Abstract
MYC deregulation, a cardinal event in Burkitt lymphoma (BL) pathogenesis, necessitates the elucidation of the molecular mechanisms governing MYC activation to devise innovative and effective therapeutic strategies. The t(8;14)(q24;q32) chromosomal translocation commonly observed in hematological malignancies results in MYC deregulation, endowing cancer cells with a competitive edge through heightened cell proliferation, cell cycle progression, apoptosis evasion, and metabolic reprogramming. Recent discoveries of recurrent MYC mutations in BL underscore the potential of precision medicine, employing tailored therapeutics to specifically inhibit MYC activity. However, the intricate genetic landscape of BL, featuring additional alterations, such as mutations in TP53, TCF3, and ID3, may necessitate a combinatorial approach targeting multiple oncogenic pathways for effective intervention. Despite significant strides in hematological malignancy treatment, a comprehensive understanding of the molecular mechanisms underpinning MYC’s oncogenic properties remains crucial for the potential development of highly potent and selective MYC-directed cancer therapies. This review offers an in-depth analysis of MYC translocation and its implications in Burkitt lymphoma, with a spotlight on cutting-edge advances in research and emerging therapeutic paradigms.
1. Introduction
Burkitt lymphoma (BL) is a highly aggressive and rapidly advancing subtype of non-Hodgkin’s lymphoma, hallmarked by elevated proliferation rates and substantial genetic instability. Burkitt lymphoma (BL) has several notable distinctions in cancer research. It was the first human tumor found to be linked with a virus [1] and one of the earliest tumors identified to have a chromosomal translocation activating an oncogene [2]. Additionally, it was the first lymphoma reported in association with HIV infection [3]. Moreover, it was the first pediatric tumor to show a response to chemotherapy alone [4]. In regions with endemic malaria, such as equatorial Africa, Brazil, and Papua New Guinea, BL is the most prevalent childhood cancer [5,6].
A defining feature of BL is the presence of chromosomal translocations involving the MYC gene, which encodes a pivotal transcription factor that orchestrates cell growth, differentiation, and apoptosis [7,8]. The MYC oncogene significantly contributes to many human cancers by regulating myriad genes. Its influence extends beyond cellular biology to impact host immunity and the tumor microenvironment [9,10]. It is activated through various methods—genetic, epigenetic, and post-translational—leading to altered expression, and thus influences different types of cancer [11]. Common genomic changes, such as gene amplification, chromosomal translocations, and mutations, can escalate MYC expression [12,13]. The gene further influences cancer cells by managing processes, such as growth, differentiation, and metabolism [14,15,16]. Although MYC expression is typically restrained in healthy cells, its heightened levels in cancer cells can trigger apoptosis. Nevertheless, its activation bypasses these physiological checks, thereby promoting cancer via several interconnected mechanisms [9,17,18]. The regulation of MYC and the diverse range of its targets is depicted in Figure 1. MYC overexpression in Burkitt lymphoma is commonly facilitated by chromosomal translocations, such as the MYC-immunoglobulin heavy chain (IgH) [19,20]. This translocation culminates in unrestrained MYC activation, fostering cell proliferation and impeding apoptosis through intricate and diverse mechanisms that entail interactions with a plethora of proteins and regulatory factors [21].
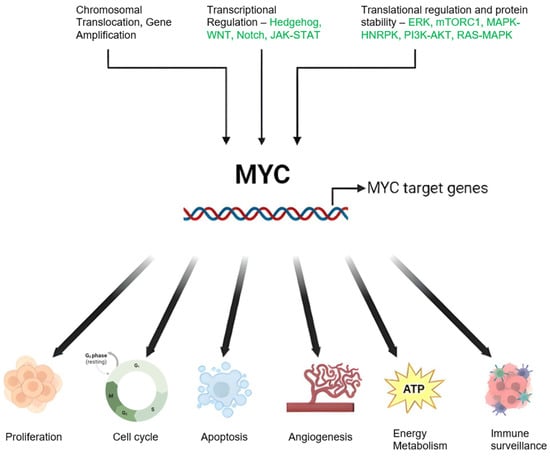
Figure 1.
Schematic Representation of MYC Regulation and Function in Cancer: Genetic alterations including chromosomal translocations and genomic amplifications result in elevated MYC mRNA expression. Modifications in upstream regulatory pathways can also influence MYC oncogene transcription. MYC protein stability is enhanced through post-translational modifications, notably preferential phosphorylation at serine 62 (S62) over threonine 58 (T58), inhibiting degradation and stimulating the MYC pathway. MYC orchestrates a variety of cancer cell-intrinsic and host-dependent pathways to foster cancer cell growth and survival, with key processes including proliferation, metabolism, protein, and ribosomal biosynthesis. Conversely, MYC suppresses cellular protective mechanisms, such as immune surveillance, and thus aids in cancer progression. Notably, MYC can paradoxically trigger cellular processes, such as apoptosis that potentially threaten cancer cell survival. The ultimate cell fate is determined by the intricate equilibrium between these events and the specific cellular context. Created with BioRender.com, accessed on 20 May 2023.
In addition to chromosomal translocations, MYC point mutations have been identified in BL. Present in approximately 30% of BL cases [22], these mutations lead to increased MYC protein stability and activity, correlating with poor prognosis. Given the central role MYC occupies in BL development and progression, there is a burgeoning interest in devising MYC-targeted therapies. However, due to MYC’s transcription factor nature and the absence of a well-defined active region, the development of small compounds targeting MYC presents a formidable challenge [8]. This review delves into the intricate roles of MYC translocation and mutations in BL pathogenesis and progression, while discussing the potential therapeutic avenues of targeting MYC in this devastating disease.
2. Epidemiology and Challenges in Treatment Approach
Burkitt lymphoma is named after Denis Burkitt [23], who first identified this disease among children in Africa. The epidemiological data of Burkitt lymphoma (Table 1) provide a fascinating insight into the geographical distribution of this disease. It is noteworthy that the incidence rates are not uniform across the globe, indicating the influence of various factors, such as genetics, environmental factors, and exposure to certain viruses, such as the Epstein–Barr virus. The data provide a comprehensive view of the disease prevalence, helping researchers and healthcare professionals in understanding the global impact of Burkitt lymphoma, and aiding in the development of targeted strategies for its prevention and treatment.
Advancements in understanding the biology of Burkitt lymphoma (BL) have led to improved outcomes for children with the disease in high-income countries (HICs). Collaborative efforts by pediatric cancer cooperative groups have contributed to these improvements through research aimed at enhancing diagnostic precision, refining staging classifications, optimizing risk stratification, and providing better supportive care [24,25,26,27]. Although high-income countries (HICs) have made significant progress in treating Burkitt lymphoma (BL), these advancements may not be directly applicable to the healthcare landscape in Africa or to the biology of endemic BL. Consequently, the majority of children with BL worldwide have not benefited from this progress [28,29]. Current estimates indicate that long-term survival rates for pediatric BL in sub-Saharan Africa (SSA) remain between 30% and 50%, unchanged since the 1970s when BL was discovered and pediatric oncology emerged as a field [6,29]. This disparity can be attributed to limited resources, late diagnoses, and treatment abandonment [30].

Table 1.
Global incidence rates of Burkitt lymphoma. Data represent the incidence rate of Burkitt lymphoma per million years. Adapted from [31].
Table 1.
Global incidence rates of Burkitt lymphoma. Data represent the incidence rate of Burkitt lymphoma per million years. Adapted from [31].
| Region/Country | Burkitt Lymphoma Incidence |
|---|---|
| Asia | |
| Israel | 3.77 |
| Saudi Arabia | 2.41 |
| Turkey | 2.30 |
| Republic of Korea | 1.72 |
| Japan | 1.55 |
| Jordan | 1.17 |
| Thailand | 0.88 |
| India | 0.59 |
| China | 0.45 |
| Europe | |
| Estonia | 5.67 |
| Switzerland | 4.12 |
| Belgium | 3.72 |
| Norway | 3.49 |
| Spain | 3.34 |
| Italy | 3.23 |
| The Netherlands | 3.16 |
| France | 3.13 |
| Lithuania | 3.00 |
| Denmark | 2.92 |
| United Kingdom | 2.68 |
| Ireland | 2.64 |
| Austria | 2.44 |
| Germany | 2.43 |
| Czech Republic | 1.97 |
| Belarus | 1.96 |
| Ukraine | 1.52 |
| Poland | 0.88 |
| Russian Federation | 0.72 |
| Oceania | |
| New Zealand | 3.21 |
| Australia | 3.12 |
| Africa | |
| Malawi | 19.3 |
| Uganda | 4.8 |
| Zambia | 4.2 |
| Rwanda | 3.5 |
| Burundi | 3.4 |
| South Sudan | 2.5 |
| Tanzania | 2.2 |
| Madagascar | 2.1 |
| Kenya | 1.7 |
| Mozambique | 1.7 |
| Ethiopia | 0.4 |
| Cameroon | 8.0 |
| Congo, Democratic People Republic of | 2.9 |
| Angola | 2.1 |
| Chad | 1.7 |
| Sudan | 2.0 |
| Egypt | 1.7 |
| Morocco | 1.7 |
| Algeria | 0.9 |
| South Africa | 1.6 |
| Cote d’Ivoire | 4.6 |
| Nigeria | 2.8 |
| Ghana | 2.4 |
| Senegal | 2.4 |
| Burkina Faso | 2.2 |
| Mali | 1.4 |
| Niger | 1.2 |
| Central/South America | |
| Puerto Rico | 5.32 |
| Colombia | 5.10 |
| North America | |
| US white people | 4.94 |
| United States (overall) | 4.75 |
| US black people | 4.04 |
| Canada | 2.35 |
To develop strategies that can increase cure rates for children with endemic BL, it is crucial to thoroughly examine the evolution of pediatric BL treatments and outcomes in SSA. This involves analyzing clinical trials and retrospective reports in parallel with the developments made in HICs. It is worth considering whether improving case detection, early diagnosis, and referral to treatment centers could significantly reduce mortality rates in Africa. Additionally, current BL treatments often cause severe side effects, complicating their use in regions with limited intensive care services [32]. The development of new drugs with fewer side effects may increase treatment uptake, avoid chemoresistance, and offer life-saving benefits to African children and immunocompromised adults.
Expanding our understanding of the genetic and poly-microbial underpinnings of BL could lead to advancements in treatment and prevention. Investigating the biological pathways involved in these infections may reveal novel approaches to diagnosis, treatment, and prevention of BL [33]. Overcoming research challenges, such as developing reliable methods to measure the prevalence and load of c-MYC translocation-positive B-cells in asymptomatic individuals, is crucial for progress in this field.
3. MYC Deregulation in Burkitt Lymphoma: Mechanisms and Implications
- (1)
- Chromosomal Translocations: In Burkitt lymphoma (BL), the most common mechanism of MYC deregulation involves chromosomal translocations that place the MYC gene under the control of immunoglobulin (Ig) enhancer elements, leading to constitutive activation of MYC expression in B-cells [34,35,36,37]. The translocation t(8;14)(q24;q32), found in approximately 80% of BL cases, is the most frequent MYC translocation in this disease [38,39]. Additional MYC translocations in BL include t(2;8)(p12;q24) and t(8;22)(q24;q11) [39,40,41]. Constitutive MYC overexpression drives uncontrolled cell proliferation, a defining feature of BL.
- (2)
- Point Mutations: In addition to chromosomal translocations, point mutations in MYC have been identified in BL. The T58A mutation increases MYC protein stability by inhibiting its degradation via the ubiquitin–proteasome pathway and enhances MYC transcriptional activity by increasing its association with transcriptional co-activators, such as TRRAP and p300/CBP [42,43].
- (3)
- Genomic Instability: MYC translocation also contributes to the genomic instability of BL cells. MYC-induced DNA replication stress can cause DNA double-strand breaks, leading to chromosomal rearrangements and mutations [44,45]. This instability can contribute to the clonal evolution of BL and the acquisition of additional mutations that drive tumor progression and resistance to therapy [46].
- (4)
- Signaling Pathways: The molecular mechanisms of MYC-induced transformation in BL are complex and involve the deregulation of multiple signaling pathways. MYC regulates the expression of genes involved in several growth/proliferation regulating signaling pathways, including the Wnt/β-catenin, NF-κB, and PI3K/Akt/mTOR pathways [47,48]. Deregulation of these pathways can contribute to the aggressive behavior of BL and resistance to therapy.
- (5)
- MYC and Cancer Metabolism: MYC has been implicated in cancer metabolism, particularly the Warburg effect, where it promotes glucose metabolism and aerobic glycolysis in cancer cells [49,50]. This metabolic adaptation enables cancer cells to withstand nutrient scarcity and hypoxic environments. MYC also plays a role in mitochondrial biogenesis and regulation in response to growth signals and cell cycle progression [51,52,53]. This provides an opportunity for therapeutic targeting of mitochondrial factors regulated by MYC in the context of the Warburg effect in cancer.
- (6)
- MYC and Immune Regulation: Studies on human patient samples and transgenic mouse models have provided evidence that MYC is involved in the regulation of innate immune regulator CD-47 and the well-known adaptive immune checkpoint PD-L1 [54,55,56]. Saravia et. al [57] found that MYC also promotes the differentiation and activation of regulatory T-cells, which suppress immune responses and promote tumor growth. MYC influences the expression of genes involved in T-cell activation while suppressing the expression of genes involved in T-cell differentiation and function [57].
Figure 2 depicts data obtained from cBio Cancer Genomics Portal, where MYC altered lymphomas are shown to have a significantly worse prognosis than patients with wild-type MYC. Since MYC translocations are found in nearly all Burkitt lymphoma cases, it is difficult to establish a direct correlation between MYC alterations and survival outcomes. Instead, other factors, such as disease stage at diagnosis, patient age, performance status, lactate dehydrogenase levels, and response to treatment, are more commonly used to assess prognosis and survival in Burkitt lymphoma. Table 2 lists the various mechanisms by which MYC could be deregulated followed by detailed discussions on these topics.
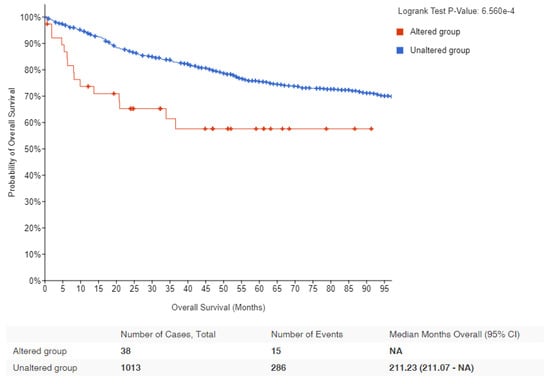
Figure 2.
Kaplan–Meier survival estimates in lymphomas with MYC alteration: Data obtained from cBioPortal for lymphoma patients showing the probability of overall survival in MYC altered (red) and unaltered (blue) groups. Logrank p-value and number of samples analyzed for each group are indicated [58,59].
Table 2.
Summary of various mechanisms by which MYC could be deregulated.
3.1. Mechanism of MYC Translocation and Its Significance in Accumulating Mutations on the Translocated Allele
3.1.1. Mechanism of IgH-MYC Translocation
Translocation occurs when chromosomes break and reattach to different chromosomes. In Burkitt lymphoma, one end of chromosome 8, where the MYC gene resides, translocates to chromosome 14, where the IgH gene is located. This leads to the fusion of the two genes. Normally, the MYC gene regulates cell division, but when it is fused with the IgH gene, it is then placed under the control of IgH’s regulatory elements. This results in overexpression of the MYC gene, leading to uncontrolled cell growth and proliferation, a key feature of lymphoma [11,60].
This translocation is often linked to errors in the somatic hypermutation process, a mechanism where B-cells modify their antibody genes to produce high-affinity antibodies. Normally, this process is tightly regulated, but errors can lead to off-target mutations, including the IgH-MYC translocation.
In the context of chromosomal rearrangements leading to the translocation of proto-oncogenes into transcription-active regions, the study by Stasevich et al. [61] highlighted the role of non-coding enhancer RNAs (eRNAs) as regulatory mechanisms affecting the oncogenes upon translocation [61]. This study emphasizes the role of non-coding enhancer RNAs (eRNAs) in regulating the activity of oncogenes following translocation. The researchers sought to identify eRNAs that might influence MYC transcription during IgH-MYC translocation in Burkitt lymphoma, with an emphasis on eRNAs that are potentially oncogenic, located at the IgH locus, and predominantly expressed in B-cells. The results from this study demonstrated that eRNA AL928768.3 is a critical player in Burkitt lymphoma development. Importantly, the study’s findings also suggest that AL928768.3, along with other yet-to-be-discovered eRNAs, could potentially serve as tissue-specific targets for cancer therapeutics [61]. This opens up a promising new avenue for the development of targeted treatments for lymphomas, particularly Burkitt lymphoma.
3.1.2. Risk Factors for Burkitt Lymphoma and IgH-MYC Translocation
Risk factors for Burkitt lymphoma, and thus the IgH-MYC translocation, include:
- Infection with the Epstein–Barr virus (EBV): Most people are infected with EBV at some point in their lives, but in rare cases, it can increase the risk of Burkitt lymphoma. It is believed that the virus may contribute to the occurrence of the IgH-MYC translocation [62].
- Malaria: In endemic regions (such as sub-Saharan Africa), chronic malaria infection weakens the immune system and is thought to contribute to the higher incidence of Burkitt lymphoma [63].
- Immune suppression: Individuals with weakened immune systems (such as those with HIV/AIDS or organ transplant recipients) have an increased risk [64].
3.1.3. Mutations Accumulate in the Translocated MYC Allele
The translocation of the c-MYC gene into the immunoglobulin heavy chain (IgH) locus in Burkitt lymphoma (BL) has been reported to result in a high number of mutations due to its exposure to the antibody hypermutation mechanism [65,66,67,68,69]. Bemark and Neuberger [65] demonstrated that the translocated c-MYC undergoes constitutive hypermutation in the Ramos BL cell line at a rate comparable to that of immunoglobulin variable (IgV) genes. This study found that the non-random distribution of mutations stems from the intrinsic bias of the mutational process, rather than a skewing effect of selection, and provided insights into the cis-acting sequences required for mutability. Specifically, IgH sequences downstream of the Sμ region were sufficient, and the IgH intronic enhancer was not necessary.
Nearly two decades later, ref. [70] examined the effect of Phorbol 12-myristate 13-acetate (PMA) stimulation on the expression of the MYC gene in Ramos BL wild-type (WT) and ADP-dependent glucokinase (ADPGK) knock-out (KO) cells. The researchers discovered a larger accumulation of mutations in MYC transcripts in Ramos WT cells compared to ADPGK KO cells upon PMA stimulation. These mutations displayed a preference for AGC triplets, indicating the influence of immunoglobulin hypermutation and an organized mutational machinery. Interestingly, sequencing of stimulated Ramos BL cells revealed the recovery of the wild-type MYC sequence in nearly half of the cases, which contrasts the findings of [65]. Additionally, MYC expression increased during activation and decreased during differentiation as B-cells transitioned from an activated state to a plasma-cell-like differentiation phase, suggesting a functional role for MYC in providing energy stimulation to differentiating B-cells.
Tandon et al. [70] also found that PMA stimulation increased MYC transcript levels in Ramos cells, but ADPGK KO cells showed a smaller elevation in MYC expression compared to WT cells. The study revealed that the accumulation of mutations is dependent on cell transcriptional activity rather than activation-induced cytidine deaminase (AID) expression levels. This finding points to a role for MYC in offering an evolutionary growth advantage to cancer cells in vitro, characterized by a higher rate of proliferation and increased transcriptional activity at the translocated MYC locus.
In a separate study, ref. [71] investigated the mutational landscape of BL using high-throughput sequencing. They identified recurrent mutations in genes associated with the B-cell receptor signaling pathway, chromatin remodeling, and DNA damage response. This research not only provided valuable insights into the genetic mechanisms underlying MYC-driven lymphomagenesis in BL, but also highlighted potential therapeutic targets for future treatment strategies.
3.2. Additional Genetic Alterations in Burkitt Lymphoma
Although MYC deregulation is a hallmark of Burkitt lymphoma (BL), it alone is insufficient to cause the disease. Additional genetic modifications are necessary for the transformation of B-cells into malignant lymphomas [72]. These additional mutations are generally thought to occur independently of the IgH-MYC translocation, namely, the IgH-MYC translocation does not directly cause these additional mutations to occur. However, it is worth noting that the overexpression of MYC might indirectly contribute to a higher mutation rate. MYC overexpression can lead to increased cell proliferation and DNA replication, which can increase the chance of errors occurring during DNA replication. Furthermore, MYC can induce a state of “replicative stress”, which can lead to DNA damage and genomic instability, further increasing the potential for additional mutations.
3.2.1. TP53 Mutations and Its Prognostic Importance in BL
TP53 is a tumor suppressor gene that encodes a transcription factor that regulates cell cycle arrest, apoptosis, and DNA repair in response to cellular stress. TP53 is frequently altered in various cancers, including BL, where it is one of the most common and important co-alterations, but its role and impact on BL biology and clinical outcome are not fully understood [73,74].
Several studies have investigated the frequency, type, and prognostic value of TP53 abnormalities (mutations, deletions, and/or copy number neutral loss of heterozygosity) in BL, using different methods, such as sequencing, fluorescence in situ hybridization (FISH), immunohistochemistry, and gene expression analysis [73,74,75]. The reported prevalence of TP53 abnormalities in BL ranges from 20% to 70%, depending on the cohort, subtype, and detection method [73,74,75]. TP53 abnormalities are more common in adult than in pediatric BL, and in immunodeficiency-associated than in endemic or sporadic BL [39] TP53 abnormalities are also more frequent in cases with high-risk features, such as high lactate dehydrogenase (LDH), bone marrow or central nervous system involvement, or double-hit or triple-hit status (concurrent rearrangements of MYC, BCL2, and/or BCL6) [73].
In a comprehensive study conducted by Burkhardt et al. [76], they analyzed a large cohort of pediatric and adult Burkitt lymphoma (BL) cases. They discovered that the mutational profile of these cancers varies by age group. A critical finding was the significant prognostic impact of recurrent mutations in the TP53 gene, which are associated with diminished expression of TP53wt protein in lymphoma samples. This lack of TP53wt expression and presence of TP53 mutations were linked to decreased survival rates, suggesting the central role of TP53 in the disease’s progression. The study further implies that new therapeutic strategies could be developed to target this dysfunctional TP53, potentially by inhibiting its upstream regulators, MDM2 and MDM4. This could decrease the ubiquitination and subsequent proteasomal degradation of TP53wt. Additionally, the use of drugs, such as eprenetapopt, which have been shown to restore TP53wt levels, could be a promising approach [76]
The identification of TP53 as a key molecular marker and driver of BL has important implications for diagnosis, prognosis, and treatment. TP53 testing should thus be incorporated into routine clinical practice to stratify patients according to their risk and response to therapy. Moreover, novel therapeutic strategies that target TP53 or its regulators may offer new opportunities to improve the outcomes of BL patients, especially those with relapsed or refractory disease.
In addition to TP53 abnormalities, other factors that can affect the TP53 pathway in BL include the expression of MDM2 and MDM4, two negative regulators of TP53 that can bind to and inhibit its activity. MDM2 overexpression has been reported in some cases of BL, especially those with immunodeficiency or double-hit status [75,77]. MDM4 overexpression has also been identified as the only TP53 pathway abnormality in a subset of BL cases without TP53 or MDM2 abnormalities, suggesting a potential alternative mechanism of TP53 inhibition [75].
3.2.2. TCF3 and ID3 Mutations in BL
Schmitz et al. [71] found mutations in transcription factor 3 (TCF3) in BL. TCF3 is expressed at high levels during B-cell development and is inhibited by the helix-loop-helix protein ID3. Another gene shown to be mutated in BL is cell cycle regulating Cyclin D3 (CCND3), which is a direct target of TCF3, refs. [71,78,79] confirmed these mutations in an independent study, analyzing their frequency and clinical relevance. Their findings revealed that CCND3 and ID3 mutations are correlated with an advanced stage of the disease, representing secondary hits after MYC deregulation, and are essential for lymphomagenesis. Mutations in ID3 have been identified in a subset of BL cases and are thought to contribute to the development of the disease by inhibiting B-cell differentiation and promoting cell proliferation [78]
In a groundbreaking study, ref. [22] sequenced the genome of a BL tumor and the germline DNA from the same individual, as well as the exomes of over 50 BL tumors, comparing them to diffuse large B-cell lymphoma (DLBCL) tumor samples. They found differentially mutated genes in BL samples, such as ID3, PIK3R1, RET, GNA13, ARID1A, and SMARCA4. Importantly, they identified ID3 mutations occurring specifically in Burkitt lymphoma samples (34% of tumors) and not in DLBCLs. The study was the first to show that ID3 mutations are responsible for promoting cell cycle progression and proliferation in Burkitt lymphoma and identifying ID3 as a new tumor suppressor gene.
3.2.3. MYC Translocation and 11q Alterations
Notably, MYC translocation serves as both a diagnostic hallmark and a critical prognostic factor for BL [80]. Patients afflicted with MYC-positive lymphomas face poorer prognoses and necessitate more aggressive treatment regimens compared to their MYC-negative counterparts [37,81] MYC-negative lymphomas, such as those harboring 11q alterations, are associated with a subtype of high-grade B-cell lymphoma, previously referred to as Burkitt-like lymphoma [82]. The lymphomas with 11q aberrations do not have the characteristic MYC translocations seen in Burkitt lymphoma. Instead, they exhibit chromosomal abnormalities involving the loss of a region on the long arm of chromosome 11 (11q). This region contains several tumor suppressor genes, such as ATM and BIRC3, which play roles in DNA repair and apoptosis. Loss of these tumor suppressor genes can contribute to the development of lymphoma. Although both MYC-positive and -negative subtypes share some genomic features, the presence or absence of MYC translocations and 11q alterations help in distinguishing these two distinct molecular subtypes of high-grade B-cell lymphomas. The grim prognosis of MYC-positive lymphomas (such as BL) is predominantly attributed to the intensified cellular proliferation and genetic instability driven by MYC’s activation of target genes governing cell cycle regulation, DNA replication, and DNA repair [83]. This genomic destabilization ultimately fosters the accumulation of additional mutations, accelerating disease progression and heightening resistance to treatment.
3.3. Evolutionary Growth Advantage as an Implication of MYC Translocation in Cancer
Multiple studies have investigated the role of MYC translocation in providing an evolutionary growth advantage to Burkitt lymphoma (BL) cells. Mlynarczyk et al. [84] proposed that BL cells exploit the germinal center reaction to gain a competitive advantage. This complex process generates high-affinity B-cell receptors through somatic hypermutation and selection. MYC translocation drives the expansion of BL cells within the germinal center, promoting their survival and proliferation, facilitated by the interaction with the microenvironment, including T-cells and stromal cells [84]
Epigenetic regulation of MYC in BL expression and progression has also been the focus of many studies [85,86,87,88]. Fernández-Serrano et al. [89] reviewed the role of epigenetic modifications, such as histone modifications H3K4me3 and H3K27ac, in regulating MYC expression in BL cells. Li et al. [90] demonstrated that MYC mutations in lymphomas are associated with changes in chromatin structure, increased levels of H3K27ac histone marks, and altered gene expression patterns, promoting lymphoma cell proliferation and survival. Cowling et al. [91] reported enhanced histone acetylation of MYC-target genes in lymphomas. Ref. [92] investigated the role of epigenetic mechanisms in MYC-driven lymphomagenesis, revealing alterations in DNA methylation patterns and their impact on specific genes involved in immunological regulation, cell adhesion, and cancer-related pathways; overall indicating a MYC-dominated growth advantage in these cells.
Recent studies have also explored the critical role of microRNAs (miRNAs) and mutations in MYC and its translocated partner genes. Ref. [93] identified several microRNAs that regulate MYC expression, such as miR-150, miR-155, and miR-143, whose deregulation in BL leads to MYC overexpression. Several miRNAs, including miR-34a, miR-145, and the let-7 family, have also been shown to directly target MYC mRNA and negatively regulate its expression [94]. On the other hand, it has also been suggested that c-MYC modulates miRNA expression, revealing a complex interplay between c-MYC and miRNAs [95,96]. One study found 211 differentially expressed genes and 49 differentially expressed miRNAs in BL [97]. A significant finding was the downregulation of ATM and NLK genes, important regulators in response to DNA damage in BL tumor cells. These tumor suppressors were targeted by multiple upregulated miRNAs which could account for their aberrant expression in BL. The combined loss of p53 induction and function due to miRNA-mediated regulation of ATM and NLK, along with the upregulation of TFAP4, may be central for human miRNAs in BL oncogenesis. This allows for the survival of BL tumor cells with the IgH-MYC chromosomal translocation and promotes MYC-induced cell cycle progression, initiating BL lymphomagenesis [97].
Klanova and Klener [98] found that mutations in MYC-translocated partner gene, BCL2, promote the growth and survival of lymphoma cells by increasing the expression of MYC and other oncogenes. MYC mutations are also found to impact the tumor microenvironment, a determining factor for chemotherapeutic potency [99,100,101] These studies showed that MYC mutations in lymphomas lead to increased recruitment of immune-suppressive cells, promoting tumor growth, angiogenesis, and metastasis.
Indeed, the marked influence of MYC on the evolutionary growth advantage of cancer cells positions it as an appealing target for innovative therapies. Therefore, by honing in on MYC as a potential treatment target, we may be able to craft therapeutic strategies that effectively counteract the survival advantages accrued by these cancer cells, opening a promising avenue toward more effective lymphoma treatment options.
4. Challenges in Developing Successful Therapies against MYC-Deregulated Burkitt Lymphoma
The development of targeted therapies against MYC-deregulated Burkitt lymphoma (BL) has proven to be challenging due to several factors, including the complexity of MYC’s roles in cellular processes and the difficulty of selectively targeting MYC without affecting normal cellular functions.
Despite advances in cancer therapy, targeted therapies against MYC-deregulated BL have remained elusive due to several factors:
- (1)
- Heterogeneity of MYC-deregulated BL: BL tumors display significant heterogeneity, both at the genetic and epigenetic levels, which contributes to variations in treatment response [66]. This diversity requires the development of therapies that can effectively target the range of molecular alterations in MYC-deregulated BL.
- (2)
- Tumor microenvironment: The tumor microenvironment, consisting of non-cancerous cells and extracellular matrix components, can influence tumor growth and therapy resistance in BL. The interplay between tumor cells and the microenvironment may contribute to the challenges in developing effective treatments against MYC-deregulated BL [102].
- (3)
- Off-target effects: MYC plays a crucial role in normal cell function, and its inhibition may lead to undesirable off-target effects that can cause cytotoxicity in normal cells. This further complicates the development of targeted therapies against MYC [103].
- (4)
- Complexity of MYC’s roles in cellular processes: MYC is a transcription factor that regulates a myriad of cellular processes, including cell cycle progression, metabolism, and protein synthesis [66]. This multifaceted role makes it difficult to selectively target MYC without disrupting normal cellular functions.
- (5)
- Paradoxical roles of MYC: A significantly high level of MYC can induce apoptosis or programmed cell death, as a protective mechanism against uncontrolled proliferation. This phenomenon, known as “MYC-induced apoptosis”, has been well-documented in scientific literature. For example, in a study by Evan et al. [104] it was observed that overexpression of MYC leads to both cell proliferation and apoptosis. This seemingly paradoxical effect is due to the fact that while MYC drives cell cycle progression, it also sensitizes cells to a variety of apoptotic signals. Therefore, unless a second mutation occurs that inhibits apoptosis, excess MYC can lead to cell death [104].
- (6)
- MYC’s “undruggable” nature: MYC has been considered an “undruggable” target due to the lack of well-defined small-molecule binding pockets and its intrinsically disordered structure, which hampers the development of small-molecule inhibitors [20].
- (7)
- The development of resistance: Targeted therapies often face the challenge of acquired resistance, which may emerge through the activation of alternative signaling pathways or the selection of resistant clones [105].
5. Potential Strategies to Overcome Challenges
- (1)
- Patient stratification and precision medicine: An enhanced approach to patient stratification could significantly improve therapeutic efficacy in MYC-deregulated Burkitt lymphoma. Stratification informed by molecular profiling, geographical variations (e.g., HIC vs. SSA regions), and age-related criteria can identify patient subgroups likely to benefit from targeted therapies. This holistic approach could optimize personalized treatment strategies, and help in mitigating the impact of tumor heterogeneity [106].
- (2)
- Targeting the tumor microenvironment: Therapeutic strategies that modulate the tumor microenvironment can potentially enhance the effectiveness of MYC-targeted therapies. For example, immunotherapies, such as immune checkpoint inhibitors and chimeric antigen receptor (CAR) T-cell therapy, can be combined with MYC-targeted treatments to improve antitumor immune responses [107].
- (3)
- Development of selective MYC inhibitors: The development of selective MYC inhibitors with reduced off-target effects can help in minimizing cytotoxicity in normal cells. Recent advancements in drug discovery technologies, such as structure-based drug design and high-throughput screening, can aid in identifying these selective inhibitors [108].
- (4)
- Indirect targeting of MYC: One approach to overcome MYC’s “undruggable” nature is to indirectly target it by modulating its transcription, translation, or protein stability [109]. Small molecules that inhibit MYC-MAX dimerization or target upstream signaling pathways regulating MYC have shown promise in preclinical studies [8].
- (5)
- Combination therapy: Combining targeted therapies against MYC with other treatment modalities, such as chemotherapy or immunotherapy, can potentially improve treatment outcomes and overcome resistance mechanisms [107].
While numerous obstacles persist in the development of successful therapies against MYC-deregulated Burkitt lymphoma, recent advances have paved the way for novel strategies to confront these challenges. The multifaceted nature of MYC necessitates a multifaceted approach to therapy. Stratification of patients, targeting of the tumor microenvironment, advancement of selective MYC inhibitors, indirect targeting of MYC, and the potential of combination therapies all show considerable promise. The journey toward effective targeted therapies is complex and arduous, but as our understanding of MYC and its role in lymphomas continues to deepen, our capacity to innovate and improve therapeutic options deepens, as well.
6. Implications for Treatment
6.1. Background
Burkitt lymphoma (BL), an aggressive lymphoid neoplasm, is characterized by its extraordinary chemosensitivity, stemming from the high proliferation rate of tumor cells that render them susceptible to cytotoxic agents [39,110,111]. For pediatric patients, modern chemoimmunotherapy regimens have produced cure rates reaching 90% or more, as substantiated by Thomas et al.’s study in 2006 using hyper-CVAD and rituximab [111]. However, the overall survival rate for adults and immunocompromised patients, or those in low-income countries, is not as promising due to challenges in tolerating intensive chemotherapy or lack of access to effective treatments. Therefore, alternative therapeutic approaches, notably therapies targeting MYC, are being developed to improve outcomes.
6.2. Strategies for Targeting MYC in Burkitt Lymphoma
6.2.1. Small-Molecule Inhibitors and Targeted Therapies
Several therapeutic strategies have emerged for targeting MYC deregulation in BL. These include small-molecule inhibitors of MYC transcriptional activity and targeted therapies aiming to disrupt MYC protein–protein interactions. The work by Llombart and Mansour [112] in 2022 underscored these promising strategies, such as BET inhibitors targeting MYC transcriptional activity and small-molecule inhibitors disrupting MYC protein–protein interactions. Several inhibitors, including JQ1 [109], MYCi975 [113], and 10058-F4 [114], have been developed and tested in preclinical studies, showing promising results in reducing MYC levels and inhibiting tumor growth in various cancer types, including BL. However, owing to the challenges in developing targeted MYC therapies, as mentioned earlier in this review, the development of these inhibitors has been largely unsuccessful commercially.
6.2.2. Targeting MYC Deregulation via Cellular Pathways
Other therapeutic targets for MYC-positive BL: In addition to targeting MYC directly, numerous other therapeutic targets for MYC-positive BL have been identified:
- CDK9: CDK9, a component of the positive transcription elongation factor b (P-TEFb) complex, is necessary for transcriptional elongation by RNA polymerase II and is one of the prospective therapeutic targets. MYC-positive BL cells were found to be highly sensitive to CDK9 inhibition, leading to decreased cell viability and increased apoptosis [115].
- MCL-1 Inhibition: Inhibition of MCL-1, an anti-apoptotic protein that is often overexpressed in cancer cells, was found to be another potential therapeutic target for MYC-positive BL [116].
- AKT Pathway: MYC translocation activates the AKT pathway, which plays a crucial role in regulating cell survival, metabolism, and proliferation. Targeting the AKT pathway was suggested as a promising therapeutic approach for MYC-positive BL [117]. The combination of an AKT inhibitor and an mTOR inhibitor can synergistically induce apoptosis in MYC-driven lymphoma cells [117].
- Restoration of Wild-Type p53 Expression: Restoring wild-type p53 expression can inhibit their proliferation and induce apoptosis [77].
- Cell Proliferation and Survival Genes: Genes, such as CDK6, CCND2, and BCL2 could serve as potential therapeutic targets for MYC-positive BL [118]. Venetoclax, a BCL2 inhibitor, was found to be efficient in inducing apoptosis in MYC-positive lymphoma cells.
- XIAP Targeting: MYC translocation can also increase the expression of a protein known as X-linked inhibitor of apoptosis protein (XIAP), which inhibits caspase-dependent apoptosis. Targeting XIAP may increase the sensitivity of MYC-positive BL cells to apoptosis-inducing drugs [119].
- Notch and MAPK Pathways: Notch pathway was also found to be involved in MYC-driven lymphomagenesis, with MYC translocation upregulating the expression of Notch receptors and ligands, leading to the activation of the Notch pathway [120]. Therefore, targeting the mitogen-activated protein kinase (MAPK) pathway, which is active in MYC-driven malignancies, has shown therapeutic promise [121].
6.2.3. Synthetic Lethality Strategies
MYC has been linked to synthetic lethality, a process in which the loss of function of two genes causes cell death but not either gene alone [122]. Therefore, targeting genes that are synthetically lethal with MYC may provide a novel approach to selectively kill MYC-dependent cancer cells. A recent study identified several genes that are synthetically lethal with MYC, including BCL2L1, MCL-1, and PIM1 [123]. Targeting these genes may provide a new avenue for therapeutic intervention in MYC-driven cancers.
6.2.4. Epigenetic Modifiers
Epigenetic modifiers have also been studied as potential treatment targets for MYC-driven malignancies. HDAC inhibitors, for example, have been demonstrated to stimulate MYC degradation and reduce the growth of MYC-dependent malignancies [124,125,126] Similarly, inhibitors of bromodomain and extra-terminal (BET) proteins, which are involved in chromatin remodeling and transcriptional regulation, have been shown to inhibit MYC expression and have promising preclinical results [60].
6.2.5. Leveraging MYC’s Role in Metabolism
MYC can also regulate cellular metabolism. For instance, oncogenic MYC can increase the expression of glutamine transporters and alter mitochondrial metabolism, making the cell dependent on exogenous glutamine for survival [8,127,128]. Therefore, inhibiting glutamine metabolism can selectively induce apoptosis in MYC-overexpressing tumor cells [129,130]. CB-839, a potent and selective inhibitor of glutaminase (GLS), an enzyme that converts glutamine to glutamate, is currently in clinical trials for treating cancers with MYC deregulation [131].
6.2.6. Investigation of MYC Inhibitor OmoMYC
The action mechanism of OmoMYC, a MYC inhibitor, was investigated by [132] to provide insights into targeting MYC for cancer therapy. OmoMYC mitigates the activation and repression actions of MYC by competing for chromatin binding with MYC/MAX, effectively reducing MYC/MAX’s binding efficacy (Figure 3b). It significantly reduces promoter occupancy and alters the expression of genes influenced by oncogenic MYC levels [133]. Remarkably, OmoMYC has shown substantial therapeutic potential in the in vivo mouse cancer models. Its effect differs from gene knockout or RNA interference techniques, which aim to obstruct all functions of a gene product. Instead, OmoMYC selectively disrupts certain protein interactions within the MYC node while preserving others, thereby reconfiguring the MYC transcriptome. Given OmoMYC’s profound therapeutic effect in animal models, these findings suggest that a successful cancer therapy targeting MYC may need a dual approach—inhibiting MYC/MAX from binding to E-boxes, while continuing to repress genes that MYC would naturally repress [132].
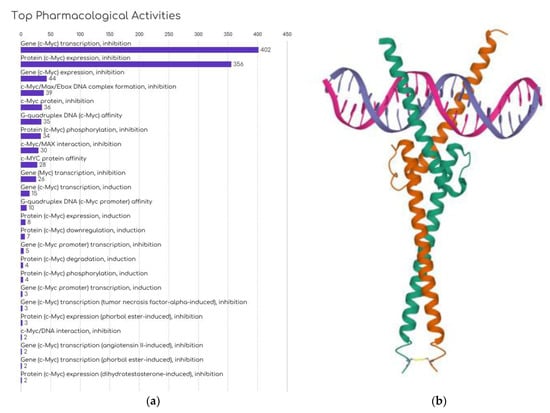
Figure 3.
Current development approaches against MYC: (a) Number of compounds by MYC inhibition strategy in development, obtained from Cortellis Drug Discovery Intelligence (CDDI—Clarivate). (b) Structure of OmoMYC bound to double-stranded DNA. PDB ID: 5I50; Jung L.A. et al. [133]; https://doi.org/10.2210/pdb5I50/pdb, accessed on 20 May 2023.
7. Challenges in Targeting MYC
Despite the potential benefits of targeting MYC, there are challenges and obstacles that researchers have encountered. MYC is considered an “undruggable” target since it lacks enzymatic activity, making the development of traditional small-molecule inhibitors challenging. Additionally, the broad role that MYC plays in cellular function indicates that strategies aimed at inhibiting MYC activity can potentially affect a variety of normal cellular processes and can lead to toxicity in healthy cells.
8. Future Directions
Given these challenges, ongoing research is aimed at identifying strategies to more selectively target MYC, thereby reducing potential toxicity. For instance, the use of targeted delivery systems, such as nanoparticles, to specifically deliver MYC inhibitors to tumor cells is one potential approach that is being explored. Moreover, the utilization of synthetic lethality strategies or combination therapies that incorporate MYC inhibition alongside other treatments may offer a more effective and tolerable therapeutic approach. A comprehensive list of therapies in development against MYC is provided in Table 3.
Table 3.
Therapies by mechanism of action: A list of therapies in development and completed for targeting MYC. Links to drug data available for each category are provided as a link to the source portal. Data obtained from Cortellis Drug Discovery Intelligence (CDDI–Clarivate).
9. Conclusions
Our understanding of the critical role of MYC deregulation in Burkitt lymphoma has advanced significantly in recent years. Through various mechanisms, such as chromosomal translocations, amplifications, and point mutations, MYC becomes deregulated, leading to the initiation and progression of Burkitt lymphoma. This deregulation is crucial for the diagnosis, prognosis, and therapeutic interventions for this aggressive malignancy.
The complex interplay between MYC, its downstream pathways, and the tumor microenvironment has been a focal point of many recent studies. As highlighted in our discussion, MYC deregulation in Burkitt lymphoma can involve epigenetic modifications, the germinal center reaction, and the deregulation of microRNAs, among other molecular mechanisms. This knowledge not only enhances our understanding of the disease, but also opens the door to potential therapeutic targets.
Despite the challenges in targeting MYC, numerous promising strategies have emerged. These include the use of small-molecule inhibitors, targeting protein–protein interactions, and exploiting synthetic lethality with MYC. Additionally, the identification of other molecular pathways, such as AKT, Notch, and MAPK, that are implicated in MYC-driven lymphomagenesis presents further therapeutic possibilities.
Furthermore, the potential role of epigenetic modifiers, such as HDAC inhibitors and BET inhibitors, in the treatment of MYC-driven malignancies highlights the necessity of understanding the broader chromatin landscape in Burkitt lymphoma. These advances in knowledge present a multifaceted approach to combating this devastating disease.
In light of these findings, it is crucial to continue expanding our knowledge of the molecular processes underlying MYC deregulation in Burkitt lymphoma. Further research is needed to develop novel, effective, and personalized therapeutic approaches to improve patient outcomes. By integrating insights from genetic, epigenetic, and signaling pathway studies, we can advance toward a comprehensive understanding of MYC-driven cancer signaling and devise innovative treatment strategies to combat Burkitt lymphoma.
Author Contributions
A.T. conceptualized and wrote the manuscript; J.A.K. helped with revisions and provided access to Cortellis Drug Discovery; V.D. critically reviewed the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
No funding was obtained for the study.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Epstein, M.A.; Achong, B.G.; Barr, Y.M. Virus Particles in Cultured Lymphoblasts from Burkitt’s Lymphoma. Lancet 1964, 1, 702–703. [Google Scholar] [CrossRef] [PubMed]
- Zech, L.; Haglund, U.; Nilsson, K.; Klein, G. Characteristic chromosomal abnormalities in biopsies and lymphoid-cell lines from patients with Burkitt and non-Burkitt lymphomas. Int. J. Cancer 1976, 17, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Schulz, T.F.; Boshoff, C.H.; Weiss, R.A. HIV infection and neoplasia. Lancet 1996, 348, 587–591. [Google Scholar] [CrossRef] [PubMed]
- Burkitt, D.P. Etiology of Burkitt’s lymphoma—An alternative hypothesis to a vectored virus. J. Natl. Cancer Inst. 1969, 42, 19–28. [Google Scholar]
- Parkin, D.M.; Ferlay, J.; Hamdi-Chérif, M.; Sitas, F.; Thomas, J.O.; Wabinga, H.; Whelan, S.L. Cancer in Africa: Epidemiology and Prevention; IARC Press: Lyon, France, 2003. [Google Scholar]
- Orem, J.; Mbidde, E.K.; Lambert, B.; de Sanjose, S.; Weiderpass, E. Burkitt’s lymphoma in Africa, a review of the epidemiology and etiology. Afr. Health Sci. 2007, 7, 166–175. [Google Scholar]
- Chang, D.W.; Claassen, G.F.; Hann, S.R.; Cole, M.D. The c-Myc transactivation domain is a direct modulator of apoptotic versus proliferative signals. Mol. Cell. Biol. 2000, 20, 4309–4319. [Google Scholar] [CrossRef]
- Chen, H.; Liu, H.; Qing, G. Targeting oncogenic Myc as a strategy for cancer treatment. Signal Transduct. Target. Ther. 2018, 3, 5. [Google Scholar] [CrossRef]
- Dhanasekaran, R.; Deutzmann, A.; Mahauad-Fernandez, W.D.; Hansen, A.S.; Gouw, A.M.; Felsher, D.W. The MYC oncogene—The grand orchestrator of cancer growth and immune evasion. Nat. Rev. Clin. Oncol. 2022, 19, 23–36. [Google Scholar] [CrossRef]
- Kress, T.R.; Sabò, A.; Amati, B. MYC: Connecting selective transcriptional control to global RNA production. Nat. Rev. Cancer 2015, 15, 593–607. [Google Scholar] [CrossRef]
- Kalkat, M.; De Melo, J.; Hickman, K.A.; Lourenco, C.; Redel, C.; Resetca, D.; Tamachi, A.; Tu, W.B.; Penn, L.Z. MYC Deregulation in Primary Human Cancers. Genes 2017, 8, 151. [Google Scholar] [CrossRef]
- Beroukhim, R.; Mermel, C.H.; Porter, D.; Wei, G.; Raychaudhuri, S.; Donovan, J.; Barretina, J.; Boehm, J.S.; Dobson, J.; Urashima, M.; et al. The landscape of somatic copy-number alteration across human cancers. Nature 2010, 463, 899–905. [Google Scholar] [CrossRef] [PubMed]
- Boxer, L.M.; Dang, C.V. Translocations involving c-myc and c-myc function. Oncogene 2001, 20, 5595–5610. [Google Scholar] [CrossRef] [PubMed]
- Pelengaris, S.; Khan, M. The many faces of c-MYC. Arch. Biochem. Biophys. 2003, 416, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Bettess, M.D.; Dubois, N.; Murphy, M.J.; Dubey, C.; Roger, C.; Robine, S.; Trumpp, A. c-Myc is required for the formation of intestinal crypts but dispensable for homeostasis of the adult intestinal epithelium. Mol. Cell. Biol. 2005, 25, 7868–7878. [Google Scholar] [CrossRef]
- De la Cova, C.; Abril, M.; Bellosta, P.; Gallant, P.; Johnston, L.A. Drosophila myc regulates organ size by inducing cell competition. Cell 2004, 117, 107–116. [Google Scholar] [CrossRef]
- Shi, Y.; Glynn, J.M.; Guilbert, L.J.; Cotter, T.G.; Bissonnette, R.P.; Green, D.R. Role for c-myc in activation-induced apoptotic cell death in T cell hybridomas. Science 1992, 257, 212–214. [Google Scholar] [CrossRef]
- Kaczmarek, L.; Hyland, J.K.; Watt, R.; Rosenberg, M.; Baserga, R. Microinjected c-myc as a competence factor. Science 1985, 228, 1313–1315. [Google Scholar] [CrossRef]
- Johnston, J.M.; Carroll, W.L. c-myc hypermutation in Burkitt’s lymphoma. Leuk. Lymphoma 1992, 8, 431–439. [Google Scholar] [CrossRef]
- Dang, C.V. MYC on the path to cancer. Cell 2012, 149, 22–35. [Google Scholar] [CrossRef]
- Vecchio, E.; Fiume, G.; Correnti, S.; Romano, S.; Iaccino, E.; Mimmi, S.; Maisano, D.; Nisticò, N.; Quinto, I. Insights about MYC and Apoptosis in B-Lymphomagenesis: An Update from Murine Models. Int. J. Mol. Sci. 2020, 21, 4265. [Google Scholar] [CrossRef]
- Love, C.; Sun, Z.; Jima, D.; Li, G.; Zhang, J.; Miles, R.; Richards, K.L.; Dunphy, C.H.; Choi, W.W.; Srivastava, G.; et al. The genetic landscape of mutations in Burkitt lymphoma. Nat. Genet. 2012, 44, 1321–1325. [Google Scholar] [CrossRef] [PubMed]
- Burkitt, D. A sarcoma involving the jaws in African children. Br. J. Surg. 1958, 46, 218–223. [Google Scholar] [CrossRef] [PubMed]
- Gerrard, M.; Cairo, M.S.; Weston, C.; Auperin, A.; Pinkerton, R.; Lambilliote, A.; Sposto, R.; McCarthy, K.; Lacombe, M.J.; Perkins, S.L.; et al. Excellent survival following two courses of COPAD chemotherapy in children and adolescents with resected localized B-cell non-Hodgkin’s lymphoma: Results of the FAB/LMB 96 international study. Br. J. Haematol. 2008, 141, 840–847. [Google Scholar] [CrossRef] [PubMed]
- Burkhardt, B.; Oschlies, I.; Klapper, W.; Zimmermann, M.; Woessmann, W.; Meinhardt, A.; Landmann, E.; Attarbaschi, A.; Niggli, F.; Schrappe, M.; et al. Non-Hodgkin’s lymphoma in adolescents: Experiences in 378 adolescent NHL patients treated according to pediatric NHL-BFM protocols. Leukemia 2011, 25, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Goldman, S.; Smith, L.; Galardy, P.; Perkins, S.L.; Frazer, J.K.; Sanger, W.; Anderson, J.R.; Gross, T.G.; Weinstein, H.; Harrison, L.; et al. Rituximab with chemotherapy in children and adolescents with central nervous system and/or bone marrow-positive Burkitt lymphoma/leukaemia: A Children’s Oncology Group Report. Br. J. Haematol. 2014, 167, 394–401. [Google Scholar] [CrossRef] [PubMed]
- Minard-Colin, V.; Aupérin, A.; Pillon, M.; Burke, G.A.A.; Barkauskas, D.A.; Wheatley, K.; Delgado, R.F.; Alexander, S.; Uyttebroeck, A.; Bollard, C.M.; et al. Rituximab for High-Risk, Mature B-Cell Non-Hodgkin’s Lymphoma in Children. N. Engl. J. Med. 2020, 382, 2207–2219. [Google Scholar] [CrossRef]
- Patte, C.; Auperin, A.; Michon, J.; Behrendt, H.; Leverger, G.; Frappaz, D.; Lutz, P.; Coze, C.; Perel, Y.; Raphaël, M.; et al. The Société Française d’Oncologie Pédiatrique LMB89 protocol: Highly effective multiagent chemotherapy tailored to the tumor burden and initial response in 561 unselected children with B-cell lymphomas and L3 leukemia. Blood 2001, 97, 3370–3379. [Google Scholar] [CrossRef]
- Ozuah, N.W.; Lubega, J.; Allen, C.E.; El-Mallawany, N.K. Five decades of low intensity and low survival: Adapting intensified regimens to cure pediatric Burkitt lymphoma in Africa. Blood Adv. 2020, 4, 4007–4019. [Google Scholar] [CrossRef]
- Mbulaiteye, S.M.; Talisuna, A.O.; Ogwang, M.D.; McKenzie, F.E.; Ziegler, J.L.; Parkin, D.M. African Burkitt’s lymphoma: Could collaboration with HIV-1 and malaria programmes reduce the high mortality rate? Lancet 2010, 375, 1661–1663. [Google Scholar] [CrossRef] [PubMed]
- Mbulaiteye, S.M.; Devesa, S.S. Burkitt Lymphoma Incidence in Five Continents. Hemato 2022, 3, 434–453. [Google Scholar] [CrossRef]
- Molyneux, E.M.; Rochford, R.; Griffin, B.; Newton, R.; Jackson, G.; Menon, G.; Harrison, C.J.; Israels, T.; Bailey, S. Burkitt’s lymphoma. Lancet 2012, 379, 1234–1244. [Google Scholar] [CrossRef]
- Bornkamm, G.W. Epstein-Barr virus and its role in the pathogenesis of Burkitt’s lymphoma: An unresolved issue. Semin. Cancer Biol. 2009, 19, 351–365. [Google Scholar] [CrossRef] [PubMed]
- Dalla-Favera, R.; Bregni, M.; Erikson, J.; Patterson, D.; Gallo, R.C.; Croce, C.M. Human c-myc onc gene is located on the region of chromosome 8 that is translocated in Burkitt lymphoma cells. Proc. Natl. Acad. Sci. USA 1982, 79, 7824–7827. [Google Scholar] [CrossRef]
- Taub, R.; Kirsch, I.; Morton, C.; Lenoir, G.; Swan, D.; Tronick, S.; Aaronson, S.; Leder, P. Translocation of the c-myc gene into the immunoglobulin heavy chain locus in human Burkitt lymphoma and murine plasmacytoma cells. Proc. Natl. Acad. Sci. USA 1982, 79, 7837–7841. [Google Scholar] [CrossRef] [PubMed]
- Hamlyn, P.H.; Rabbitts, T.H. Translocation joins c-myc and immunoglobulin gamma 1 genes in a Burkitt lymphoma revealing a third exon in the c-myc oncogene. Nature 1983, 304, 135–139. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.; Papenhausen, P.; Shao, H. The Role of c-MYC in B-Cell Lymphomas: Diagnostic and Molecular Aspects. Genes 2017, 8, 116. [Google Scholar] [CrossRef]
- Basso, K.; Frascella, E.; Zanesco, L.; Rosolen, A. Improved long-distance polymerase chain reaction for the detection of t(8;14)(q24;q32) in Burkitt’s lymphomas. Am. J. Pathol. 1999, 155, 1479–1485. [Google Scholar] [CrossRef] [PubMed]
- Blum, K.A.; Lozanski, G.; Byrd, J.C. Adult Burkitt leukemia and lymphoma. Blood 2004, 104, 3009–3020. [Google Scholar] [CrossRef]
- Hecht, J.L.; Aster, J.C. Molecular biology of Burkitt’s lymphoma. J. Clin. Oncol. 2000, 18, 3707–3721. [Google Scholar] [CrossRef] [PubMed]
- Neri, A.; Barriga, F.; Knowles, D.M.; Magrath, I.T.; Dalla-Favera, R. Different regions of the immunoglobulin heavy-chain locus are involved in chromosomal translocations in distinct pathogenetic forms of Burkitt lymphoma. Proc. Natl. Acad. Sci. USA 1988, 85, 2748–2752. [Google Scholar] [CrossRef] [PubMed]
- Devaiah, B.N.; Mu, J.; Akman, B.; Uppal, S.; Weissman, J.D.; Cheng, D.; Baranello, L.; Nie, Z.; Levens, D.; Singer, D.S. MYC protein stability is negatively regulated by BRD4. Proc. Natl. Acad. Sci. USA 2020, 117, 13457–13467. [Google Scholar] [CrossRef] [PubMed]
- Hinds, J.W.; Feris, E.J.; Wilkins, O.M.; Deary, L.T.; Wang, X.; Cole, M.D. S146L in MYC is a context-dependent activating substitution in cancer development. PLoS ONE 2022, 17, e0272771. [Google Scholar] [CrossRef] [PubMed]
- Kuzyk, A.; Mai, S. c-MYC-induced genomic instability. Cold Spring Harb. Perspect. Med. 2014, 4, a014373. [Google Scholar] [CrossRef]
- Kumari, A.; Folk, W.P.; Sakamuro, D. The Dual Roles of MYC in Genomic Instability and Cancer Chemoresistance. Genes 2017, 8, 158. [Google Scholar] [CrossRef] [PubMed]
- Curti, L.; Campaner, S. MYC-Induced Replicative Stress: A Double-Edged Sword for Cancer Development and Treatment. Int. J. Mol. Sci. 2021, 22, 6168. [Google Scholar] [CrossRef]
- Shortt, J.; Martin, B.P.; Newbold, A.; Hannan, K.M.; Devlin, J.R.; Baker, A.J.; Ralli, R.; Cullinane, C.; Schmitt, C.A.; Reimann, M.; et al. Combined inhibition of PI3K-related DNA damage response kinases and mTORC1 induces apoptosis in MYC-driven B-cell lymphomas. Blood 2013, 121, 2964–2974. [Google Scholar] [CrossRef]
- Han, S.S.; Yun, H.; Son, D.J.; Tompkins, V.S.; Peng, L.; Chung, S.T.; Kim, J.S.; Park, E.S.; Janz, S. NF-kappaB/STAT3/PI3K signaling crosstalk in iMyc E mu B lymphoma. Mol. Cancer 2010, 9, 97. [Google Scholar] [CrossRef]
- Dang, C.V. The interplay between MYC and HIF in the Warburg effect. In Ernst Schering Foundation Symposium Proceedings; Springer: Berlin/Heidelberg, Germany, 2007; pp. 35–53. [Google Scholar]
- Dang, C.V. MYC, metabolism, cell growth, and tumorigenesis. Cold Spring Harb. Perspect. Med. 2013, 3, a014217. [Google Scholar] [CrossRef]
- Kim, J.; Lee, J.H.; Iyer, V.R. Global identification of Myc target genes reveals its direct role in mitochondrial biogenesis and its E-box usage in vivo. PLoS ONE 2008, 3, e1798. [Google Scholar] [CrossRef]
- Popay, T.M.; Wang, J.; Adams, C.M.; Howard, G.C.; Codreanu, S.G.; Sherrod, S.D.; McLean, J.A.; Thomas, L.R.; Lorey, S.L.; Machida, Y.J.; et al. MYC regulates ribosome biogenesis and mitochondrial gene expression programs through its interaction with host cell factor-1. eLife 2021, 10, e60191. [Google Scholar] [CrossRef]
- Morrish, F.; Hockenbery, D. MYC and mitochondrial biogenesis. Cold Spring Harb. Perspect. Med. 2014, 4, a014225. [Google Scholar] [CrossRef] [PubMed]
- Casey, S.C.; Baylot, V.; Felsher, D.W. The MYC oncogene is a global regulator of the immune response. Blood 2018, 131, 2007–2015. [Google Scholar] [CrossRef] [PubMed]
- Casey, S.C.; Tong, L.; Li, Y.; Do, R.; Walz, S.; Fitzgerald, K.N.; Gouw, A.M.; Baylot, V.; Gütgemann, I.; Eilers, M.; et al. MYC regulates the antitumor immune response through CD47 and PD-L1. Science 2016, 352, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.Y.; Kim, A.; Kim, S.K.; Chang, Y.S. MYC expression correlates with PD-L1 expression in non-small cell lung cancer. Lung Cancer 2017, 110, 63–67. [Google Scholar] [CrossRef]
- Saravia, J.; Zeng, H.; Dhungana, Y.; Bastardo Blanco, D.; Nguyen, T.M.; Chapman, N.M.; Wang, Y.; Kanneganti, A.; Liu, S.; Raynor, J.L.; et al. Homeostasis and transitional activation of regulatory T cells require c-Myc. Sci. Adv. 2020, 6, eaaw6443. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef]
- Meyer, N.; Penn, L.Z. Reflecting on 25 years with MYC. Nat. Rev. Cancer 2008, 8, 976–990. [Google Scholar] [CrossRef]
- Stasevich, E.M.; Uvarova, A.N.; Murashko, M.M.; Khabusheva, E.R.; Sheetikov, S.A.; Prassolov, V.S.; Kuprash, D.V.; Demin, D.E.; Schwartz, A.M. Enhancer RNA AL928768.3 from the IGH Locus Regulates MYC Expression and Controls the Proliferation and Chemoresistance of Burkitt Lymphoma Cells with IGH/MYC Translocation. Int. J. Mol. Sci. 2022, 23, 4624. [Google Scholar] [CrossRef]
- Brady, G.; MacArthur, G.J.; Farrell, P.J. Epstein-Barr virus and Burkitt lymphoma. J. Clin. Pathol. 2007, 60, 1397–1402. [Google Scholar] [CrossRef]
- Broen, K.; Dickens, J.; Trangucci, R.; Ogwang, M.D.; Tenge, C.N.; Masalu, N.; Reynolds, S.J.; Kawira, E.; Kerchan, P.; Were, P.A.; et al. Burkitt lymphoma risk shows geographic and temporal associations with Plasmodium falciparum infections in Uganda, Tanzania, and Kenya. Proc. Natl. Acad. Sci. USA 2023, 120, e2211055120. [Google Scholar] [CrossRef] [PubMed]
- Blinder, V.S.; Chadburn, A.; Furman, R.R.; Mathew, S.; Leonard, J.P. Improving outcomes for patients with Burkitt lymphoma and HIV. AIDS Patient Care STDs 2008, 22, 175–187. [Google Scholar] [CrossRef] [PubMed]
- Bemark, M.; Neuberger, M.S. The c-MYC allele that is translocated into the IgH locus undergoes constitutive hypermutation in a Burkitt’s lymphoma line. Oncogene 2000, 19, 3404–3410. [Google Scholar] [CrossRef]
- Schmitz, R.; Ceribelli, M.; Pittaluga, S.; Wright, G.; Staudt, L.M. Oncogenic mechanisms in Burkitt lymphoma. Cold Spring Harb. Perspect. Med. 2014, 4, a014282. [Google Scholar] [CrossRef] [PubMed]
- Hummel, M.; Bentink, S.; Berger, H.; Klapper, W.; Wessendorf, S.; Barth, T.F.; Bernd, H.W.; Cogliatti, S.B.; Dierlamm, J.; Feller, A.C.; et al. A biologic definition of Burkitt’s lymphoma from transcriptional and genomic profiling. N. Engl. J. Med. 2006, 354, 2419–2430. [Google Scholar] [CrossRef] [PubMed]
- López, C.; Kleinheinz, K.; Aukema, S.M.; Rohde, M.; Bernhart, S.H.; Hübschmann, D.; Wagener, R.; Toprak, U.H.; Raimondi, F.; Kreuz, M.; et al. Genomic and transcriptomic changes complement each other in the pathogenesis of sporadic Burkitt lymphoma. Nat. Commun. 2019, 10, 1459. [Google Scholar] [CrossRef] [PubMed]
- Küppers, R.; Dalla-Favera, R. Mechanisms of chromosomal translocations in B cell lymphomas. Oncogene 2001, 20, 5580–5594. [Google Scholar] [CrossRef]
- Tandon, A.; Birkenhagen, J.; Nagalla, D.; Kölker, S.; Sauer, S.W. ADP-dependent glucokinase as a novel onco-target for haematological malignancies. Sci. Rep. 2020, 10, 13584. [Google Scholar] [CrossRef]
- Schmitz, R.; Young, R.M.; Ceribelli, M.; Jhavar, S.; Xiao, W.; Zhang, M.; Wright, G.; Shaffer, A.L.; Hodson, D.J.; Buras, E.; et al. Burkitt lymphoma pathogenesis and therapeutic targets from structural and functional genomics. Nature 2012, 490, 116–120. [Google Scholar] [CrossRef]
- Sánchez-Beato, M.; Sánchez-Aguilera, A.; Piris, M.A. Cell cycle deregulation in B-cell lymphomas. Blood 2003, 101, 1220–1235. [Google Scholar] [CrossRef]
- Newman, A.M.; Zaka, M.; Zhou, P.; Blain, A.E.; Erhorn, A.; Barnard, A.; Crossland, R.E.; Wilkinson, S.; Enshaei, A.; De Zordi, J.; et al. Genomic abnormalities of TP53 define distinct risk groups of paediatric B-cell non-Hodgkin lymphoma. Leukemia 2022, 36, 781–789. [Google Scholar] [CrossRef] [PubMed]
- López, C.; Burkhardt, B.; Chan, J.K.C.; Leoncini, L.; Mbulaiteye, S.M.; Ogwang, M.D.; Orem, J.; Rochford, R.; Roschewski, M.; Siebert, R. Burkitt lymphoma. Nat. Rev. Dis. Prim. 2022, 8, 78. [Google Scholar] [CrossRef] [PubMed]
- Leventaki, V.; Rodic, V.; Tripp, S.R.; Bayerl, M.G.; Perkins, S.L.; Barnette, P.; Schiffman, J.D.; Miles, R.R. TP53 pathway analysis in paediatric Burkitt lymphoma reveals increased MDM4 expression as the only TP53 pathway abnormality detected in a subset of cases. Br. J. Haematol. 2012, 158, 763–771. [Google Scholar] [CrossRef] [PubMed]
- Burkhardt, B.; Michgehl, U.; Rohde, J.; Erdmann, T.; Berning, P.; Reutter, K.; Rohde, M.; Borkhardt, A.; Burmeister, T.; Dave, S.; et al. Clinical relevance of molecular characteristics in Burkitt lymphoma differs according to age. Nat. Commun. 2022, 13, 3881. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.; Carroll, M.; Thomas-Tikhonenko, A. p53 status dictates responses of B lymphomas to monotherapy with proteasome inhibitors. Blood 2007, 109, 4936–4943. [Google Scholar] [CrossRef] [PubMed]
- Richter, J.; Schlesner, M.; Hoffmann, S.; Kreuz, M.; Leich, E.; Burkhardt, B.; Rosolowski, M.; Ammerpohl, O.; Wagener, R.; Bernhart, S.H.; et al. Recurrent mutation of the ID3 gene in Burkitt lymphoma identified by integrated genome, exome and transcriptome sequencing. Nat. Genet. 2012, 44, 1316–1320. [Google Scholar] [CrossRef] [PubMed]
- Rohde, M.; Bonn, B.R.; Zimmermann, M.; Lange, J.; Möricke, A.; Klapper, W.; Oschlies, I.; Szczepanowski, M.; Nagel, I.; Schrappe, M.; et al. Relevance of ID3-TCF3-CCND3 pathway mutations in pediatric aggressive B-cell lymphoma treated according to the non-Hodgkin Lymphoma Berlin-Frankfurt-Münster protocols. Haematologica 2017, 102, 1091–1098. [Google Scholar] [CrossRef]
- Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Harris, N.L.; Stein, H.; Siebert, R.; Advani, R.; Ghielmini, M.; Salles, G.A.; Zelenetz, A.D.; et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016, 127, 2375–2390. [Google Scholar] [CrossRef]
- Lin, P.; Dickason, T.J.; Fayad, L.E.; Lennon, P.A.; Hu, P.; Garcia, M.; Routbort, M.J.; Miranda, R.; Wang, X.; Qiao, W.; et al. Prognostic value of MYC rearrangement in cases of B-cell lymphoma, unclassifiable, with features intermediate between diffuse large B-cell lymphoma and Burkitt lymphoma. Cancer 2012, 118, 1566–1573. [Google Scholar] [CrossRef]
- Salaverria, I.; Martin-Guerrero, I.; Wagener, R.; Kreuz, M.; Kohler, C.W.; Richter, J.; Pienkowska-Grela, B.; Adam, P.; Burkhardt, B.; Claviez, A.; et al. A recurrent 11q aberration pattern characterizes a subset of MYC-negative high-grade B-cell lymphomas resembling Burkitt lymphoma. Blood 2014, 123, 1187–1198. [Google Scholar] [CrossRef]
- García-Gutiérrez, L.; Delgado, M.D.; León, J. MYC Oncogene Contributions to Release of Cell Cycle Brakes. Genes 2019, 10, 244. [Google Scholar] [CrossRef]
- Mlynarczyk, C.; Fontán, L.; Melnick, A. Germinal center-derived lymphomas: The darkest side of humoral immunity. Immunol. Rev. 2019, 288, 214–239. [Google Scholar] [CrossRef]
- Lindström, M.S.; Wiman, K.G. Role of genetic and epigenetic changes in Burkitt lymphoma. Semin. Cancer Biol. 2002, 12, 381–387. [Google Scholar] [CrossRef] [PubMed]
- McMahon, S.B.; Wood, M.A.; Cole, M.D. The essential cofactor TRRAP recruits the histone acetyltransferase hGCN5 to c-Myc. Mol. Cell. Biol. 2000, 20, 556–562. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Kunjibettu, S.; McMahon, S.B.; Cole, M.D. The ATM-related domain of TRRAP is required for histone acetyltransferase recruitment and Myc-dependent oncogenesis. Genes Dev. 2001, 15, 1619–1624. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, M.L.; Reyes-Garau, D.; Armengol, M.; Fernández-Serrano, M.; Roué, G. Recent Advances in the Targeting of Epigenetic Regulators in B-Cell Non-Hodgkin Lymphoma. Front. Genet. 2019, 10, 986. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Serrano, M.; Winkler, R.; Santos, J.C.; Le Pannérer, M.M.; Buschbeck, M.; Roué, G. Histone Modifications and Their Targeting in Lymphoid Malignancies. Int. J. Mol. Sci. 2021, 23, 253. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Van Calcar, S.; Qu, C.; Cavenee, W.K.; Zhang, M.Q.; Ren, B. A global transcriptional regulatory role for c-Myc in Burkitt’s lymphoma cells. Proc. Natl. Acad. Sci. USA 2003, 100, 8164–8169. [Google Scholar] [CrossRef] [PubMed]
- Cowling, V.H.; Turner, S.A.; Cole, M.D. Burkitt’s lymphoma-associated c-Myc mutations converge on a dramatically altered target gene response and implicate Nol5a/Nop56 in oncogenesis. Oncogene 2014, 33, 3519–3527. [Google Scholar] [CrossRef]
- Poole, C.J.; Zheng, W.; Lodh, A.; Yevtodiyenko, A.; Liefwalker, D.; Li, H.; Felsher, D.W.; van Riggelen, J. DNMT3B overexpression contributes to aberrant DNA methylation and MYC-driven tumor maintenance in T-ALL and Burkitt’s lymphoma. Oncotarget 2017, 8, 76898–76920. [Google Scholar] [CrossRef]
- Videtta, A.D.; Malagnino, V.; De Falco, G. Current understanding of the role and regulation of miRNAs in Burkitt lymphoma. Blood Lymphat. Cancer Targets Ther. 2018, 8, 33–45. [Google Scholar] [CrossRef] [PubMed]
- Shams, R.; Asadzadeh Aghdaei, H.; Behmanesh, A.; Sadeghi, A.; Zali, M.; Salari, S.; Padrón, J.M. MicroRNAs Targeting MYC Expression: Trace of Hope for Pancreatic Cancer Therapy. A Systematic Review. Cancer Manag. Res. 2020, 12, 2393–2404. [Google Scholar] [CrossRef] [PubMed]
- Onnis, A.; De Falco, G.; Antonicelli, G.; Onorati, M.; Bellan, C.; Sherman, O.; Sayed, S.; Leoncini, L. Alteration of microRNAs regulated by c-Myc in Burkitt lymphoma. PLoS ONE 2010, 5, e12960. [Google Scholar] [CrossRef] [PubMed]
- Bui, T.V.; Mendell, J.T. Myc: Maestro of microRNAs. Genes Cancer 2010, 1, 568–575. [Google Scholar] [CrossRef]
- Oduor, C.I.; Kaymaz, Y.; Chelimo, K.; Otieno, J.A.; Ong’echa, J.M.; Moormann, A.M.; Bailey, J.A. Integrative microRNA and mRNA deep-sequencing expression profiling in endemic Burkitt lymphoma. BMC Cancer 2017, 17, 761. [Google Scholar] [CrossRef]
- Klanova, M.; Klener, P. BCL-2 Proteins in Pathogenesis and Therapy of B-Cell Non-Hodgkin Lymphomas. Cancers 2020, 12, 938. [Google Scholar] [CrossRef]
- Meškytė, E.M.; Keskas, S.; Ciribilli, Y. MYC as a Multifaceted Regulator of Tumor Microenvironment Leading to Metastasis. Int. J. Mol. Sci. 2020, 21, 7710. [Google Scholar] [CrossRef]
- Dews, M.; Homayouni, A.; Yu, D.; Murphy, D.; Sevignani, C.; Wentzel, E.; Furth, E.E.; Lee, W.M.; Enders, G.H.; Mendell, J.T.; et al. Augmentation of tumor angiogenesis by a Myc-activated microRNA cluster. Nat. Genet. 2006, 38, 1060–1065. [Google Scholar] [CrossRef]
- Whitfield, J.R.; Soucek, L. Tumor microenvironment: Becoming sick of Myc. Cell. Mol. Life Sci. 2012, 69, 931–934. [Google Scholar] [CrossRef]
- Balkwill, F.R.; Capasso, M.; Hagemann, T. The tumor microenvironment at a glance. J. Cell Sci. 2012, 125 Pt 23, 5591–5596. [Google Scholar] [CrossRef]
- Soucek, L.; Whitfield, J.; Martins, C.P.; Finch, A.J.; Murphy, D.J.; Sodir, N.M.; Karnezis, A.N.; Swigart, L.B.; Nasi, S.; Evan, G.I. Modelling Myc inhibition as a cancer therapy. Nature 2008, 455, 679–683. [Google Scholar] [CrossRef] [PubMed]
- Evan, G.I.; Wyllie, A.H.; Gilbert, C.S.; Littlewood, T.D.; Land, H.; Brooks, M.; Waters, C.M.; Penn, L.Z.; Hancock, D.C. Induction of apoptosis in fibroblasts by c-myc protein. Cell 1992, 69, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Sabnis, A.J.; Bivona, T.G. Principles of Resistance to Targeted Cancer Therapy: Lessons from Basic and Translational Cancer Biology. Trends Mol. Med. 2019, 25, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Math, M.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef] [PubMed]
- Armand, P. Immune checkpoint blockade in hematologic malignancies. Blood 2015, 125, 3393–3400. [Google Scholar] [CrossRef] [PubMed]
- Koh, C.M.; Sabò, A.; Guccione, E. Targeting MYC in cancer therapy: RNA processing offers new opportunities. BioEssays News Rev. Mol. Cell. Dev. Biol. 2016, 38, 266–275. [Google Scholar] [CrossRef]
- Delmore, J.E.; Issa, G.C.; Lemieux, M.E.; Rahl, P.B.; Shi, J.; Jacobs, H.M.; Kastritis, E.; Gilpatrick, T.; Paranal, R.M.; Qi, J.; et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 2011, 146, 904–917. [Google Scholar] [CrossRef]
- Pagano, L.; Caira, M.; Valentini, C.G.; Fianchi, L. Clinical aspects and therapy of sporadic burkitt lymphoma. Mediterr. J. Hematol. Infect. Dis. 2009, 1, e2009030. [Google Scholar] [CrossRef]
- Thomas, D.A.; Faderl, S.; O’Brien, S.; Bueso-Ramos, C.; Cortes, J.; Garcia-Manero, G.; Giles, F.J.; Verstovsek, S.; Wierda, W.G.; Pierce, S.A.; et al. Chemoimmunotherapy with hyper-CVAD plus rituximab for the treatment of adult Burkitt and Burkitt-type lymphoma or acute lymphoblastic leukemia. Cancer 2006, 106, 1569–1580. [Google Scholar] [CrossRef]
- Llombart, V.; Mansour, M.R. Therapeutic targeting of “undruggable” MYC. eBioMedicine 2022, 75, 103756. [Google Scholar] [CrossRef]
- Tang, M.; O’Grady, S.; Crown, J.; Duffy, M.J. MYC as a therapeutic target for the treatment of triple-negative breast cancer: Preclinical investigations with the novel MYC inhibitor, MYCi975. Breast Cancer Res. Treat. 2022, 195, 105–115. [Google Scholar] [CrossRef]
- Huang, M.J.; Cheng, Y.C.; Liu, C.R.; Lin, S.; Liu, H.E. A small-molecule c-Myc inhibitor, 10058-F4, induces cell-cycle arrest, apoptosis, and myeloid differentiation of human acute myeloid leukemia. Exp. Hematol. 2006, 34, 1480–1489. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, T.; Ajiro, M.; Watanabe, A.; Matsushima, S.; Ueda, K.; Hagiwara, M. Application of the CDK9 inhibitor FIT-039 for the treatment of KSHV-associated malignancy. BMC Cancer 2023, 23, 71. [Google Scholar] [CrossRef] [PubMed]
- Daly, B.T.; Ippolito, M.T.; Gu, J.J.; Mavis, M.C.; Torka, P.; Hernandez-Ilizaliturri, F.J.; Barth, M.J. MCL-1 Inhibition by the Selective MCL-1 Inhibitor AMG-176 Induces in Vitro Activity Against Burkitt Lymphoma Cell Lines and Synergistically Enhances the Cytotoxic Effect of Chemotherapy and BH3 Mimetics. Blood 2019, 134 (Suppl. 1), 5303. [Google Scholar] [CrossRef]
- Ahmadi, S.E.; Rahimi, S.; Zarandi, B.; Chegeni, R.; Safa, M. MYC: A multipurpose oncogene with prognostic and therapeutic implications in blood malignancies. J. Hematol. Oncol. 2021, 14, 121. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Medeiros, L.J.; Young, K.H. Diagnostic and predictive biomarkers for lymphoma diagnosis and treatment in the era of precision medicine. Mod. Pathol. 2016, 29, 1118–1142. [Google Scholar] [CrossRef] [PubMed]
- Satta, T.; Grant, S. Enhancing venetoclax activity in hematological malignancies. Expert Opin. Investig. Drugs 2020, 29, 697–708. [Google Scholar] [CrossRef]
- Pozzo, F.; Bittolo, T.; Vendramini, E.; Bomben, R.; Bulian, P.; Rossi, F.M.; Zucchetto, A.; Tissino, E.; Degan, M.; D’Arena, G.; et al. NOTCH1-mutated chronic lymphocytic leukemia cells are characterized by a MYC-related overexpression of nucleophosmin 1 and ribosome-associated components. Leukemia 2017, 31, 2407–2415. [Google Scholar] [CrossRef]
- Lovec, H.; Grzeschiczek, A.; Kowalski, M.B.; Möröy, T. Cyclin D1/bcl-1 cooperates with myc genes in the generation of B-cell lymphoma in transgenic mice. EMBO J. 1994, 13, 3487–3495. [Google Scholar] [CrossRef]
- Thng, D.K.H.; Toh, T.B.; Chow, E.K. Capitalizing on Synthetic Lethality of MYC to Treat Cancer in the Digital Age. Trends Pharmacol. Sci. 2021, 42, 166–182. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, S.; Shi, Q.; Yang, D.; Allen, T.D. Pro-Survival Bcl-2 Proteins Are Modifiers of MYC-VX-680 Synthetic Lethality. J. Cell. Immunol. 2020, 2, 227–232. [Google Scholar] [CrossRef]
- Mertz, J.A.; Conery, A.R.; Bryant, B.M.; Sandy, P.; Balasubramanian, S.; Mele, D.A.; Bergeron, L.; Sims, R.J. Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proc. Natl. Acad. Sci. USA 2011, 108, 16669–16674. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Li, D.; Zhou, J. Histone Deacetylase 6 as a Therapeutic Target in B Cell-Associated Hematological Malignancies. Front. Pharmacol. 2020, 11, 971. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.-G.; Kong, C.-Y.; Wu, H.-M.; Song, P.; Zhang, X.; Yuan, Y.-P.; Deng, W.; Tang, Q.-Z. Toll-like Receptor 5 Deficiency Diminishes Doxorubicin-Induced Acute Cardiotoxicity in Mice. Theranostics 2020, 10, 11013–11025. [Google Scholar] [CrossRef]
- Yuneva, M.; Zamboni, N.; Oefner, P.; Sachidanandam, R.; Lazebnik, Y. Deficiency in glutamine but not glucose induces MYC-dependent apoptosis in human cells. J. Cell Biol. 2007, 178, 93–105. [Google Scholar] [CrossRef]
- Gao, P.; Tchernyshyov, I.; Chang, T.-C.; Lee, Y.-S.; Kita, K.; Ochi, T.; Zeller, K.I.; De Marzo, A.M.; Van Eyk, J.E.; Mendell, J.T.; et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 2009, 458, 762–765. [Google Scholar] [CrossRef]
- Mullen, A.R.; Wheaton, W.W.; Jin, E.S.; Chen, P.-H.; Sullivan, L.B.; Cheng, T.; Yang, Y.; Linehan, W.M.; Chandel, N.S.; DeBerardinis, R.J. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature 2011, 481, 385–388. [Google Scholar] [CrossRef]
- Metallo, C.M.; Gameiro, P.A.; Bell, E.L.; Mattaini, K.R.; Yang, J.; Hiller, K.; Jewell, C.M.; Johnson, Z.R.; Irvine, D.J.; Guarente, L.; et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 2011, 481, 380–384. [Google Scholar] [CrossRef]
- Jacque, N.; Ronchetti, A.M.; Larrue, C.; Meunier, G.; Birsen, R.; Willems, L.; Saland, E.; Decroocq, J.; Maciel, T.T.; Lambert, M.; et al. Targeting glutaminolysis has antileukemic activity in acute myeloid leukemia and synergizes with BCL-2 inhibition. Blood 2015, 126, 1346–1356. [Google Scholar] [CrossRef]
- Savino, M.; Annibali, D.; Carucci, N.; Favuzzi, E.; Cole, M.D.; Evan, G.I.; Soucek, L.; Nasi, S. The action mechanism of the Myc inhibitor termed Omomyc may give clues on how to target Myc for cancer therapy. PLoS ONE 2011, 6, e22284. [Google Scholar] [CrossRef]
- Jung, L.A.; Gebhardt, A.; Koelmel, W.; Ade, C.P.; Walz, S.; Kuper, J.; von Eyss, B.; Letschert, S.; Redel, C.; d’Artista, L.; et al. OmoMYC blunts promoter invasion by oncogenic MYC to inhibit gene expression characteristic of MYC-dependent tumors. Oncogene 2017, 36, 1911–1924. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).