Structural Influence and Interactive Binding Behavior of Dopamine and Norepinephrine on the Greek-Key-Like Core of α-Synuclein Protofibril Revealed by Molecular Dynamics Simulations


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
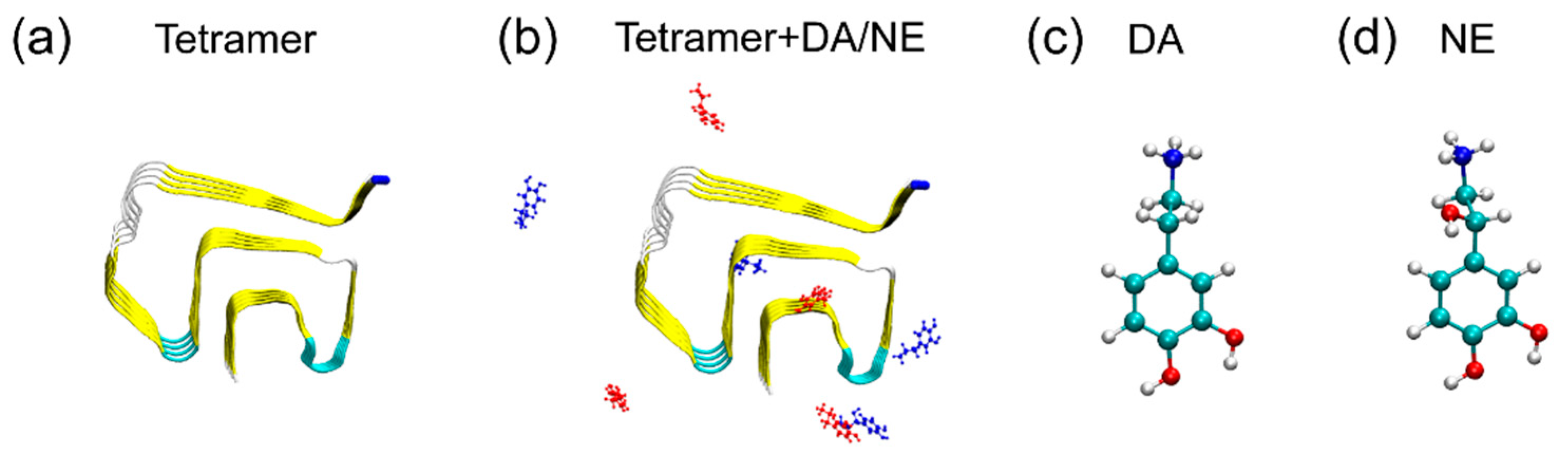
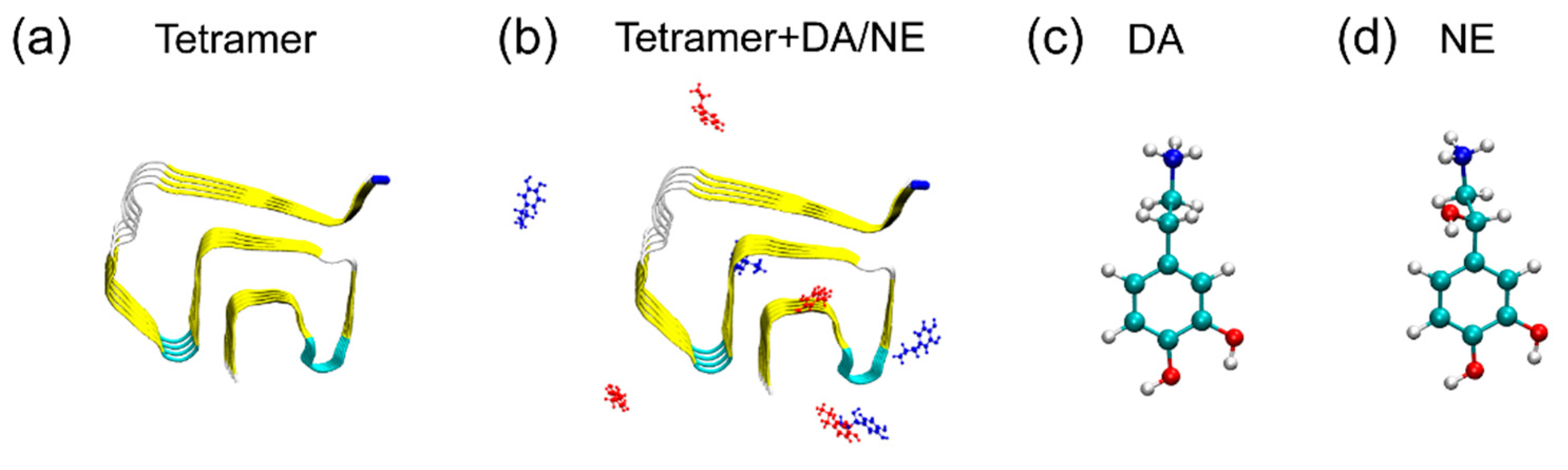
2.1. αS44-–96 Tetramer and DA/NE Molecules
2.2. MD Simulation Details
2.3. Analysis
3. Results and Discussion
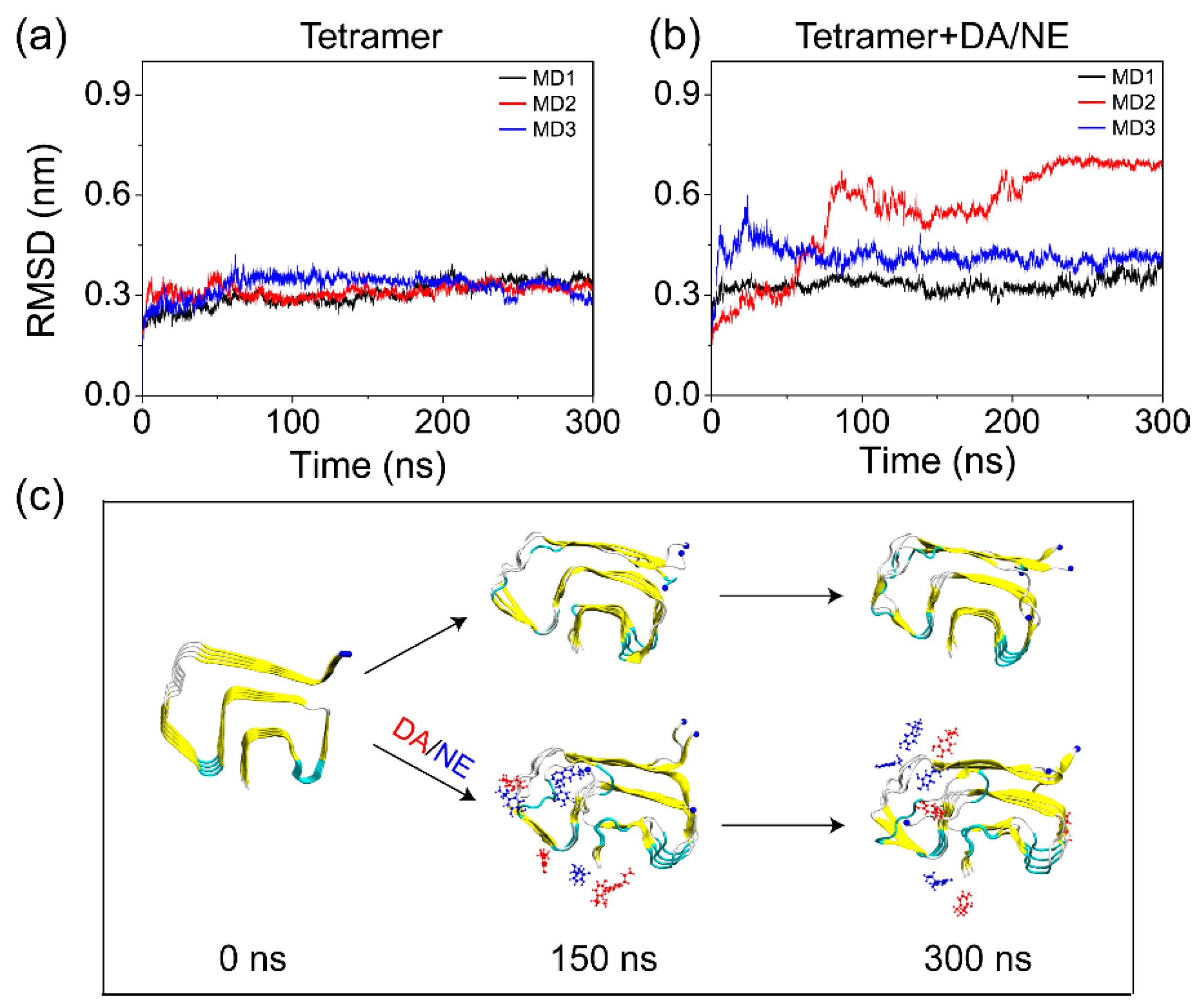
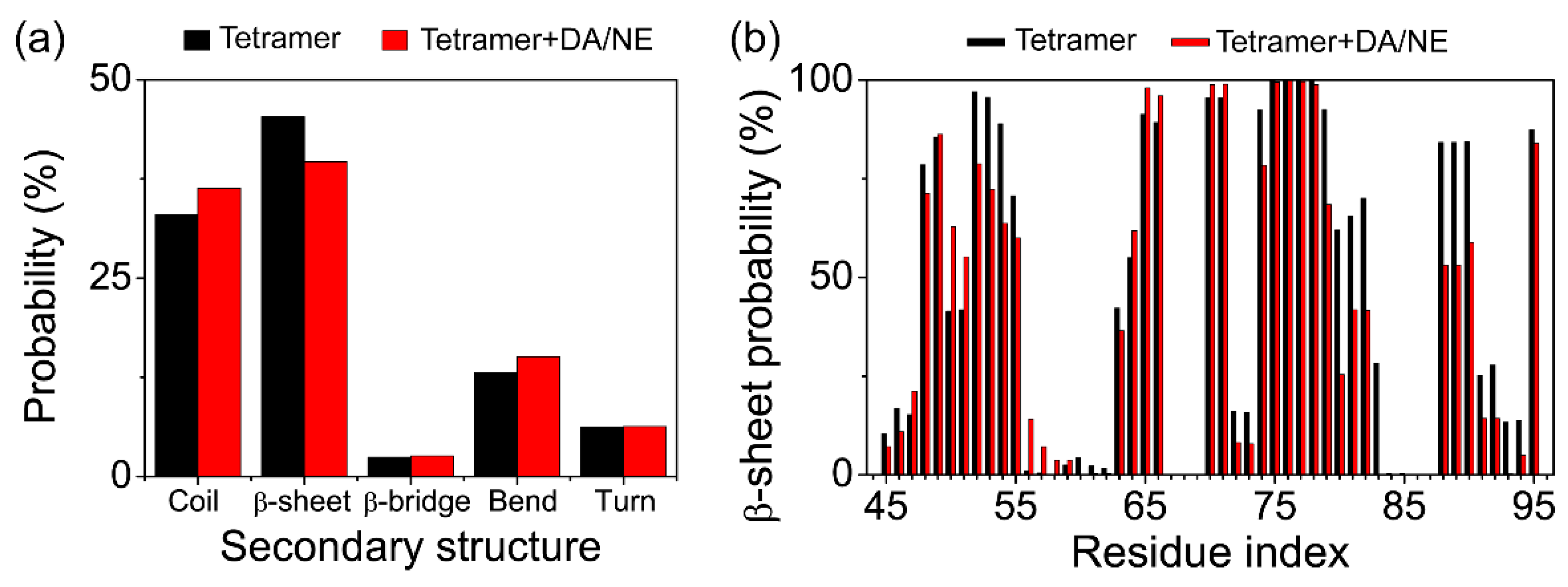
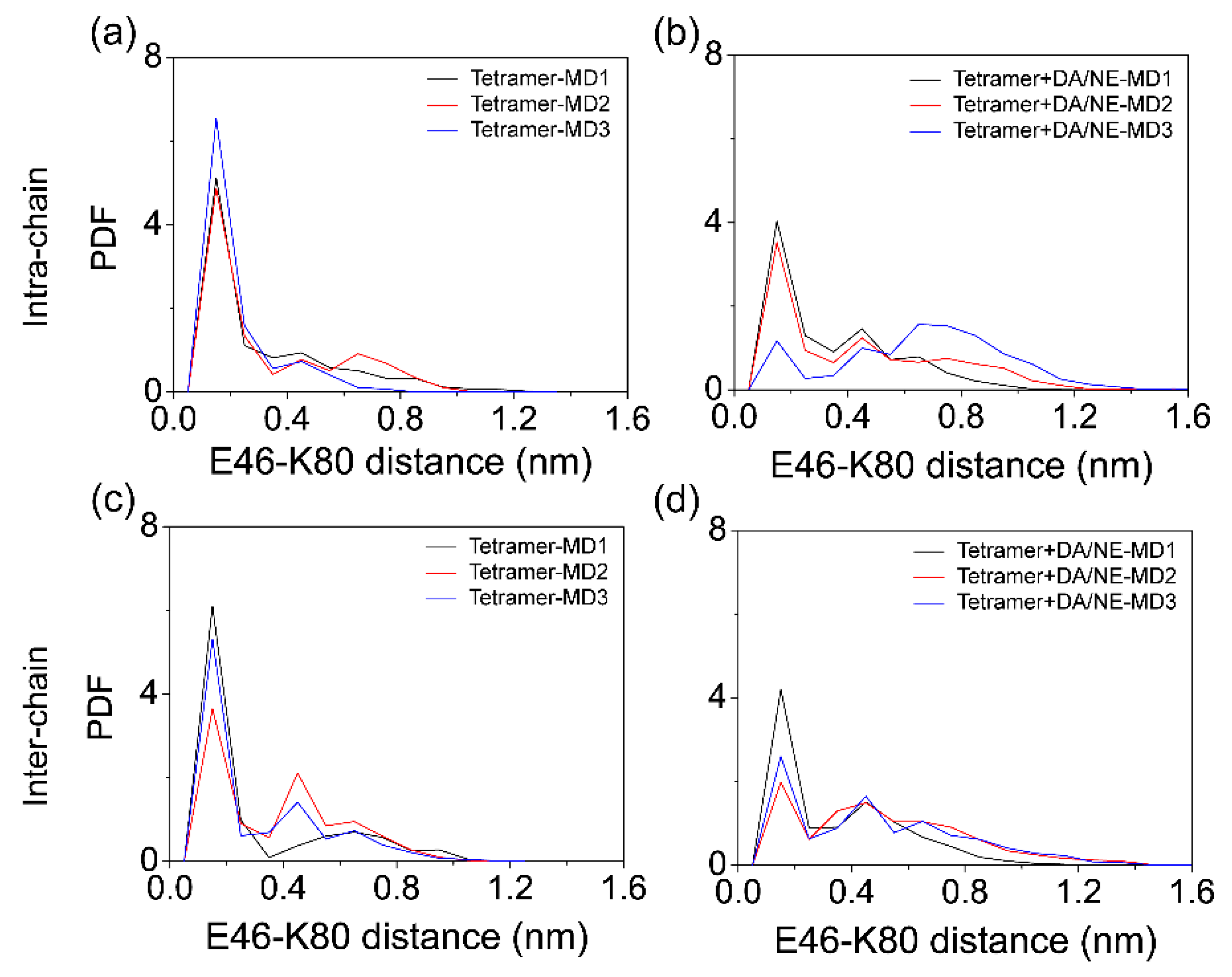
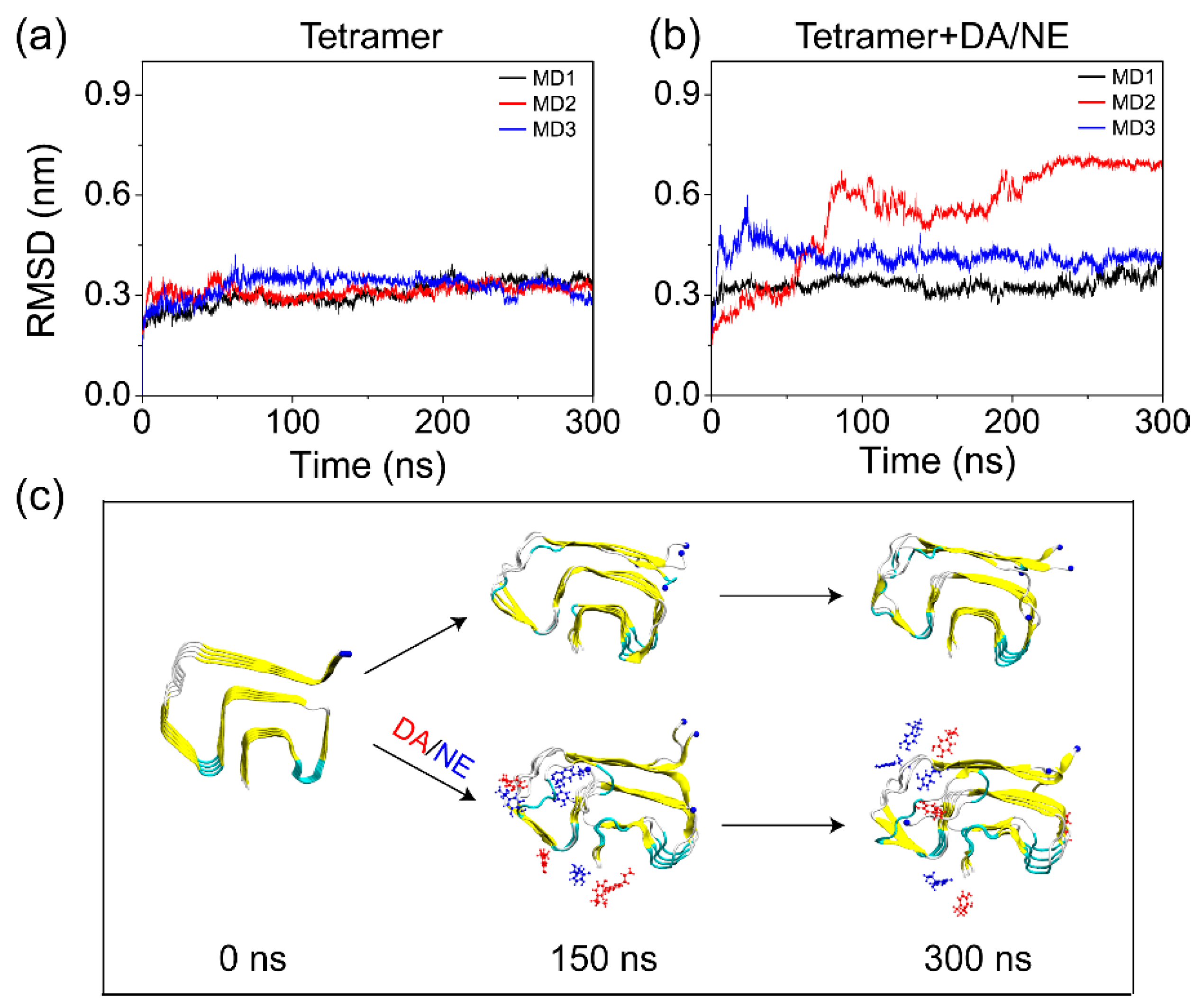
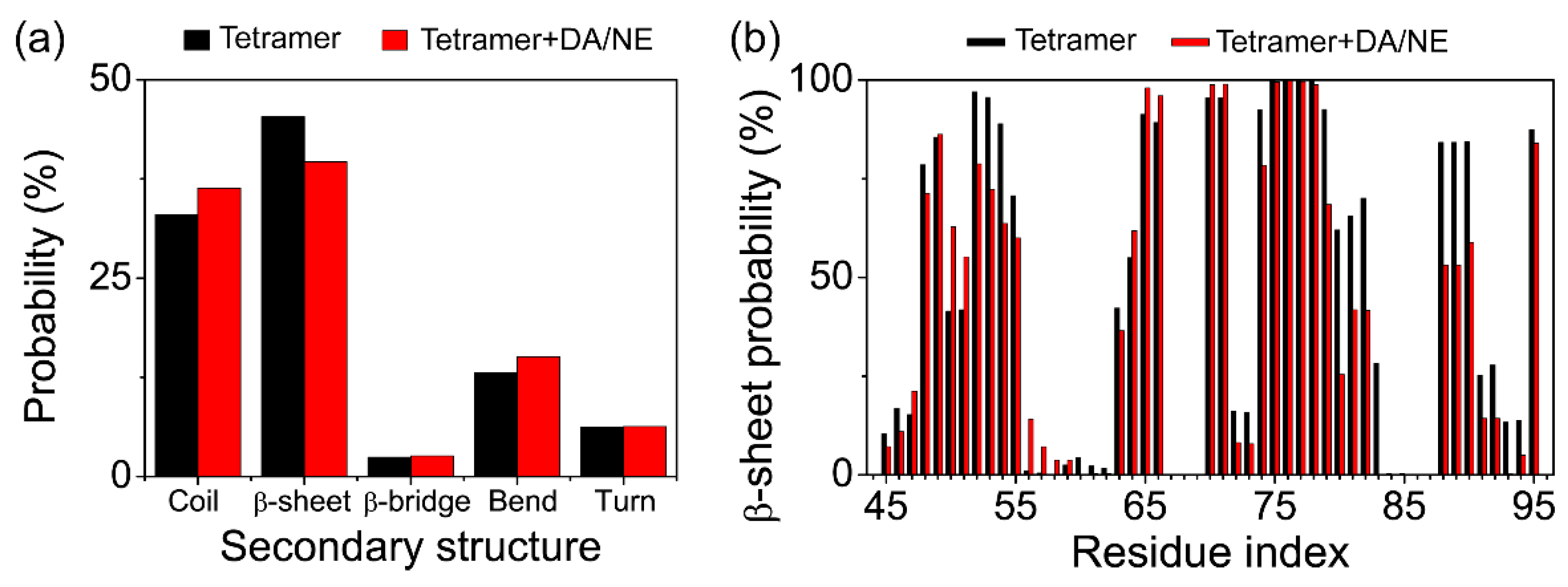
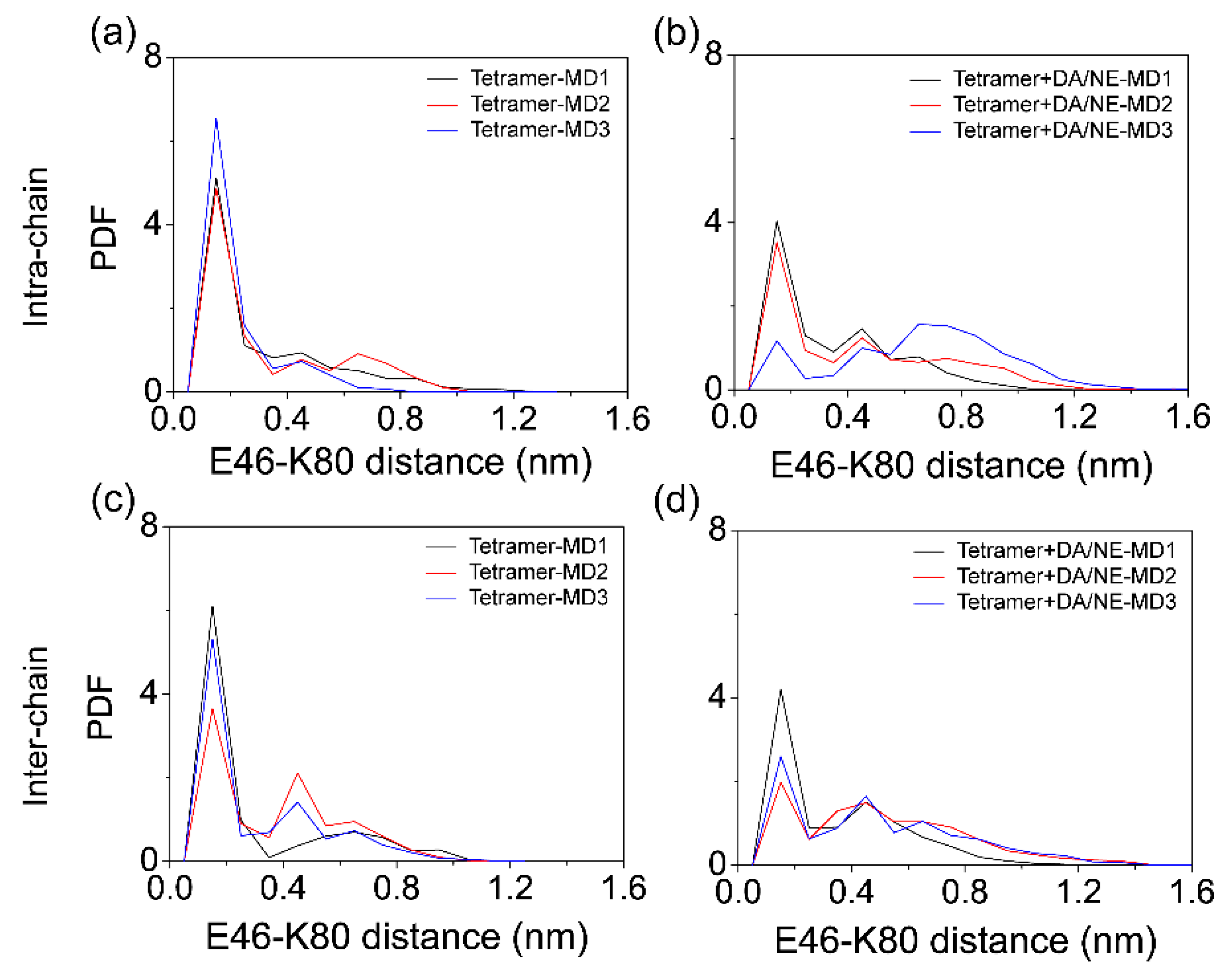
3.1. DA/NE Molecules Destabilize αS Protofibril by Disrupting the β-Sheet Structure and Destroying the Intra- and Inter-Peptide E46-K80 Salt Bridges
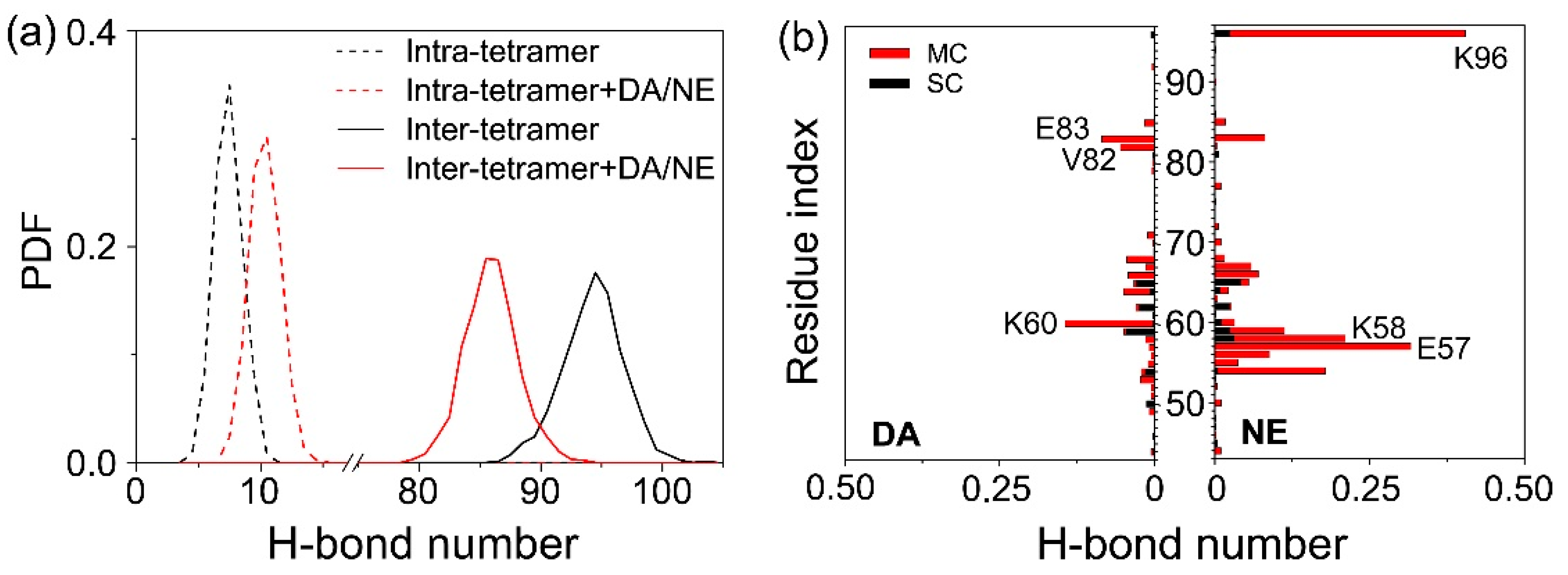
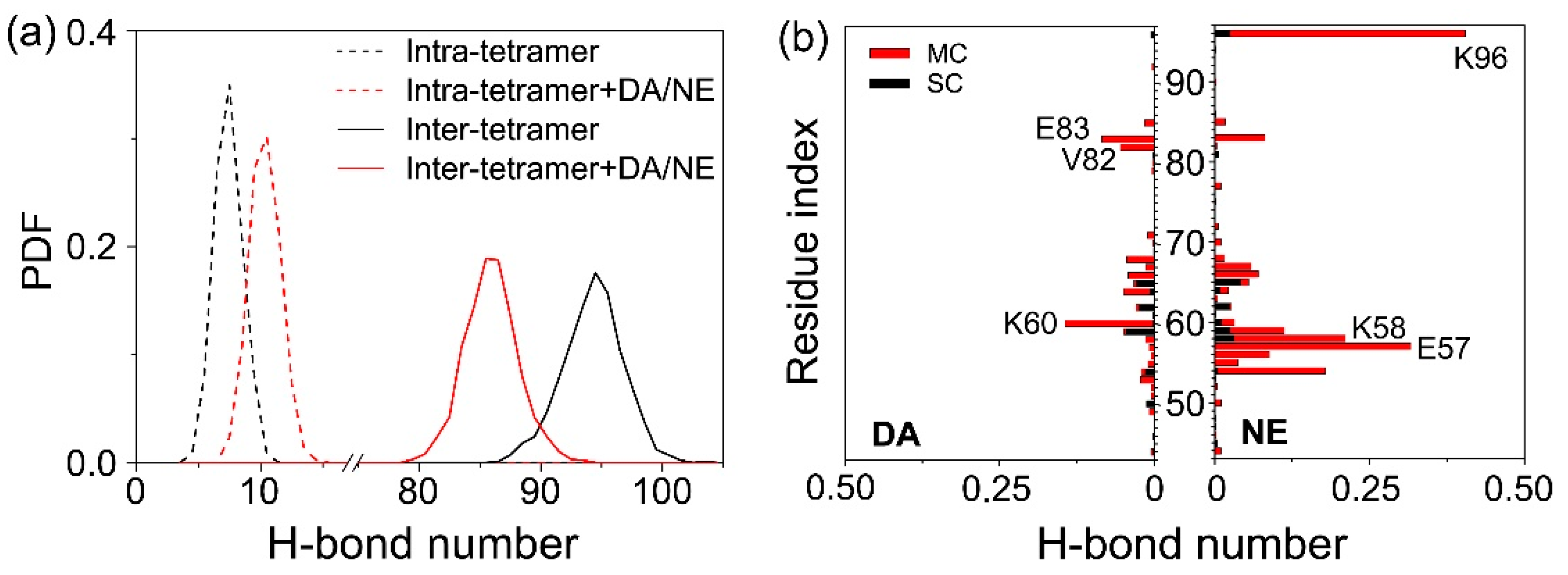
3.2. DA/NE Molecules Destroy the Inter-Chain Backbone Hydrogen Bonds, and NE Molecules Form More H-Bonds with αS Tetramer Than DA Molecules
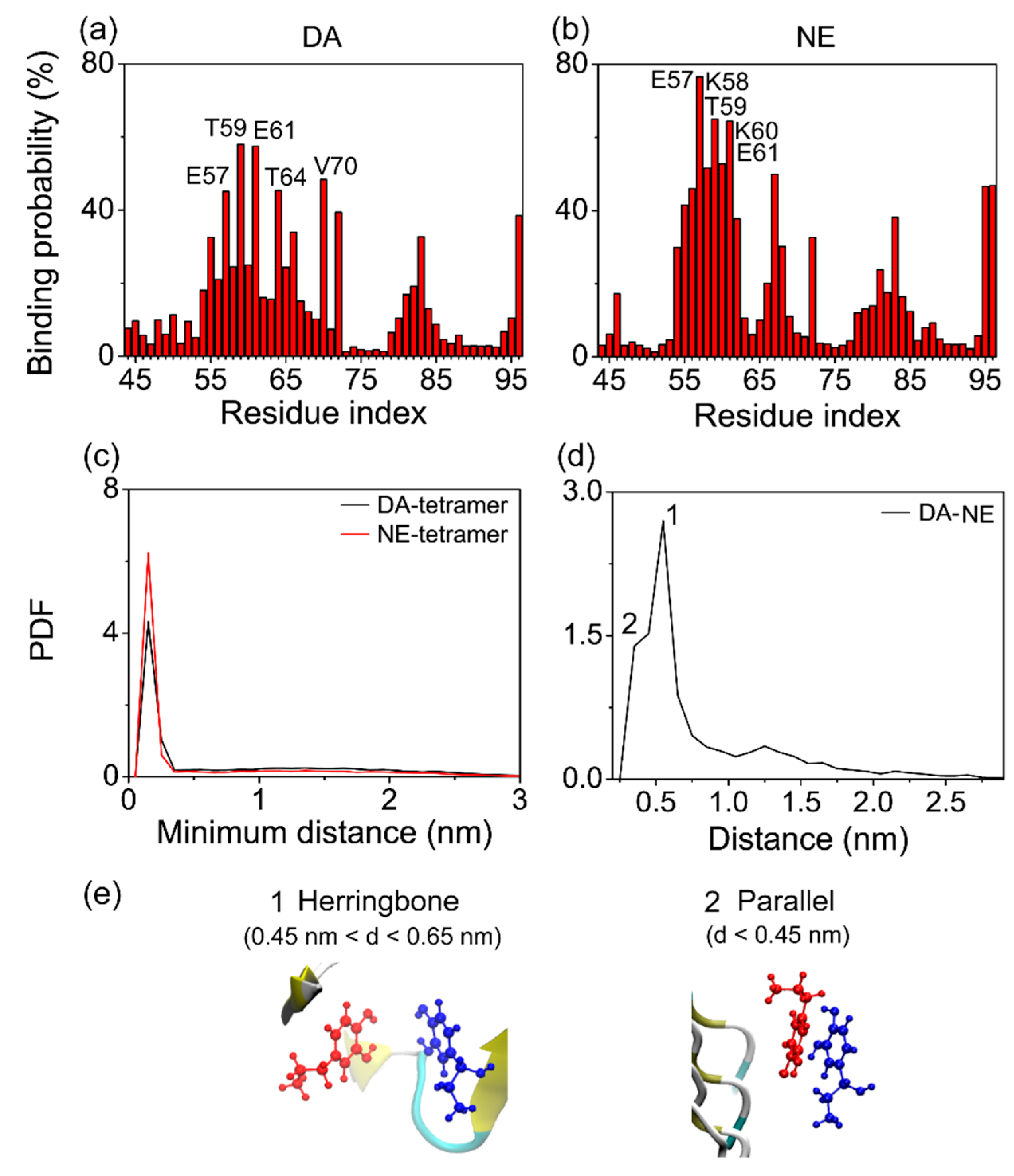
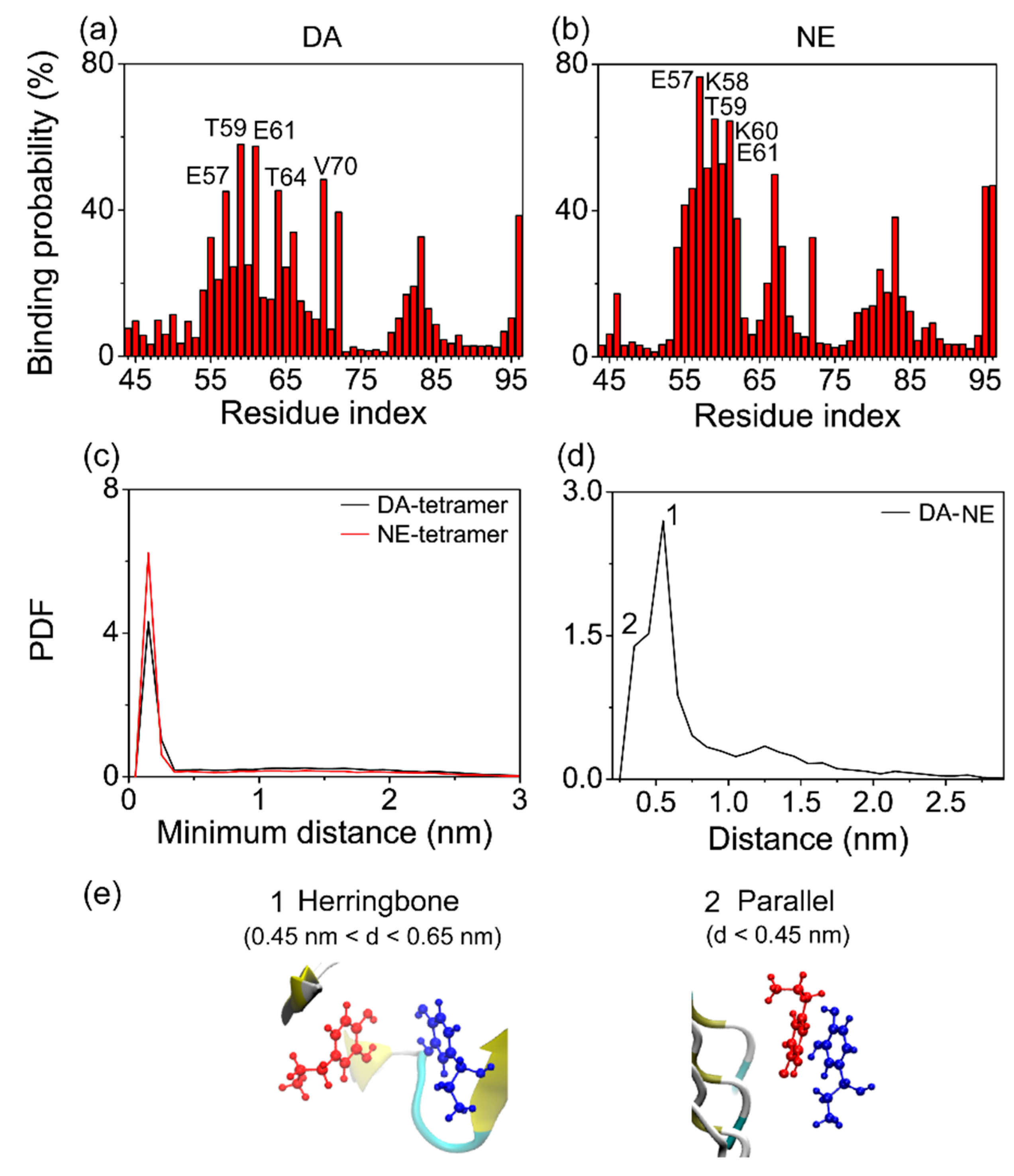
3.3. The Binding Sites and Interactive Binding Behavior of DA/NE Molecules on αS Tetramer
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chiti, F.; Dobson, C.M. Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 2006, 75, 333–366. [Google Scholar] [CrossRef] [PubMed]
- Chiti, F.; Dobson, C.M. Protein misfolding, amyloid formation, and human disease: A summary of progress over the last decade. Annu. Rev. Biochem. 2017, 86, 27–68. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg, D.S.; Sawaya, M.R. Structural studies of amyloid proteins at the molecular level. Annu. Rev. Biochem. 2017, 86, 69–95. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. Alpha-synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef] [PubMed]
- Baba, M.; Nakajo, S.; Tu, P.H.; Tomita, T.; Nakaya, K.; Lee, V.M.; Trojanowski, J.Q.; Iwatsubo, T. Aggregation of alpha-synuclein in Lewy bodies of sporadic Parkinson’s disease and dementia with Lewy bodies. Am. J. Pathol. 1998, 152, 879–884. [Google Scholar] [PubMed]
- Serpell, L.C.; Berriman, J.; Jakes, R.; Goedert, M.; Crowther, R.A. Fiber diffraction of synthetic alpha-synuclein filaments shows amyloid-like cross-beta conformation. Proc. Natl. Acad. Sci. USA 2000, 97, 4897–4902. [Google Scholar] [CrossRef]
- Nelson, R.; Sawaya, M.R.; Balbirnie, M.; Madsen, A.O.; Riekel, C.; Grothe, R.; Eisenberg, D. Structure of the cross-beta spine of amyloid-like fibrils. Nature 2005, 435, 773–778. [Google Scholar] [CrossRef]
- Stefani, M.; Dobson, C.M. Protein aggregation and aggregate toxicity: New insights into protein folding, misfolding diseases and biological evolution. J. Mol. Med. 2003, 81, 678–699. [Google Scholar] [CrossRef]
- Gillam, J.E.; MacPhee, C.E. Modelling amyloid fibril formation kinetics: Mechanisms of nucleation and growth. J. Phys. Condens. Matter 2013, 25, 373101. [Google Scholar] [CrossRef]
- Lashuel, H.A.; Petre, B.M.; Wall, J.; Simon, M.; Nowak, R.J.; Walz, T.; Lansbury, P.T., Jr. Alpha-synuclein, especially the Parkinson’s disease-associated mutants, forms pore-like annular and tubular protofibrils. J. Mol. Biol. 2002, 322, 1089–1102. [Google Scholar] [CrossRef]
- Ingelsson, M. Alpha-synuclein oligomers-neurotoxic molecules in Parkinson’s disease and other Lewy body disorders. Front. Neurosci. 2016, 10, 408. [Google Scholar] [CrossRef] [PubMed]
- El-Agnaf, O.M.; Paleologou, K.E.; Greer, B.; Abogrein, A.M.; King, J.E.; Salem, S.A.; Fullwood, N.J.; Benson, F.E.; Hewitt, R.; Ford, K.J.; et al. A strategy for designing inhibitors of alpha-synuclein aggregation and toxicity as a novel treatment for Parkinson’s disease and related disorders. FASEB J. 2004, 18, 1315–1317. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.S.; Lim, D.; Kim, J.Y.; Kang, S.J.; Kim, Y.H.; Im, H. Beta-sheet-breaking peptides inhibit the fibrillation of human alpha-synuclein. Biochem. Biophys. Res. Commun. 2009, 387, 682–687. [Google Scholar] [CrossRef] [PubMed]
- Bieschke, J.; Russ, J.; Friedrich, R.P.; Ehrnhoefer, D.E.; Wobst, H.; Neugebauer, K.; Wanker, E.E. EGCG remodels mature alpha-synuclein and amyloid-beta fibrils and reduces cellular toxicity. Proc. Natl. Acad. Sci. USA 2010, 107, 7710–7715. [Google Scholar] [CrossRef]
- Ahsan, N.; Mishra, S.; Jain, M.K.; Surolia, A.; Gupta, S. Curcumin pyrazole and its derivative (N-(3-Nitrophenylpyrazole) curcumin inhibit aggregation, disrupt fibrils and modulate toxicity of wild type and mutant alpha-synuclein. Sci. Rep. 2015, 5, 9862. [Google Scholar] [CrossRef]
- Iljina, M.; Hong, L.; Horrocks, M.H.; Ludtmann, M.H.; Choi, M.L.; Hughes, C.D.; Ruggeri, F.S.; Guilliams, T.; Buell, A.K.; Lee, J.E.; et al. Nanobodies raised against monomeric a-synuclein inhibit fibril formation and destabilize toxic oligomeric species. BMC Biol. 2017, 15, 57. [Google Scholar] [CrossRef]
- Li, X.; Koudstaal, W.; Fletcher, L.; Costa, M.; van Winsen, M.; Siregar, B.; Inganas, H.; Kim, J.; Keogh, E.; Macedo, J.; et al. Naturally occurring antibodies isolated from PD patients inhibit synuclein seeding in vitro and recognize Lewy pathology. Acta Neuropathol. 2019, 137, 825–836. [Google Scholar] [CrossRef]
- Kim, D.; Yoo, J.M.; Hwang, H.; Lee, J.; Lee, S.H.; Yun, S.P.; Park, M.J.; Lee, M.; Choi, S.; Kwon, S.H.; et al. Graphene quantum dots prevent alpha-synucleinopathy in Parkinson’s disease. Nat. Nanotechnol. 2018, 13, 812–818. [Google Scholar] [CrossRef]
- Sun, Y.; Kakinen, A.; Zhang, C.; Yang, Y.; Faridi, A.; Davis, T.P.; Cao, W.; Ke, P.C.; Ding, F. Amphiphilic surface chemistry of fullerenols is necessary for inhibiting the amyloid aggregation of alpha-synuclein NACore. Nanoscale 2019, 11, 11933–11945. [Google Scholar] [CrossRef]
- Venda, L.L.; Cragg, S.J.; Buchman, V.L.; Wade-Martins, R. Alpha-synuclein and dopamine at the crossroads of Parkinson’s disease. Trends Neurosci. 2010, 33, 559–568. [Google Scholar] [CrossRef]
- Bisaglia, M.; Filograna, R.; Beltramini, M.; Bubacco, L. Are dopamine derivatives implicated in the pathogenesis of Parkinson’s disease? Ageing Res. Rev. 2014, 13, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Delaville, C.; Deurwaerdere, P.D.; Benazzouz, A. Noradrenaline and Parkinson’s disease. Front. Syst. Neurosci. 2011, 5, 31. [Google Scholar] [CrossRef] [PubMed]
- Espay, A.J.; LeWitt, P.A.; Kaufmann, H. Norepinephrine deficiency in Parkinson’s disease: The case for noradrenergic enhancement. Mov. Disord. 2014, 29, 1710–1719. [Google Scholar] [CrossRef] [PubMed]
- Conway, K.A.; Rochet, J.C.; Bieganski, R.M.; Lansbury, P.T., Jr. Kinetic stabilization of the alpha-synuclein protofibril by a dopamine-alpha-synuclein adduct. Science 2001, 294, 1346–1349. [Google Scholar] [CrossRef]
- Li, J.; Zhu, M.; Manning-Bog, A.B.; Di Monte, D.A.; Fink, A.L. Dopamine and L-dopa disaggregate amyloid fibrils: Implications for Parkinson’s and Alzheimer’s disease. FASEB J. 2004, 18, 962–964. [Google Scholar] [CrossRef]
- Cappai, R.; Leck, S.L.; Tew, D.J.; Williamson, N.A.; Smith, D.P.; Galatis, D.; Sharples, R.A.; Curtain, C.C.; Ali, F.E.; Cherny, R.A.; et al. Dopamine promotes alpha-synuclein aggregation into SDS-resistant soluble oligomers via a distinct folding pathway. FASEB J. 2005, 19, 1377–1379. [Google Scholar] [CrossRef]
- Herrera, F.E.; Chesi, A.; Paleologou, K.E.; Schmid, A.; Munoz, A.; Vendruscolo, M.; Gustincich, S.; Lashuel, H.A.; Carloni, P. Inhibition of alpha-synuclein fibrillization by dopamine is mediated by interactions with five C-terminal residues and with E83 in the NAC region. PLoS ONE 2008, 3, e3394. [Google Scholar] [CrossRef]
- Latawiec, D.; Herrera, F.; Bek, A.; Losasso, V.; Candotti, M.; Benetti, F.; Carlino, E.; Kranjc, A.; Lazzarino, M.; Gustincich, S.; et al. Modulation of alpha-synuclein aggregation by dopamine analogs. PLoS ONE 2010, 5, e9234. [Google Scholar] [CrossRef]
- Ono, K.; Takasaki, J.; Takahashi, R.; Ikeda, T.; Yamada, M. Effects of antiparkinsonian agents on beta-amyloid and alpha-synuclein oligomer formation in vitro. J. Neurosci. Res. 2013, 91, 1371–1381. [Google Scholar] [CrossRef]
- Tavassoly, O.; Nokhrin, S.; Dmitriev, O.Y.; Lee, J.S. Cu(II) and dopamine bind to alpha-synuclein and cause large conformational changes. FEBS J. 2014, 281, 2738–2753. [Google Scholar] [CrossRef]
- Jain, M.K.; Bhat, R. Modulation of human alpha-synuclein aggregation by a combined effect of calcium and dopamine. Neurobiol. Dis. 2014, 63, 115–128. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A.F.; Matera, K.M. Stabilization of alpha-synuclein oligomers in vitro by the neurotransmitters, dopamine and norepinephrine: The effect of oxidized catecholamines. Neurochem. Res. 2015, 40, 1341–1349. [Google Scholar] [CrossRef] [PubMed]
- Jha, N.N.; Kumar, R.; Panigrahi, R.; Navalkar, A.; Ghosh, D.; Sahay, S.; Mondal, M.; Kumar, A.; Maji, S.K. Comparison of alpha-synuclein fibril inhibition by four different amyloid inhibitors. ACS Chem. Neurosci. 2017, 8, 2722–2733. [Google Scholar] [CrossRef]
- Fink, A.L. The aggregation and fibrillation of alpha-synuclein. Acc. Chem. Res. 2006, 39, 628–634. [Google Scholar] [CrossRef]
- Illes-Toth, E.; Dalton, C.F.; Smith, D.P. Binding of dopamine to alpha-synuclein is mediated by specific conformational states. J. Am. Soc. Mass Spectrom. 2013, 24, 1346–1354. [Google Scholar] [CrossRef] [Green Version]
- Pham, C.L.; Cappai, R. The interplay between lipids and dopamine on alpha-synuclein oligomerization and membrane binding. Biosci. Rep. 2013, 33, e00074. [Google Scholar] [CrossRef]
- Singh, P.; Bhat, R. Binding of noradrenaline to native and intermediate states during the fibrillation of alpha-synuclein leads to the formation of stable and structured cytotoxic species. ACS Chem. Neurosci. 2019, 10, 2741–2755. [Google Scholar] [CrossRef]
- Khalife, M.; Morshedi, D.; Aliakbari, F.; Tayaranian Marvian, A.; Mohammad Beigi, H.; Azimzadeh Jamalkandi, S.; Pan-Montojo, F. Alpha-synuclein fibrils interact with dopamine reducing its cytotoxicity on PC12 cells. Protein J. 2015, 34, 291–303. [Google Scholar] [CrossRef]
- Tuttle, M.D.; Comellas, G.; Nieuwkoop, A.J.; Covell, D.J.; Berthold, D.A.; Kloepper, K.D.; Courtney, J.M.; Kim, J.K.; Barclay, A.M.; Kendall, A.; et al. Solid-state NMR structure of a pathogenic fibril of full-length human alpha-synuclein. Nat. Struct. Mol. Biol. 2016, 23, 409–415. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Masuda, M.; Sasakawa, H.; Nonaka, T.; Hanashima, S.; Hisanaga, S.; Kato, K.; Hasegawa, M. Characterization of inhibitor-bound alpha-synuclein dimer: Role of alpha-synuclein N-terminal region in dimerization and inhibitor binding. J. Mol. Biol. 2010, 395, 445–456. [Google Scholar] [CrossRef]
- Dibenedetto, D.; Rossetti, G.; Caliandro, R.; Carloni, P. A molecular dynamics simulation-based interpretation of nuclear magnetic resonance multidimensional heteronuclear spectra of alpha-synuclein.dopamine adducts. Biochemistry 2013, 52, 6672–6683. [Google Scholar] [CrossRef] [PubMed]
- Corvaglia, S.; Sanavio, B.; Hong Enriquez, R.P.; Sorce, B.; Bosco, A.; Scaini, D.; Sabella, S.; Pompa, P.P.; Scoles, G.; Casalis, L. Atomic force microscopy based nanoassay: A new method to study alpha-Synuclein-dopamine bioaffinity interactions. Sci. Rep. 2014, 4, 5366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ponzio, F.; Hallman, H.; Jonsson, G. Noradrenaline and dopamine interaction in rat brain during development. Med. Biol. 1981, 59, 161–169. [Google Scholar] [PubMed]
- Colpaert, F.C. Pharmacological characteristics of tremor, rigidity and hypokinesia induced by reserpine in rat. Neuropharmacology 1987, 26, 1431–1440. [Google Scholar] [CrossRef]
- Taylor, T.N.; Caudle, W.M.; Shepherd, K.R.; Noorian, A.; Jackson, C.R.; Iuvone, P.M.; Weinshenker, D.; Greene, J.G.; Miller, G.W. Nonmotor symptoms of Parkinson’s disease revealed in an animal model with reduced monoamine storage capacity. J. Neurosci. 2009, 29, 8103–8113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schüttelkopf, A.W.; Van Aalten, D.M.F. PRODRG: A tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 1355–1363. [Google Scholar] [CrossRef] [Green Version]
- Shao, Y.; Molnar, L.; Jung, Y.; Kussmann, J.; Ochsenfeld, C.; Brown, S.; Gilbert, A.; Slipchenko, L.; Levchenko, S.; O’Neill, D.; et al. Advances in methods and algorithms in a modern quantum chemistry program package. Phys. Chem. Chem. Phys. 2006, 8, 3172–3191. [Google Scholar] [CrossRef]
- Schmidt, M.W.; Baldridge, K.K.; Boatz, J.A.; Elbert, S.T.; Gordon, M.S.; Jensen, J.H.; Koseki, S.; Matsunaga, N.; Nguyen, K.A.; Su, S. General atomic and molecular electronic structure system. J. Comput. Chem. 1993, 14, 1347–1363. [Google Scholar] [CrossRef]
- François-Yves, D.; Adrien, P.; Thomas, Z.; Corentin, S.; Rodolphe, L.; Nicolas, G.; Dimitri, L.; Wilfried, R.; Piotr, C. The R.E.D. tools: Advances in RESP and ESP charge derivation and force field library building. Phys. Chem. Chem. Phys. 2010, 12, 7821–7839. [Google Scholar]
- Corona-Avendano, S.; Alarcon-Angeles, G.; Rosquete-Pina, G.A.; Rojas-Hernandez, A.; Gutierrez, A.; Ramirez-Silva, M.T.; Romero-Romo, M.; Palomar-Pardave, M. New insights on the nature of the chemical species involved during the process of dopamine deprotonation in aqueous solution: Theoretical and experimental study. J. Phys. Chem. B 2007, 111, 1640–1647. [Google Scholar] [CrossRef]
- Farley, I.J.; Hornykiewicz, O. Noradrenaline distribution insubcortical areas of the human brain. Brain Res. 1977, 126, 53–62. [Google Scholar] [CrossRef]
- Hardy, J.A.; Wester, P.; Backstrom, I.; Gottfries, J.; Oreland, L.; Stenstrom, A.; Winblad, B. The regional distribution of dopamine and serotonin uptake and transmitter concentrations in the human brain. Neurochem. Int. 1987, 10, 445–450. [Google Scholar] [CrossRef]
- Van, D.S.D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins 2010, 78, 1950–1958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.; Fraaije, J.G. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Miyamoto, S.; Kollman, P.A. Settle: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 1998, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef] [PubMed]
- Barlow, D.J.; Thornton, J.M. Ion-pairs in proteins. J. Mol. Biol. 1983, 168, 867–885. [Google Scholar] [CrossRef]
- Kumar, S.; Nussinov, R. Salt bridge stability in monomeric proteins. J. Mol. Biol. 1999, 293, 1241–1255. [Google Scholar] [CrossRef]
- Kumar, S.; Nussinov, R. Close-range electrostatic interactions in proteins. ChemBioChem 2002, 3, 604–617. [Google Scholar] [CrossRef]
- Luo, Y.; Ma, B.; Nussinov, R.; Wei, G. Structural insight into tau protein’s paradox of intrinsically disordered behavior, self-acetylation activity, and aggregation. J. Phys. Chem. Lett. 2014, 5, 3026–3031. [Google Scholar] [CrossRef] [PubMed]
- Qian, Z.; Zou, Y.; Zhang, Q.; Chen, P.; Ma, B.; Wei, G.; Nussinov, R. Atomistic-level study of the interactions between hIAPP protofibrils and membranes: Influence of pH and lipid composition. Biochim. Biophys. Acta Biomembr. 2018, 1860, 1818–1825. [Google Scholar] [CrossRef] [PubMed]
- Lei, J.; Qi, R.; Tang, Y.; Wang, W.; Wei, G.; Nussinov, R.; Ma, B. Conformational stability and dynamics of the cancer-associated isoform Delta133p53beta are modulated by p53 peptides and p53-specific DNA. FASEB J. 2019, 33, 4225–4235. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Sun, Y.; Chen, Y.; Lei, J.; Wei, G. Molecular dynamics simulations reveal the mechanism of graphene oxide nanosheet inhibition of Abeta1-42 peptide aggregation. Phys. Chem. Chem. Phys. 2019, 21, 10981–10991. [Google Scholar] [CrossRef]
- Liu, Z.; Zou, Y.; Zhang, Q.; Chen, P.; Liu, Y.; Qian, Z. Distinct binding dynamics, sites and interactions of fullerene and fullerenols with amyloid-beta peptides revealed by molecular dynamics simulations. Int. J. Mol. Sci. 2019, 20, 2048. [Google Scholar] [CrossRef] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. Model. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Xu, L.; Nussinov, R.; Ma, B. Coupling of the non-amyloid-component (NAC) domain and the KTK(E/Q)GV repeats stabilize the alpha-synuclein fibrils. Eur. J. Med. Chem. 2016, 121, 841–850. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Bhattacharya, S.; Thompson, D. The fold preference and thermodynamic stability of alpha-synuclein fibrils is encoded in the non-amyloid-beta component region. Phys. Chem. Chem. Phys. 2018, 20, 4502–4512. [Google Scholar] [CrossRef] [PubMed]
- Romo, T.D.; Lewis, A.K.; Braun, A.R.; Grossfield, A.; Sachs, J.N. Minimal Nucleation State of alpha-Synuclein Is Stabilized by Dynamic Threonine-Water Networks. ACS Chem. Neurosci. 2017, 8, 1859–1864. [Google Scholar] [CrossRef] [PubMed]
- Giasson, B.I.; Murray, I.V.; Trojanowski, J.Q.; Lee, V.M. A hydrophobic stretch of 12 amino acid residues in the middle of alpha-synuclein is essential for filament assembly. J. Biol. Chem. 2001, 276, 2380–2386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bedard, L.; Lefevre, T.; Morin-Michaud, E.; Auger, M. Besides fibrillization: Putative role of the peptide fragment 71-82 on the structural and assembly behavior of alpha-synuclein. Biochemistry 2014, 53, 6463–6472. [Google Scholar] [CrossRef] [PubMed]
- Sanjeev, A.; Mattaparthi, V.S.K. Investigation on the molecular interactions stabilizing the structure of alpha-synuclein fibril: An in silico study. Cent. Nerv. Syst. Agents Med. Chem. 2017, 17, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Qian, Z.; Zhang, Q.; Liu, Y.; Chen, P. Assemblies of amyloid-beta30-36 hexamer and its G33V/L34T mutants by replica-exchange molecular dynamics simulation. PLoS ONE 2017, 12, e0188794. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Zhou, S.; Shi, D.; Bai, Q.; Liu, H.; Yao, X. Influence of EGCG on alpha-synuclein (alphaS) aggregation and identification of their possible binding mode: A computational study using molecular dynamics simulation. Chem. Biol. Drug Des. 2018, 91, 162–171. [Google Scholar] [CrossRef]
- Mo, Y.; Brahmachari, S.; Lei, J.; Gilead, S.; Tang, Y.; Gazit, E.; Wei, G. The inhibitory effect of hydroxylated carbon nanotubes on the aggregation of human islet amyloid polypeptide revealed by a combined computational and experimental study. ACS Chem. Neurosci. 2018, 9, 2741–2752. [Google Scholar] [CrossRef]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zou, Y.; Liu, Z.; Zhu, Z.; Qian, Z. Structural Influence and Interactive Binding Behavior of Dopamine and Norepinephrine on the Greek-Key-Like Core of α-Synuclein Protofibril Revealed by Molecular Dynamics Simulations. Processes 2019, 7, 850. https://doi.org/10.3390/pr7110850
Zou Y, Liu Z, Zhu Z, Qian Z. Structural Influence and Interactive Binding Behavior of Dopamine and Norepinephrine on the Greek-Key-Like Core of α-Synuclein Protofibril Revealed by Molecular Dynamics Simulations. Processes. 2019; 7(11):850. https://doi.org/10.3390/pr7110850
Chicago/Turabian StyleZou, Yu, Zhiwei Liu, Zhiqiang Zhu, and Zhenyu Qian. 2019. "Structural Influence and Interactive Binding Behavior of Dopamine and Norepinephrine on the Greek-Key-Like Core of α-Synuclein Protofibril Revealed by Molecular Dynamics Simulations" Processes 7, no. 11: 850. https://doi.org/10.3390/pr7110850