
The Combination of Oncolytic Virus and Antibody Blockade of TGF-β Enhances the Efficacy of αvβ6-Targeting CAR T Cells Against Pancreatic Cancer in an Immunocompetent Model

 ,
,  ,
,  , , , and
, , , and
Simple Summary
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cells and Cell Culture
2.2. Cloning of a Murine αvβ6-Targeted CAR into the MSGV Gammaretroviral Vector
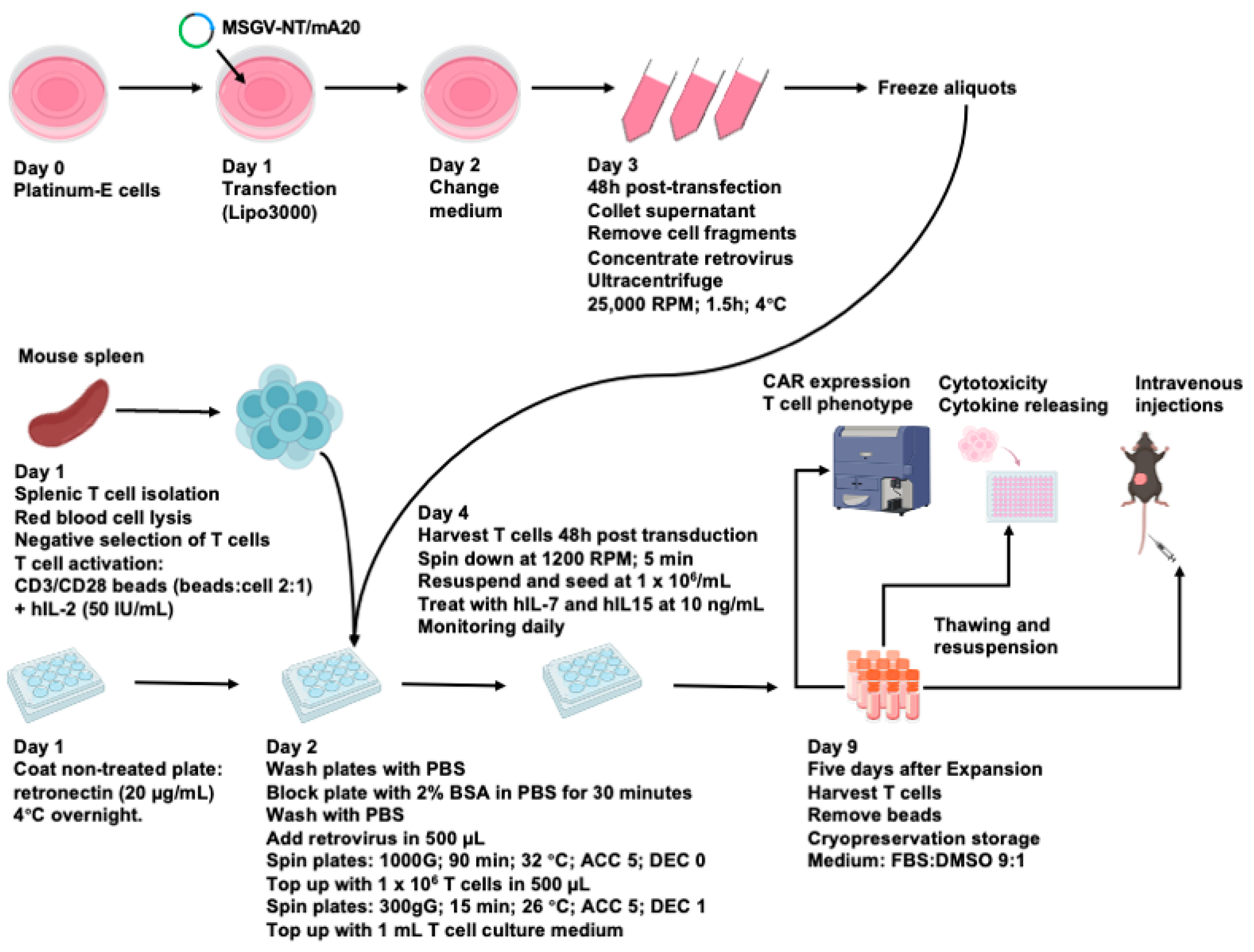
2.3. Murine CAR T Cells Production
2.4. Flow Cytometry
2.5. Cytotoxicity Assay of the CAR-T Cells
2.6. Cytokine Release Analysis
2.7. Cytotoxicity Assay of the Virus
2.8. Viral Infection and Replication Assay
2.9. Animal Experiments
2.10. IVIS Bioluminescence Imaging
2.11. Tissue Harvesting and Immunohistochemistry
3. Results
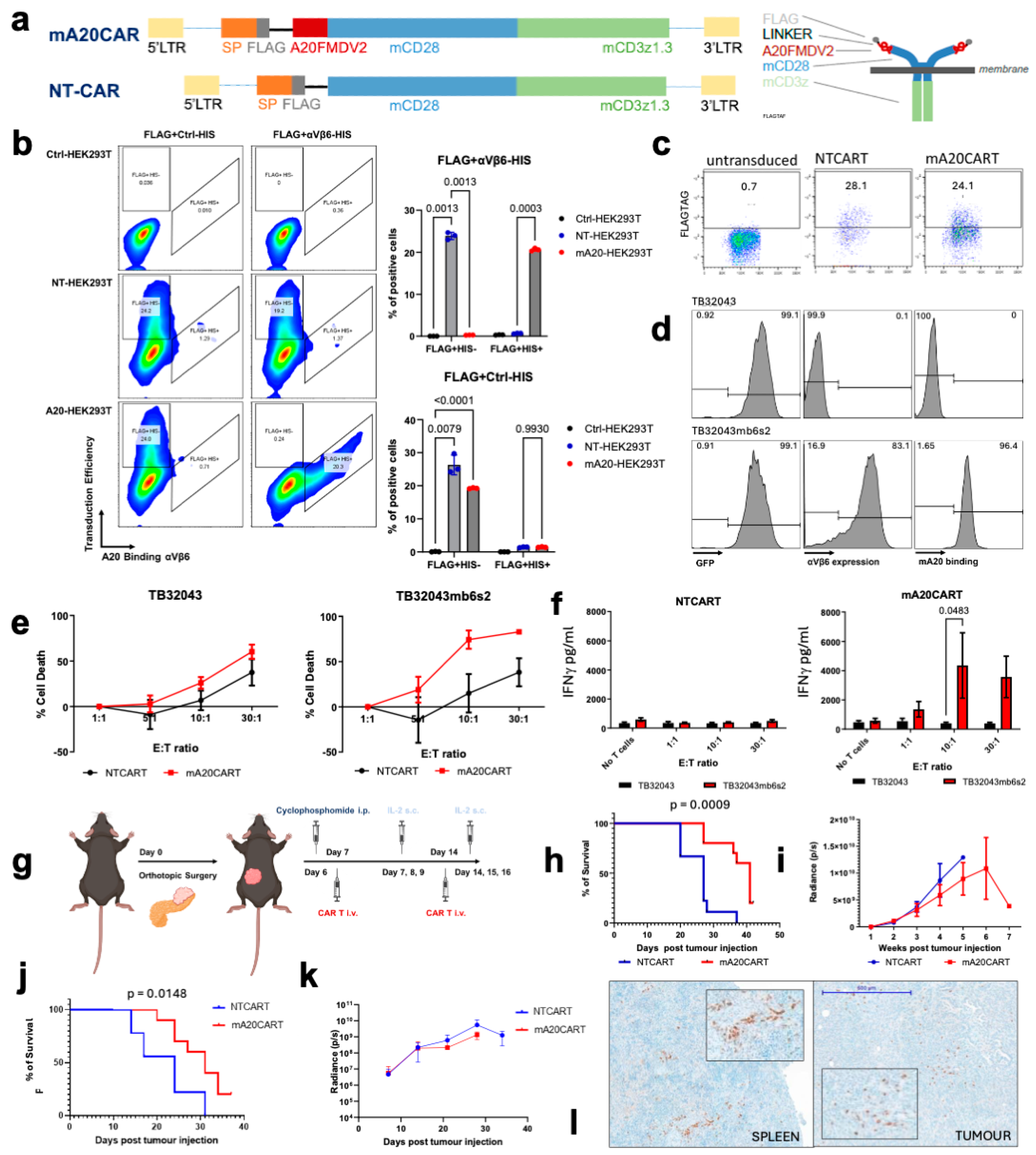
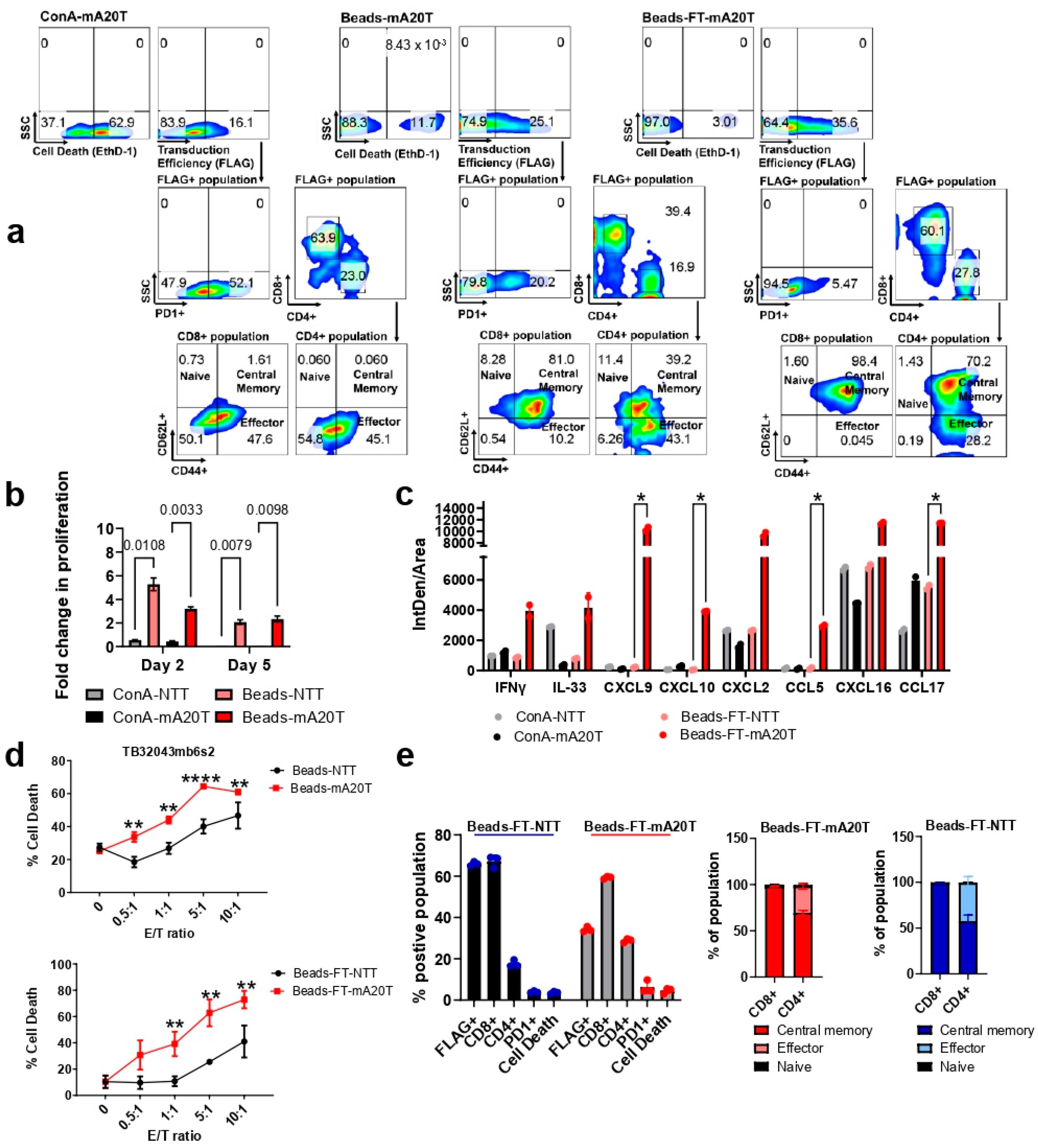
3.1. Murine A20FMDV2 Peptide-Based CAR T Cells Demonstrate a Specific Anti-Tumour Effect Toward Pancreatic Cancer Cells Overexpressing Integrin αvβ6
3.2. An Optimized Process for the Production and Long-Term Cryopreservation Storage of Murine CAR T Cells
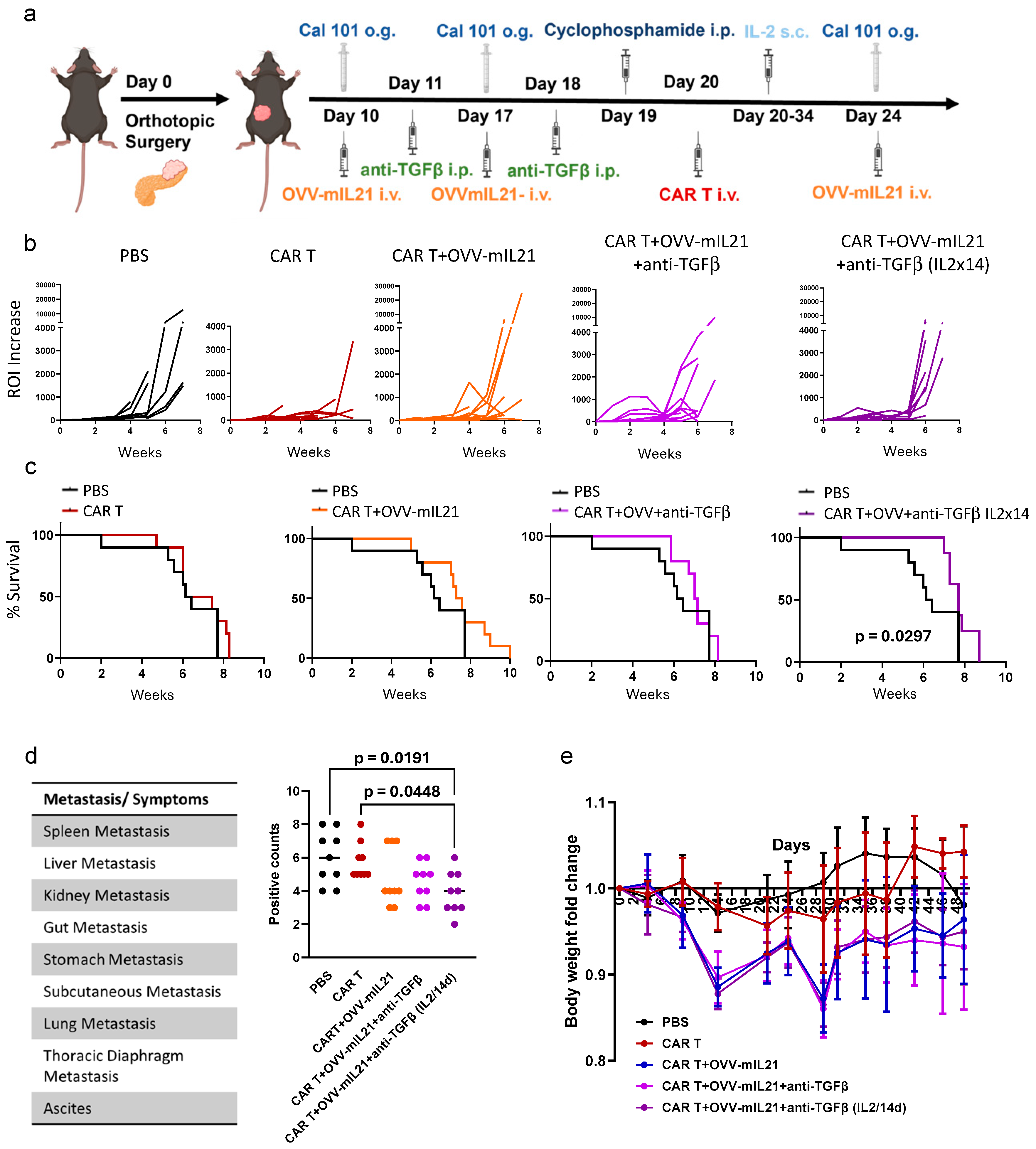
3.3. Oncolytic Vaccinia Virus Combined with TGF-β Antibody Improves the Efficacy of CAR T Cell Therapy in an Immunocompetent Pancreatic Cancer Model
3.4. Triple Combination Therapy Controls Orthotopic Tumour Growth and Attenuates Metastasis in Pancreatic Cancer Within an Immunocompetent Model When Extending IL-2 Supplementation
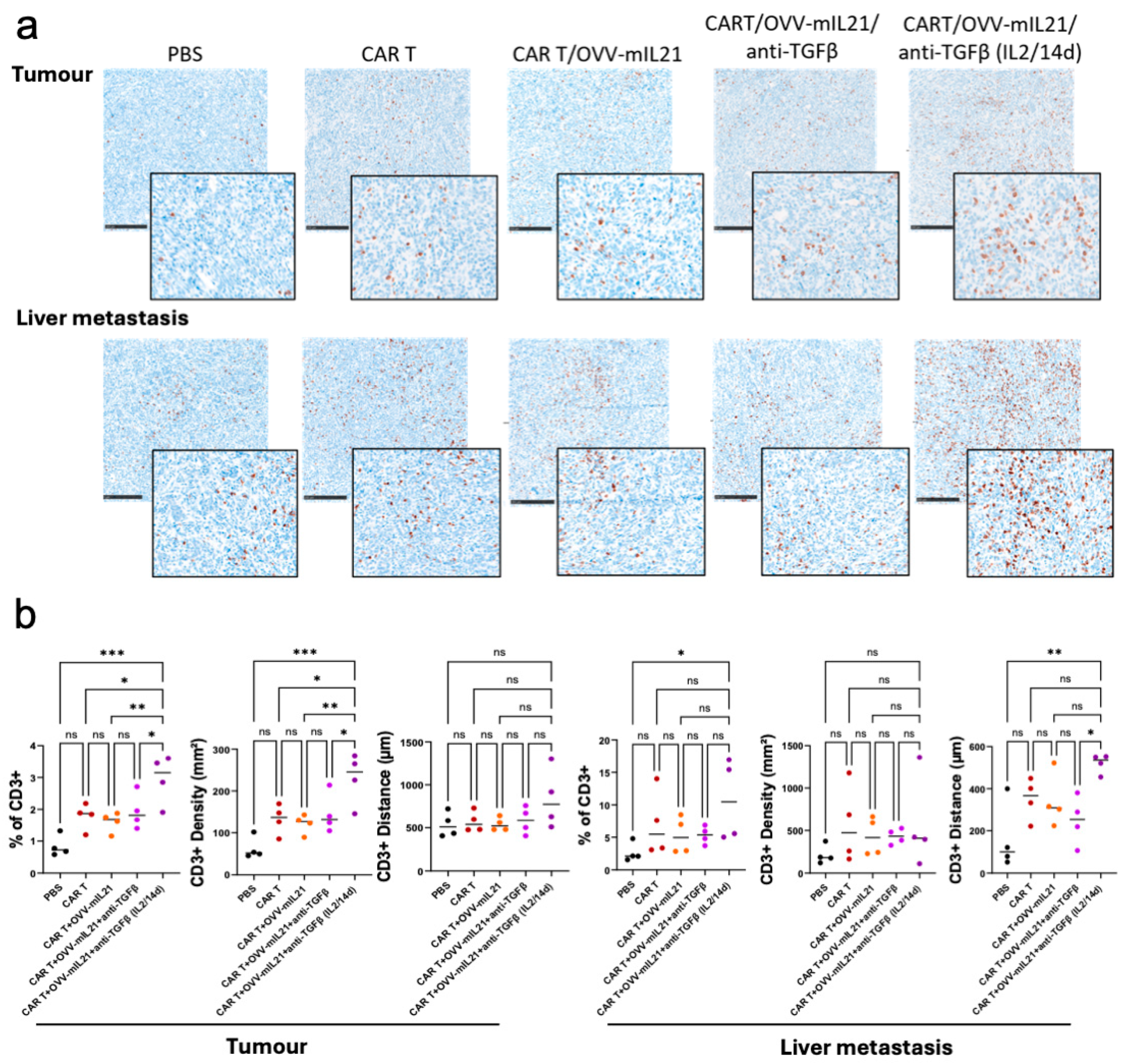
3.5. The Triple Therapy Enhances CAR T Cell Efficacy by Improving T Cell Infiltration and Persistence in Pancreatic Cancer
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Capasso, M.; Franceschi, M.; Rodriguez-Castro, K.I.; Crafa, P.; Cambie, G.; Miraglia, C.; Barchi, A.; Nouvenne, A.; Leandro, G.; Meschi, T.; et al. Epidemiology and risk factors of pancreatic cancer. Acta Biomed. 2018, 89, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Quaresma, M.; Coleman, M.P.; Rachet, B. 40-year trends in an index of survival for all cancers combined and survival adjusted for age and sex for each cancer in England and Wales, 1971–2011: A population-based study. Lancet 2015, 385, 1206–1218. [Google Scholar] [CrossRef] [PubMed]
- Davila, M.L.; Brentjens, R.J. CD19-Targeted CAR T cells as novel cancer immunotherapy for relapsed or refractory B-cell acute lymphoblastic leukemia. Clin. Adv. Hematol. Oncol. 2016, 14, 802–808. [Google Scholar]
- Davila, M.L.; Brentjens, R.J. CAR T cell therapy: Looking back and looking forward. Nat. Cancer 2022, 3, 1418–1419. [Google Scholar] [CrossRef]
- D’Aloia, M.M.; Zizzari, I.G.; Sacchetti, B.; Pierelli, L.; Alimandi, M. CAR-T cells: The long and winding road to solid tumors. Cell Death Dis. 2018, 9, 282. [Google Scholar] [CrossRef]
- Flugel, C.L.; Majzner, R.G.; Krenciute, G.; Dotti, G.; Riddell, S.R.; Wagner, D.L.; Abou-El-Enein, M. Overcoming on-target, off-tumour toxicity of CAR T cell therapy for solid tumours. Nat. Rev. Clin. Oncol. 2023, 20, 49–62. [Google Scholar] [CrossRef]
- Sipos, B.; Hahn, D.; Carceller, A.; Piulats, J.; Hedderich, J.; Kalthoff, H.; Goodman, S.L.; Kosmahl, M.; Kloppel, G. Immunohistochemical screening for beta6-integrin subunit expression in adenocarcinomas using a novel monoclonal antibody reveals strong up-regulation in pancreatic ductal adenocarcinomas in vivo and in vitro. Histopathology 2004, 45, 226–236. [Google Scholar] [CrossRef]
- Reader, C.S.; Vallath, S.; Steele, C.W.; Haider, S.; Brentnall, A.; Desai, A.; Moore, K.M.; Jamieson, N.B.; Chang, D.; Bailey, P.; et al. The integrin alphavbeta6 drives pancreatic cancer through diverse mechanisms and represents an effective target for therapy. J. Pathol. 2019, 249, 332–342. [Google Scholar] [CrossRef]
- Bates, R.C.; Bellovin, D.I.; Brown, C.; Maynard, E.; Wu, B.; Kawakatsu, H.; Sheppard, D.; Oettgen, P.; Mercurio, A.M. Transcriptional activation of integrin beta6 during the epithelial-mesenchymal transition defines a novel prognostic indicator of aggressive colon carcinoma. J. Clin. Investig. 2005, 115, 339–347. [Google Scholar] [CrossRef]
- Elayadi, A.N.; Samli, K.N.; Prudkin, L.; Liu, Y.H.; Bian, A.; Xie, X.J.; Wistuba, I.I.; Roth, J.A.; McGuire, M.J.; Brown, K.C. A peptide selected by biopanning identifies the integrin alphavbeta6 as a prognostic biomarker for nonsmall cell lung cancer. Cancer Res. 2007, 67, 5889–5895. [Google Scholar] [CrossRef] [PubMed]
- Bandyopadhyay, A.; Raghavan, S. Defining the role of integrin alphavbeta6 in cancer. Curr. Drug Targets 2009, 10, 645–652. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Wu, Q.; Dong, Z.; Liu, K. Integrins in cancer: Emerging mechanisms and therapeutic opportunities. Pharmacol. Ther. 2023, 247, 108458. [Google Scholar] [CrossRef] [PubMed]
- Meecham, A.; Marshall, J. Harnessing the power of foot-and-mouth-disease virus for targeting integrin alpha-v beta-6 for the therapy of cancer. Expert. Opin. Drug Discov. 2021, 16, 737–744. [Google Scholar] [CrossRef]
- Whilding, L.M.; Parente-Pereira, A.C.; Zabinski, T.; Davies, D.M.; Petrovic, R.M.G.; Kao, Y.V.; Saxena, S.A.; Romain, A.; Costa-Guerra, J.A.; Violette, S.; et al. Targeting of Aberrant alphavbeta6 Integrin Expression in Solid Tumors Using Chimeric Antigen Receptor-Engineered T Cells. Mol. Ther. 2017, 25, 259–273. [Google Scholar] [CrossRef]
- Brown, N.F.; Murray, E.R.; Cutmore, L.C.; Howard, P.; Masterson, L.; Zammarchi, F.; Hartley, J.A.; van Berkel, P.H.; Marshall, J.F. Integrin-αvβ6 targeted peptide-toxin therapy in a novel αvβ6-expressing immunocompetent model of pancreatic cancer. Pancreatology 2024, 24, 445–455. [Google Scholar] [CrossRef]
- Moore, K.M.; Desai, A.; Delgado, B.L.; Trabulo, S.M.D.; Reader, C.; Brown, N.F.; Murray, E.R.; Brentnall, A.; Howard, P.; Masterson, L.; et al. Integrin alphavbeta6-specific therapy for pancreatic cancer developed from foot-and-mouth-disease virus. Theranostics 2020, 10, 2930–2942. [Google Scholar] [CrossRef]
- Slack, R.J.; Hafeji, M.; Rogers, R.; Ludbrook, S.B.; Marshall, J.F.; Flint, D.J.; Pyne, S.; Denyer, J.C. Pharmacological Characterization of the alphavbeta6 Integrin Binding and Internalization Kinetics of the Foot-and-Mouth Disease Virus Derived Peptide A20FMDV2. Pharmacology 2016, 97, 114–125. [Google Scholar] [CrossRef]
- Hausner, S.H.; DiCara, D.; Marik, J.; Marshall, J.F.; Sutcliffe, J.L. Use of a peptide derived from foot-and-mouth disease virus for the noninvasive imaging of human cancer: Generation and evaluation of 4-[18F]fluorobenzoyl A20FMDV2 for in vivo imaging of integrin alphavbeta6 expression with positron emission tomography. Cancer Res. 2007, 67, 7833–7840. [Google Scholar] [CrossRef]
- Whilding, L.M.; Halim, L.; Draper, B.; Parente-Pereira, A.C.; Zabinski, T.; Davies, D.M.; Maher, J. CAR T-Cells Targeting the Integrin alphavbeta6 and Co-Expressing the Chemokine Receptor CXCR2 Demonstrate Enhanced Homing and Efficacy against Several Solid Malignancies. Cancers 2019, 11, 674. [Google Scholar] [CrossRef]
- Lee, J.W.; Komar, C.A.; Bengsch, F.; Graham, K.; Beatty, G.L. Genetically Engineered Mouse Models of Pancreatic Cancer: The KPC Model (LSL-Kras(G12D/+);LSL-Trp53(R172H/+);Pdx-1-Cre), Its Variants, and Their Application in Immuno-oncology Drug Discovery. Curr. Protoc. Pharmacol. 2016, 73, 14.39.1–14.39.20. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, E.N.; Cutmore, L.C.; Marshall, J.F. Syngeneic Mouse Models for Pre-Clinical Evaluation of CAR T Cells. Cancers 2024, 16, 3186. [Google Scholar] [CrossRef] [PubMed]
- Schober, M.; Jesenofsky, R.; Faissner, R.; Weidenauer, C.; Hagmann, W.; Michl, P.; Heuchel, R.L.; Haas, S.L.; Lohr, J.M. Desmoplasia and chemoresistance in pancreatic cancer. Cancers 2014, 6, 2137–2154. [Google Scholar] [CrossRef] [PubMed]
- Siegler, E.L.; Wang, P. Preclinical Models in Chimeric Antigen Receptor-Engineered T-Cell Therapy. Hum. Gene Ther. 2018, 29, 534–546. [Google Scholar] [CrossRef]
- Sterner, R.C.; Sterner, R.M. CAR-T cell therapy: Current limitations and potential strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef]
- Lin, D.; Shen, Y.; Liang, T. Oncolytic virotherapy: Basic principles, recent advances and future directions. Signal Transduct. Target. Ther. 2023, 8, 156. [Google Scholar] [CrossRef]
- Rezaei, R.; Esmaeili Gouvarchin Ghaleh, H.; Farzanehpour, M.; Dorostkar, R.; Ranjbar, R.; Bolandian, M.; Mirzaei Nodooshan, M.; Ghorbani Alvanegh, A. Combination therapy with CAR T cells and oncolytic viruses: A new era in cancer immunotherapy. Cancer Gene Ther. 2022, 29, 647–660. [Google Scholar] [CrossRef]
- Marelli, G.; Chard Dunmall, L.S.; Yuan, M.; Di Gioia, C.; Miao, J.; Cheng, Z.; Zhang, Z.; Liu, P.; Ahmed, J.; Gangeswaran, R.; et al. A systemically deliverable Vaccinia virus with increased capacity for intertumoral and intratumoral spread effectively treats pancreatic cancer. J. Immunother. Cancer 2021, 9, e001624. [Google Scholar] [CrossRef]
- Wang, N.; Wang, J.; Zhang, Z.; Cao, H.; Yan, W.; Chu, Y.; Chard Dunmall, L.S.; Wang, Y. A novel vaccinia virus enhances anti-tumor efficacy and promotes a long-term anti-tumor response in a murine model of colorectal cancer. Mol. Ther. Oncolytics 2021, 20, 71–81. [Google Scholar] [CrossRef]
- Chakravarthy, A.; Khan, L.; Bensler, N.P.; Bose, P.; De Carvalho, D.D. TGF-beta-associated extracellular matrix genes link cancer-associated fibroblasts to immune evasion and immunotherapy failure. Nat. Commun. 2018, 9, 4692. [Google Scholar] [CrossRef]
- Castro, F.; Cardoso, A.P.; Goncalves, R.M.; Serre, K.; Oliveira, M.J. Interferon-Gamma at the Crossroads of Tumor Immune Surveillance or Evasion. Front. Immunol. 2018, 9, 847. [Google Scholar] [CrossRef] [PubMed]
- Liyanage, U.K.; Moore, T.T.; Joo, H.G.; Tanaka, Y.; Herrmann, V.; Doherty, G.; Drebin, J.A.; Strasberg, S.M.; Eberlein, T.J.; Goedegebuure, P.S.; et al. Prevalence of regulatory T cells is increased in peripheral blood and tumor microenvironment of patients with pancreas or breast adenocarcinoma. J. Immunol. 2002, 169, 2756–2761. [Google Scholar] [CrossRef] [PubMed]
- Principe, D.R.; Park, A.; Dorman, M.J.; Kumar, S.; Viswakarma, N.; Rubin, J.; Torres, C.; McKinney, R.; Munshi, H.G.; Grippo, P.J.; et al. TGFbeta Blockade Augments PD-1 Inhibition to Promote T-Cell-Mediated Regression of Pancreatic Cancer. Mol. Cancer Ther. 2019, 18, 613–620. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Lang, M.; Zhao, T.; Feng, X.; Zheng, C.; Huang, C.; Hao, J.; Dong, J.; Luo, L.; Li, X.; et al. Cancer-FOXP3 directly activated CCL5 to recruit FOXP3(+)Treg cells in pancreatic ductal adenocarcinoma. Oncogene 2017, 36, 3048–3058. [Google Scholar] [CrossRef]
- Brown, N.F.; Marshall, J.F. Integrin-Mediated TGFbeta Activation Modulates the Tumour Microenvironment. Cancers 2019, 11, 1221. [Google Scholar] [CrossRef]
- Xu, J.; Lamouille, S.; Derynck, R. TGF-beta-induced epithelial to mesenchymal transition. Cell Res. 2009, 19, 156–172. [Google Scholar] [CrossRef]
- Dong, X.; Zhao, B.; Iacob, R.E.; Zhu, J.; Koksal, A.C.; Lu, C.; Engen, J.R.; Springer, T.A. Force interacts with macromolecular structure in activation of TGF-beta. Nature 2017, 542, 55–59. [Google Scholar] [CrossRef]
- Breuss, J.M.; Gallo, J.; DeLisser, H.M.; Klimanskaya, I.V.; Folkesson, H.G.; Pittet, J.F.; Nishimura, S.L.; Aldape, K.; Landers, D.V.; Carpenter, W.; et al. Expression of the beta 6 integrin subunit in development, neoplasia and tissue repair suggests a role in epithelial remodeling. J. Cell Sci. 1995, 108 Pt 6, 2241–2251. [Google Scholar] [CrossRef]
- Munger, J.S.; Huang, X.; Kawakatsu, H.; Griffiths, M.J.; Dalton, S.L.; Wu, J.; Pittet, J.F.; Kaminski, N.; Garat, C.; Matthay, M.A.; et al. The integrin alpha v beta 6 binds and activates latent TGF beta 1: A mechanism for regulating pulmonary inflammation and fibrosis. Cell 1999, 96, 319–328. [Google Scholar] [CrossRef]
- Worthington, J.J.; Kelly, A.; Smedley, C.; Bauche, D.; Campbell, S.; Marie, J.C.; Travis, M.A. Integrin alphavbeta8-Mediated TGF-beta Activation by Effector Regulatory T Cells Is Essential for Suppression of T-Cell-Mediated Inflammation. Immunity 2015, 42, 903–915. [Google Scholar] [CrossRef]
- Seed, R.I.; Kobayashi, K.; Ito, S.; Takasaka, N.; Cormier, A.; Jespersen, J.M.; Publicover, J.; Trilok, S.; Combes, A.J.; Chew, N.W.; et al. A tumor-specific mechanism of T(reg) enrichment mediated by the integrin alphavbeta8. Sci. Immunol. 2021, 6, eabf0558. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, P.; Moisini, I.; Geiger, T.L. Identification of a murine CD28 dileucine motif that suppresses single-chain chimeric T-cell receptor expression and function. Blood 2003, 102, 4320–4325. [Google Scholar] [CrossRef] [PubMed]
- Kochenderfer, J.N.; Yu, Z.; Frasheri, D.; Restifo, N.P.; Rosenberg, S.A. Adoptive transfer of syngeneic T cells transduced with a chimeric antigen receptor that recognizes murine CD19 can eradicate lymphoma and normal B cells. Blood 2010, 116, 3875–3886. [Google Scholar] [CrossRef] [PubMed]
- Lanitis, E.; Rota, G.; Kosti, P.; Ronet, C.; Spill, A.; Seijo, B.; Romero, P.; Dangaj, D.; Coukos, G.; Irving, M. Optimized gene engineering of murine CAR-T cells reveals the beneficial effects of IL-15 coexpression. J. Exp. Med. 2021, 218, e20192203. [Google Scholar] [CrossRef]
- Blank, C.U.; Haining, W.N.; Held, W.; Hogan, P.G.; Kallies, A.; Lugli, E.; Lynn, R.C.; Philip, M.; Rao, A.; Restifo, N.P.; et al. Defining ‘T cell exhaustion’. Nat. Rev. Immunol. 2019, 19, 665–674. [Google Scholar] [CrossRef]
- Liu, Q.; Sun, Z.; Chen, L. Memory T cells: Strategies for optimizing tumor immunotherapy. Protein Cell 2020, 11, 549–564. [Google Scholar] [CrossRef]
- Bhat, P.; Leggatt, G.; Waterhouse, N.; Frazer, I.H. Interferon-gamma derived from cytotoxic lymphocytes directly enhances their motility and cytotoxicity. Cell Death Dis. 2017, 8, e2836. [Google Scholar] [CrossRef]
- Larson, R.C.; Kann, M.C.; Bailey, S.R.; Haradhvala, N.J.; Llopis, P.M.; Bouffard, A.A.; Scarfo, I.; Leick, M.B.; Grauwet, K.; Berger, T.R.; et al. CAR T cell killing requires the IFNgammaR pathway in solid but not liquid tumours. Nature 2022, 604, 563–570. [Google Scholar] [CrossRef]
- Chen, L.; Sun, R.; Xu, J.; Zhai, W.; Zhang, D.; Yang, M.; Yue, C.; Chen, Y.; Li, S.; Turnquist, H.; et al. Tumor-Derived IL33 Promotes Tissue-Resident CD8(+) T Cells and Is Required for Checkpoint Blockade Tumor Immunotherapy. Cancer Immunol. Res. 2020, 8, 1381–1392. [Google Scholar] [CrossRef]
- Kuhn, N.F.; Lopez, A.V.; Li, X.; Cai, W.; Daniyan, A.F.; Brentjens, R.J. CD103(+) cDC1 and endogenous CD8(+) T cells are necessary for improved CD40L-overexpressing CAR T cell antitumor function. Nat. Commun. 2020, 11, 6171. [Google Scholar] [CrossRef]
- Kohli, K.; Pillarisetty, V.G.; Kim, T.S. Key chemokines direct migration of immune cells in solid tumors. Cancer Gene Ther. 2022, 29, 10–21. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Conejo-Garcia, J.R.; Katsaros, D.; Gimotty, P.A.; Massobrio, M.; Regnani, G.; Makrigiannakis, A.; Gray, H.; Schlienger, K.; Liebman, M.N.; et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N. Engl. J. Med. 2003, 348, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Steele, C.W.; Karim, S.A.; Leach, J.D.G.; Bailey, P.; Upstill-Goddard, R.; Rishi, L.; Foth, M.; Bryson, S.; McDaid, K.; Wilson, Z.; et al. CXCR2 Inhibition Profoundly Suppresses Metastases and Augments Immunotherapy in Pancreatic Ductal Adenocarcinoma. Cancer Cell 2016, 29, 832–845. [Google Scholar] [CrossRef] [PubMed]
- Sanford, D.E.; Belt, B.A.; Panni, R.Z.; Mayer, A.; Deshpande, A.D.; Carpenter, D.; Mitchem, J.B.; Plambeck-Suess, S.M.; Worley, L.A.; Goetz, B.D.; et al. Inflammatory monocyte mobilization decreases patient survival in pancreatic cancer: A role for targeting the CCL2/CCR2 axis. Clin. Cancer Res. 2013, 19, 3404–3415. [Google Scholar] [CrossRef]
- Marshall, L.A.; Marubayashi, S.; Jorapur, A.; Jacobson, S.; Zibinsky, M.; Robles, O.; Hu, D.X.; Jackson, J.J.; Pookot, D.; Sanchez, J.; et al. Tumors establish resistance to immunotherapy by regulating T(reg) recruitment via CCR4. J. Immunother. Cancer 2020, 8, e000764. [Google Scholar] [CrossRef]
- Chen, Y.; Yu, F.; Jiang, Y.; Chen, J.; Wu, K.; Chen, X.; Lin, Y.; Zhang, H.; Li, L.; Zhang, Y. Adoptive Transfer of Interleukin-21-stimulated Human CD8+ T Memory Stem Cells Efficiently Inhibits Tumor Growth. J. Immunother. 2018, 41, 274–283. [Google Scholar] [CrossRef]
- Topchyan, P.; Xin, G.; Chen, Y.; Zheng, S.; Burns, R.; Shen, J.; Kasmani, M.Y.; Kudek, M.; Yang, N.; Cui, W. Harnessing the IL-21-BATF Pathway in the CD8(+) T Cell Anti-Tumor Response. Cancers 2021, 13, 1263. [Google Scholar] [CrossRef]
- Ferguson, M.S.; Chard Dunmall, L.S.; Gangeswaran, R.; Marelli, G.; Tysome, J.R.; Burns, E.; Whitehead, M.A.; Aksoy, E.; Alusi, G.; Hiley, C.; et al. Transient Inhibition of PI3Kdelta Enhances the Therapeutic Effect of Intravenous Delivery of Oncolytic Vaccinia Virus. Mol. Ther. 2020, 28, 1263–1275. [Google Scholar] [CrossRef]
- Murad, J.P.; Tilakawardane, D.; Park, A.K.; Lopez, L.S.; Young, C.A.; Gibson, J.; Yamaguchi, Y.; Lee, H.J.; Kennewick, K.T.; Gittins, B.J.; et al. Pre-conditioning modifies the TME to enhance solid tumor CAR T cell efficacy and endogenous protective immunity. Mol. Ther. 2021, 29, 2335–2349. [Google Scholar] [CrossRef]
- Kankeu Fonkoua, L.A.; Sirpilla, O.; Sakemura, R.; Siegler, E.L.; Kenderian, S.S. CAR T cell therapy and the tumor microenvironment: Current challenges and opportunities. Mol. Ther. Oncolytics 2022, 25, 69–77. [Google Scholar] [CrossRef]
- McGuigan, A.; Kelly, P.; Turkington, R.C.; Jones, C.; Coleman, H.G.; McCain, R.S. Pancreatic cancer: A review of clinical diagnosis, epidemiology, treatment and outcomes. World J. Gastroenterol. 2018, 24, 4846–4861. [Google Scholar] [CrossRef] [PubMed]
- Martinez, M.; Moon, E.K. CAR T Cells for Solid Tumors: New Strategies for Finding, Infiltrating, and Surviving in the Tumor Microenvironment. Front. Immunol. 2019, 10, 128. [Google Scholar] [CrossRef] [PubMed]
- June, C.H.; O’Connor, R.S.; Kawalekar, O.U.; Ghassemi, S.; Milone, M.C. CAR T cell immunotherapy for human cancer. Science 2018, 359, 1361–1365. [Google Scholar] [CrossRef] [PubMed]
- Newick, K.; O’Brien, S.; Moon, E.; Albelda, S.M. CAR T Cell Therapy for Solid Tumors. Annu. Rev. Med. 2017, 68, 139–152. [Google Scholar] [CrossRef]
- Mahadevan, D.; Von Hoff, D.D. Tumor-stroma interactions in pancreatic ductal adenocarcinoma. Mol. Cancer Ther. 2007, 6, 1186–1197. [Google Scholar] [CrossRef]
- Whatcott, C.J.; Diep, C.H.; Jiang, P.; Watanabe, A.; LoBello, J.; Sima, C.; Hostetter, G.; Shepard, H.M.; Von Hoff, D.D.; Han, H. Desmoplasia in Primary Tumors and Metastatic Lesions of Pancreatic Cancer. Clin. Cancer Res. 2015, 21, 3561–3568. [Google Scholar] [CrossRef]
- Feig, C.; Gopinathan, A.; Neesse, A.; Chan, D.S.; Cook, N.; Tuveson, D.A. The pancreas cancer microenvironment. Clin. Cancer Res. 2012, 18, 4266–4276. [Google Scholar] [CrossRef]
- Clark, C.E.; Hingorani, S.R.; Mick, R.; Combs, C.; Tuveson, D.A.; Vonderheide, R.H. Dynamics of the immune reaction to pancreatic cancer from inception to invasion. Cancer Res. 2007, 67, 9518–9527. [Google Scholar] [CrossRef]
- Stromnes, I.M.; Hulbert, A.; Pierce, R.H.; Greenberg, P.D.; Hingorani, S.R. T-cell Localization, Activation, and Clonal Expansion in Human Pancreatic Ductal Adenocarcinoma. Cancer Immunol. Res. 2017, 5, 978–991. [Google Scholar] [CrossRef]
- Cutmore, L.C.; Brown, N.F.; Raj, D.; Chauduri, S.; Wang, P.; Maher, J.; Wang, Y.; Lemoine, N.R.; Marshall, J.F. Pancreatic Cancer UK Grand Challenge: Developments and challenges for effective CAR T cell therapy for pancreatic ductal adenocarcinoma. Pancreatology 2020, 20, 394–408. [Google Scholar] [CrossRef]
- Marofi, F.; Motavalli, R.; Safonov, V.A.; Thangavelu, L.; Yumashev, A.V.; Alexander, M.; Shomali, N.; Chartrand, M.S.; Pathak, Y.; Jarahian, M.; et al. CAR T cells in solid tumors: Challenges and opportunities. Stem Cell Res. Ther. 2021, 12, 81. [Google Scholar] [CrossRef] [PubMed]
- Vignali, D.A.; Collison, L.W.; Workman, C.J. How regulatory T cells work. Nat. Rev. Immunol. 2008, 8, 523–532. [Google Scholar] [CrossRef] [PubMed]
- Pandiyan, P.; Zhu, J. Origin and functions of pro-inflammatory cytokine producing Foxp3+ regulatory T cells. Cytokine 2015, 76, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Toomer, K.H.; Malek, T.R. Cytokine Signaling in the Development and Homeostasis of Regulatory T cells. Cold Spring Harb. Perspect. Biol. 2018, 10, a028597. [Google Scholar] [CrossRef]
- de Geus, S.W.; Boogerd, L.S.; Swijnenburg, R.J.; Mieog, J.S.; Tummers, W.S.; Prevoo, H.A.; Sier, C.F.; Morreau, H.; Bonsing, B.A.; van de Velde, C.J.; et al. Selecting Tumor-Specific Molecular Targets in Pancreatic Adenocarcinoma: Paving the Way for Image-Guided Pancreatic Surgery. Mol. Imaging Biol. 2016, 18, 807–819. [Google Scholar] [CrossRef]
- Li, Z.; Lin, P.; Gao, C.; Peng, C.; Liu, S.; Gao, H.; Wang, B.; Wang, J.; Niu, J.; Niu, W. Integrin beta6 acts as an unfavorable prognostic indicator and promotes cellular malignant behaviors via ERK-ETS1 pathway in pancreatic ductal adenocarcinoma (PDAC). Tumor Biol. 2016, 37, 5117–5131. [Google Scholar] [CrossRef]
- Dicara, D.; Burman, A.; Clark, S.; Berryman, S.; Howard, M.J.; Hart, I.R.; Marshall, J.F.; Jackson, T. Foot-and-mouth disease virus forms a highly stable, EDTA-resistant complex with its principal receptor, integrin alphavbeta6: Implications for infectiousness. J. Virol. 2008, 82, 1537–1546. [Google Scholar] [CrossRef]
- Matsushita, K.; Toiyama, Y.; Tanaka, K.; Saigusa, S.; Hiro, J.; Uchida, K.; Inoue, Y.; Kusunoki, M. Soluble CXCL16 in preoperative serum is a novel prognostic marker and predicts recurrence of liver metastases in colorectal cancer patients. Ann. Surg. Oncol. 2012, 19 (Suppl. S3), S518–S527. [Google Scholar] [CrossRef]
- Wang, J.; Liu, C.; Chang, X.; Qi, Y.; Zhu, Z.; Yang, X. Fibrosis of mesothelial cell-induced peritoneal implantation of ovarian cancer cells. Cancer Manag. Res. 2018, 10, 6641–6647. [Google Scholar] [CrossRef]
- Mizukami, Y.; Kono, K.; Kawaguchi, Y.; Akaike, H.; Kamimura, K.; Sugai, H.; Fujii, H. CCL17 and CCL22 chemokines within tumor microenvironment are related to accumulation of Foxp3+ regulatory T cells in gastric cancer. Int. J. Cancer 2008, 122, 2286–2293. [Google Scholar] [CrossRef]
- Berlato, C.; Khan, M.N.; Schioppa, T.; Thompson, R.; Maniati, E.; Montfort, A.; Jangani, M.; Canosa, M.; Kulbe, H.; Hagemann, U.B.; et al. A CCR4 antagonist reverses the tumor-promoting microenvironment of renal cancer. J. Clin. Investig. 2017, 127, 801–813. [Google Scholar] [CrossRef] [PubMed]
- Evgin, L.; Kottke, T.; Tonne, J.; Thompson, J.; Huff, A.L.; van Vloten, J.; Moore, M.; Michael, J.; Driscoll, C.; Pulido, J.; et al. Oncolytic virus-mediated expansion of dual-specific CAR T cells improves efficacy against solid tumors in mice. Sci. Transl. Med. 2022, 14, eabn2231. [Google Scholar] [CrossRef] [PubMed]
- Nishio, N.; Diaconu, I.; Liu, H.; Cerullo, V.; Caruana, I.; Hoyos, V.; Bouchier-Hayes, L.; Savoldo, B.; Dotti, G. Armed oncolytic virus enhances immune functions of chimeric antigen receptor-modified T cells in solid tumors. Cancer Res. 2014, 74, 5195–5205. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.; Luo, Y.; Da, T.; Guedan, S.; Ruella, M.; Scholler, J.; Keith, B.; Young, R.M.; Engels, B.; Sorsa, S.; et al. Pancreatic cancer therapy with combined mesothelin-redirected chimeric antigen receptor T cells and cytokine-armed oncolytic adenoviruses. JCI Insight 2018, 3, e99573. [Google Scholar] [CrossRef]
- Tanoue, K.; Rosewell Shaw, A.; Watanabe, N.; Porter, C.; Rana, B.; Gottschalk, S.; Brenner, M.; Suzuki, M. Armed Oncolytic Adenovirus-Expressing PD-L1 Mini-Body Enhances Antitumor Effects of Chimeric Antigen Receptor T Cells in Solid Tumors. Cancer Res. 2017, 77, 2040–2051. [Google Scholar] [CrossRef]
- Rosewell Shaw, A.; Porter, C.E.; Watanabe, N.; Tanoue, K.; Sikora, A.; Gottschalk, S.; Brenner, M.K.; Suzuki, M. Adenovirotherapy Delivering Cytokine and Checkpoint Inhibitor Augments CAR T Cells against Metastatic Head and Neck Cancer. Mol. Ther. 2017, 25, 2440–2451. [Google Scholar] [CrossRef]
- Porter, C.E.; Rosewell Shaw, A.; Jung, Y.; Yip, T.; Castro, P.D.; Sandulache, V.C.; Sikora, A.; Gottschalk, S.; Ittman, M.M.; Brenner, M.K.; et al. Oncolytic Adenovirus Armed with BiTE, Cytokine, and Checkpoint Inhibitor Enables CAR T Cells to Control the Growth of Heterogeneous Tumors. Mol. Ther. 2020, 28, 1251–1262. [Google Scholar] [CrossRef]
- Wing, A.; Fajardo, C.A.; Posey, A.D., Jr.; Shaw, C.; Da, T.; Young, R.M.; Alemany, R.; June, C.H.; Guedan, S. Improving CART-Cell Therapy of Solid Tumors with Oncolytic Virus-Driven Production of a Bispecific T-cell Engager. Cancer Immunol. Res. 2018, 6, 605–616. [Google Scholar] [CrossRef]
- Li, Y.; Bleakley, M.; Yee, C. IL-21 influences the frequency, phenotype, and affinity of the antigen-specific CD8 T cell response. J. Immunol. 2005, 175, 2261–2269. [Google Scholar] [CrossRef]
- Brady, J.; Hayakawa, Y.; Smyth, M.J.; Nutt, S.L. IL-21 induces the functional maturation of murine NK cells. J. Immunol. 2004, 172, 2048–2058. [Google Scholar] [CrossRef]
- Coquet, J.M.; Kyparissoudis, K.; Pellicci, D.G.; Besra, G.; Berzins, S.P.; Smyth, M.J.; Godfrey, D.I. IL-21 is produced by NKT cells and modulates NKT cell activation and cytokine production. J. Immunol. 2007, 178, 2827–2834. [Google Scholar] [CrossRef] [PubMed]
- Nakano, H.; Kishida, T.; Asada, H.; Shin-Ya, M.; Shinomiya, T.; Imanishi, J.; Shimada, T.; Nakai, S.; Takeuchi, M.; Hisa, Y.; et al. Interleukin-21 triggers both cellular and humoral immune responses leading to therapeutic antitumor effects against head and neck squamous cell carcinoma. J. Gene Med. 2006, 8, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Nurieva, R.; Yang, X.O.; Martinez, G.; Zhang, Y.; Panopoulos, A.D.; Ma, L.; Schluns, K.; Tian, Q.; Watowich, S.S.; Jetten, A.M.; et al. Essential autocrine regulation by IL-21 in the generation of inflammatory T cells. Nature 2007, 448, 480–483. [Google Scholar] [CrossRef]
- Castermans, K.; Tabruyn, S.P.; Zeng, R.; van Beijnum, J.R.; Eppolito, C.; Leonard, W.J.; Shrikant, P.A.; Griffioen, A.W. Angiostatic activity of the antitumor cytokine interleukin-21. Blood 2008, 112, 4940–4947. [Google Scholar] [CrossRef]
- Croce, M.; Rigo, V.; Ferrini, S. IL-21: A pleiotropic cytokine with potential applications in oncology. J. Immunol. Res. 2015, 2015, 696578. [Google Scholar] [CrossRef]
- Heo, J.; Reid, T.; Ruo, L.; Breitbach, C.J.; Rose, S.; Bloomston, M.; Cho, M.; Lim, H.Y.; Chung, H.C.; Kim, C.W.; et al. Randomized dose-finding clinical trial of oncolytic immunotherapeutic vaccinia JX-594 in liver cancer. Nat. Med. 2013, 19, 329–336. [Google Scholar] [CrossRef]
- Mastrangelo, M.J.; Maguire, H.C.; Eisenlohr, L.C.; Laughlin, C.E.; Monken, C.E.; McCue, P.A.; Kovatich, A.J.; Lattime, E.C. Intratumoral recombinant GM-CSF-encoding virus as gene therapy in patients with cutaneous melanoma. Cancer Gene Ther. 1999, 6, 409–422. [Google Scholar] [CrossRef]
- Giampieri, S.; Manning, C.; Hooper, S.; Jones, L.; Hill, C.S.; Sahai, E. Localized and reversible TGFbeta signalling switches breast cancer cells from cohesive to single cell motility. Nat. Cell Biol. 2009, 11, 1287–1296. [Google Scholar] [CrossRef]
- Principe, D.R.; DeCant, B.; Mascarinas, E.; Wayne, E.A.; Diaz, A.M.; Akagi, N.; Hwang, R.; Pasche, B.; Dawson, D.W.; Fang, D.; et al. TGFbeta Signaling in the Pancreatic Tumor Microenvironment Promotes Fibrosis and Immune Evasion to Facilitate Tumorigenesis. Cancer Res. 2016, 76, 2525–2539. [Google Scholar] [CrossRef]
- Principe, D.R.; Doll, J.A.; Bauer, J.; Jung, B.; Munshi, H.G.; Bartholin, L.; Pasche, B.; Lee, C.; Grippo, P.J. TGF-beta: Duality of function between tumor prevention and carcinogenesis. J. Natl. Cancer Inst. 2014, 106, djt369. [Google Scholar] [CrossRef]
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Kadel, E.E., III; Koeppen, H.; Astarita, J.L.; Cubas, R.; et al. TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018, 554, 544–548. [Google Scholar] [CrossRef] [PubMed]
- Seoane, J.; Gomis, R.R. TGF-beta Family Signaling in Tumor Suppression and Cancer Progression. Cold Spring Harb. Perspect. Biol. 2017, 9, a022277. [Google Scholar] [CrossRef] [PubMed]
- Vanpouille-Box, C.; Formenti, S.C. Dual Transforming Growth Factor-beta and Programmed Death-1 Blockade: A Strategy for Immune-Excluded Tumors? Trends Immunol. 2018, 39, 435–437. [Google Scholar] [CrossRef] [PubMed]
- Tauriello, D.V.F.; Palomo-Ponce, S.; Stork, D.; Berenguer-Llergo, A.; Badia-Ramentol, J.; Iglesias, M.; Sevillano, M.; Ibiza, S.; Canellas, A.; Hernando-Momblona, X.; et al. TGFbeta drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 2018, 554, 538–543. [Google Scholar] [CrossRef]
- Tang, N.; Cheng, C.; Zhang, X.; Qiao, M.; Li, N.; Mu, W.; Wei, X.F.; Han, W.; Wang, H. TGF-beta inhibition via CRISPR promotes the long-term efficacy of CAR T cells against solid tumors. JCI Insight 2020, 5, e133977. [Google Scholar] [CrossRef]
- Kloss, C.C.; Lee, J.; Zhang, A.; Chen, F.; Melenhorst, J.J.; Lacey, S.F.; Maus, M.V.; Fraietta, J.A.; Zhao, Y.; June, C.H. Dominant-Negative TGF-beta Receptor Enhances PSMA-Targeted Human CAR T Cell Proliferation And Augments Prostate Cancer Eradication. Mol. Ther. 2018, 26, 1855–1866. [Google Scholar] [CrossRef]
- Narayan, V.; Barber-Rotenberg, J.S.; Jung, I.Y.; Lacey, S.F.; Rech, A.J.; Davis, M.M.; Hwang, W.T.; Lal, P.; Carpenter, E.L.; Maude, S.L.; et al. PSMA-targeting TGFbeta-insensitive armored CAR T cells in metastatic castration-resistant prostate cancer: A phase 1 trial. Nat. Med. 2022, 28, 724–734. [Google Scholar] [CrossRef]
- Foster, A.E.; Dotti, G.; Lu, A.; Khalil, M.; Brenner, M.K.; Heslop, H.E.; Rooney, C.M.; Bollard, C.M. Antitumor activity of EBV-specific T lymphocytes transduced with a dominant negative TGF-beta receptor. J. Immunother. 2008, 31, 500–505. [Google Scholar] [CrossRef]
- Stuber, T.; Monjezi, R.; Wallstabe, L.; Kuhnemundt, J.; Nietzer, S.L.; Dandekar, G.; Wockel, A.; Einsele, H.; Wischhusen, J.; Hudecek, M. Inhibition of TGF-beta-receptor signaling augments the antitumor function of ROR1-specific CAR T-cells against triple-negative breast cancer. J. Immunother. Cancer 2020, 8, e000676. [Google Scholar] [CrossRef]
- Zhang, L.; Yu, Z.; Muranski, P.; Palmer, D.C.; Restifo, N.P.; Rosenberg, S.A.; Morgan, R.A. Inhibition of TGF-beta signaling in genetically engineered tumor antigen-reactive T cells significantly enhances tumor treatment efficacy. Gene Ther. 2013, 20, 575–580. [Google Scholar] [CrossRef]
- Martin, C.J.; Datta, A.; Littlefield, C.; Kalra, A.; Chapron, C.; Wawersik, S.; Dagbay, K.B.; Brueckner, C.T.; Nikiforov, A.; Danehy, F.T., Jr.; et al. Selective inhibition of TGFbeta1 activation overcomes primary resistance to checkpoint blockade therapy by altering tumor immune landscape. Sci. Transl. Med. 2020, 12, eaay8456. [Google Scholar] [CrossRef] [PubMed]
- Sanjabi, S.; Oh, S.A.; Li, M.O. Regulation of the Immune Response by TGF-beta: From Conception to Autoimmunity and Infection. Cold Spring Harb. Perspect. Biol. 2017, 9, a022236. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Zheng, L.; Yuan, Q.; Zhen, G.; Crane, J.L.; Zhou, X.; Cao, X. Transforming growth factor-beta in stem cells and tissue homeostasis. Bone Res. 2018, 6, 2. [Google Scholar] [CrossRef] [PubMed]
- Herbertz, S.; Sawyer, J.S.; Stauber, A.J.; Gueorguieva, I.; Driscoll, K.E.; Estrem, S.T.; Cleverly, A.L.; Desaiah, D.; Guba, S.C.; Benhadji, K.A.; et al. Clinical development of galunisertib (LY2157299 monohydrate), a small molecule inhibitor of transforming growth factor-beta signaling pathway. Drug Des. Devel Ther. 2015, 9, 4479–4499. [Google Scholar] [CrossRef]
- Ciardiello, D.; Elez, E.; Tabernero, J.; Seoane, J. Clinical development of therapies targeting TGFbeta: Current knowledge and future perspectives. Ann. Oncol. 2020, 31, 1336–1349. [Google Scholar] [CrossRef]
- Teixeira, A.F.; Ten Dijke, P.; Zhu, H.J. On-Target Anti-TGF-beta Therapies Are Not Succeeding in Clinical Cancer Treatments: What Are Remaining Challenges? Front. Cell Dev. Biol. 2020, 8, 605. [Google Scholar] [CrossRef]
- Wang, Y. Pre-Clinical Development for IND Application of a Novel Systemically Deliverable Oncolytic Vaccinia Virus for Treatment of Pancreatic Cancer. Available online: https://gtr.ukri.org/projects?ref=MR%2FV006053%2F1#/tabOverview (accessed on 28 April 2025).

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, Z.; Cutmore, L.C.; Baleeiro, R.B.; Hartlebury, J.J.; Brown, N.; Chard-Dunmall, L.; Lemoine, N.; Wang, Y.; Marshall, J.F. The Combination of Oncolytic Virus and Antibody Blockade of TGF-β Enhances the Efficacy of αvβ6-Targeting CAR T Cells Against Pancreatic Cancer in an Immunocompetent Model. Cancers 2025, 17, 1534. https://doi.org/10.3390/cancers17091534
Zhao Z, Cutmore LC, Baleeiro RB, Hartlebury JJ, Brown N, Chard-Dunmall L, Lemoine N, Wang Y, Marshall JF. The Combination of Oncolytic Virus and Antibody Blockade of TGF-β Enhances the Efficacy of αvβ6-Targeting CAR T Cells Against Pancreatic Cancer in an Immunocompetent Model. Cancers. 2025; 17(9):1534. https://doi.org/10.3390/cancers17091534
Chicago/Turabian StyleZhao, Zuoyi, Lauren C. Cutmore, Renato B. Baleeiro, Joseph J. Hartlebury, Nicholas Brown, Louisa Chard-Dunmall, Nicholas Lemoine, Yaohe Wang, and John F. Marshall. 2025. "The Combination of Oncolytic Virus and Antibody Blockade of TGF-β Enhances the Efficacy of αvβ6-Targeting CAR T Cells Against Pancreatic Cancer in an Immunocompetent Model" Cancers 17, no. 9: 1534. https://doi.org/10.3390/cancers17091534
APA StyleZhao, Z., Cutmore, L. C., Baleeiro, R. B., Hartlebury, J. J., Brown, N., Chard-Dunmall, L., Lemoine, N., Wang, Y., & Marshall, J. F. (2025). The Combination of Oncolytic Virus and Antibody Blockade of TGF-β Enhances the Efficacy of αvβ6-Targeting CAR T Cells Against Pancreatic Cancer in an Immunocompetent Model. Cancers, 17(9), 1534. https://doi.org/10.3390/cancers17091534