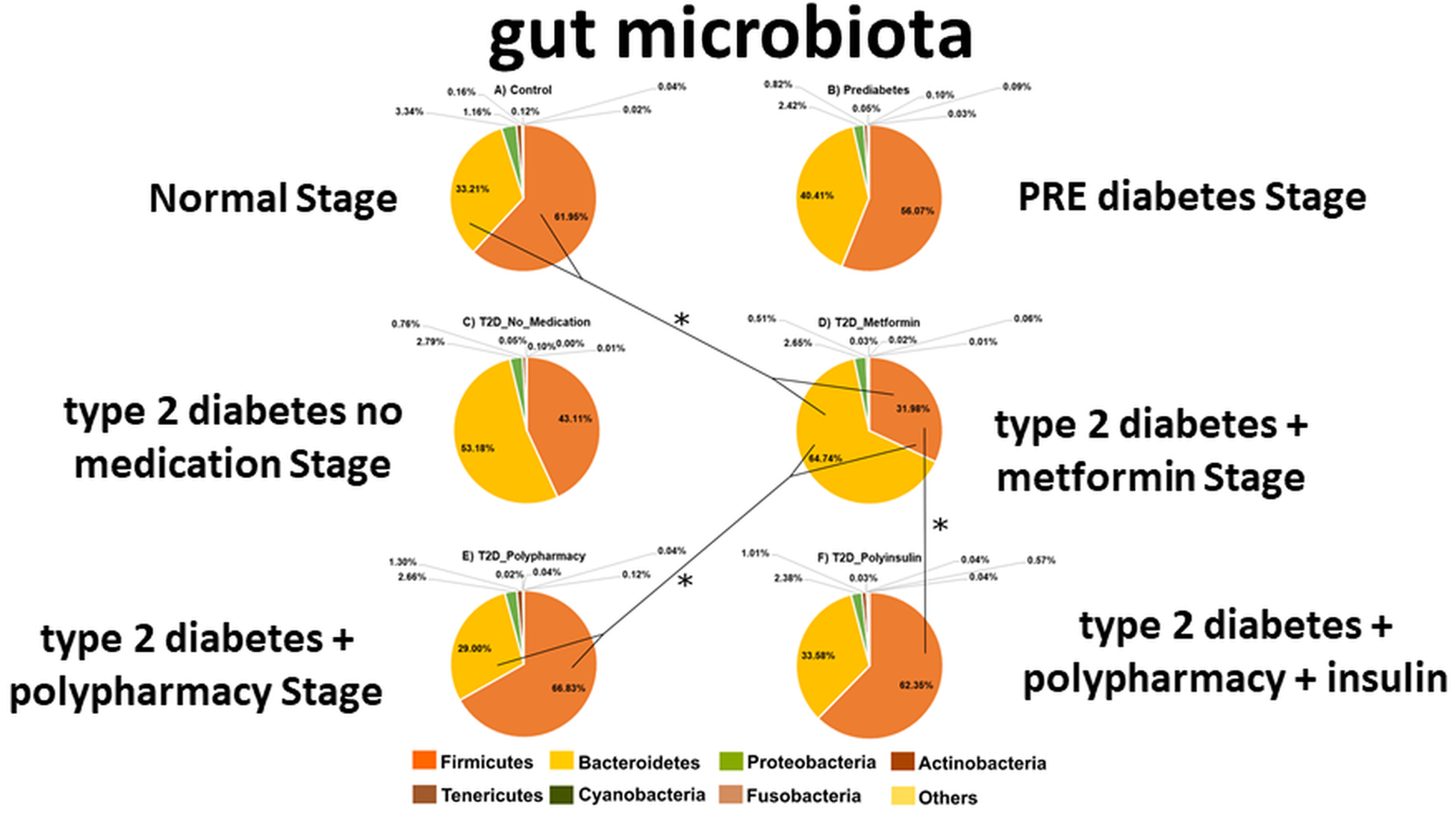
3.2. Firmicutes and Bacteroidetes Abundance Outline Major Changes in Gut Microbiota Diversity of CO, PRE and T2D Conditions
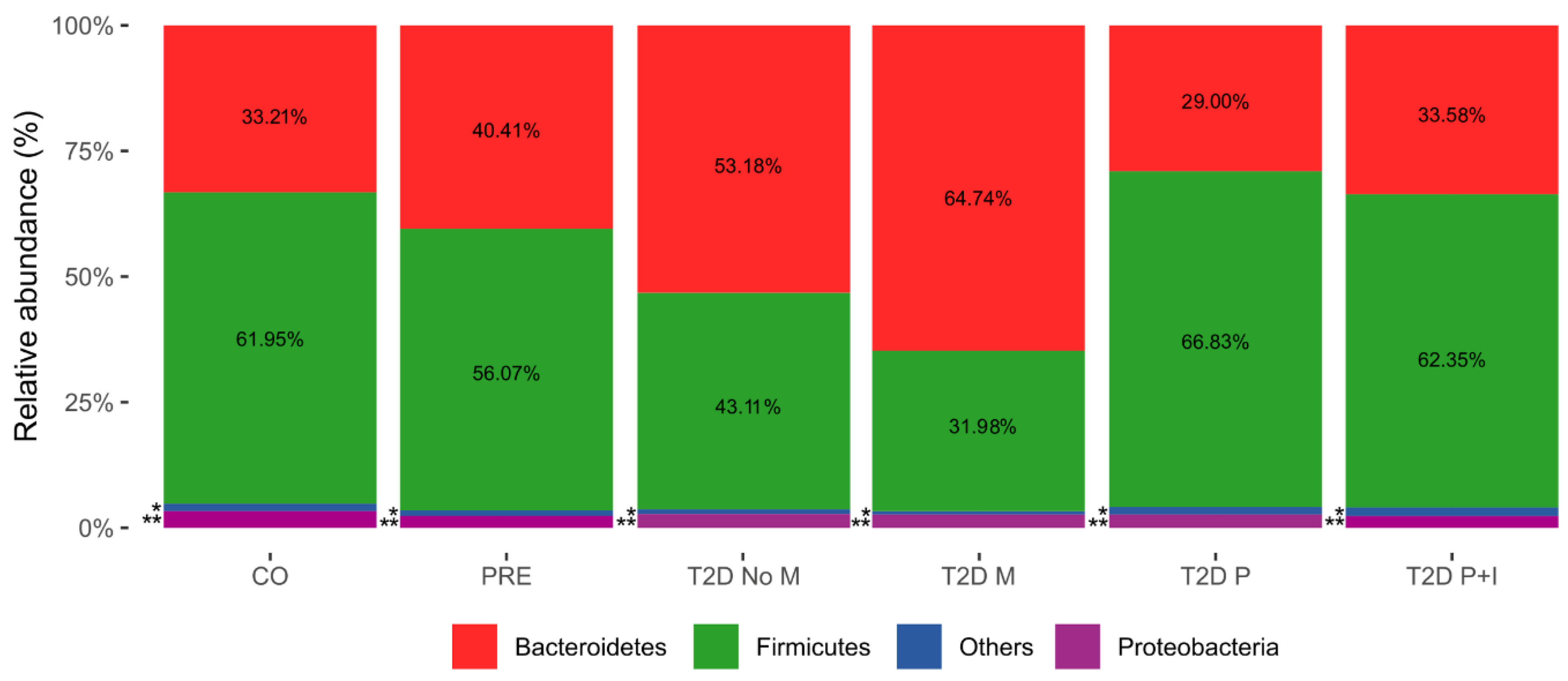
3.3. The Gut Bacterial Richness and Diversity in the T2D−No−M Group is Lower than the Rest of the Groups
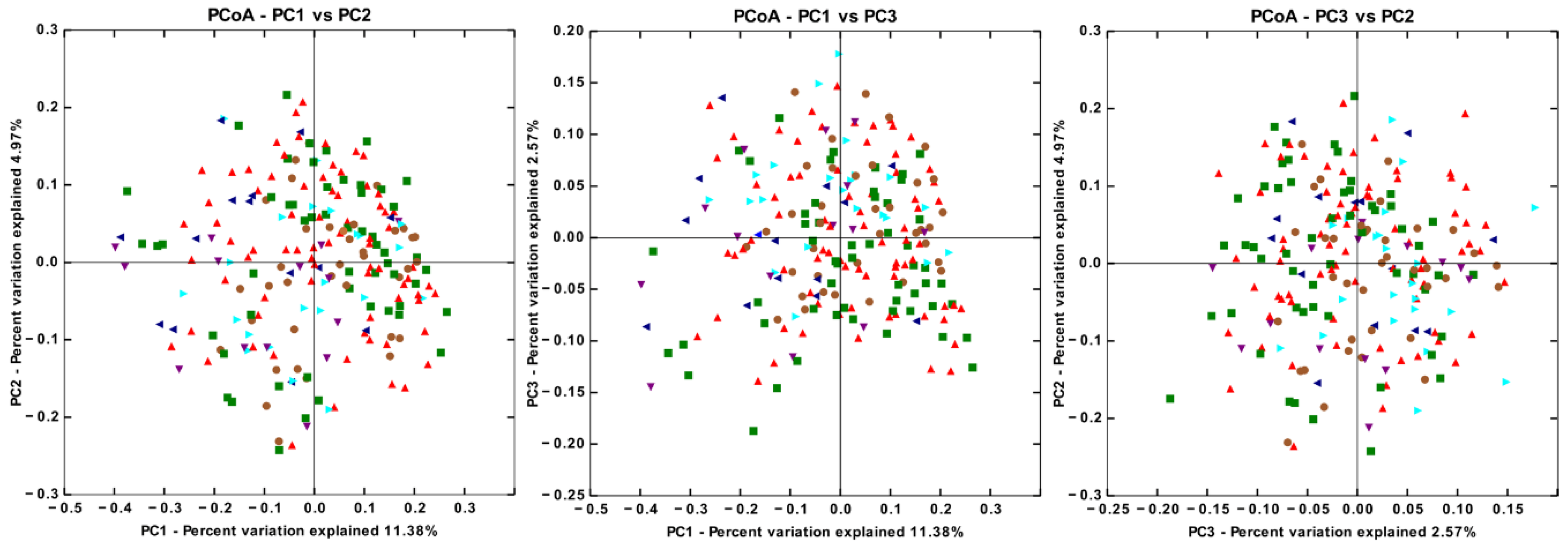
3.4. There are Significant Differences in Gut Microbiota Beta Diversity between Groups
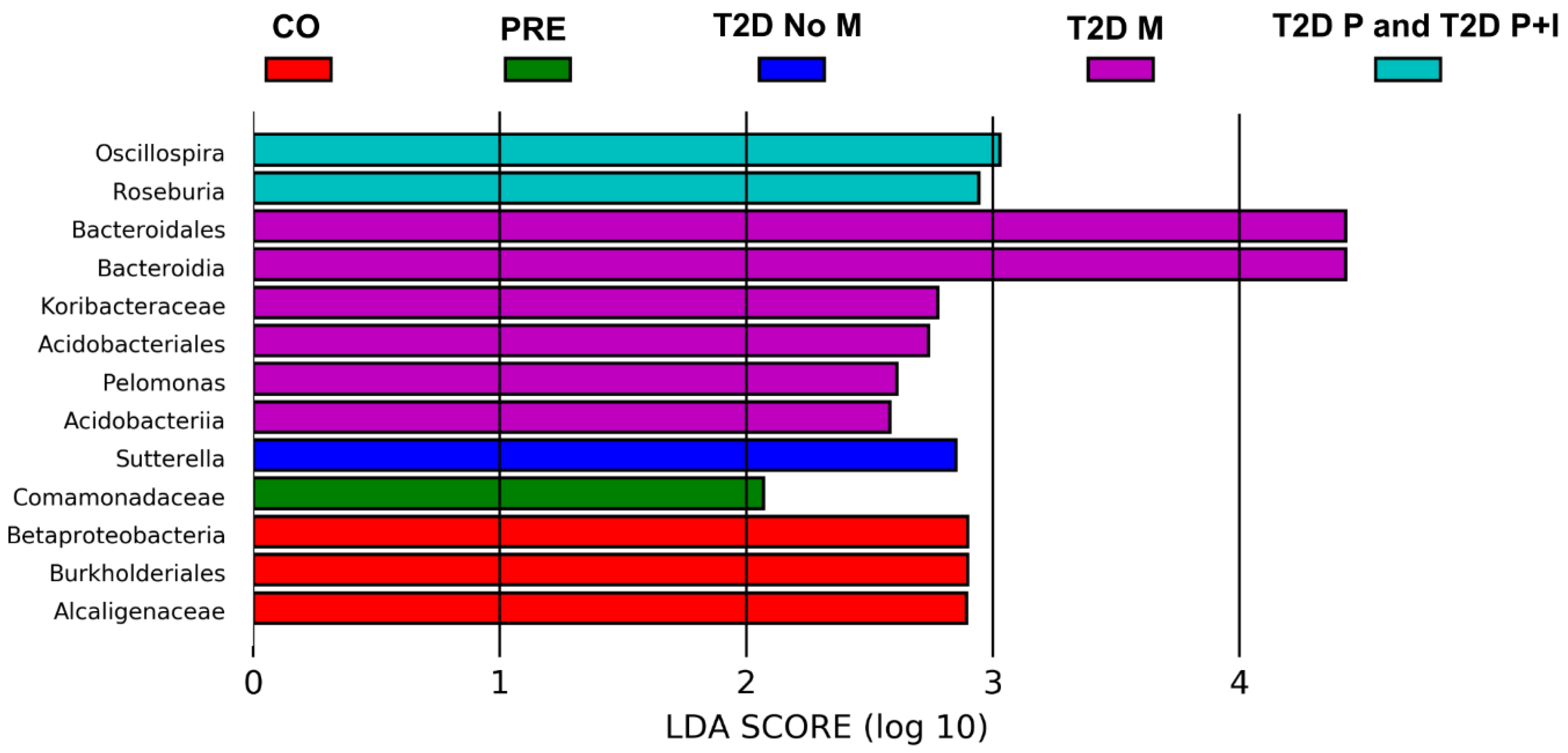
3.5. Differences in Gut Bacterial Abundance are Observed in All Studied Groups
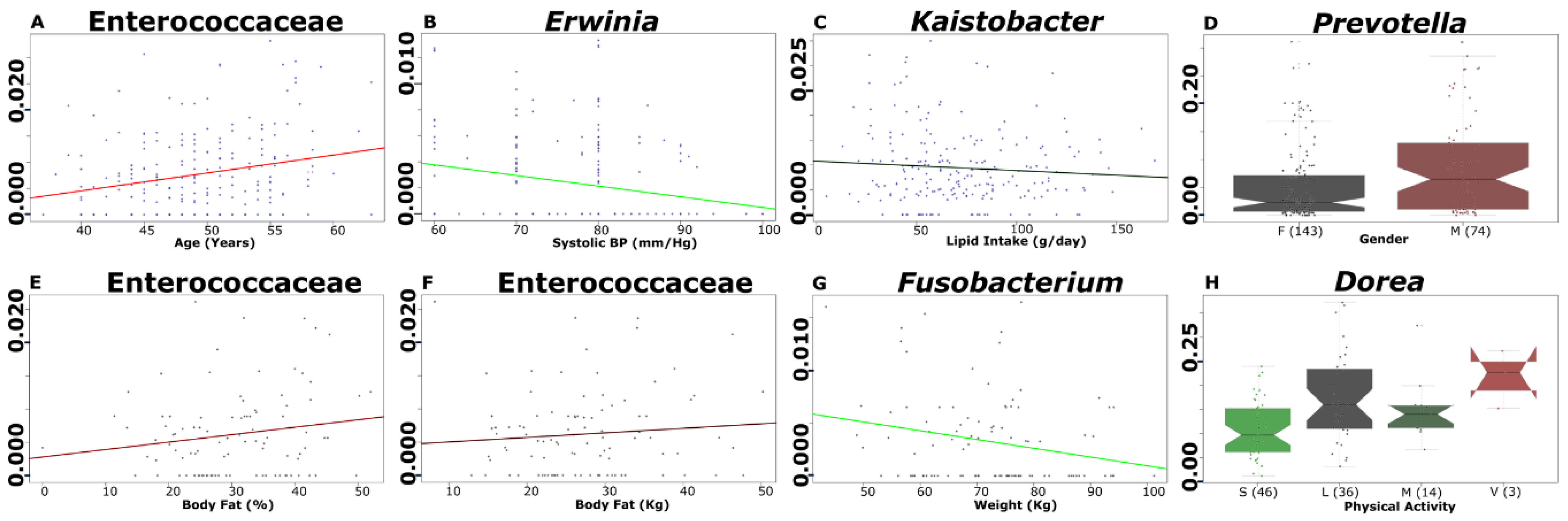
3.6. Selected Microbial Taxa Are Significantly Associated with Clinical, Anthropometrical, Dietary Intake and Physical Activity Parameters
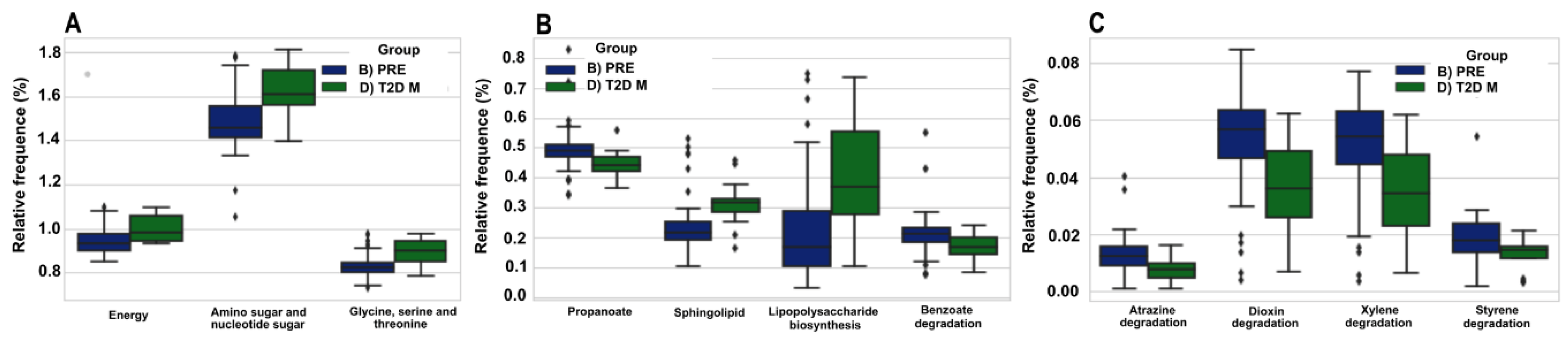
3.7. The Predicted Functional Metagenome of the Gut Microbiota Shows Differences in PRE and T2D−M Groups
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- World Health Organization. 2017. Available online: http://www.who.int/mediacentre/factsheets/fs312/en/ (accessed on 1 April 2018).
- Global Report on Diabetes. 2016. Available online: http://apps.who.int/iris/bitstream/handle/10665/204871/9789241565257_eng.pdf?sequence=1 (accessed on 9 March 2018).
- Instituto Nacional de Estadística y Geografía. Causas de Mortalidad, Base Interactiva de Datos. 2017. Available online: https://www.inegi.org.mx/sistemas/olap/registros/vitales/mortalidad/tabulados/ConsultaMortalidad.asp (accessed on 31 July 2019).
- Human Microbiome Project Consortium. A framework for human microbiome research. Nature 2012, 486, 215–221. [Google Scholar] [CrossRef]
- Belizário, J.E.; Napolitano, M. Human microbiomes and their roles in dysbiosis, common diseases, and novel therapeutic approaches. Front. Microbiol. 2015, 6, 1050. [Google Scholar] [CrossRef]
- Cani, P.D.; Delzenne, N.M. The role of the gut microbiota in energy metabolism and metabolic disease. Curr. Pharm. Des. 2009, 15, 1546–1558. [Google Scholar] [CrossRef]
- Han, J.L.; Lin, H.L. Intestinal microbiota and type 2 diabetes: From mechanism insights to therapeutic perspective. World J. Gastroenterol. 2014, 20, 17737–17745. [Google Scholar] [CrossRef]
- Lau, E.; Carvalho, D.; Pina-Vaz, C.; Barbosa, J.A.; Freitas, P. Beyond gut microbiota: Understanding obesity and type 2 diabetes. Hormones (Athens) 2015, 14, 358–369. [Google Scholar] [CrossRef]
- Sato, J.; Kanazawa, A.; Ikeda, F.; Yoshihara, T.; Goto, H.; Abe, H.; Komiya, K.; Kawaguchi, M.; Shimizu, T.; Ogihara, T.; et al. Gut dysbiosis and detection of “live gut bacteria” in blood of Japanese patients with type 2 diabetes. Diabetes Care 2014, 37, 2343–2350. [Google Scholar] [CrossRef]
- Qin, J.; Li, Y.; Cai, Z.; Li, S.; Zhu, J.; Zhang, F.; Liang, S.; Zhang, W.; Guan, Y.; Shen, D.; et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 2012, 490, 55–60. [Google Scholar] [CrossRef]
- Karlsson, F.H.; Tremaroli, V.; Nookaew, I.; Bergström, G.; Behre, C.J.; Fagerberg, B.; Nielsen, J.; Bäckhed, F.l. Gut metagenome in European women with normal, impaired and diabetic glucose control. Nature 2013, 498, 99–103. [Google Scholar] [CrossRef]
- Ross, M.C.; Muzny, D.M.; McCormick, J.B.; Gibbs, R.A.; Fisher-Hoch, S.P.; Petrosino, J.F. 16S gut community of the Cameron County Hispanic Cohort. Microbiome 2015, 3, 7. [Google Scholar] [CrossRef]
- Sepp, E.; Kolk, H.; Lõivukene, K.; Mikelsaar, M. Higher blood glucose level associated with body mass index and gut microbiota in elderly people. Microb. Ecol. Health Dis. 2014, 3, 25. [Google Scholar] [CrossRef]
- Wu, H.; Esteve, E.; Tremaroli, V.; Khan, M.T.; Caesar, R.; Mannerås-Holm, L.; Ståhlman, M.; Olsson, L.M.; Serino, M.; Planas-Fèlix, M.; et al. Metformin alters the gut microbiome of individuals with treatment-naive type 2 diabetes, contributing to the therapeutic effects of the drug. Nat. Med. 2017, 23, 850–858. [Google Scholar] [CrossRef] [PubMed]
- InBody. Application. Diabetes. InBody: A Non-Invasive, Painless Solution. 2018. Available online: https://inbodyusa.com/pages/diabetes (accessed on 23 March 2018).
- Petroni, M.L.; Bertoli, S.; Maggioni, M.; Morini, P.; Battezzati, A.; Tagliaferri, M.A.; Liuzzi, A.; Testolin, G. Feasibility of air plethysmography (BOD POD) in morbid obesity: A pilot study. Acta Diabetol. 2003, 40, 59–62. [Google Scholar] [CrossRef] [PubMed]
- Pérez Lizaur, A.B.; Palacios González, B.; Castro Becerra, A.L.; Flores Galicia, I. Sistema Mexicano de Alimentos Equivalentes, 4th ed.; México Press: Mexico City, Mexico, 2014. [Google Scholar]
- World Health Organization. Global Physical Activity Surveillance. 2018. Available online: http://www.who.int/ncds/surveillance/steps/GPAQ/en/ (accessed on 1 March 2018).
- Ainsworth, B.E.; Haskell, W.L.; Herrmann, S.D.; Meckes, N.; Bassett, D.R., Jr.; Tudor-Locke, C.; Greer, J.L.; Vezina, J.; Whitt-Glover, M.C.; Leon, A.S. Compendium of Physical Activities: A second update of codes and MET values. Med. Sci. Sports Exerc. 2011, 43, 1575–1581. [Google Scholar] [CrossRef]
- Murugesan, S.; Ulloa-Martínez, M.; Martínez-Rojano, H.; Galván-Rodríguez, F.M.; Miranda-Brito, C.; Romano, M.C.; Piña-Escobedo, A.; Pizano-Zárate, M.L.; Hoyo-Vadillo, C.; García-Mena, J. Study of the diversity and short-chain fatty acids production by the bacterial community in overweight and obese Mexican children. Eur. J. Clin. Microbiol. Infect. Dis. 2015, 34, 1337–1346. [Google Scholar] [CrossRef] [PubMed]
- McMurdie, P.J.; Holmes, S. Phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [PubMed]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef]
- Thorsen, J.; Brejnrod, A.; Mortensen, M.; Rasmussen, M.A.; Stokholm, J.; Al-Soud, W.A.; Sørensen, S.; Bisgaard, H.; Waage, J. Large-scale benchmarking reveals false discoveries and count transformation sensitivity in 16S rRNA gene amplicon data analysis methods used in microbiome studies. Microbiome 2016, 4, 62. [Google Scholar] [CrossRef]
- Hawinkel, S.; Mattiello, F.; Bijnens, L.; Thas, O. A broken promise: Microbiome differential abundance methods do not control the false discovery rate. Brief. Bioinform. 2019, 20, 210–221. [Google Scholar] [CrossRef]
- Langille, M.G.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Thurber, R.L.V.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef]
- Parks, D.H.; Tyson, G.W.; Hugenholtz, P.; Beiko, R.G. STAMP: Statistical analysis of taxonomic and functional profiles. Bioinformatics 2014, 30, 3123–3124. [Google Scholar] [CrossRef]
- Morgan, X.C.; Tickle, T.L.; Sokol, H.; Gevers, D.; Devaney, K.L.; Ward, D.V.; Reyes, J.A.; Shah, S.A.; LeLeiko, N.; Snapper, S.B.; et al. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol. 2012, 13, R79. [Google Scholar] [CrossRef]
- Shin, N.R.; Bose, S.; Wang, J.H.; Ansari, A.; Lim, S.K.; Chin, Y.W.; Choi, H.S.; Kim, H. Flos Lonicera Combined with Metformin Ameliorates Hepatosteatosis and Glucose Intolerance in Association with Gut Microbiota Modulation. Front. Microbiol. 2017, 8, 2271. [Google Scholar] [CrossRef]
- Horie, M.; Miura, T.; Hirakata, S.; Hosoyama, A.; Sugino, S.; Umeno, A.; Murotomi, K.; Yoshida, Y.; Koike, T. Comparative analysis of the intestinal flora in type 2 diabetes and nondiabetic mice. Exp. Anim. 2017, 66, 405–416. [Google Scholar] [CrossRef]
- Dedysh, S.N.; Yilmaz, P. Refining the taxonomic structure of the phylum Acidobacteria. Int. J. Syst. Evol. Microbiol. 2018, 68, 3796–3806. [Google Scholar] [CrossRef]
- Long, C.L.; Geiger, J.W.; Kinney, J.M. Absorption of glucose from the colon and rectum. Metabolism 1967, 16, 413–418. [Google Scholar] [CrossRef]
- Wu, T.; Xie, C.; Wu, H.; Jones, K.L.; Horowitz, M.; Rayner, C. Metformin reduces the rate of small intestinal glucose absorption in type 2 diabetes. Diabetes Obes. Metab. 2017, 19, 290–293. [Google Scholar] [CrossRef]
- Stinson, L.F.; Boyce, M.C.; Payne, M.S.; Keelan, J.A. The Not-so-Sterile Womb: Evidence That the Human Fetus Is Exposed to Bacteria Prior to Birth. Front. Microbiol. 2019, 10, 1124. [Google Scholar] [CrossRef]
- Allin, K.H.; Tremaroli, V.; Caesar, R.; Jensen, B.A.H.; Damgaard, M.T.F.; Bahl, M.I.; Licht, T.R.; Hansen, T.H.; Nielsen, T.; Dantoft, T.M.; et al. Aberrant intestinal microbiota in individuals with prediabetes. Diabetologia 2018, 61, 810–820. [Google Scholar] [CrossRef]
- Wang, Y.; Luo, X.; Mao, X.; Tao, Y.; Ran, X.; Zhao, H.; Xiong, J.; Li, L. Gut microbiome analysis of type 2 diabetic patients from the Chinese minority ethnic groups the Uygurs and Kazaks. PLoS ONE 2017, 12, e0172774. [Google Scholar] [CrossRef]
- Rajilić-Stojanović, M.; Smidt, H.; de Vos, W.M. Diversity of the human gastrointestinal tract microbiota revisited. Environ. Microbiol. 2007, 9, 2125–2136. [Google Scholar] [CrossRef]
- Wu, X.; Ma, C.; Han, L.; Nawaz, M.; Gao, F.; Zhang, X.; Yu, P.; Zhao, C.; Li, L.; Zhou, A.; et al. Molecular characterisation of the faecal microbiota in patients with type II diabetes. Curr. Microbiol. 2010, 61, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Bo, T.; Shao, S.; Wu, D.; Niu, S.; Zhao, J.; Gao, L. Relative variations of gut microbiota in disordered cholesterol metabolism caused by high-cholesterol diet and host genetics. Microbiologyopen 2017, 6, e491. [Google Scholar] [CrossRef] [PubMed]
- Murtaza, N.; Burke, L.M.; Vlahovich, N.; Charlesson, B.; O’Neill, H.; Ross, M.L.; Campbell, K.L.; Krause, L.; Morrison, M. The Effects of Dietary Pattern during Intensified Training on Stool Microbiota of Elite Race Walkers. Nutrients 2019, 11, 261. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.J.; Eum, S.Y.; Rampersaud, E.; Daunert, S.; Abreu, M.T.; Toborek, M. Exercise attenuates PCB-induced changes in the mouse gut microbiome. Environ. Health Perspect. 2013, 121, 725–730. [Google Scholar] [CrossRef]
- Rodriguez, J.; Hiel, S.; Delzenne, N.M. Metformin: Old friend, new ways of action-implication of the gut microbiome? Curr. Opin. Clin. Nutr. Metab. Care 2018, 21, 294–301. [Google Scholar] [CrossRef]
- Boutagy, N.E.; McMillan, R.P.; Frisard, M.I.; Hulver, M.W. Metabolic endotoxemia with obesity: Is it real and is it relevant? Biochimie 2016, 124, 11–20. [Google Scholar] [CrossRef]
- Pussinen, P.J.; Havulinna, A.S.; Lehto, M.; Sundvall, J.; Salomaa, V. Endotoxemia is associated with an increased risk of incident diabetes. Diabetes Care 2011, 34, 392–397. [Google Scholar] [CrossRef]
- Forslund, K.; Hildebrand, F.; Nielsen, T.; Falony, G.; Le Chatelier, E.; Sunagawa, S.; Prifti, E.; Vieira-Silva, S.; Gudmundsdottir, V.; Pedersen, H.K.; et al. Disentangling type 2 diabetes and metformin treatment signatures in the human gut microbiota. Nature 2015, 528, 262–266. [Google Scholar] [CrossRef]
- Jourova, L.; Anzenbacher, P.; Anzenbacherova, E. Human gut microbiota plays a role in the metabolism of drugs. Biomed. Pap. Med. Fac. Univ. Palacky Olomouc Czech Repub. 2016, 160, 317–326. [Google Scholar] [CrossRef]
- Carmody, R.N.; Turnbaugh, P.J. Host-microbial interactions in the metabolism of therapeutic and diet-derived xenobiotics. J. Clin. Investig. 2014, 124, 4173–4181. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CO | PRE | T2D−No−M | T2D−M | T2D−P | T2D−P+I | p-Value | |
|---|---|---|---|---|---|---|---|
| Number of subjects | 76 | 54 | 14 | 14 | 22 | 37 | NA |
| Gender (women/men) | 50/26 | 36/18 | 7/7 | 12/2 | 14/8 | 24/13 | NA |
| Age (years) | 48.0 ± 5.4 | 50.2 ± 5.0 | 48.1 ± 4.7 | 48.1 ± 4.6 | 51.3 ± 5.6 | 50.5 ± 4.5 | 0.237 |
| Blood Pressure | |||||||
| SBP (mm/Hg) | 75.6 ± 8.3 | 77.9 ± 9.9 | 80.4 ± 8.6 | 77.9 ± 9.6 | 76.7 ± 7.2 | 79.0 ± 10.8 | 0.256 |
| DBP (mm/Hg) | 114.5 ± 13.4 | 117.5 ± 11.3 | 122.1 ± 10.2 | 120.5 ± 12.3 | 117.7 ± 10.3 | 124.2 ± 28.9 | 0.078 |
| Anthropometric Profile | |||||||
| Weight (kg) | 71.8 ± 12.9 | 71.6 ± 11.9 | 78.9 ± 12.1 | 73.9 ± 13.6 | 76.9 ± 15.4 | 79.4 ± 18.6 | 0.078 |
| BMI (kg/m2) | 27.7 ± 3.7 | 28.4 ± 4.1 | 30.8 ± 4.6 | 30.1 ± 4.8 | 30.3 ± 5.0 | 31.5 ± 7.8 | 0.012 |
| Waist Circumference (cm) | 89.7 ± 14.0 | 92.0 ± 11.1 | 99.3 ± 11.6 | 97.3 ± 8.3 | 94.8 ± 20.0 | 99.8 ± 15.7 | 0.002 |
| Body Fat Bod Pod (%) | 29.6 ± 8.2 β | 30.6 ± 9.5 δ | 30.8 ± 10.8 τ | 34.0 ± 7.8 τ | ND | ND | 0.464 |
| Biochemical profile | |||||||
| HbA1c (%) | 5.4 ± 0.2 | 5.9 ± 0.2 | 8.4 ± 2.2 | 7.5 ± 1.0 | 8.1 ± 2.7 | 9.4 ± 2.0 | <0.001 |
| Fasting glucose (mg/dL) | 92.9 ± 10.1 | 97.2 ± 11.0 | 199.3 ± 108.4 | 148.6 ± 40.9 | 147.7 ± 68.7 | 186.6 ± 77.5 | <0.001 |
| Cholesterol (mg/dL) | 195.6 ± 29.3 | 204.9 ± 53.1 | 271.0 ± 160.8 | 205.2 ± 34.3 | 169.3 ± 31.1 Ω | 182.8 ± 35.2 β | <0.001 |
| Triglycerides (mg/dL) | 159.6 ± 89.9 | 154.2 ± 63.2 | 274.2 ± 191.0 | 226.6 ± 125.3 | 148.7 ± 51.1 Ω | 206.3 ± 104.6 β | 0.002 |
| HDL (mg/dL) | 47.5 ± 10.2 € | 47.6 ± 9.4 ¥ | 46.9 ± 8.6 | 45.7 ± 9.5 | 40.0 ± 7.5 Ω | 42.0 ± 8.6 π | 0.005 |
| LDL (mg/dL) | 114.8 ± 26.2 £ | 127.4 ± 51.4 ¥ | 137.9 ± 37.5 ∞ | 115.5 ± 29.5 α | 102.2 ± 28.2 Ω | 105.3 ± 27.9 π | 0.019 |
| Dietary profile | |||||||
| Kcal (kcal/day) | 1903.3 ± 678.0 | 2027.1 ± 608.7 | 1983.0 ± 610.8 | 1975.6 ± 780.6 | 2487.3 ± 811.4 | 2597.3 ± 887.7 | <0.001 |
| Protein (g/day) | 90.0 ± 36.1 | 93.7 ± 31.6 | 98.1 ± 38.4 | 98.1 ± 36.0 | 123.8 ± 45.0 | 114.6 ± 43.2 | 0.001 |
| Lipids (g/day) | 65.3 ± 29.0 | 66.6 ± 30.1 | 63.1 ± 24.9 | 65.5 ± 23.7 | 88.5 ± 34.2 | 92.1 ± 38.5 | <0.001 |
| Carbohydrates (g/day) | 241.8 ± 94.8 | 262.8 ± 79.2 | 256.0 ± 105.4 | 248.4 ± 122.1 | 294.2 ± 115.4 | 317.0 ± 110.4 | 0.021 |
| Sugar from beverage (g/day) | 14.3 ± 22.4 | 12.4 ± 16.3 | 7.6 ± 11.4 | 18.5 ± 19.4 | 15.7 ± 15.4 | 15.4 ± 13.8 | 0.241 |
| Sugar (g/day) | 43.8 ± 37.6 | 34.2 ± 23.9 | 18.2 ± 18.7 | 18.5 ± 27.6 | 30.0 ± 28.9 | 24.8 ± 23.3 | 0.011 |
| Physical Activity | |||||||
| METs | 2.4 ± 2.1 | 2.1 ± 1.9 | 1.8 ± 0.7 | 1.4 ± 0.6 | 1.8 ± 1.3 | 2.1 ± 2.7 | 0.526 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chávez-Carbajal, A.; Pizano-Zárate, M.L.; Hernández-Quiroz, F.; Ortiz-Luna, G.F.; Morales-Hernández, R.M.; De Sales-Millán, A.; Hernández-Trejo, M.; García-Vite, A.; Beltrán-Lagunes, L.; Hoyo-Vadillo, C.; et al. Characterization of the Gut Microbiota of Individuals at Different T2D Stages Reveals a Complex Relationship with the Host. Microorganisms 2020, 8, 94. https://doi.org/10.3390/microorganisms8010094
Chávez-Carbajal A, Pizano-Zárate ML, Hernández-Quiroz F, Ortiz-Luna GF, Morales-Hernández RM, De Sales-Millán A, Hernández-Trejo M, García-Vite A, Beltrán-Lagunes L, Hoyo-Vadillo C, et al. Characterization of the Gut Microbiota of Individuals at Different T2D Stages Reveals a Complex Relationship with the Host. Microorganisms. 2020; 8(1):94. https://doi.org/10.3390/microorganisms8010094
Chicago/Turabian StyleChávez-Carbajal, Alejandra, María Luisa Pizano-Zárate, Fernando Hernández-Quiroz, Guillermo Federico Ortiz-Luna, Rosa María Morales-Hernández, Amapola De Sales-Millán, María Hernández-Trejo, Angelina García-Vite, Luis Beltrán-Lagunes, Carlos Hoyo-Vadillo, and et al. 2020. "Characterization of the Gut Microbiota of Individuals at Different T2D Stages Reveals a Complex Relationship with the Host" Microorganisms 8, no. 1: 94. https://doi.org/10.3390/microorganisms8010094
APA StyleChávez-Carbajal, A., Pizano-Zárate, M. L., Hernández-Quiroz, F., Ortiz-Luna, G. F., Morales-Hernández, R. M., De Sales-Millán, A., Hernández-Trejo, M., García-Vite, A., Beltrán-Lagunes, L., Hoyo-Vadillo, C., & García-Mena, J. (2020). Characterization of the Gut Microbiota of Individuals at Different T2D Stages Reveals a Complex Relationship with the Host. Microorganisms, 8(1), 94. https://doi.org/10.3390/microorganisms8010094