

Combined Omipalisib and MAPK Inhibition Suppress PDAC Growth
 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Reagents
2.2. Western Blot and Antibodies
2.3. EdU Proliferation Assay
2.4. Migration Assay
2.5. Colony Formation Assay
2.6. Histology
2.7. Mice
2.8. Seahorse Extracellular Flux Analysis
2.9. Statistical Analysis
3. Results
3.1. Inhibition of MAPK or PI3K Signaling Maintains Alternative Mitogenic Signaling Pathway Activation
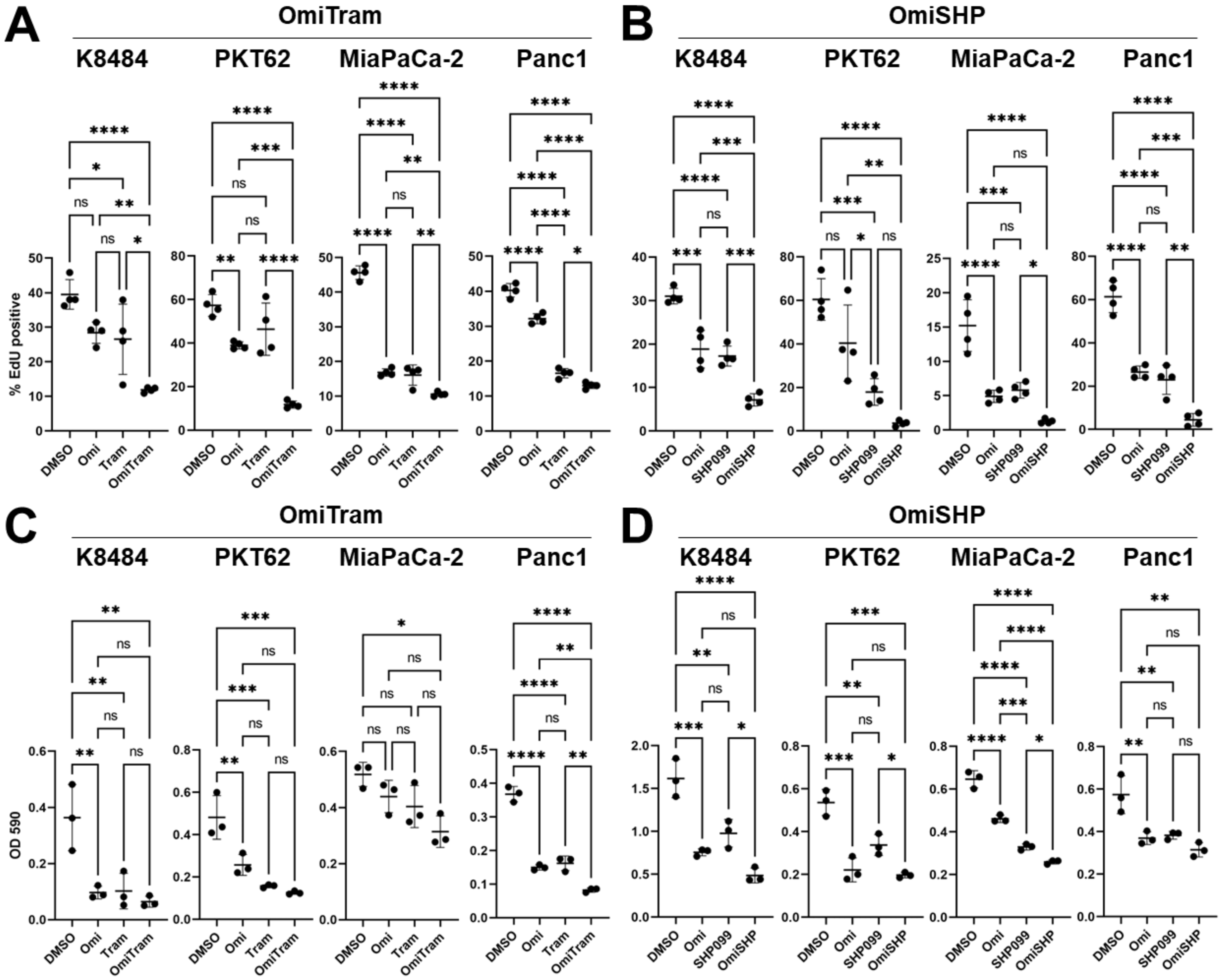
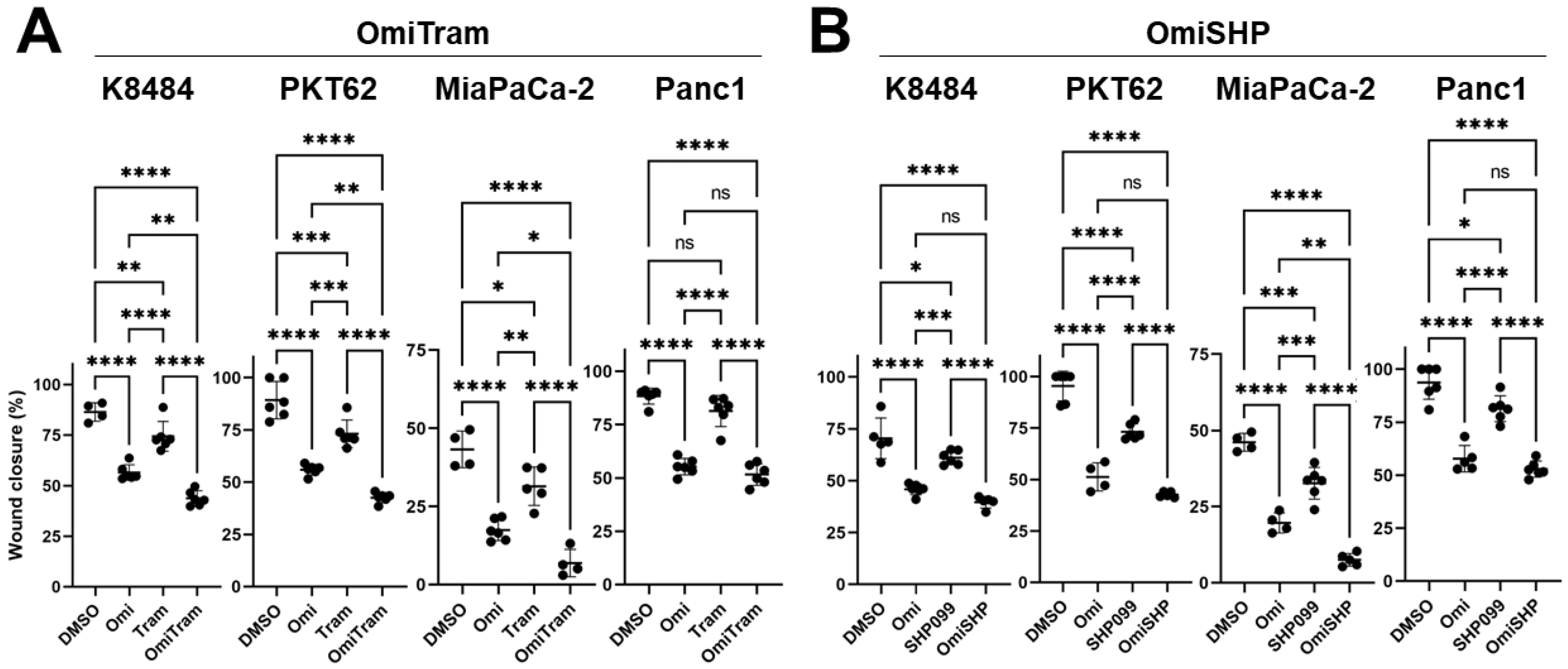
3.2. Combined Targeting of PI3K and MAPK Pathways Inhibits Proliferation, Colony-Forming Ability, and Migration of PDAC Cells In Vitro
3.3. Effects of PI3K and MAPK Pathway Inhibition on Tumor Metabolism
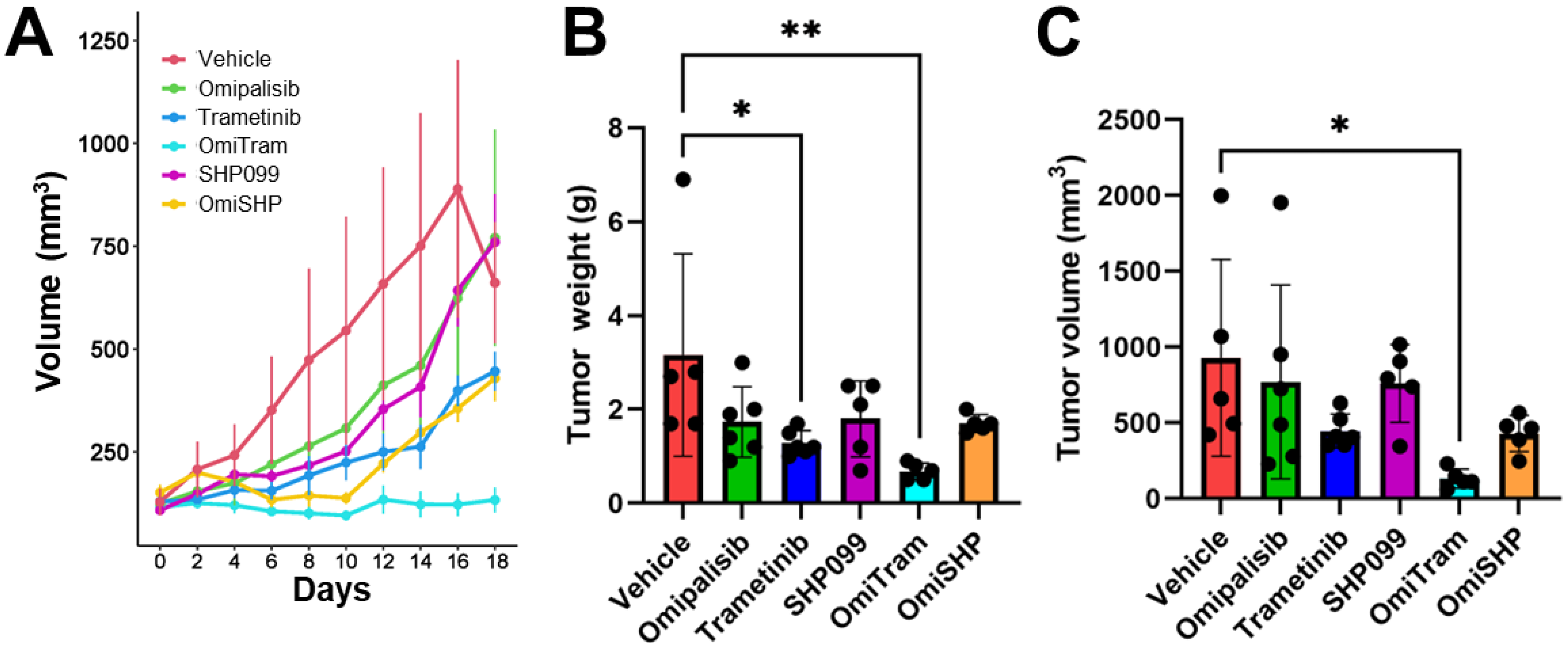
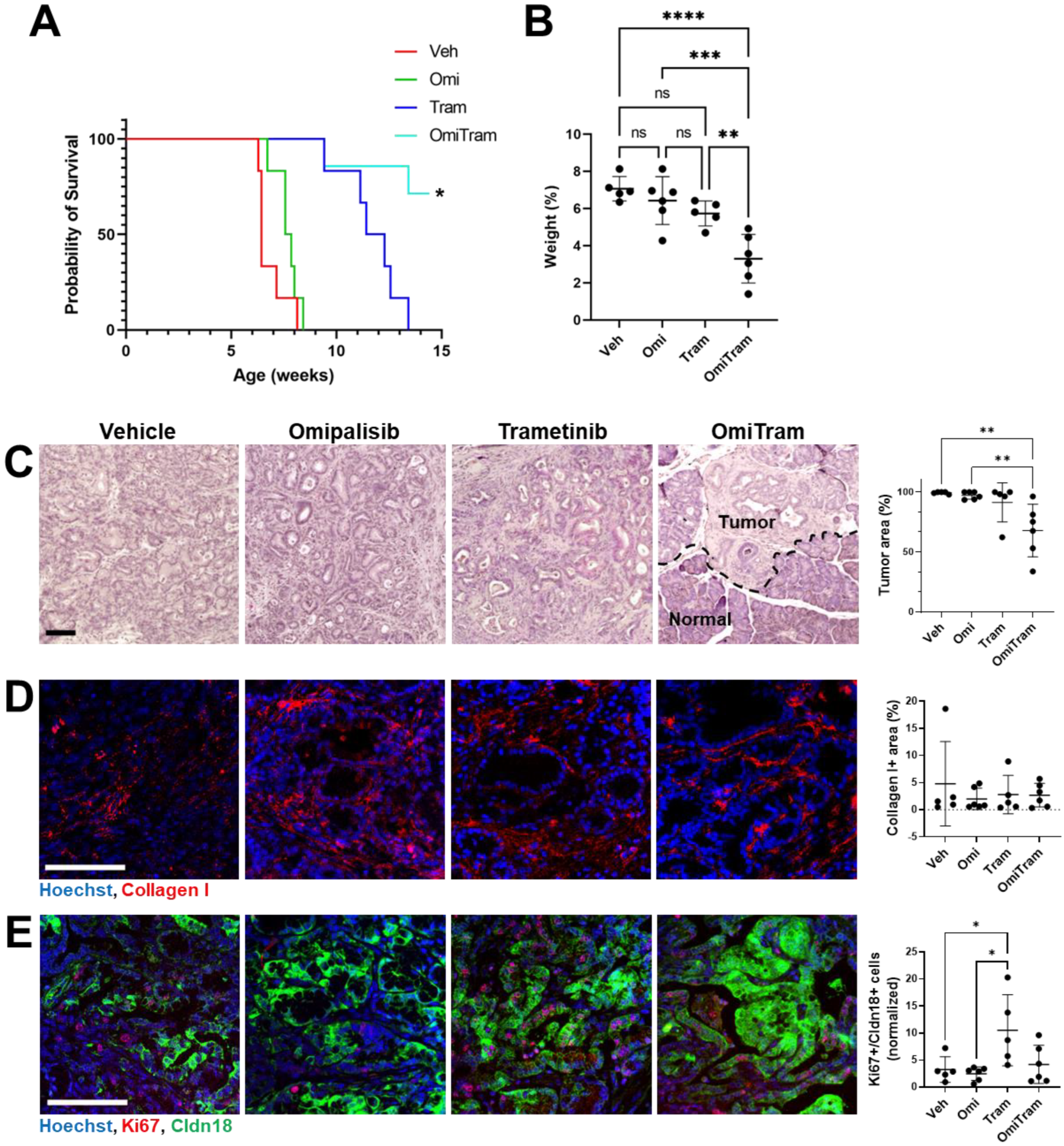
3.4. Combined PI3K and MAPK Pathway Inhibition Suppresses Tumor Growth In Vivo
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Giaquinto, A.N.; Jemal, A. Cancer statistics, 2024. CA Cancer J. Clin. 2024, 74, 12–49. [Google Scholar] [CrossRef] [PubMed]
- Burris, H.A., 3rd; Moore, M.J.; Andersen, J.; Green, M.R.; Rothenberg, M.L.; Modiano, M.R.; Cripps, M.C.; Portenoy, R.K.; Storniolo, A.M.; Tarassoff, P.; et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: A randomized trial. J. Clin. Oncol. 1997, 15, 2403–2413. [Google Scholar] [CrossRef]
- Conroy, T.; Bachet, J.B.; Ayav, A.; Huguet, F.; Lambert, A.; Caramella, C.; Marechal, R.; Van Laethem, J.L.; Ducreux, M. Current standards and new innovative approaches for treatment of pancreatic cancer. Eur. J. Cancer 2016, 57, 10–22. [Google Scholar]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouche, O.; Guimbaud, R.; Becouarn, Y.; Adenis, A.; Raoul, J.L.; Gourgou-Bourgade, S.; de la Fouchardiere, C.; et al. FOLFIRINOX versus Gemcitabine for Metastatic Pancreatic Cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [PubMed]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.M.; Gingras, M.C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef]
- Mustachio, L.M.; Chelariu-Raicu, A.; Szekvolgyi, L.; Roszik, J. Targeting KRAS in Cancer: Promising Therapeutic Strategies. Cancers 2021, 13, 1204. [Google Scholar] [CrossRef]
- Singhi, A.D.; George, B.; Greenbowe, J.R.; Chung, J.; Suh, J.; Maitra, A.; Klempner, S.J.; Hendifar, A.; Milind, J.M.; Golan, T.; et al. Real-Time Targeted Genome Profile Analysis of Pancreatic Ductal Adenocarcinomas Identifies Genetic Alterations That Might Be Targeted with Existing Drugs or Used as Biomarkers. Gastroenterology 2019, 156, 2242–2253.e4. [Google Scholar]
- Chan-Seng-Yue, M.; Kim, J.C.; Wilson, G.W.; Ng, K.; Figueroa, E.F.; O’Kane, G.M.; Connor, A.A.; Denroche, R.E.; Grant, R.C.; McLeod, J.; et al. Transcription phenotypes of pancreatic cancer are driven by genomic events during tumor evolution. Nat. Genet. 2020, 52, 231–240. [Google Scholar] [PubMed]
- Huang, L.; Guo, Z.; Wang, F.; Fu, L. KRAS mutation: From undruggable to druggable in cancer. Signal Transduct. Target Ther. 2021, 6, 386. [Google Scholar] [CrossRef]
- Vatansever, S.; Erman, B.; Gümüş, Z.H. Oncogenic G12D mutation alters local conformations and dynamics of K-Ras. Sci. Rep. 2019, 9, 11730. [Google Scholar] [CrossRef]
- Hunter, J.C.; Manandhar, A.; Carrasco, M.A.; Gurbani, D.; Gondi, S.; Westover, K.D. Biochemical and Structural Analysis of Common Cancer-Associated KRAS Mutations. Mol. Cancer Res. 2015, 13, 1325–1335. [Google Scholar]
- Nakajima, E.C.; Drezner, N.; Li, X.; Mishra-Kalyani, P.S.; Liu, Y.; Zhao, H.; Bi, Y.; Liu, J.; Rahman, A.; Wearne, E.; et al. FDA Approval Summary: Sotorasib for KRAS G12C-Mutated Metastatic NSCLC. Clin. Cancer Res. 2022, 28, 1482–1486. [Google Scholar] [CrossRef]
- Bannoura, S.F.; Khan, H.Y.; Azmi, A.S. KRAS G12D targeted therapies for pancreatic cancer: Has the fortress been conquered? Front. Oncol. 2022, 12, 1013902. [Google Scholar] [CrossRef]
- Linehan, A.; O’Reilly, M.; McDermott, R.; O’Kane, G.M. Targeting KRAS mutations in pancreatic cancer: Opportunities for future strategies. Front. Med. 2024, 11, 1369136. [Google Scholar] [CrossRef]
- Mendoza, M.C.; Er, E.E.; Blenis, J. The Ras-ERK and PI3K-mTOR pathways: Cross-talk and compensation. Trends Biochem. Sci. 2011, 36, 320–328. [Google Scholar] [CrossRef]
- Britten, C.D. PI3K and MEK inhibitor combinations: Examining the evidence in selected tumor types. Cancer Chemother. Pharmacol. 2013, 71, 1395–1409. [Google Scholar] [CrossRef] [PubMed]
- Muzumdar, M.D.; Chen, P.Y.; Dorans, K.J.; Chung, K.M.; Bhutkar, A.; Hong, E.; Noll, E.M.; Sprick, M.R.; Trumpp, A.; Jacks, T. Survival of pancreatic cancer cells lacking KRAS function. Nat. Commun. 2017, 8, 1090. [Google Scholar] [CrossRef] [PubMed]
- Subbiah, V.; Kreitman, R.J.; Wainberg, Z.A.; Gazzah, A.; Lassen, U.; Stein, A.; Wen, P.Y.; Dietrich, S.; de Jonge, M.J.A.; Blay, J.Y.; et al. Dabrafenib plus trametinib in BRAFV600E-mutated rare cancers: The phase 2 ROAR trial. Nat. Med. 2023, 29, 1103–1112. [Google Scholar] [CrossRef] [PubMed]
- Long, G.V.; Hauschild, A.; Santinami, M.; Atkinson, V.; Mandalà, M.; Chiarion-Sileni, V.; Larkin, J.; Nyakas, M.; Dutriaux, C.; Haydon, A.; et al. Adjuvant Dabrafenib plus Trametinib in Stage III BRAF-Mutated Melanoma. N. Engl. J. Med. 2017, 377, 1813–1823. [Google Scholar] [CrossRef]
- Odogwu, L.; Mathieu, L.; Blumenthal, G.; Larkins, E.; Goldberg, K.B.; Griffin, N.; Bijwaard, K.; Lee, E.Y.; Philip, R.; Jiang, X.; et al. FDA Approval Summary: Dabrafenib and Trametinib for the Treatment of Metastatic Non-Small Cell Lung Cancers Harboring BRAF V600E Mutations. Oncologist 2018, 23, 740–745. [Google Scholar] [CrossRef]
- Barbato, M.I.; Nashed, J.; Bradford, D.; Ren, Y.; Khasar, S.; Miller, C.P.; Zolnik, B.S.; Zhao, H.; Li, Y.; Bi, Y.; et al. FDA Approval Summary: Dabrafenib in combination with trametinib for BRAF V600E mutation-positive low-grade glioma. Clin. Cancer Res. 2023, 30, 263–268. [Google Scholar] [CrossRef] [PubMed]
- FDA Grants Accelerated Approval to Dabrafenib in Combination with Trametinib for Unresectable or Metastatic Solid Tumors with BRAF V600E Mutation. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-dabrafenib-combination-trametinib-unresectable-or-metastatic-solid (accessed on 10 November 2023).
- Brown, W.S.; McDonald, P.C.; Nemirovsky, O.; Awrey, S.; Chafe, S.C.; Schaeffer, D.F.; Li, J.; Renouf, D.J.; Stanger, B.Z.; Dedhar, S. Overcoming Adaptive Resistance to KRAS and MEK Inhibitors by Co-targeting mTORC1/2 Complexes in Pancreatic Cancer. Cell Rep. Med. 2020, 1, 100131. [Google Scholar]
- Pettazzoni, P.; Viale, A.; Shah, P.; Carugo, A.; Ying, H.; Wang, H.; Genovese, G.; Seth, S.; Minelli, R.; Green, T.; et al. Genetic events that limit the efficacy of MEK and RTK inhibitor therapies in a mouse model of KRAS-driven pancreatic cancer. Cancer Res. 2015, 75, 1091–1101. [Google Scholar]
- Brauswetter, D.; Gurbi, B.; Varga, A.; Várkondi, E.; Schwab, R.; Bánhegyi, G.; Fábián, O.; Kéri, G.; Vályi-Nagy, I.; Peták, I. Molecular subtype specific efficacy of MEK inhibitors in pancreatic cancers. PLoS ONE 2017, 12, e0185687. [Google Scholar]
- Infante, J.R.; Somer, B.G.; Park, J.O.; Li, C.P.; Scheulen, M.E.; Kasubhai, S.M.; Oh, D.Y.; Liu, Y.; Redhu, S.; Steplewski, K.; et al. A randomised, double-blind, placebo-controlled trial of trametinib, an oral MEK inhibitor, in combination with gemcitabine for patients with untreated metastatic adenocarcinoma of the pancreas. Eur. J. Cancer 2014, 50, 2072–2081. [Google Scholar] [PubMed]
- Kun, E.; Tsang, Y.T.M.; Ng, C.W.; Gershenson, D.M.; Wong, K.K. MEK inhibitor resistance mechanisms and recent developments in combination trials. Cancer Treat. Rev. 2021, 92, 102137. [Google Scholar]
- Hosein, A.N.; Dougan, S.K.; Aguirre, A.J.; Maitra, A. Translational advances in pancreatic ductal adenocarcinoma therapy. Nat. Cancer 2022, 3, 272–286. [Google Scholar]
- Munster, P.; Aggarwal, R.; Hong, D.; Schellens, J.H.; van der Noll, R.; Specht, J.; Witteveen, P.O.; Werner, T.L.; Dees, E.C.; Bergsland, E.; et al. First-in-Human Phase I Study of GSK2126458, an Oral Pan-Class I Phosphatidylinositol-3-Kinase Inhibitor, in Patients with Advanced Solid Tumor Malignancies. Clin. Cancer Res. 2016, 22, 1932–1939. [Google Scholar]
- Weisner, J.; Landel, I.; Reintjes, C.; Uhlenbrock, N.; Trajkovic-Arsic, M.; Dienstbier, N.; Hardick, J.; Ladigan, S.; Lindemann, M.; Smith, S.; et al. Preclinical Efficacy of Covalent-Allosteric AKT Inhibitor Borussertib in Combination with Trametinib in KRAS-Mutant Pancreatic and Colorectal Cancer. Cancer Res. 2019, 79, 2367–2378. [Google Scholar]
- Engelman, J.A.; Chen, L.; Tan, X.; Crosby, K.; Guimaraes, A.R.; Upadhyay, R.; Maira, M.; McNamara, K.; Perera, S.A.; Song, Y.; et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat. Med. 2008, 14, 1351–1356. [Google Scholar] [CrossRef]
- Carracedo, A.; Ma, L.; Teruya-Feldstein, J.; Rojo, F.; Salmena, L.; Alimonti, A.; Egia, A.; Sasaki, A.T.; Thomas, G.; Kozma, S.C.; et al. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J. Clin. Investig. 2008, 118, 3065–3074. [Google Scholar] [CrossRef]
- Roy, S.K.; Srivastava, R.K.; Shankar, S. Inhibition of PI3K/AKT and MAPK/ERK pathways causes activation of FOXO transcription factor, leading to cell cycle arrest and apoptosis in pancreatic cancer. J. Mol. Signal 2010, 5, 10. [Google Scholar] [CrossRef] [PubMed]
- Kandil, E.; Tsumagari, K.; Ma, J.; Abd Elmageed, Z.Y.; Li, X.; Slakey, D.; Mondal, D.; Abdel-Mageed, A.B. Synergistic inhibition of thyroid cancer by suppressing MAPK/PI3K/AKT pathways. J. Surg. Res. 2013, 184, 898–906. [Google Scholar]
- Alagesan, B.; Contino, G.; Guimaraes, A.R.; Corcoran, R.B.; Deshpande, V.; Wojtkiewicz, G.R.; Hezel, A.F.; Wong, K.K.; Loda, M.; Weissleder, R.; et al. Combined MEK and PI3K inhibition in a mouse model of pancreatic cancer. Clin. Cancer Res. 2015, 21, 396–404. [Google Scholar] [CrossRef] [PubMed]
- Awasthi, N.; Kronenberger, D.; Stefaniak, A.; Hassan, M.S.; von Holzen, U.; Schwarz, M.A.; Schwarz, R.E. Dual inhibition of the PI3K and MAPK pathways enhances nab-paclitaxel/gemcitabine chemotherapy response in preclinical models of pancreatic cancer. Cancer Lett. 2019, 459, 41–49. [Google Scholar] [PubMed]
- Junttila, M.R.; Devasthali, V.; Cheng, J.H.; Castillo, J.; Metcalfe, C.; Clermont, A.C.; Otter, D.D.; Chan, E.; Bou-Reslan, H.; Cao, T.; et al. Modeling targeted inhibition of MEK and PI3 kinase in human pancreatic cancer. Mol. Cancer Ther. 2015, 14, 40–47. [Google Scholar]
- Bedard, P.L.; Tabernero, J.; Janku, F.; Wainberg, Z.A.; Paz-Ares, L.; Vansteenkiste, J.; Van Cutsem, E.; Pérez-García, J.; Stathis, A.; Britten, C.D.; et al. A phase Ib dose-escalation study of the oral pan-PI3K inhibitor buparlisib (BKM120) in combination with the oral MEK1/2 inhibitor trametinib (GSK1120212) in patients with selected advanced solid tumors. Clin. Cancer Res. 2015, 21, 730–738. [Google Scholar]
- Grilley-Olson, J.E.; Bedard, P.L.; Fasolo, A.; Cornfeld, M.; Cartee, L.; Razak, A.R.; Stayner, L.A.; Wu, Y.; Greenwood, R.; Singh, R.; et al. A phase Ib dose-escalation study of the MEK inhibitor trametinib in combination with the PI3K/mTOR inhibitor GSK2126458 in patients with advanced solid tumors. Investig. New Drugs 2016, 34, 740–749. [Google Scholar] [CrossRef]
- Chan, G.; Kalaitzidis, D.; Neel, B.G. The tyrosine phosphatase Shp2 (PTPN11) in cancer. Cancer Metastasis Rev. 2008, 27, 179–192. [Google Scholar] [CrossRef]
- Chan, R.J.; Feng, G.S. PTPN11 is the first identified proto-oncogene that encodes a tyrosine phosphatase. Blood 2007, 109, 862–867. [Google Scholar] [CrossRef]
- Ruess, D.A.; Heynen, G.J.; Ciecielski, K.J.; Ai, J.; Berninger, A.; Kabacaoglu, D.; Görgülü, K.; Dantes, Z.; Wörmann, S.M.; Diakopoulos, K.N.; et al. Mutant KRAS-driven cancers depend on PTPN11/SHP2 phosphatase. Nat. Med. 2018, 24, 954–960. [Google Scholar] [CrossRef]
- Garcia Fortanet, J.; Chen, C.H.; Chen, Y.N.; Chen, Z.; Deng, Z.; Firestone, B.; Fekkes, P.; Fodor, M.; Fortin, P.D.; Fridrich, C.; et al. Allosteric Inhibition of SHP2: Identification of a Potent, Selective, and Orally Efficacious Phosphatase Inhibitor. J. Med. Chem. 2016, 59, 7773–7782. [Google Scholar] [CrossRef] [PubMed]
- Fedele, C.; Ran, H.; Diskin, B.; Wei, W.; Jen, J.; Geer, M.J.; Araki, K.; Ozerdem, U.; Simeone, D.M.; Miller, G.; et al. SHP2 Inhibition Prevents Adaptive Resistance to MEK Inhibitors in Multiple Cancer Models. Cancer Discov. 2018, 8, 1237–1249. [Google Scholar] [CrossRef] [PubMed]
- Fedele, C.; Li, S.; Teng, K.W.; Foster, C.J.R.; Peng, D.; Ran, H.; Mita, P.; Geer, M.J.; Hattori, T.; Koide, A.; et al. SHP2 inhibition diminishes KRASG12C cycling and promotes tumor microenvironment remodeling. J. Exp. Med. 2021, 218, e20201414. [Google Scholar] [CrossRef]
- Chen, Y.N.; LaMarche, M.J.; Chan, H.M.; Fekkes, P.; Garcia-Fortanet, J.; Acker, M.G.; Antonakos, B.; Chen, C.H.; Chen, Z.; Cooke, V.G.; et al. Allosteric inhibition of SHP2 phosphatase inhibits cancers driven by receptor tyrosine kinases. Nature 2016, 535, 148–152. [Google Scholar] [CrossRef] [PubMed]
- Frank, K.J.; Mulero-Sánchez, A.; Berninger, A.; Ruiz-Cañas, L.; Bosma, A.; Görgülü, K.; Wu, N.; Diakopoulos, K.N.; Kaya-Aksoy, E.; Ruess, D.A.; et al. Extensive preclinical validation of combined RMC-4550 and LY3214996 supports clinical investigation for KRAS mutant pancreatic cancer. Cell Rep. Med. 2022, 3, 100815. [Google Scholar] [CrossRef]
- Amante, R.J.; Jehanno, C.; De Silva, D.; Coissieux, M.M.; Ackerknecht, M.; Romanet, V.; Sethi, A.; Hamelin, B.; Preca, B.T.; Piscuoglio, S.; et al. PI3K inhibition circumvents resistance to SHP2 blockade in metastatic triple-negative breast cancer. J. Mammary Gland. Biol. Neoplasia 2023, 28, 13. [Google Scholar] [CrossRef]
- Sun, B.; Jensen, N.R.; Chung, D.; Yang, M.; LaRue, A.C.; Cheung, H.W.; Wang, Q. Synergistic effects of SHP2 and PI3K pathway inhibitors in GAB2-overexpressing ovarian cancer. Am. J. Cancer Res. 2019, 9, 145–159. [Google Scholar]
- Mulero-Sánchez, A.; Ramirez, C.F.A.; du Chatinier, A.; Wang, H.; Koomen, S.J.I.; Song, J.Y.; de Groot, M.H.P.; Lieftink, C.; Bosma, A.; Burylo, A.; et al. Rational combination of SHP2 and mTOR inhibition for the treatment of hepatocellular carcinoma. Mol. Oncol. 2023, 17, 964–980. [Google Scholar] [CrossRef]
- Heynen, G.; Lisek, K.; Vogel, R.; Wulf-Goldenberg, A.; Alcaniz, J.; Montaudon, E.; Marangoni, E.; Birchmeier, W. Targeting SHP2 phosphatase in breast cancer overcomes RTK-mediated resistance to PI3K inhibitors. Breast Cancer Res. 2022, 24, 23. [Google Scholar] [CrossRef]
- Olive, K.P.; Jacobetz, M.A.; Davidson, C.J.; Gopinathan, A.; McIntyre, D.; Honess, D.; Madhu, B.; Goldgraben, M.A.; Caldwell, M.E.; Allard, D.; et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 2009, 324, 1457–1461. [Google Scholar] [PubMed]
- Messaggio, F.; Mendonsa, A.M.; Castellanos, J.; Nagathihalli, N.S.; Gorden, L.; Merchant, N.B.; VanSaun, M.N. Adiponectin receptor agonists inhibit leptin induced pSTAT3 and in vivo pancreatic tumor growth. Oncotarget 2017, 8, 85378–85391. [Google Scholar] [PubMed]
- Manley, S.J.; Olou, A.A.; Jack, J.L.; Ruckert, M.T.; Walsh, R.M.; Eades, A.E.; Bye, B.A.; Ambrose, J.; Messaggio, F.; Anant, S.; et al. Synthetic adiponectin-receptor agonist, AdipoRon, induces glycolytic dependence in pancreatic cancer cells. Cell Death Dis. 2022, 13, 114. [Google Scholar] [PubMed]
- Nussinov, R.; Tsai, C.J.; Jang, H. A New View of Pathway-Driven Drug Resistance in Tumor Proliferation. Trends Pharmacol. Sci. 2017, 38, 427–437. [Google Scholar]
- Conway, J.R.; Herrmann, D.; Evans, T.J.; Morton, J.P.; Timpson, P. Combating pancreatic cancer with PI3K pathway inhibitors in the era of personalised medicine. Gut 2019, 68, 742–758. [Google Scholar] [CrossRef]
- Xue, G.; Hemmings, B.A. PKB/Akt-dependent regulation of cell motility. J. Natl. Cancer Inst. 2013, 105, 393–404. [Google Scholar]
- Kong, B.; Wu, W.; Cheng, T.; Schlitter, A.M.; Qian, C.; Bruns, P.; Jian, Z.; Jäger, C.; Regel, I.; Raulefs, S.; et al. A subset of metastatic pancreatic ductal adenocarcinomas depends quantitatively on oncogenic Kras/Mek/Erk-induced hyperactive mTOR signalling. Gut 2016, 65, 647–657. [Google Scholar]
- Ying, H.; Kimmelman, A.C.; Lyssiotis, C.A.; Hua, S.; Chu, G.C.; Fletcher-Sananikone, E.; Locasale, J.W.; Son, J.; Zhang, H.; Coloff, J.L.; et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 2012, 149, 656–670. [Google Scholar]
- Ijichi, H.; Chytil, A.; Gorska, A.E.; Aakre, M.E.; Fujitani, Y.; Fujitani, S.; Wright, C.V.; Moses, H.L. Aggressive pancreatic ductal adenocarcinoma in mice caused by pancreas-specific blockade of transforming growth factor-beta signaling in cooperation with active Kras expression. Genes Dev. 2006, 20, 3147–3160. [Google Scholar] [CrossRef]
- Peng, J.; Sun, B.F.; Chen, C.Y.; Zhou, J.Y.; Chen, Y.S.; Chen, H.; Liu, L.; Huang, D.; Jiang, J.; Cui, G.S.; et al. Single-cell RNA-seq highlights intra-tumoral heterogeneity and malignant progression in pancreatic ductal adenocarcinoma. Cell Res. 2019, 29, 725–738. [Google Scholar]
- Yang, K.; Yang, T.; Yu, J.; Li, F.; Zhao, X. Integrated transcriptional analysis reveals macrophage heterogeneity and macrophage-tumor cell interactions in the progression of pancreatic ductal adenocarcinoma. BMC Cancer 2023, 23, 199. [Google Scholar] [CrossRef] [PubMed]
- Bu, L.; Yonemura, A.; Yasuda-Yoshihara, N.; Uchihara, T.; Ismagulov, G.; Takasugi, S.; Yasuda, T.; Okamoto, Y.; Kitamura, F.; Akiyama, T.; et al. Tumor microenvironmental 15-PGDH depletion promotes fibrotic tumor formation and angiogenesis in pancreatic cancer. Cancer Sci. 2022, 113, 3579–3592. [Google Scholar] [CrossRef]
- Panebianco, C.; Villani, A.; Pisati, F.; Orsenigo, F.; Ulaszewska, M.; Latiano, T.P.; Potenza, A.; Andolfo, A.; Terracciano, F.; Tripodo, C.; et al. Butyrate, a postbiotic of intestinal bacteria, affects pancreatic cancer and gemcitabine response in in vitro and in vivo models. Biomed. Pharmacother. 2022, 151, 113163. [Google Scholar] [CrossRef]
- Novoplansky, O.; Shnerb, A.B.; Marripati, D.; Jagadeeshan, S.; Shareb, R.A.; Conde-López, C.; Zorea, J.; Prasad, M.; Lulu, T.B.; Yegodayev, K.M.; et al. Activation of the EGFR/PI3K/AKT pathway limits the efficacy of trametinib treatment in head and neck cancer. Mol. Oncol. 2023, 17, 2618–2636. [Google Scholar] [CrossRef] [PubMed]
- Wells, V.; Downward, J.; Mallucci, L. Functional inhibition of PI3K by the betaGBP molecule suppresses Ras-MAPK signalling to block cell proliferation. Oncogene 2007, 26, 7709–7714. [Google Scholar] [CrossRef]
- Ebi, H.; Costa, C.; Faber, A.C.; Nishtala, M.; Kotani, H.; Juric, D.; Della Pelle, P.; Song, Y.; Yano, S.; Mino-Kenudson, M.; et al. PI3K regulates MEK/ERK signaling in breast cancer via the Rac-GEF, P-Rex1. Proc. Natl. Acad. Sci. USA 2013, 110, 21124–21129. [Google Scholar] [CrossRef] [PubMed]
- Kemp, S.B.; Cheng, N.; Markosyan, N.; Sor, R.; Kim, I.K.; Hallin, J.; Shoush, J.; Quinones, L.; Brown, N.V.; Bassett, J.B.; et al. Efficacy of a Small-Molecule Inhibitor of KrasG12D in Immunocompetent Models of Pancreatic Cancer. Cancer Discov. 2023, 13, 298–311. [Google Scholar] [CrossRef]
- Baer, R.; Cintas, C.; Therville, N.; Guillermet-Guibert, J. Implication of PI3K/Akt pathway in pancreatic cancer: When PI3K isoforms matter? Adv. Biol. Regul. 2015, 59, 19–35. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bye, B.A.; Jack, J.L.; Pierce, A.; Walsh, R.M.; Eades, A.E.; Chalise, P.; Olou, A.; VanSaun, M.N. Combined Omipalisib and MAPK Inhibition Suppress PDAC Growth. Cancers 2025, 17, 1152. https://doi.org/10.3390/cancers17071152
Bye BA, Jack JL, Pierce A, Walsh RM, Eades AE, Chalise P, Olou A, VanSaun MN. Combined Omipalisib and MAPK Inhibition Suppress PDAC Growth. Cancers. 2025; 17(7):1152. https://doi.org/10.3390/cancers17071152
Chicago/Turabian StyleBye, Bailey A., Jarrid L. Jack, Alexandra Pierce, Richard McKinnon Walsh, Austin E. Eades, Prabhakar Chalise, Appolinaire Olou, and Michael N. VanSaun. 2025. "Combined Omipalisib and MAPK Inhibition Suppress PDAC Growth" Cancers 17, no. 7: 1152. https://doi.org/10.3390/cancers17071152
APA StyleBye, B. A., Jack, J. L., Pierce, A., Walsh, R. M., Eades, A. E., Chalise, P., Olou, A., & VanSaun, M. N. (2025). Combined Omipalisib and MAPK Inhibition Suppress PDAC Growth. Cancers, 17(7), 1152. https://doi.org/10.3390/cancers17071152