Interaction of Diet and Ozone Exposure on Oxidative Stress Parameters within Specific Brain Regions of Male Brown Norway Rats

Abstract
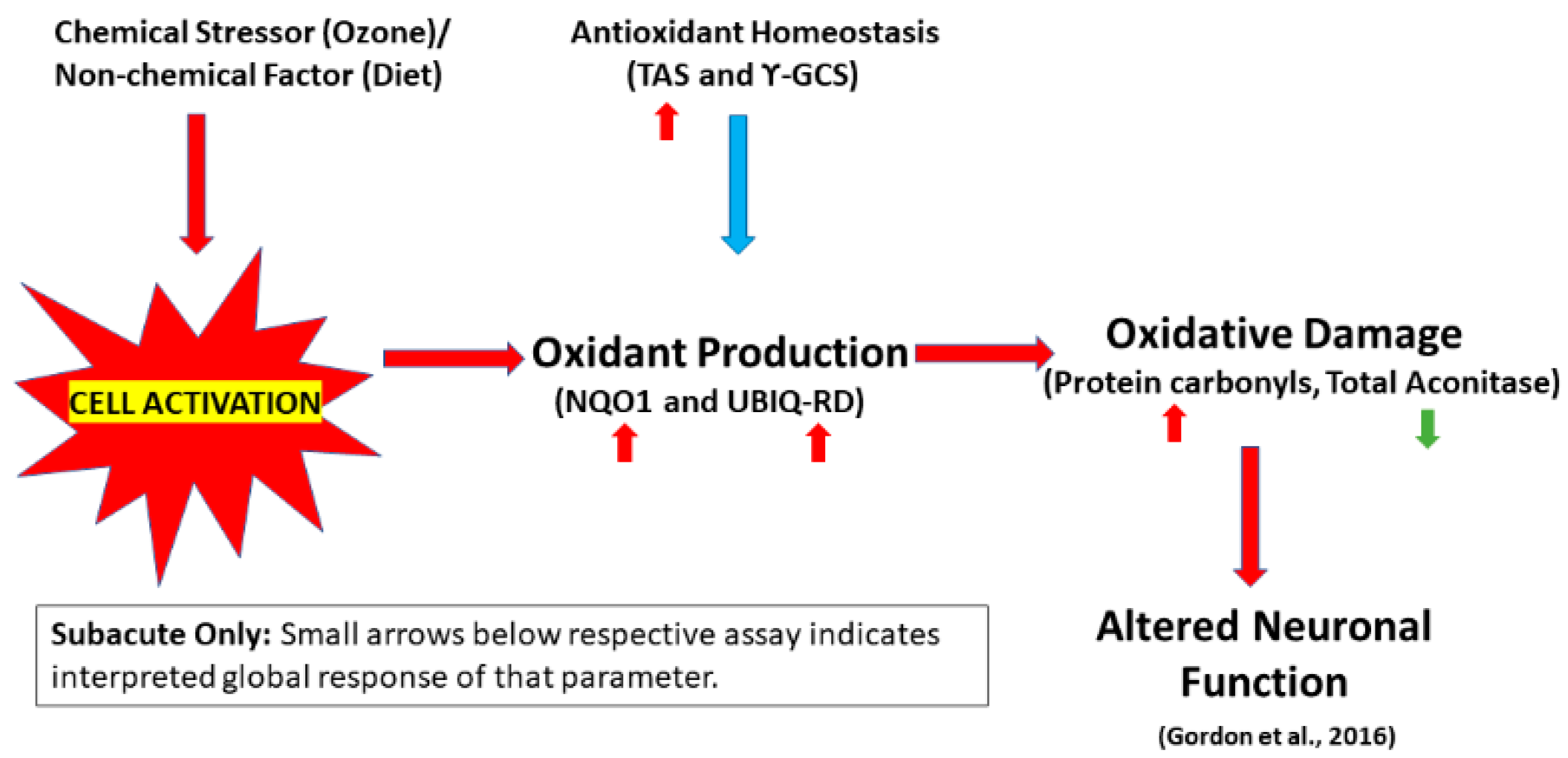
:1. Introduction
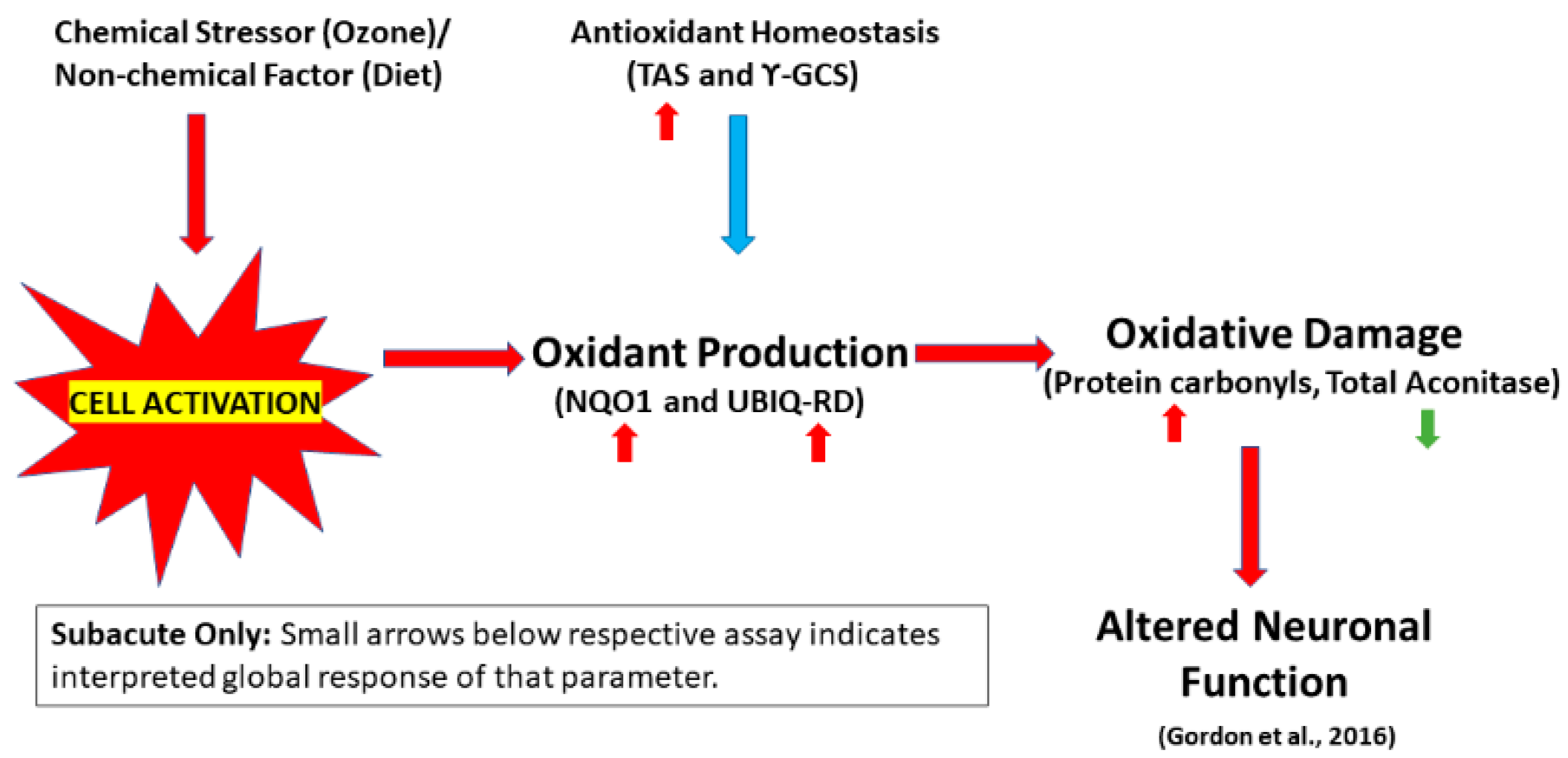
2. Results
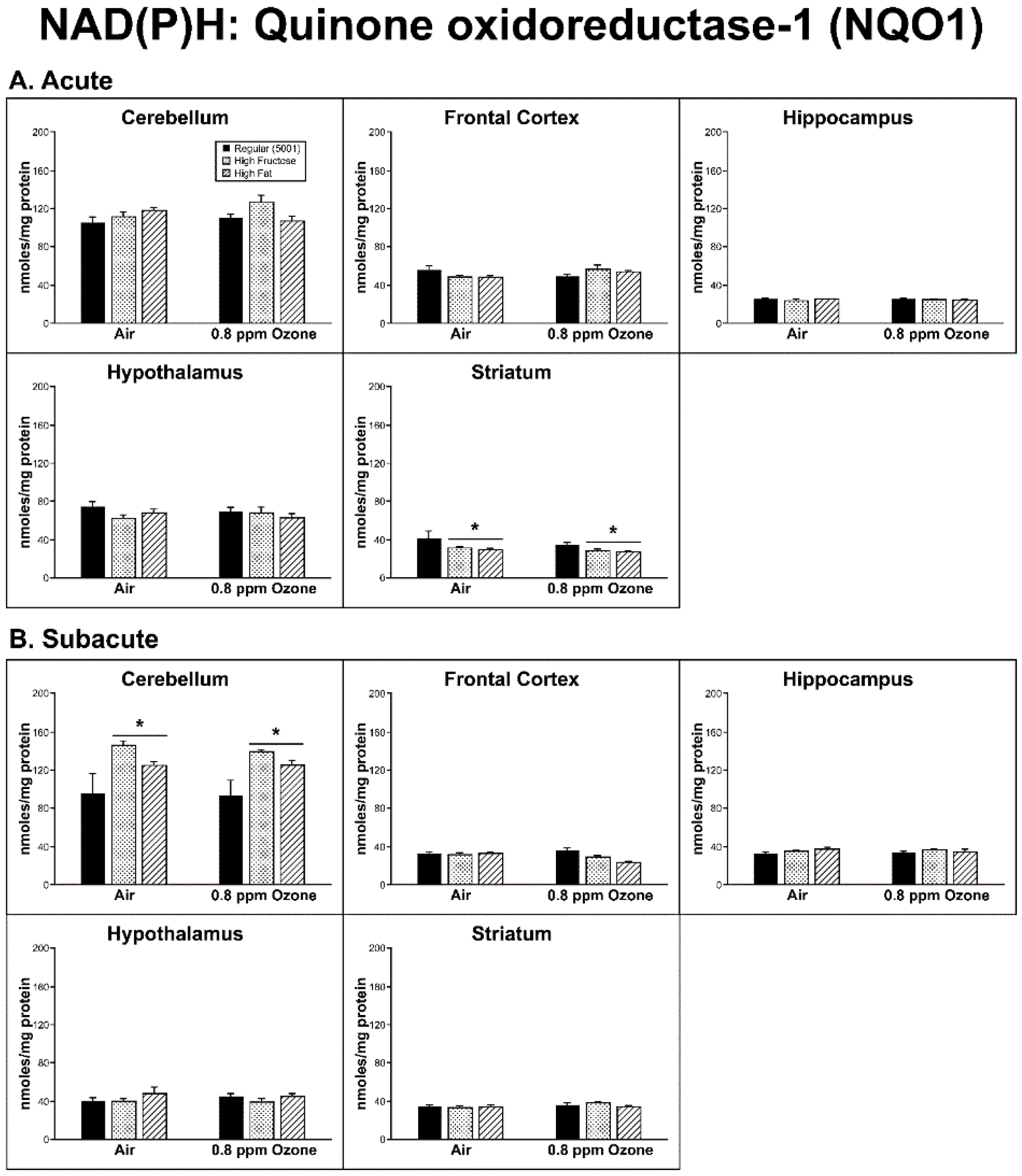
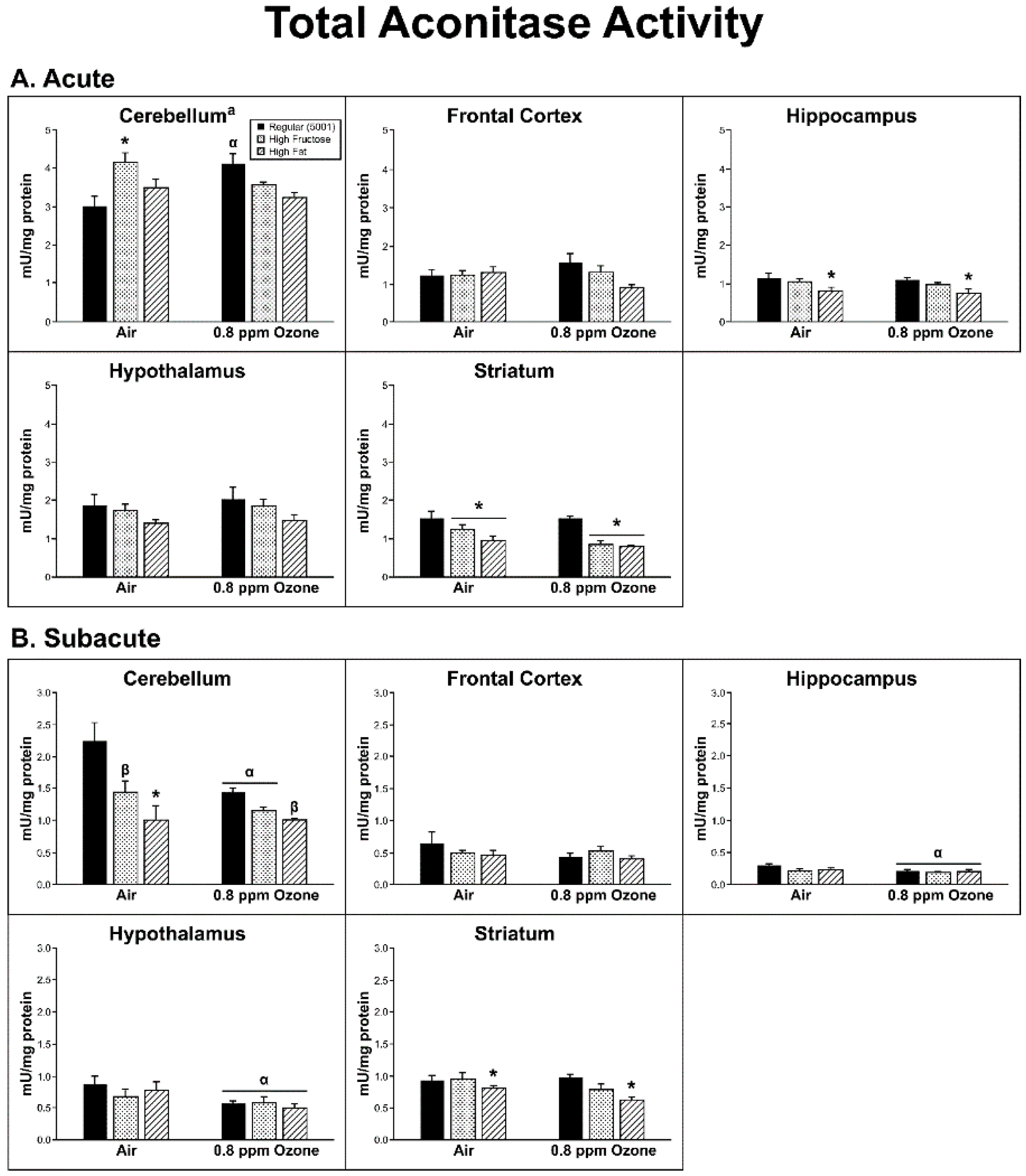
2.1. Production of Reactive Oxygen Species (ROS)
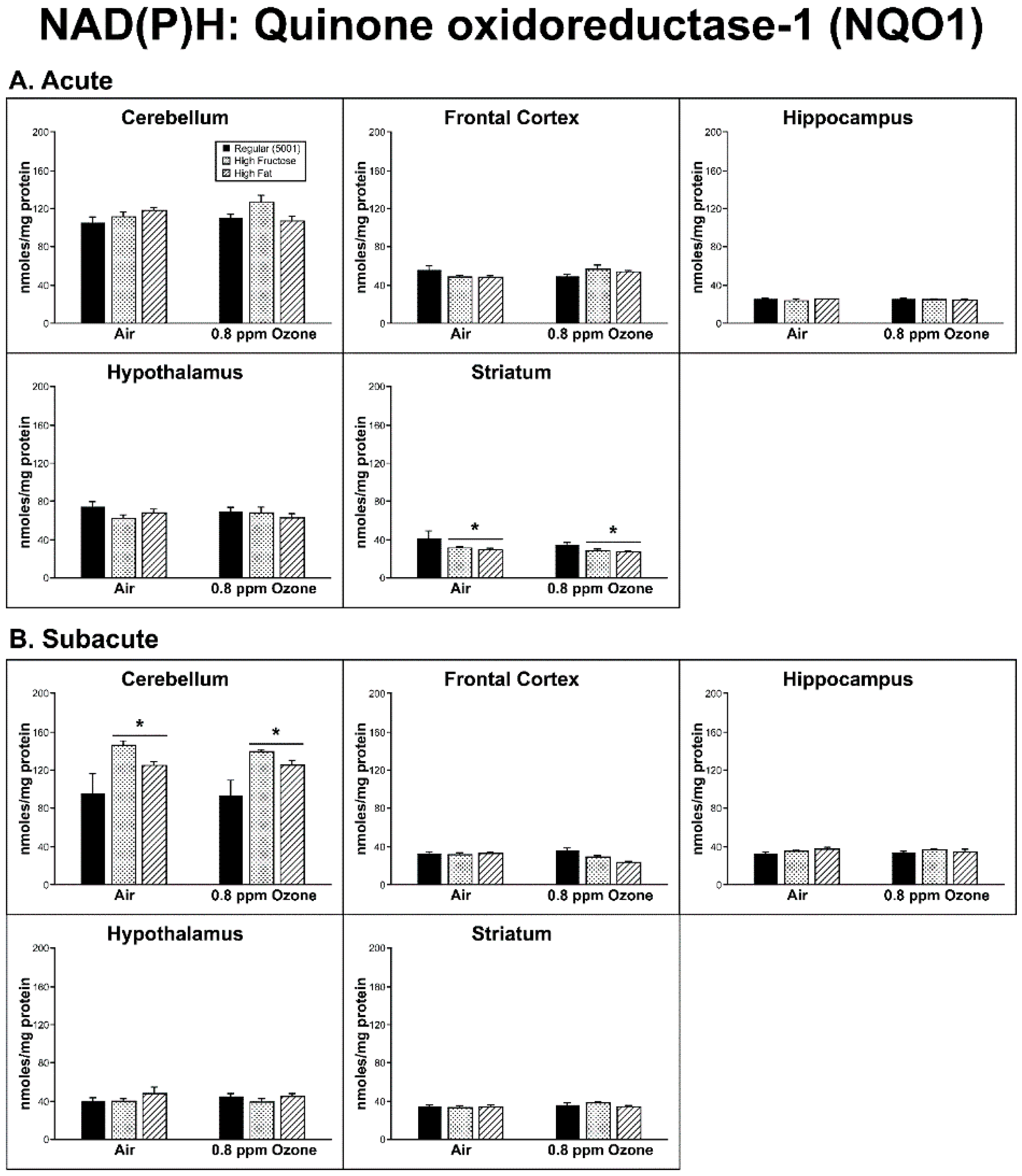
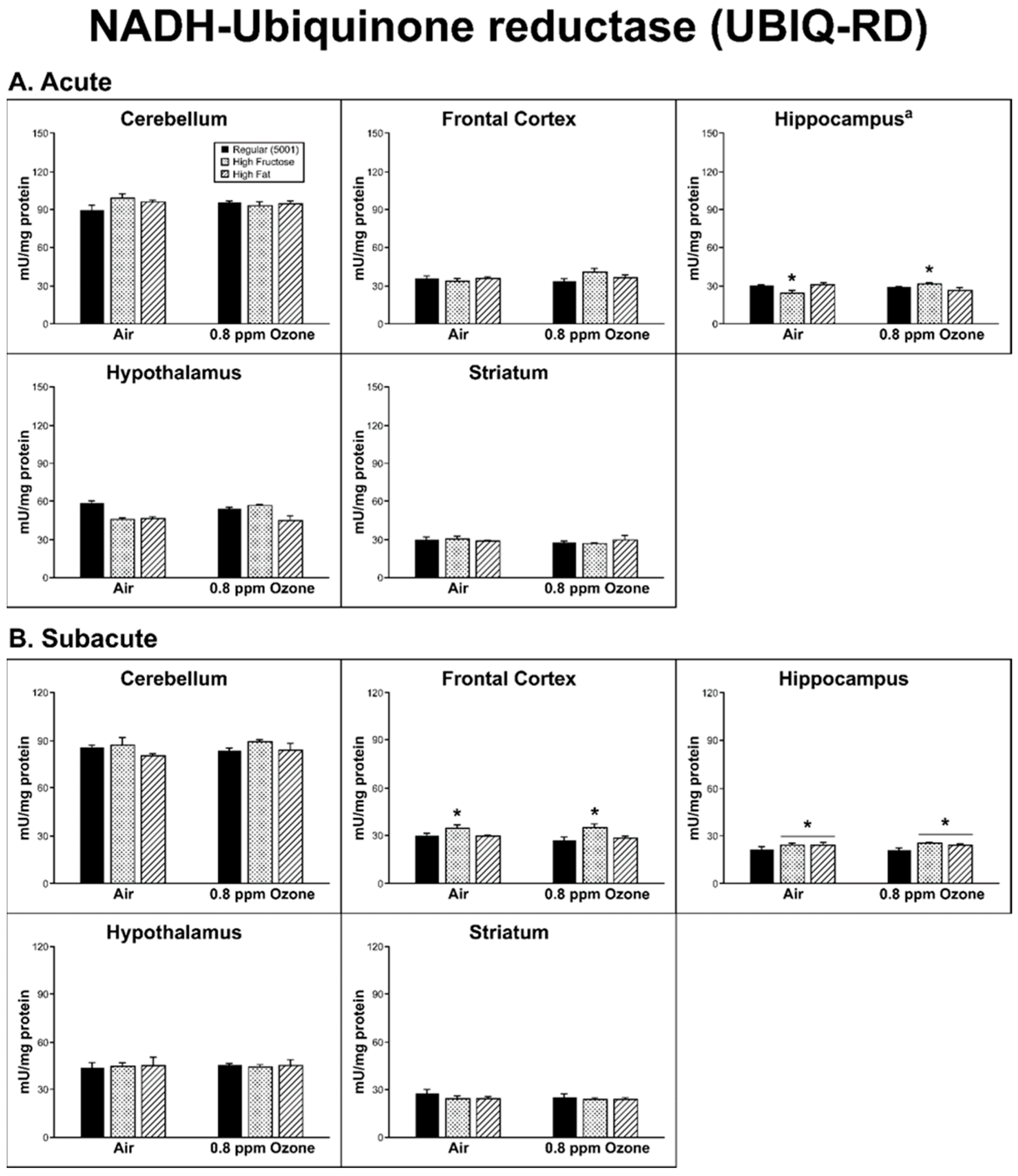
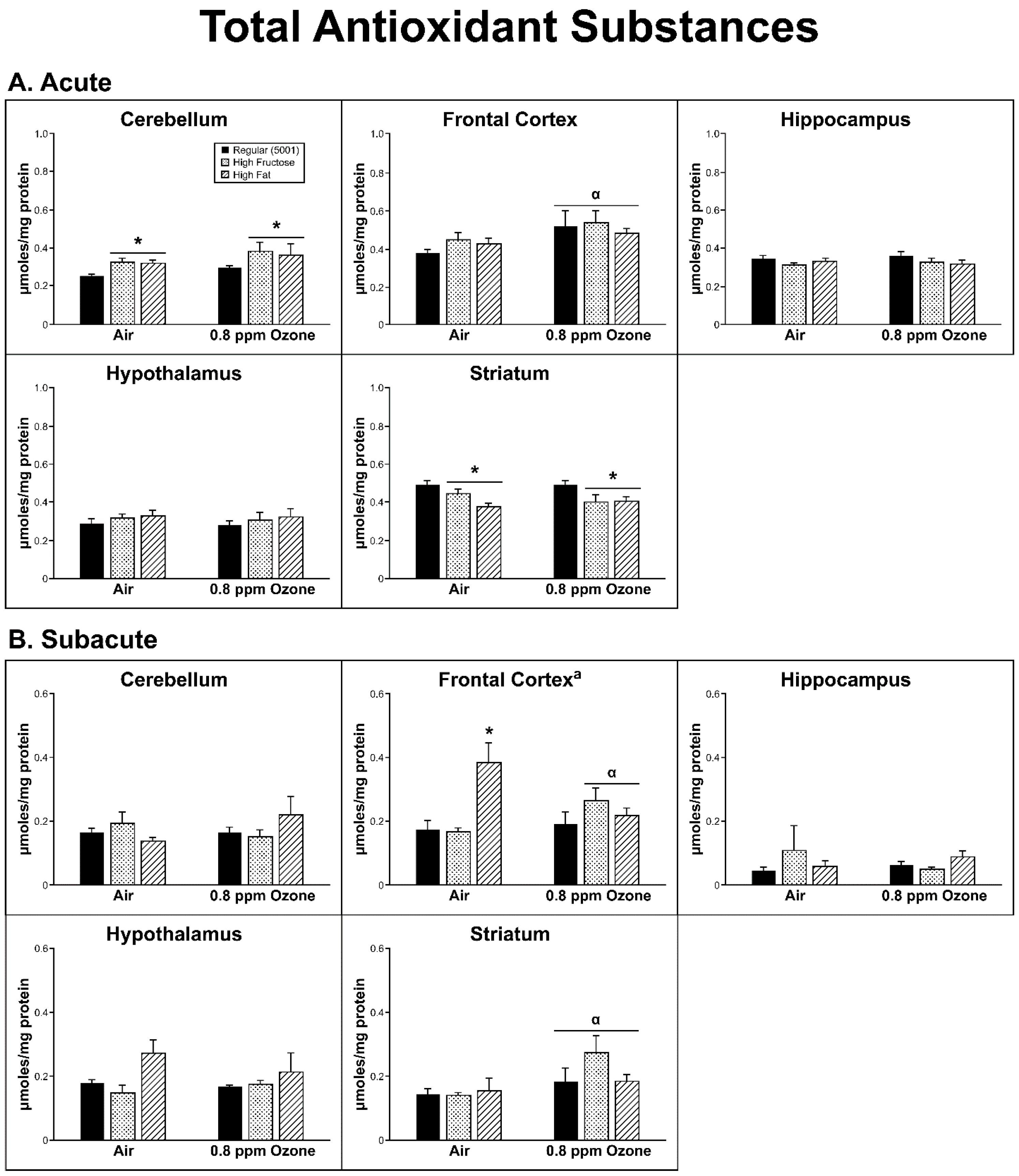
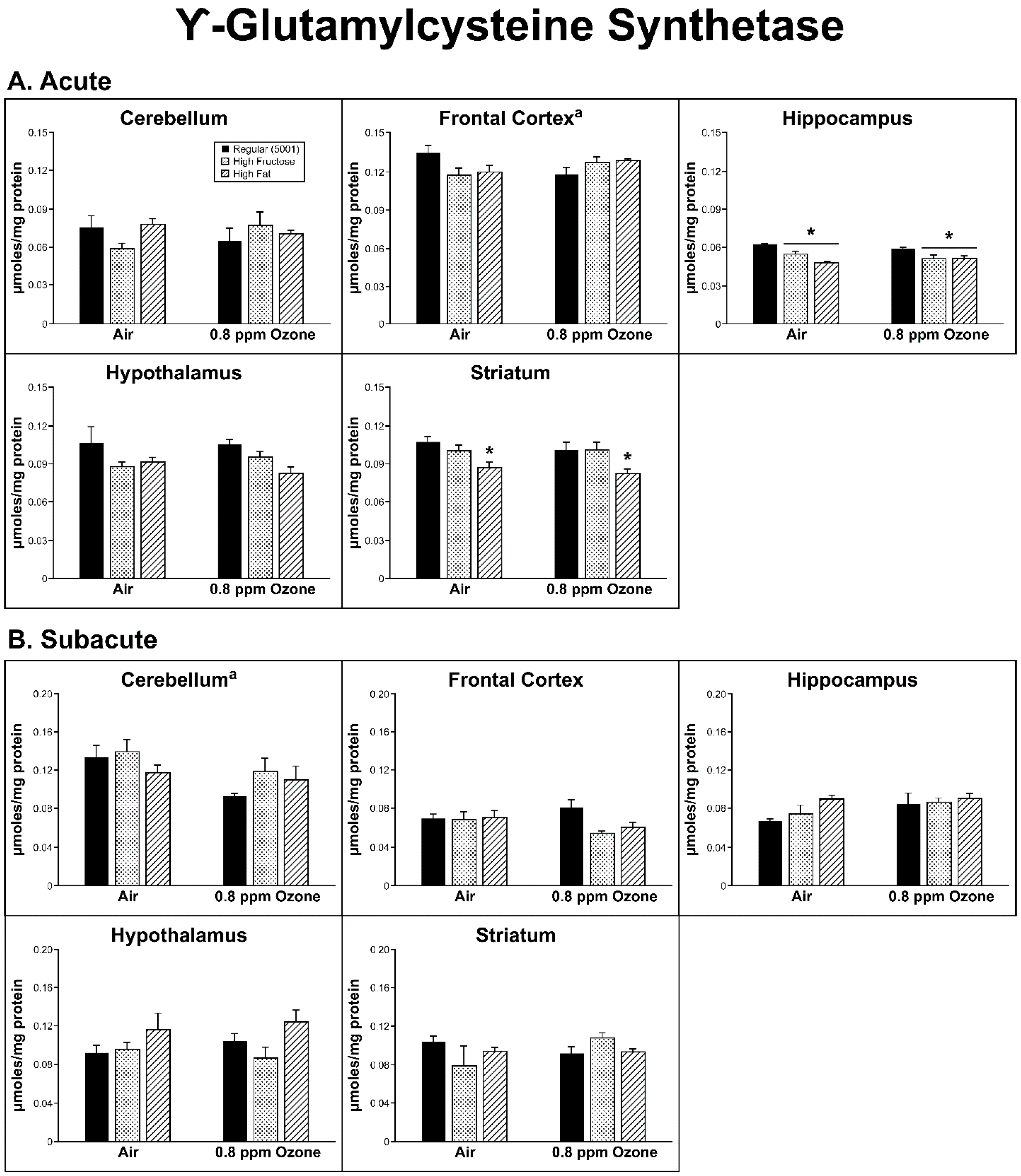
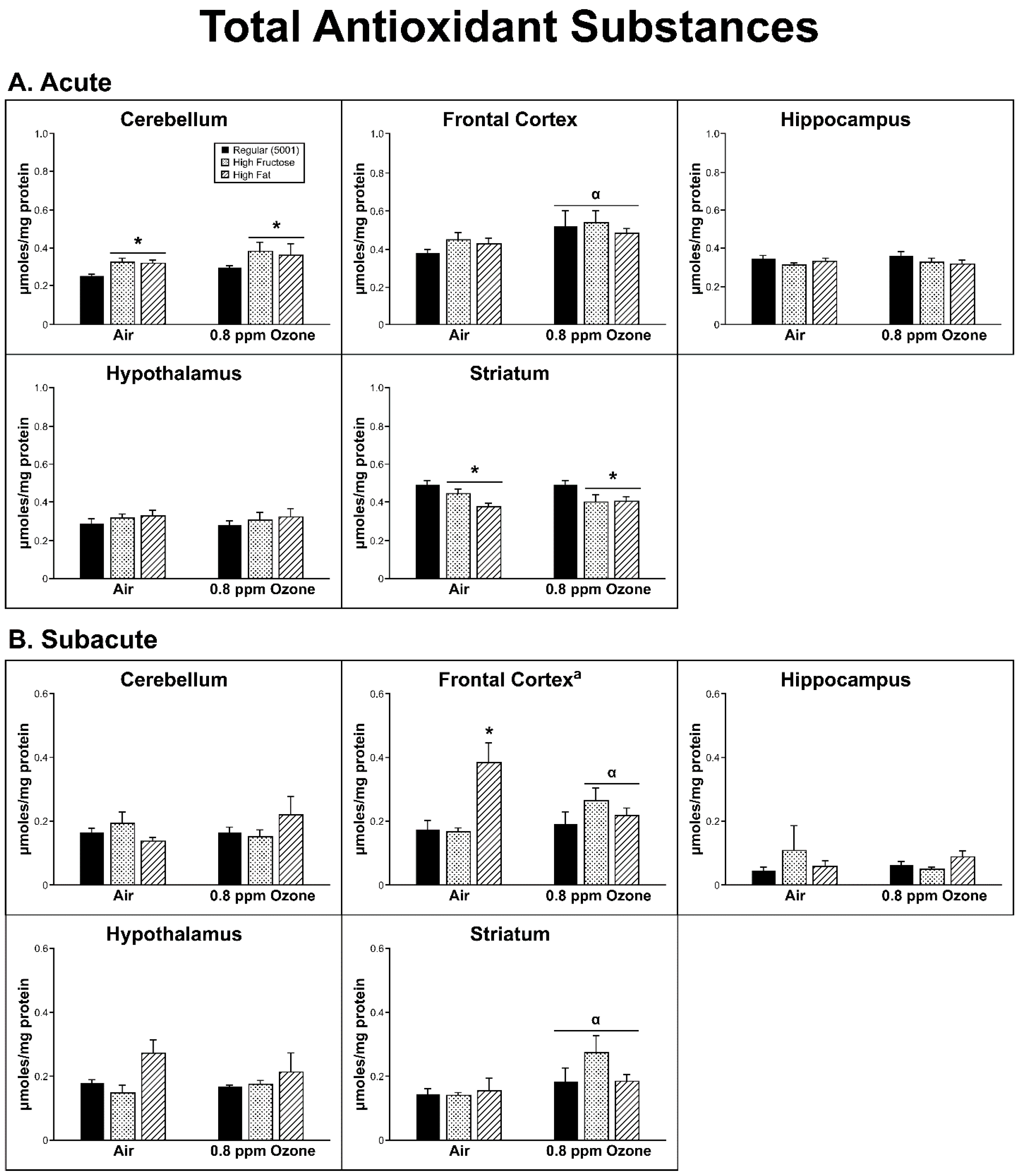
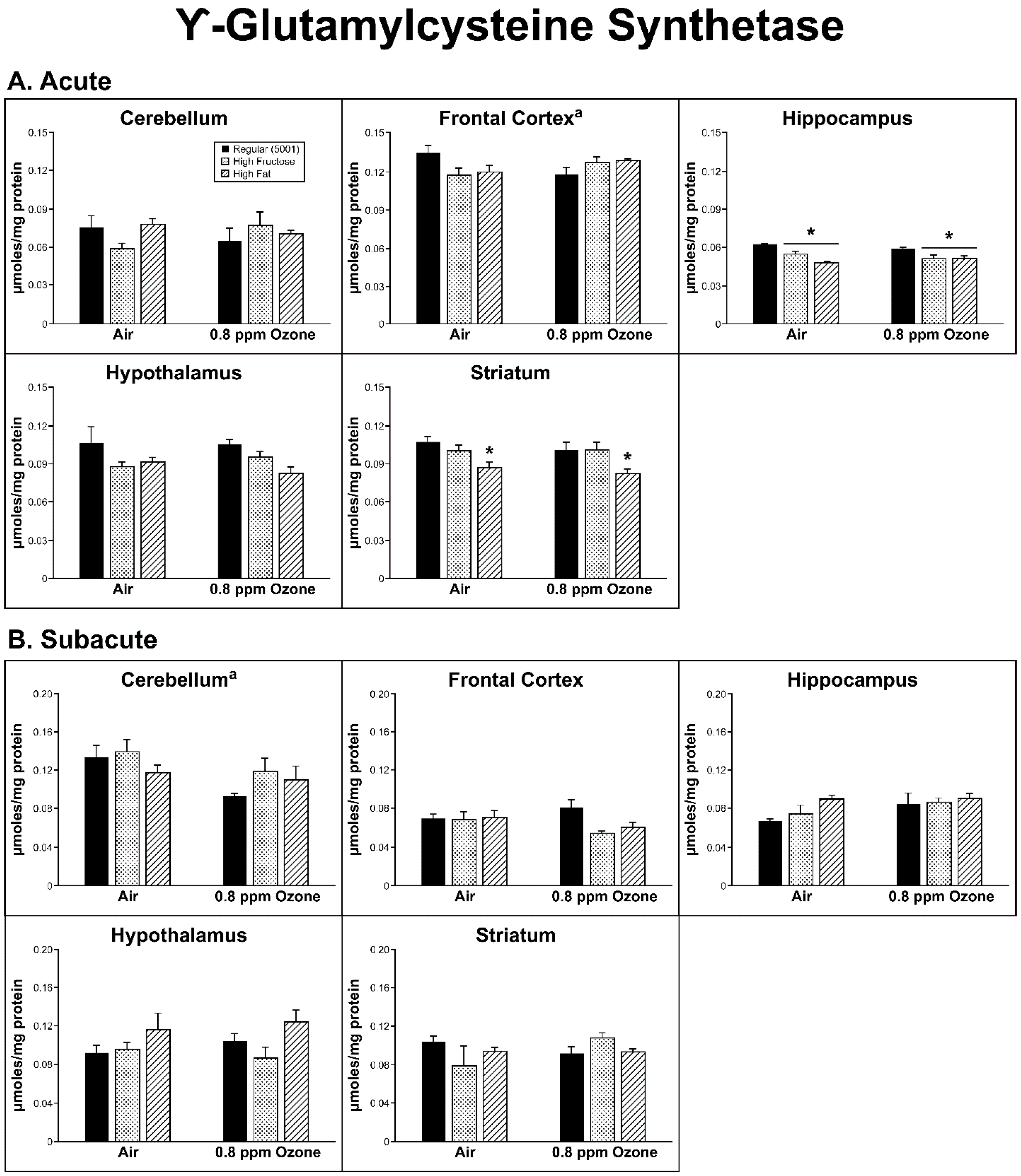
2.2. Antioxidant Homeostasis
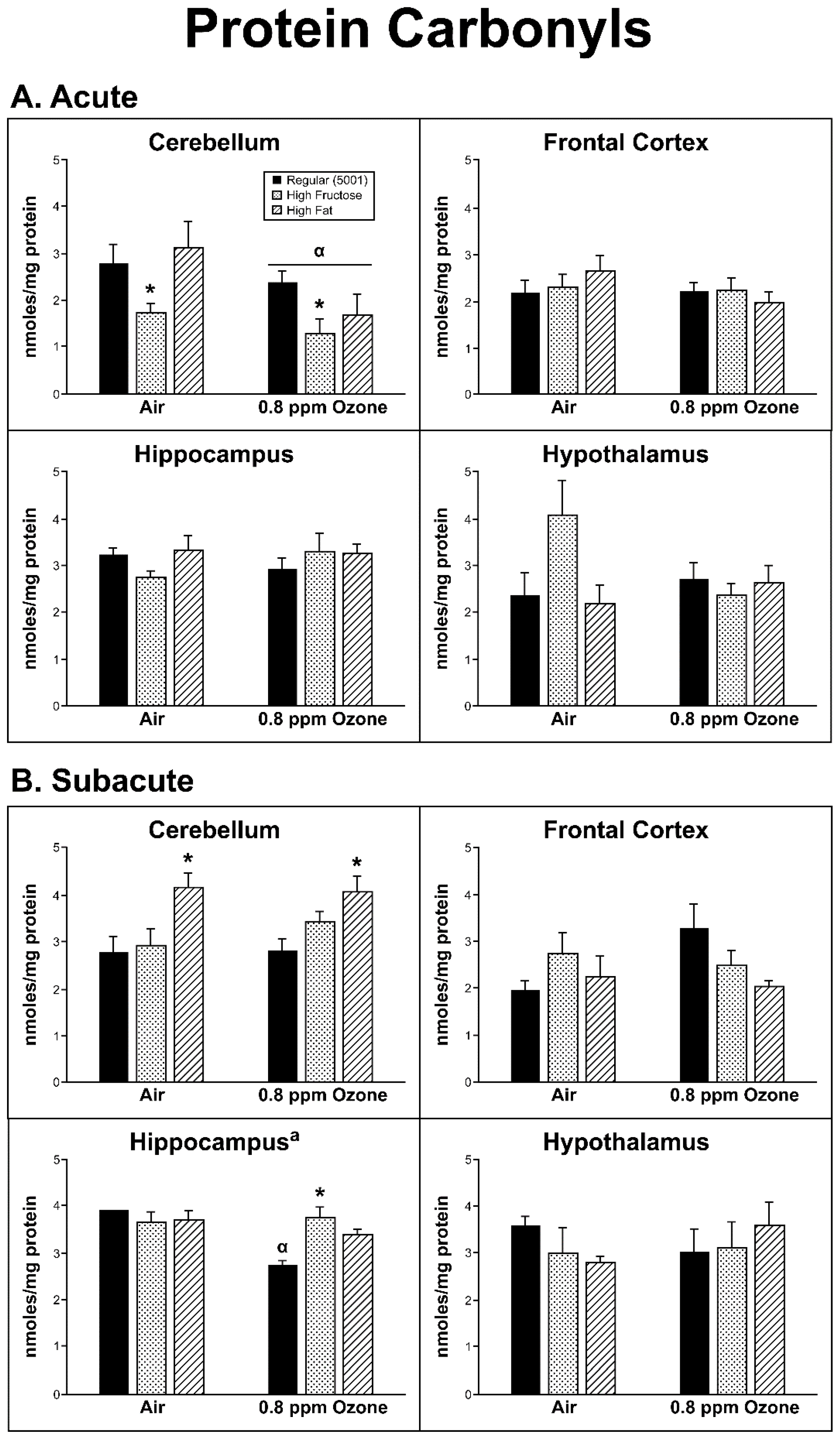
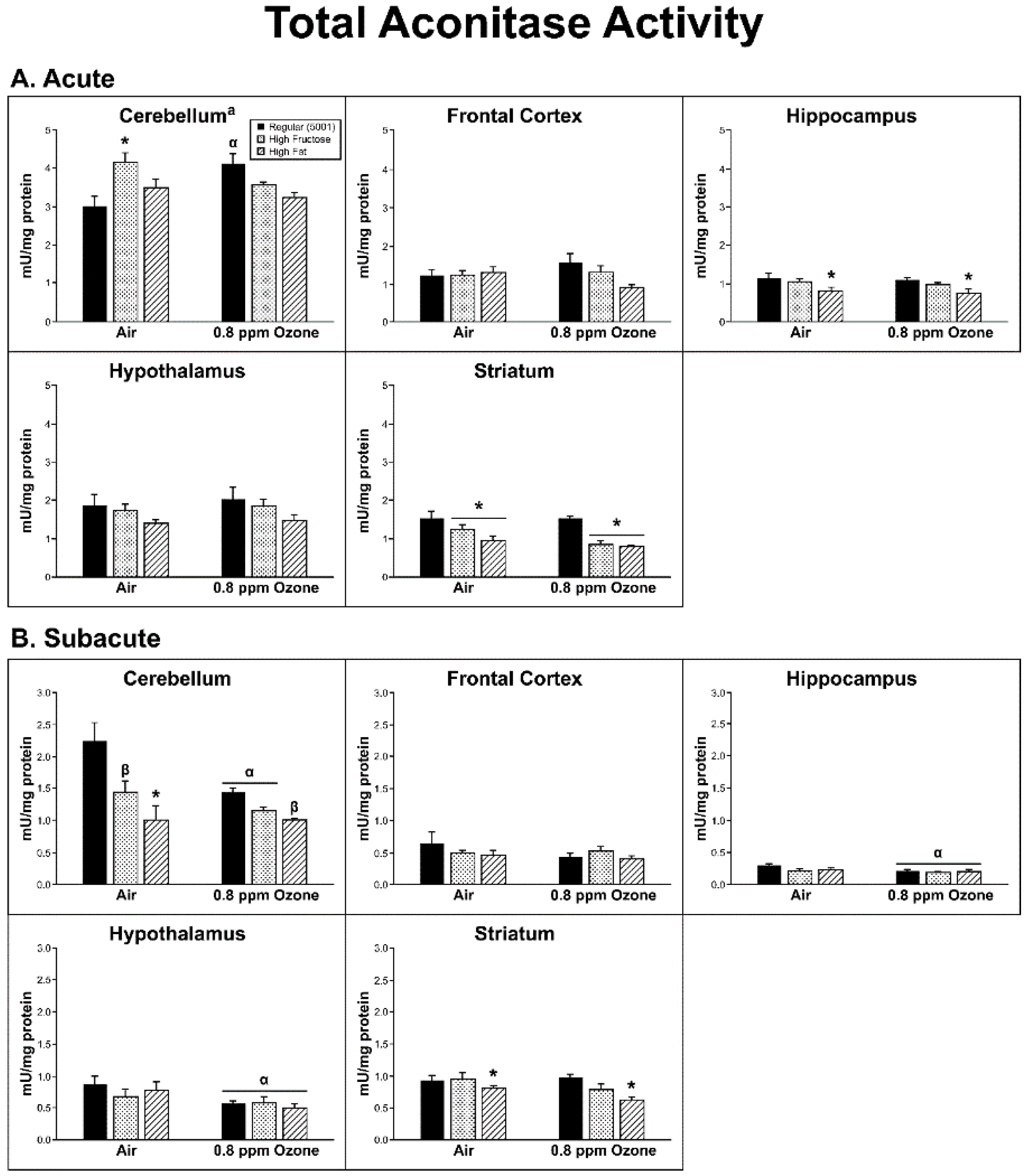
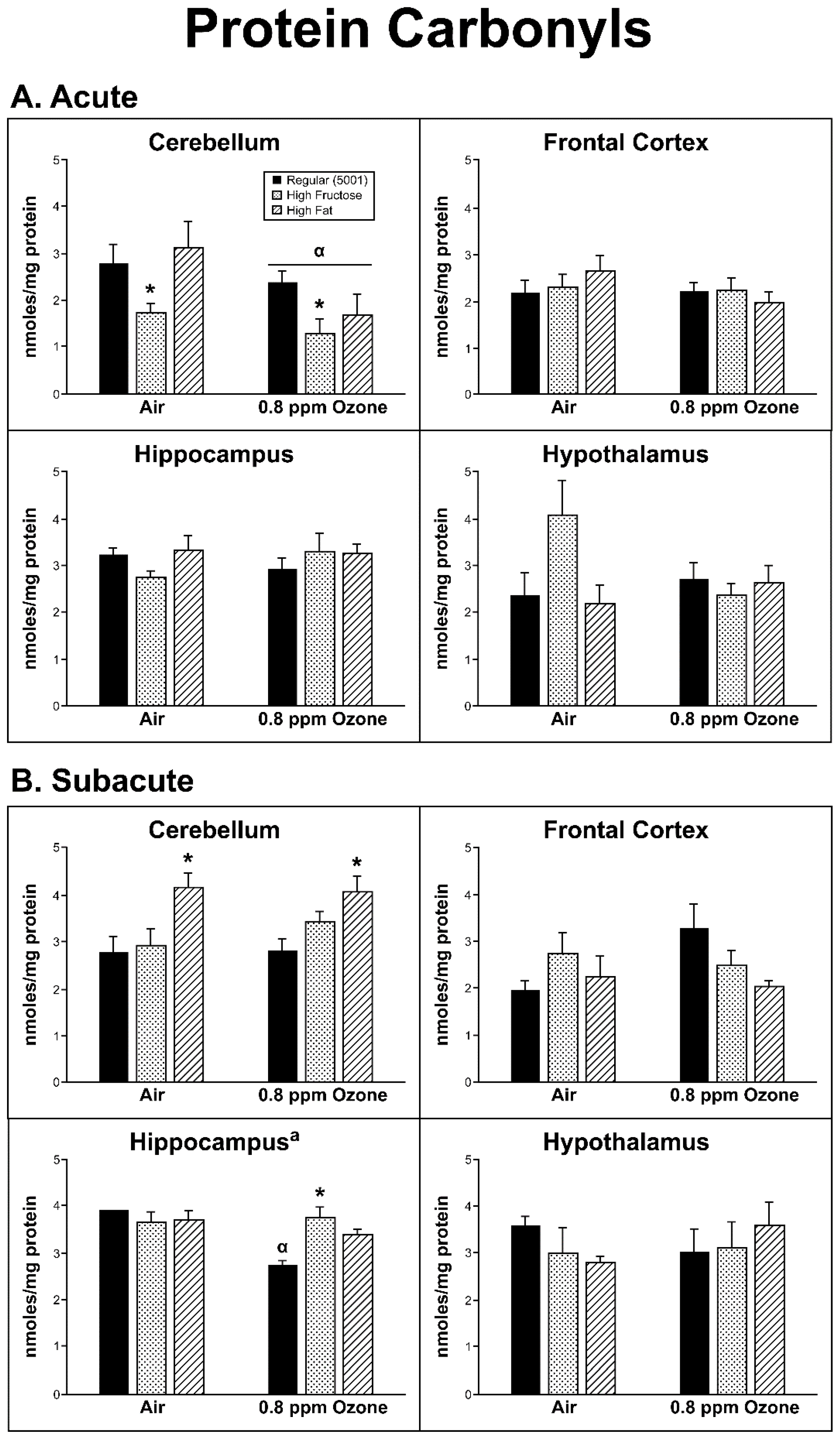
2.3. Oxidative Damage
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Dietary Regimen
4.3. O3 Generation and Exposure
4.4. Necropsy and Tissue Isolation
4.5. Tissue Preparation
4.6. Markers of ROS Production
4.7. Markers of Cellular Antioxidant Homeostasis
4.8. Markers of Oxidative Damage
4.9. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Finucane, M.M.; Stevens, G.A.; Cowan, M.J.; Danaei, G.; Lin, J.K.; Paciorek, C.J.; Singh, G.M.; Gutierrez, H.R.; Lu, Y.; Bahalim, A.N. National, regional, and global trends in body-mass index since 1980: Systematic analysis of health examination surveys and epidemiological studies with 960 country-years and 9·1 million participants. Lancet 2011, 377, 557–567. [Google Scholar] [CrossRef]
- Danaei, G.; Finucane, M.M.; Lu, Y.; Singh, G.M.; Cowan, M.J.; Paciorek, C.J.; Lin, J.K.; Frazadfar, F.; Khang, Y.H.; Setvens, G.A.; et al. National, regional, and global trends in fasting plasma glucose and diabetes prevalence since 1980: Systematic analysis of health examination surveys and epidemiological studies with 370 country-years and 2·7 million participants. Lancet 2011, 378, 31–40. [Google Scholar] [CrossRef]
- Hu, F.; Liu, S.; Dam, R. Diet and risk of Type II diabetes: The role of types of fat and carbohydrate. Diabetologia 2001, 44, 805–817. [Google Scholar] [CrossRef] [PubMed]
- Bray, G.; Nielsen, S.; Popkin, B. Consumption of high-fructose corn syrup in beverages may play a role in the epidemic of obesity. Am. J. Clin. Nutr. 2004, 79, 537–543. [Google Scholar] [CrossRef]
- Bray, G.A.; Popkin, B.M. Dietary fat intake does affect obesity! Am. J. Clin. Nutr. 1998, 68, 1157–1173. [Google Scholar] [CrossRef]
- Drewnowski, A. The Real Contribution of Added Sugars and Fats to Obesity. Epid. Rev. 2007, 29, 160–171. [Google Scholar] [CrossRef] [Green Version]
- Gross, L.S.; Li, L.; Ford, E.S.; Liu, S. Increased consumption of refined carbohydrates and the epidemic of type 2 diabetes in the United States: An ecologic assessment. Am. J. Clin. Nutr. 2004, 79, 774–779. [Google Scholar] [CrossRef]
- Yu, T.; Sheu, S.-S.; Robotham, J.; Yoon, Y. Mitochondrial fission mediates high glucose-induced cell death through elevated production of reactive oxygen species. Cardiovas. Res. 2008, 79, 341–351. [Google Scholar] [CrossRef] [Green Version]
- Milagro, F.I.I.; Campión, J.; Martínez, J.A. Weight gain induced by high-fat feeding involves increased liver oxidative stress. Obesity (Silver Spring, Md.) 2006, 14, 1118–1123. [Google Scholar] [CrossRef]
- Bass, V.; Gordon, C.J.; Jarema, K.A.; MacPhail, R.C.; Cascio, W.E.; Phillips, P.M.; Ledbetter, A.D.; Schladweiler, M.C.; Andrews, D.; Miller, D.; et al. Ozone induces glucose intolerance and systemic metabolic effects in young and aged brown Norway rats. Toxicol. Appl. Pharm. 2013, 273, 551–560. [Google Scholar] [CrossRef] [Green Version]
- Gackière, F.; Saliba, L.; Baude, A.; Bosler, O.; Strube, C. Ozone inhalation activates stress-responsive regions of the CNS. J. Neurochem. 2011, 117, 961–972. [Google Scholar] [CrossRef] [Green Version]
- Tentolouris, N.; Liatis, S.; Katsilambros, N. Sympathetic system activity in obesity and metabolic syndrome. Ann. N. Y. Acad. Sci. 2006, 1083, 129–152. [Google Scholar] [CrossRef] [PubMed]
- Tentolouris, N.; Argyrakopoulou, G.; Katsilambros, N. Perturbed autonomic nervous system function in metabolic syndrome. Neuromol. Med. 2008, 10, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Kodavanti, U.P. Stretching the stress boundary: Linking air pollution health effects to a neurohormonal stress response. Biochim. Biophys. Acta 2016, 1860, 2880–2890. [Google Scholar] [CrossRef]
- Miller, D.B.; Snow, S.J.; Henriquez, A.; Schladweiler, M.C.; Ledbetter, A.D.; Richards, J.E.; Andrews, D.L.; Kodavanti, U.P. Systemic metabolic derangement, pulmonary effects, and insulin insufficiency following subchronic ozone exposure in rats. Toxicol. Appl. Pharmacol. 2016, 306, 47–57. [Google Scholar] [CrossRef] [Green Version]
- Mumaw, C.L.; Levesque, S.; McGraw, C.; Robertson, S.; Lucas, S.; Stafflinger, J.E.; Campen, M.J.; Hall, P.; Norenberg, J.P.; Anderson, T.; et al. Microglial priming through the lung-brain axis: The role of air pollution-induced circulating factors. FASEB J. 2016, 30, 1880–1891. [Google Scholar] [CrossRef] [PubMed]
- Santiago-Lopez, D.; Bautista-Martinez, J.A.; Hernandex, C.I.; Aguilar-Martinez, M.; Rivas-Arancibia, A. Oxidative stress, progressive damage in the substantia nigra and plasma dopamine oxidation, in rats chronically exposed to ozone. Toxicol. Lett. 2010, 197, 193–200. [Google Scholar] [CrossRef]
- Snow, S.J.; Gordon, C.J.; Bass, V.L.; Schladweiler, M.C.; Ledbetter, A.D.; Jarema, K.A.; Phillips, P.M.; Johnstone, A.F.M.; Kodavanti, U.P. Age-related differences in pulmonary effects of acute and subchronic episodic ozone exposures in Brown Norway rats. Inhal. Toxicol. 2016, 28, 313–323. [Google Scholar] [CrossRef]
- Gordon, C.J.; Phillips, P.M.; Ledbetter, A.; Snow, S.J.; Schladweiler, M.C.; Johnstone, A.F.; Kodavanti, U.P. Active vs. Sedentary lifestyle from weaning to adulthood and susceptibility to ozone in rats. Am. J. Physiol. Lung Cell Mol. Physiol. 2017, 312, L100–L109. [Google Scholar] [CrossRef]
- Gordon, C.J.; Phillips, P.M.; Johnstone, A.F.M.; Beasley, T.E.; Ledbetter, T.E.; Schladweiler, M.C.; Snow, S.J.; Kodavanti, U.P. Effect of high-fructose and high-fat diets on pulmonary sensitivity, motor activity, and body composition of brown Norway rats exposed to ozone. Inhal. Toxicol. 2016, 28, 203–215. [Google Scholar] [CrossRef]
- Seigel, D.; Ross, D. Immunodetection of NAD(P)H:quinone oxidoreductase 1 (NQO1) in human tissues. Free Radic. Biol. Med. 2000, 29, 246–253. [Google Scholar] [PubMed]
- Van Muiswinkel, F.L.; de Vos, R.A.; Bol, J.G.; Andringa, G.; Jansen Steur, E.N.; Ross, D.; Siegel, D.; Drukarch, B. Expression of NAD(P)H:quinone oxidoreductase in the normal and Parkinsonian substantia nigra. Neurobiol. Aging 2004, 25, 1253–1262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.S.; Jung, J.S.; Jeong, Y.H.; Hyan, J.W.; Le, T.K.; Kim, D.H.; Choi, E.C.; Kim, H.S. Antioxidant mechanism of isoflavone metaolies in hydrogen peroxide-stimulate rat primary astrocytes: Critical role of hemoxygnase-1 and NQO1 expression. J. Neurochem. 2011, 119, 909–919. [Google Scholar] [CrossRef] [PubMed]
- Rainia, A.K.; Templeton, D.J.; Deak, J.C.; Perry, G.; Smith, M.A. Quinone reductase (NQO1), a sensitive redox indicator, is increased in Alzheimer’s disease. Redox Rep. 1999, 4, 23–27. [Google Scholar] [CrossRef] [PubMed]
- Weiss, H.; Friedrich, T.; Hofhaus, G.; Preis, D. The respiratory-chain NADH dehydrogenase (complex I.) of mitochondria. Eur. J. Biochem. 1991, 197, 563–576. [Google Scholar] [CrossRef] [PubMed]
- Schapira, A.H. Mitochondrial dysfunction in neurodegenerative disorders. Biochem. Biolphys. Acta 1998, 1366, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Schapira, A.H.; Patel, S. Targeting mitochondria for neuroprotection in Parkinson’s disease. Antioxid. Redox. Signal. 2012, 16, 965–973. [Google Scholar] [CrossRef]
- Schapira, A.H.; Cooper, J.M.; Dexter, D.; Clark, J.B.; Jenner, P.; Marsden, C.D. Mitochondrial complex I deficiency in Parkinson’s disease. J. Neurochem. 1990, 54, 823–827. [Google Scholar] [CrossRef] [PubMed]
- Giachin, G.; Bouverot, R.; Acajjaour, S.; Pantalone, S.; Soler-Lopez, M. Dynamics of Human Mitochondrial Complex I Assembly: Implications for Neurodegenerative Diseases. Front. Mol. Biosci. 2016, 3. [Google Scholar] [CrossRef]
- Pandya, J.D.; Royland, J.E.; MacPhail, R.C.; Sullivan, P.G.; Kodavanti, P.R.S. Age- and brain region-specific differences in mitochondrial bioenergetics in Brown Norway rats. Neurobiol. Aging 2016, 42, 25–34. [Google Scholar] [CrossRef]
- Halliwell, B.; Aeschbach, R.; Löliger, J.; Aruoma, O.I. The characterization of antioxidants. Food Chem. Toxicol. 1995, 33, 601–617. [Google Scholar] [CrossRef] [PubMed]
- Fariss, M.W.; Chan, C.B.; Patel, M.; Van Houten, B.; Orrenius, S. Role of mitochondria in toxic oxidative stress. Mol. Interv. 2005, 5, 94–111. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.J.; Wang, X.; Kaufman, B.A.; Butow, R.A. Aconitase couples metabolic regulation to mitochondrial DNA maintenance. Science 2005, 307, 714–717. [Google Scholar] [CrossRef] [PubMed]
- Friguet, B.; Bulteau, A.L.; Chondrogianni, N.; Conconi, M.; Petropoulos, I. Protein degradation by the proteasome and its implications in aging. Ann. N. Y. Acad. Sci. 2000, 908, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Rivas-Arancibia, S.; Guevara-Guzman, R.; Lopez-Vidal, Y.; Rodriguez-Martinez, E.; Zanardo-Gomes, M.; Angoa-Perez, M.; Raisman-Vozari, R. Oxidative stress caused by ozone exposure induces loss of brain repair in the hippocampus of adult rats. Toxicol. Sci. 2010, 113, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Longhi, R.; Almeida, R.F.; Pettenuzzo, L.F.; Souza, D.G.; Machado, L.; Quincozes-Santos, A.; Souza, D.O. Effect of trans fatty acid-enreiched diet on mitochondrial inflammatory, and oxidative stress parametes in the cortex and hippocampus of Wistar rats. Eur. J. Nutr. 2010, 57, 1913–1924. [Google Scholar] [CrossRef]
- Ma, W.; Yuan, L.; Yu, H.; Xi, Y.; Xiao, R. Mitochondrial dysfunction and oxidative damage in the brain of diet-induced obese rats but not diet-resistant rats. Life Sci. 2014, 110, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Morrison, C.D.; Pistell, P.J.; Ingram, D.K.; Johnson, W.E.; Liu, Y.; Fernandez-Kin, S.O.; White, C.L.; Pupera, M.N.; Uranga, R.M.; Bruce-Keller, A.J.; et al. High Fat Diet Increases Hippocampal Oxidative Stress and Cognitive Impairment in Aged Mice: Implications for decreased Nrf2 signaling. J. Neurochem. 2010, 114, 1581–1589. [Google Scholar] [CrossRef]
- Zhang, X.; Fong, F.; Ren, J.; Driscoll, M.J.; Culver, B. High dietary fat induces NADPH oxidase-associated oxidative stress and inflammation in rat cerebral cortex. Exp. Neurol. 2005, 191, 315–325. [Google Scholar] [CrossRef]
- Tucsek, Z.; Toth, P.; Sosnowska, D.; Gautam, T.; Mitschelen, M.; Koller, A.; Szalai, G.; Sonntag, W.E.; Ungvari, Z.; Csiszar, A. Obesity in Agining Exacerbates Blood-Brain Barrier Disruption, Neuroinflammation, and Oxidative Stress in the Mouse Hippocampus: Effects on of Genes Involved in Beta-Amyloid Generation and Alzheimer’s Disease. J. Geront. 2014, 69, 1212–1226. [Google Scholar] [CrossRef]
- Souza, C.G.; Moreira, J.D.; Siqueira, I.R.; Pereira, A.G.; Rieger, D.K.; Souza, D.O.; Souza, T.M.; Portela, L.V.; Perry, M.L.S. Highly palatable diet consumption increases protein oxidation in rat frontal cortex and anxiety-like behavior. Life Sci. 2007, 81, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Cavaliere, G.; Viggiano, E.; Trinchese, G.; De Fillipo, C.; Messina, A.; Monda, V.; Valenzano, A.; Cincione, R.I.; Zammit, C.; Cimmino, F.; et al. Long Feeding High-Fat Induces Hypothalamic Oxidative Stress and Inflammation, and Prolonged Hypothalamic AAMPK Activation in Rat Animal Model. Front. Physiol. 2018, 9, 818. [Google Scholar] [CrossRef] [PubMed]
- Amin, K.A.; Kamel, H.H.; Abd Eltawab, M.A. The relation of high fat diet, metabolic tentdisturbances and crain oxidative dysfunction: Modulation by hydroxy citric acid. Lipids Health Dis. 2011, 10, 74. [Google Scholar] [CrossRef] [PubMed]
- Gordon, C.J.; Phillips, P.M.; Johnstone, A.F.M.; Schmid, J.; Schladweiler, M.C.; Ledbetter, A.; Snow, S.J.; Kodavanti, U.P. Effects of maternal high-fat diet and sedentary lifestyle on susceptibility of adult offspring to ozone exposure in rats. Inhal. Toxicol. 2017, 29, 239–254. [Google Scholar] [CrossRef] [PubMed]
- Gordon, C.J.; Jarema, K.A.; Lehmann, J.R.; Schladweiler, M.C.; Schmid, J.E.; Ward, W.O.; Kodavanti, U.P.; Nyska, A.; MacPhail, R.C. Susceptibility of adult and senescent brown Norway rats to repeated ozone exposure: An assessment of behavior, serum biochemistry and cardiopulmonary function. Inhal. Toxicol. 2013, 25, 141–159. [Google Scholar] [CrossRef]
- Gordon, C.J.; Johnstone, A.F.M.; Aydin, C.; Phillips, P.M.; MacPhail, R.C.; Kodavanti, U.P.; Ledbetter, A.D.; Jarema, K.A. Episodic ozone exposure in adult and senescent brown Norway rats: Acute and delayed effect on heart rate, core temperature, and motor activity. Inhal. Toxicol. 2014, 26, 380–390. [Google Scholar] [CrossRef]
- Glowinski, J.; Iversen, L.L. Regional studies of catecholamines in the rat brain. 1. The disposition of [3H]-norepinephrine, [3H]-dopamine and [3H]-DOPA in various regions of the brain. J. Neurochem. 1996, 13, 655–669. [Google Scholar] [CrossRef] [PubMed]
- Bello, R.I.; Gomez-Diaz, C.; Navarro, F.; Alcain, F.J.; Villalba, J.M. Expression of NAD(P)H:quinone oxidoreductase 1 in HeLa cells: Role of hydrogen peroxide and growth phase. J. Biol. Chem. 2001, 276, 44379–44384. [Google Scholar] [CrossRef]
- Lind, C.; Cadenas, E.; Hochstein, P.; Ernster, L. DT-diaphorase: Purification, properties, and function. Meth. Enzymol. 1990, 186, 287–301. [Google Scholar] [PubMed]
- Cormier, A.; Morin, C.; Zini, R.; Tillement, J.P.; Lagrue, G. In vitro effects of nicotine on mitochondrial respiration and superoxide anion generation. Brain Res. 2001, 900, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Miller, N.J.; Rice-Evans, C.; Davies, M.J.; Gopinathan, V.; Milner, A. A novel method for measuring antioxidant capacity and its application to monitoring the antioxidant status in premature neonates. Clin. Sci. 1993, 84, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Seelig, G.F.; Meister, A. γ-Glutamylcysteine synthetase: Interactions of an essential sulfhydryl group. J. Biol. Chem. 1984, 259, 3534–3538. [Google Scholar] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Component | Regular (5001) a | High Fructose b | High Fat c |
|---|---|---|---|
| Fat | 13.5 | 5.2 | 34.3 |
| Carbohydrates | 58.0 | 60.4 | 27.3 |
| Protein | 28.5 | 18.3 | 23.5 |
| Metabolizable Energy (kcal/g) | 3.02 | 3.6 | 5.1 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Valdez, J.M.; Johnstone, A.F.M.; Richards, J.E.; Schmid, J.E.; Royland, J.E.; Kodavanti, P.R.S. Interaction of Diet and Ozone Exposure on Oxidative Stress Parameters within Specific Brain Regions of Male Brown Norway Rats. Int. J. Mol. Sci. 2019, 20, 11. https://doi.org/10.3390/ijms20010011
Valdez JM, Johnstone AFM, Richards JE, Schmid JE, Royland JE, Kodavanti PRS. Interaction of Diet and Ozone Exposure on Oxidative Stress Parameters within Specific Brain Regions of Male Brown Norway Rats. International Journal of Molecular Sciences. 2019; 20(1):11. https://doi.org/10.3390/ijms20010011
Chicago/Turabian StyleValdez, Joseph M., Andrew F. M. Johnstone, Judy E. Richards, Judith E. Schmid, Joyce E. Royland, and Prasada Rao S. Kodavanti. 2019. "Interaction of Diet and Ozone Exposure on Oxidative Stress Parameters within Specific Brain Regions of Male Brown Norway Rats" International Journal of Molecular Sciences 20, no. 1: 11. https://doi.org/10.3390/ijms20010011