Abstract
Background: Systemic sclerosis (SSc) is a rare connective tissue disease characterized by vasculopathy, autoimmunity, and fibrosis. Due to its low prevalence and heterogeneous clinical presentation, early diagnosis remains challenging, often delaying appropriate treatment. The disease progresses from microvascular dysfunction, manifesting as Raynaud’s phenomenon, to systemic fibrosis affecting multiple organs, including the lungs, gastrointestinal tract, heart, and kidneys. There have been considerable advancements in understanding the pathophysiology of the disease during the last few years and this has already resulted in the improvement of the therapeutic approaches used to control organ-specific manifestations. However, the underlying cause of the disease still remains incompletely elucidated. Methods: Here, we summarize the current knowledge on the SSc pathogenesis. Results: The pathophysiology involves an interplay of chronic inflammation, impaired vascular function, and excessive extracellular matrix deposition, leading to progressive organ damage. Endothelial dysfunction in SSc is driven by immune-mediated injury, oxidative stress, and the imbalance of vasoconstrictors and vasodilators, leading to capillary loss and chronic hypoxia. Autoantibodies against endothelial cells or other toxic factors induce apoptosis and impair angiogenesis, further exacerbating vascular damage. Despite increased angiogenic factor levels, capillary repair mechanisms are defective, resulting in progressive ischemic damage. Dysregulated immune responses involving Th2 cytokines, B cells, and macrophages contribute to fibroblast activation and excessive collagen deposition. Transforming growth factor-beta (TGF-β) plays a central role in fibrotic progression, while fibroblasts resist apoptosis, perpetuating tissue scarring. The extracellular matrix in SSc is abnormally stiff, reinforcing fibroblast activation and creating a self-perpetuating fibrotic cycle. Conclusions: Advances in molecular and cellular understanding have facilitated targeted therapies, yet effective disease-modifying treatments remain limited. Future research should focus on precision medicine approaches, integrating biomarkers and novel therapeutics to improve patient outcomes.
1. Introduction
Systemic sclerosis (SSc) is a rare, complex connective tissue disease that presents significant challenges. Its low prevalence can often lead to delays in early diagnosis and the initiation of appropriate treatment [1]. Given the multi-organ complications, SSc patients require multidisciplinary management and continuous follow-up. Nonetheless, the pathomechanisms driving disease onset and progression remain incompletely understood.
Overall, SSc is uniquely defined by its combination of vasculopathy, autoimmunity, and fibrosis. However, vasculopathy is an early trigger, often manifesting as Raynaud’s syndrome (Figure 1A), which can precede disease onset [2,3], serving as an early marker of microcirculatory dysfunction in the acral regions. As the disease progresses, digital ulceration (Figure 1B) may develop, sometimes leading to necrosis and eventual fingertip loss. Additionally, patients may experience extensive calcifications, severe pruritus, and prominent telangiectasias [2]. The clinical presentation of advanced SSc is highly characteristic and relatively easy to diagnose. However, early-stage cases are frequently overlooked, suggesting that the disease may be more prevalent than currently recognized [1]. Recent works by several groups have emphasized the need for the early detection of SSc and have developed the concept of VEDOSS (very early diagnoses of systemic sclerosis) [4]. In later stages or rapidly progressing subtypes, systemic involvement extends beyond the skin (Figure 1C), frequently affecting the internal organs. Pulmonary complications, particularly lung fibrosis and renal disease, are common. Gastrointestinal manifestations, such as reflux, gastric telangiectasia, and esophageal motility disorders, are observed in most patients, while musculoskeletal involvement is also frequent. Cardiac disease, though often underestimated, is likely more prevalent than previously thought [3,5,6,7]. Substantial progress in managing organ complications has led to an improved quality of life for many patients [8,9]. Nevertheless, developing disease-modifying therapies requires a deeper understanding of the pathophysiological events driving fibrosis and tissue damage [8,9].
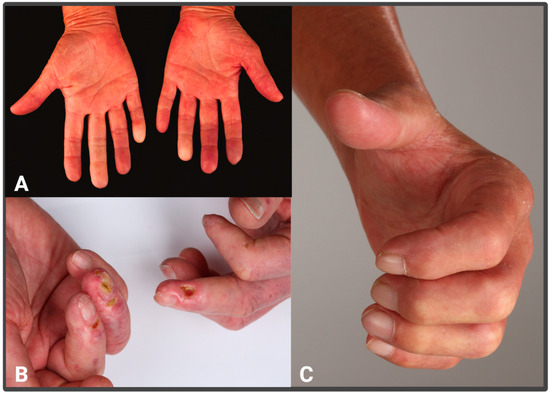
Figure 1.
Clinical features of vasculopathy and fibrosis in SSc patients: (A) Raynaud’s phenomenon; (B) digital ulcers; (C) severe stiffening of the skin leading to contractures.
In the following, we summarize the current knowledge on SSc pathogenesis and discuss how novel biomedical techniques have enhanced our understanding of fibrotic mechanisms.
2. Etiology and Risk Factors
Although the development of systemic sclerosis requires an (unknown) trigger, the involvement of genetic factors in the pathogenesis of SSc has been studied first in twin studies, analyzing human leucocyte antigen genes and more recently also in large multicenter genome-wide association studies. These have identified many genes (e.g., TNFSF4 (1q25.1), STAT4 (2q32.2-q32.3), DNASE1L3 (3p14.3), and IRF5-TNPO3 (7q32.1) or CD247) [10,11] that are involved in the control of vasculopathy and fibrosis and which are probably related to susceptibility to disease development. In addition, several environmental factors have been identified that lead to scleroderma or scleroderma-like conditions. Examples include silica dust, drugs, food contaminants, and others [12]. Although the exact mode of action of most compounds is still not understood, the data suggest that genetic susceptibility, together with external factors including potential viral infections [13], are crucial for the initial disease induction.
3. Vascular Alterations and Endothelial Damage in SSc
An interplay of autoimmune processes, vascular endothelial damage, and an overproduction of extracellular matrix (ECM) are crucial for pathophysiology and determine the clinical characteristics of this disease. In routine histology, the key stages of SSc pathogenesis can be detected: initial endothelial cell swelling followed by lympho-histiocytic inflammatory infiltration around affected blood vessels, and ultimately, dense extracellular matrix deposition with activated myofibroblasts and homogenized collagen bundles (Figure 2). Extensive research into the cellular and molecular alterations underlying these processes has facilitated the identification of novel therapeutic targets.
The Raynaud phenomenon (RP) usually manifests as a very characteristic and early clinical sign preceding sclerosis. Vascular changes can be easily detected clinically through nailfold microscopy [14], which can indicate vascular dysfunction even before the manifestation of fibrosis. These include early changes such as capillary ectasias, active patterns with megacapillaries and hemorrhages, and late changes that present as capillary bunching. The early inflammatory changes also appear histologically as prominent perivascular and periadnexial infiltrates [15,16].
Autopsy studies have shown widespread intimal proliferation affecting pulmonary, coronary, and renal arteries which is not inflammatory in nature. Early signs of vascular dysfunction include impaired permeability and tone, alongside an imbalance between vasoconstrictor endothelin (ET) [17] and vasodilator nitric oxide (NO) [18,19]. Platelet activation and coagulation abnormalities further contribute to the vasculopathy observed in SSc patients. The early stage of systemic sclerosis is also often clinically referred to as an edematous phase, as affected patients may experience swelling of the fingers (“puffy fingers”) (Figure 1A) and milk glass opacities in the lungs, which are characteristic of alveolar or interstitial edema.
The exact cause of the initial vascular injury remains unclear, with potential contributors including infectious agents, cytotoxic T cells, and autoantibodies targeting endothelial cells [7,20]. Microcirculatory changes, such as capillary dropout and altered architecture, are prominent, alongside endothelial cell injury, which is central to the pathogenesis of SSc vasculopathy. It is hypothesized that chronic circulatory disturbance, with repetitive hypoxia and the release of Vascular Endothelial Growth Factor (VEGF) and TGF-β, increases vascular permeability. In addition, endothelial cell swelling and subsequent apoptosis occur (Figure 2), leading to the altered expression of adhesion molecules [2,21]. High levels of von Willebrand factor and ET-1 indicate endothelial damage, while conflicting reports exist regarding endothelial apoptosis [22]. As a result, fluid and blood extravasation occurs, along with the influx of immune cells. It remains unclear whether vascular leakage is the primary cause of immune cell infiltration or if the presence of inflammatory cells secondarily affects vascular permeability [23].

Figure 2.
Pathophysiology in SSc: The pathogenesis of systemic sclerosis (SSc) involves a multifaceted interplay between vascular injury, immune dysregulation, and fibroblast activation, culminating in progressive fibrosis and organ dysfunction. Initial endothelial cell damage and immune cell infiltration lead to the loss of small vessels and the release of pro-inflammatory mediators, including cytokines, reactive oxygen species (ROS), and damage-associated molecular patterns (DAMPs). These activate both innate and adaptive immune cells such as Th2 lymphocytes, B cells, mast cells, and M2 macrophages. B cells generate pathogenic autoantibodies, while Th2 cytokines and TGF-β stimulate fibroblast activation and myofibroblast differentiation. Myofibroblasts, derived from multiple cellular sources including resident fibroblasts, (pre-)adipocytes, endothelial cells, and mesenchymal cells, excessively produce extracellular matrix (ECM) components such as collagens, fibronectin, and fibrillins. Mechanical tension within the ECM feeds back to further activate fibroblasts via integrin-mediated signaling. This positive feedback loop sustains a stiffened ECM environment, impairs fibroblast apoptosis, and perpetuates fibrosis. The figure illustrates the central role of myofibroblasts in ECM remodeling and the integration of immune, vascular, and fibrotic pathways in SSc progression. Created in BioRender. Al-Gburi, S. (2025) https://BioRender.com/hdz8tk7, accessed on 18 May 2025 [21].
Figure 2.
Pathophysiology in SSc: The pathogenesis of systemic sclerosis (SSc) involves a multifaceted interplay between vascular injury, immune dysregulation, and fibroblast activation, culminating in progressive fibrosis and organ dysfunction. Initial endothelial cell damage and immune cell infiltration lead to the loss of small vessels and the release of pro-inflammatory mediators, including cytokines, reactive oxygen species (ROS), and damage-associated molecular patterns (DAMPs). These activate both innate and adaptive immune cells such as Th2 lymphocytes, B cells, mast cells, and M2 macrophages. B cells generate pathogenic autoantibodies, while Th2 cytokines and TGF-β stimulate fibroblast activation and myofibroblast differentiation. Myofibroblasts, derived from multiple cellular sources including resident fibroblasts, (pre-)adipocytes, endothelial cells, and mesenchymal cells, excessively produce extracellular matrix (ECM) components such as collagens, fibronectin, and fibrillins. Mechanical tension within the ECM feeds back to further activate fibroblasts via integrin-mediated signaling. This positive feedback loop sustains a stiffened ECM environment, impairs fibroblast apoptosis, and perpetuates fibrosis. The figure illustrates the central role of myofibroblasts in ECM remodeling and the integration of immune, vascular, and fibrotic pathways in SSc progression. Created in BioRender. Al-Gburi, S. (2025) https://BioRender.com/hdz8tk7, accessed on 18 May 2025 [21].

Chronic inflammation promotes endothelial–mesenchymal transformation, which facilitates a profibrotic state (Figure 2). Endothelial cells play a key role in vasoregulation, tissue homeostasis, immune regulation, and platelet aggregation, as they act as “guards” between tissue and blood vessels, controlling various immunological processes. Endothelial dysfunction promotes a pro-inflammatory state [24]. Furthermore, many studies have shown that autoantibodies against endothelial cells (so-called anti-endothelial cell autoantibodies [AECAs]) can induce endothelial apoptosis [25]. These autoantibodies comprise a heterogeneous group of proteins that target various structures of endothelial cells and are found in approximately 22–86% of patients with systemic sclerosis. They stimulate the production of reactive oxygen species, the formation of PDGF (platelet-derived growth factor), and the expression of various adhesion molecules, such as VCAM-1, ICAM-1, and E-selectin, which facilitate leukocyte invasion [26]. Pericytes, which help stabilize blood vessels, may differentiate into various cell types and are involved in vascular changes in SSc [27].
Elevated levels of ET-1 have been linked to various SSc complications, promoting vasoconstriction and fibroblast activity [28]. Conversely, NO production is reduced, impairing vascular relaxation and contributing to enhanced platelet aggregation and oxidative injury [29].
Angiogenesis, the formation of new blood vessels [30,31], is disrupted in SSc, despite elevated levels of angiogenic factors like Vascular Endothelial Growth Factor (VEGF) [31]. This lack of response leads to significant capillary loss without significant new vessel formation. Additionally, vasculogenesis and the role of progenitor cells in vascular repair are not well understood, with conflicting evidence regarding their presence in SSc.
Altogether, endothelial dysfunction, particularly in microcirculation, appears to drive the early phases and progression of this disease. However, these vascular alterations can also be responsible for major clinical complications such as pulmonary arterial hypertension (PAH), digital ulcers, and renal crisis [32]. Conversely, vasoprotective or vasodilatory therapies, such as those with prostaglandin agonists, endothelin-1 receptor antagonists, and PDE5 inhibitors, positively influence vasculopathic complications [33].
4. Autoimmune Dysregulation in Systemic Sclerosis
Systemic sclerosis (SSc) is characterized by the dysregulated interplay between the innate and adaptive immune systems (Figure 2). Apoptotic and damaged endothelial cells release damage-associated molecular patterns (DAMPs), which recruit and activate immune cells [34]. This immune activation occurs even before overt endothelial cell damage is detectable and is driven by elevated levels of pro-inflammatory cytokines like interleukin (IL)-6 [35]. The interplay between vascular damage and immune activation perpetuates a self-sustaining cycle that exacerbates disease progression.
The adaptive immune system plays a crucial role in SSc pathogenesis (Figure 2), with type 2 helper T (Th2) cells being particularly active. These cells produce IL-4 and IL-13, which drive fibroblast proliferation, enhance extracellular matrix (ECM) production, and increase collagen synthesis. Additionally, IL-4 and IL-13 suppress matrix metalloproteinases (MMPs) [36], further contributing to ECM accumulation (Table 1). These cytokines stimulate the production of transforming growth factor-β (TGF-β), a central mediator of fibrosis that activates the SMAD and mitogen-activated protein kinase (MAPK) signaling pathways in fibroblasts [37]. This cascade promotes fibroblast proliferation and collagen deposition while inhibiting ECM degradation. Feedback loops involving TGF-β and Th2 cytokines sustain fibrosis in a vicious cycle.
B cells, including plasma cells and their precursors [38], significantly contribute to SSc by producing autoantibodies against DNA-topoisomerase 1, centromeres, endothelial cells, and other antigens [39]. Many of these autoantibodies are markers for disease development; other autoantibodies, e.g., targeting endothelial cells [25], PDGF receptors, and fibrillin-1, are thought to directly activate fibroblasts [40], stimulating collagen synthesis. Furthermore, B cells produce IL-6, which promotes Th2 differentiation (Table 1) and macrophage polarization toward the M2 phenotype. Dysregulated regulatory B cells (Bregs) exacerbate disease by reducing IL-10 production, which diminishes their immunosuppressive effects.
The innate immune system also plays a critical role in SSc (Figure 2). Macrophages, particularly M2 macrophages, contribute to both tissue repair and fibrosis by producing profibrotic cytokines, including TGF-β, IL-4, IL-13, and IL-6 [41,42,43]. These cytokines activate fibroblasts and drive ECM deposition (Table 1). Neutrophils contribute through the release of reactive oxygen species (ROS) [44] and neutrophil extracellular traps (NETs) [45,46], which liberate latent TGF-β from the ECM, amplifying fibrosis. Plasmacytoid dendritic cells (pDCs) further contribute by producing interferon-α (IFN-α) and chemokine CXCL4 [47], both of which sustain immune activation and chronic inflammation (Table 1). Enhanced Toll-like receptor-8 (TLR8) signaling in pDCs establishes a positive feedback loop that maintains this inflammatory state.
Mast cells play multifaceted roles in SSc, attracted to fibrotic lesions by local signals such as plasminogen activator inhibitor-1 (PAI-1). Upon activation, mast cells release profibrotic mediators, including TGF-β, PDGF [48], and fibronectin. Direct interactions between mast cells and fibroblasts via adhesion molecules like intercellular adhesion molecule-1 (ICAM-1) further contribute to ECM deposition [49]. However, studies suggest that fibrosis can progress independently of mast cells, highlighting the complexity of SSc pathogenesis [50].
The intricate interactions between immune cells and fibroblasts are central to SSc pathogenesis. Fibroblasts in affected tissues exhibit a profibrotic phenotype, producing excessive amounts of collagen and other ECM components. Cytokines like TGF-β, IL-4, and IL-13 [51] from immune cells reinforce this phenotype, perpetuating chronic inflammation and fibrosis (Table 1). Insights from animal models, such as tight-skin mice, have demonstrated that targeting these cytokines can reduce fibrosis [52]. Similarly, models of graft-versus-host disease (GVHD), which share features with SSc, have shown that inhibiting Th2 cytokines prevents fibrotic progression [53].
Taken together, SSc is driven by the complex interactions among endothelial dysfunction, immune dysregulation, and fibroblast activation. The crosstalk between immune cells and fibroblasts creates a self-perpetuating cycle of inflammation and fibrosis. The contributions of Th2 cells, B cells, macrophages, neutrophils, dendritic cells, and mast cells highlight the multifactorial nature of immune activation in SSc. Targeting key cytokines such as IL-4, IL-13, TGF-β, and IL-6 holds therapeutic potential (Table 1). Understanding these mechanisms provides crucial insights into developing targeted therapies aimed at modulating immune responses, reducing fibrosis, and improving vascular function in SSc patients.

Table 1.
Cytokines, chemokines, and growth factors in systemic sclerosis.
Table 1.
Cytokines, chemokines, and growth factors in systemic sclerosis.
| Cytokines/chemokines/growth factors involved in vasculopathy | ||
|---|---|---|
| Endothelin-1 (ET-1) | Potent vasoconstrictor; promotes vascular dysfunction, fibroblast activation, and is elevated in SSc patients. Involved in PAH and DU development. | [17,28] |
| Nitric Oxide (NO) | Vasodilator; its impaired production leads to vascular tone dysregulation, platelet aggregation, and oxidative injury. | [18,19,29] |
| Vascular Endothelial Growth Factor (VEGF) | Key factor for angiogenesis; elevated in SSc but ineffective, leading to defective capillary repair and progressive ischemia. | [30,31] |
| Platelet-Derived Growth Factor (PDGF) | Induces fibroblast proliferation, contributing to vascular remodeling and fibrosis; linked to vascular dysfunction in SSc. | [26,48] |
| CXCL4 | Chemokine produced by plasmacytoid dendritic cells; amplifies immune activation, vascular injury, and fibrosis. | [47] |
| Interleukin-6 (IL-6) | Elevated early; drives endothelial activation, Th2 polarization, and chronic inflammation, and contributes to vascular damage. | [35,41] |
| Cytokines/chemokines/growth factors involved in fibrosis | ||
| Transforming Growth Factor-β (TGF-β) | Master regulator of fibrosis; promotes fibroblast activation, ECM production, myofibroblast differentiation, and suppresses ECM degradation. | [37,54,55] |
| Interleukin-4 (IL-4) | Th2 cytokine; enhances fibroblast proliferation and collagen production, suppresses ECM degradation, and promotes fibrotic progression. | [36,51] |
| Interleukin-13 (IL-13) | Works alongside IL-4; boosts collagen synthesis and fibroblast proliferation; and sustains fibrotic cycles. | [36,51] |
| Interleukin-6 (IL-6) | In addition to vascular roles, promotes M2 macrophage polarization and enhances fibrotic signaling. | [41,43] |
| Interferon-α (IFN-α) | Produced by plasmacytoid dendritic cells; promotes immune activation and maintains fibrotic and inflammatory environments. | [47] |
| Oncostatin M | Produced by mononuclear cells; acts synergistically with IL-6 to stimulate fibroblast activation and fibrosis. | [41] |
| Connective Tissue Growth Factor (CTGF) | Acts downstream of TGF-β; critical in fibroblast activation and persistent ECM accumulation. | [56] |
| Osteopontin (OPN) | Pro-inflammatory glycoprotein promoting fibroblast activation, myofibroblast differentiation, ECM deposition, and chronic inflammation; linked to disease severity in SSc. | [57] |
| Interleukin-17 (IL-17) | Pro-inflammatory cytokine from Th17 cells; enhances fibroblast proliferation, collagen expression, and synergizes with TGF-β in fibrotic pathways. | [58,59] |
| Interleukin-11 (IL-11) | Promotes fibroblast activation, ECM production, and collagen deposition; implicated in lung and skin fibrosis. Revelant for cardiac and renal fibrosis. | [60,61] |
| Interleukin-31 (IL-31) | Associated with pruritus in SSc; emerging evidence suggests profibrotic roles via immune–fibroblast crosstalk. | [62,63] |
5. Fibrosis and ECM Deposition in SSc
The excessive deposition of ECM molecules is a hallmark of scleroderma and is ultimately responsible for tissue damage with all the clinical implications (Figure 1C). The persistent activation of fibroblasts and their transformation into myofibroblasts play a critical role in this pathological process. Myofibroblasts are key mediators of ECM remodeling, and their sustained presence in SSc results in uncontrolled ECM synthesis, fibrosis, and ultimately, irreversible tissue damage (Figure 2).
5.1. Activation and Origin of Fibroblasts in SSc
The origins of activated fibroblasts in SSc remain a topic of extensive research [64,65]. They may arise from multiple sources, including circulating progenitor cells, subcutaneous layers, resident tissue fibroblasts, and transdifferentiated epithelial or endothelial cells (Figure 2). Once activated, fibroblasts acquire the characteristics of myofibroblasts, which are central to wound healing and scar formation. These cells exhibit contractile properties, express alpha-smooth muscle actin (α-SMA), and contribute to ECM production [66]. Under physiological conditions, myofibroblasts facilitate tissue repair and are subsequently eliminated through apoptosis [67]. However, in SSc, myofibroblasts are thought to persist due to a dysregulation in apoptotic pathways, leading to excessive ECM deposition. This results in increased tissue stiffness, reduced mechanical stability, and progressive fibrosis, ultimately impairing organ function [68,69,70] (Figure 2).
5.2. Fibroblast Survival and Resistance to Apoptosis
Myofibroblast survival in SSc is facilitated by an imbalance between pro-apoptotic and anti-apoptotic signals. Apoptosis, a crucial mechanism for eliminating excess myofibroblasts following tissue repair, is regulated by proteins such as BAX and BIM (pro-apoptotic) and BCL-2 family proteins (anti-apoptotic). In physiological wound healing, myofibroblasts undergo apoptosis when ECM stiffness decreases, reducing BCL-2 signaling and allowing BIM-mediated cell death [71,72,73].
In SSc, however, mechanotransduction pathways alter apoptotic signaling, increasing the expression of BIM while simultaneously upregulating BCL-XL, an anti-apoptotic protein that inhibits BIM activation. This allows myofibroblasts to evade apoptosis and continue producing ECM components. Experimental studies have shown that inhibiting BCL-XL can promote myofibroblast apoptosis [74].
Transforming growth factor-beta (TGFβ) is a key profibrotic cytokine implicated in SSc pathogenesis (Table 1). Elevated levels of TGFβ are observed in SSc skin and lung tissues, where it stimulates fibroblast activation, ECM synthesis, and myofibroblast differentiation [54,55]. TGFβ also influences apoptotic pathways by modulating sphingolipid metabolism, particularly through the downregulation of acid sphingomyelinase (ASMase), a critical enzyme in Fas-mediated apoptosis. Reduced ASMase levels in SSc fibroblasts are thought to promote apoptosis resistance and enhance fibrotic signaling [75]. MicroRNAs (miRNAs) further contribute to the apoptotic imbalance in SSc. miRNA-21, which is upregulated in SSc fibroblasts, binds to and degrades the mRNA of pro-apoptotic BAX, further suppressing myofibroblast apoptosis. This has been reported to create a pro-survival environment, perpetuating fibrosis and ECM accumulation [76].
In addition to reduced apoptosis, myofibroblast populations in SSc might expand due to increased transdifferentiation and activation. TGFβ, PDGF, and connective tissue growth factor (CTGF) drive fibroblast differentiation into myofibroblasts [56] (Table 1). However, circulating fibrocytes, epithelial–mesenchymal transition (EMT), endothelial-to-mesenchymal transition (EndoMT), and pericyte differentiation [77,78,79] might also contribute to this process (Figure 2).
A notable feature of SSc is the loss of subcutaneous adipose tissue. Adipocytes are increasingly recognized as contributors to fibrotic progression through adipocyte–myofibroblast transition (AMT). TGFβ stimulation of adipocytes inhibits adipogenesis and upregulates profibrotic genes, leading to the conversion of adipocytes into fibroblast-like cells. Both adipose-derived mesenchymal stem cells and mature adipocytes undergo this transition, contributing to the fibrosis seen in SSc patients (Figure 2).
Mechanical tension is a crucial regulator of fibroblast function and myofibroblast differentiation. In SSc, increased ECM stiffness enhances fibroblast activation via integrins and focal adhesion complexes. α11β1 integrin, in particular, plays a critical role in fibrosis by transducing the mechanical and biochemical signals that sustain myofibroblast activity. Depletion of α11β1 integrin has been shown to suppress fibrosis and impair fibroblast transdifferentiation. Increased mechanical tension also promotes TGFβ activation from its latent form in the ECM (Figure 2). TGFβ is sequestered within the ECM in an inactive state, but integrin-mediated tension releases active TGFβ, perpetuating fibroblast activation and ECM deposition. These findings highlight the interplay between mechanical and biochemical cues in fibrosis progression [80,81,82,83,84].
Fibroblasts are a heterogeneous population with distinct functional properties. Recent studies utilizing single-cell and bulk RNA sequencing have identified fibroblast subtypes that are enriched in SSc skin. Profibrotic fibroblasts expressing markers such as COMP, COL11A1, MYOC, CCL19, and SFRP4 are significantly increased, while antifibrotic fibroblasts marked by CXCL12 and PI16 are reduced. The balance between these fibroblast subsets correlates with disease severity. Increased levels of profibrotic fibroblasts are associated with progressive skin fibrosis, whereas higher proportions of CXCL12+ and PI16+ fibroblasts correlate with stable disease. Machine learning models incorporating fibroblast markers have improved the classification of progressive versus stable SSc cases, highlighting their potential as diagnostic and therapeutic targets [85].
5.3. The ECM in SSc
Fibrosis in SSc is characterized by the excessive deposition of collagen types I, III, V, and VI, as well as fibronectin, elastin, glycosaminoglycans, and proteoglycans. The ECM of SSc patients also contains increased levels of damage-associated molecular patterns (DAMPs), such as fibronectin-EDA and tenascin-C, which activate profibrotic pathways through toll-like receptor 4 (TLR4) signaling. Cartilage oligomeric matrix protein (COMP) is highly expressed in SSc and other fibrotic conditions. COMP regulates collagen fibrillogenesis and is essential for the ECM’s structural integrity. It also facilitates collagen secretion, contributing to the excessive accumulation and altered macromolecular arrangement observed in fibrotic tissues [21].
Although the excessive deposition of different components of the ECM is characteristic for fibrotic processes, it also has to be noted that the stiffness of the tissue depends on the macromolecular organization of the collagens. This is determined by various factors. The so-called FACITs (fibril-associated collagens with interrupted triple helices) play a crucial role in controlling the macromolecular organization and the fibril diameter. In addition to their structural significance, these extracellular matrix (ECM) proteins can also have functional activities. It has been shown that collagen XII, a FACIT collagen in skin, significantly influences the number of myofibroblasts. Mechanistically, this can be attributed to the indirect communication between macrophages and fibroblasts, where collagen XII affects the release of fibrogenic cytokines by macrophages [86]. This is also true for other non-collagenous ECM proteins, such as COMP, which are induced during fibrotic processes [87]. All of these proteins have both structural and functional activities, alter the macromolecular organization and biomechanical properties of connective tissue, and regulate the function of fibroblasts, endothelial cells, and inflammatory cells in fibrotic processes (Figure 2).
6. Therapeutic Approaches for SSc Based on the Understanding of Its Pathophysiology
Understanding these mechanisms at a molecular level provides an insight into potential therapeutic strategies, including targeting apoptosis pathways, modulating TGFβ signaling, and disrupting mechanical tension-mediated fibroblast activation. Future research focusing on fibroblast heterogeneity and ECM dynamics will be crucial in developing effective treatments for SSc and related fibrotic diseases.
Recent technological breakthroughs and an improved understanding of disease mechanisms at the cellular and molecular levels have already led to better patient stratification and the development of novel therapeutic approaches. Clinical trials now benefit from molecular classification based on gene expression profiles in skin biopsies. While the predictive value of this classification is still being evaluated, it enhances patient selection for targeted therapies when combined with serum biomarkers and refined clinical criteria [88].
Therapeutic advancements have significantly improved the management of organ complications associated with SSc [89]. Mainly for diffuse cutaneous SSc patients, immunosuppressive agents such as mycophenolate mofetil, methotrexate, cyclophosphamide, rituximab, and tocilizumab are widely used [89,90,91,92,93,94], while patients with rapidly progressive disease may benefit from autologous hematopoietic stem cell transplantation [95]. Lung disease management has improved, with mycophenolate mofetil for SSc-ILD and antifibrotic agents like nintedanib [89,96] and possibly pirfenidone showing promise. PAH is commonly treated with combination therapy, including phosphodiesterase 5 inhibitors and endothelin receptor antagonists, sometimes supplemented with prostacyclin analogs. Most recently, sotatercept, a first-in-class activin-signaling inhibitor, has also been approved for the treatment of PAH [97]. Raynaud’s phenomenon and digital ulcers are managed using calcium channel blockers, phosphodiesterase 5 inhibitors, and intravenous iloprost, with bosentan helping to prevent new ulcer formation [98,99,100,101]. However, more research is needed to optimize treatment strategies for other disease manifestations.
Novel compounds target the microvascular alterations, the immune response (e.g., JAK inhibitors, IL-4/IL-13 inhibitors, Belimumab), and also the different steps in the activation of myofibroblasts (e.g., TGFβ inhibitors, ROCK inhibitors, LPA inhibitors) [102].
Given the pivotal role of B cells in SSc pathogenesis, CD19-targeting chimeric antigen receptor (CAR) T cell therapy has emerged as a new approach for severe diffuse SSc in patients unresponsive to conventional treatments. Recent studies demonstrate that CAR T cell therapy can halt disease progression, improve key clinical features such as skin fibrosis and lung function, and reduce autoantibody levels.
Future research must assess the durability of these therapeutic effects and compare CAR T cell therapy with other advanced treatments, such as autologous stem cell transplantation and CD20-targeting therapies. All these new approaches have been developed based on a better understanding of the pathophysiology of this complex disease and mark a promising step toward more effective and potentially curative treatments for the benefit of patients with systemic sclerosis.
7. Conclusions
At the core of SSc pathogenesis lies a dynamic and self-reinforcing interplay between vascular injury, chronic immune activation, and fibroblast dysregulation. Early endothelial dysfunction—possibly triggered by genetic predisposition, environmental exposures, or infectious insults—leads to capillary dropout, impaired vasoregulation, and hypoxia. This vascular damage is compounded by the emergence of autoantibodies and the infiltration of various immune cells that further drive inflammation and fibrosis. Immune mediators, particularly Th2 cytokines (IL-4, IL-13), TGF-β, and IL-6 (Table 1), perpetuate fibroblast activation and ECM accumulation. Notably, fibroblasts in SSc not only become resistant to apoptosis but also exhibit enhanced mechanosensing capabilities that amplify fibrotic responses in the context of increased tissue stiffness.
Recent advances have further illuminated the cellular origins and heterogeneity of fibroblasts involved in SSc, uncovering key transcriptional and functional differences that correlate with disease activity and treatment response. Technologies such as single-cell RNA sequencing and machine learning models have identified distinct profibrotic and antifibrotic fibroblast populations, offering potential biomarkers for disease stratification and new targets for therapeutic intervention.
Therapeutically, while conventional immunosuppressive regimens remain standard in managing diffuse cutaneous disease and organ involvement, novel strategies are emerging from our improved understanding of disease biology. These include antifibrotic agents like nintedanib, biologics targeting cytokine pathways (e.g., IL-4/IL-13, IL-6) (Table 1), and cellular therapies such as autologous hematopoietic stem cell transplantation. In severe refractory cases, CAR T cell therapies targeting CD19+ B cells show early promise, demonstrating potential not only to halt disease progression but to reverse key pathological features.
However, despite these advancements, there is still no universally effective disease-modifying therapy for SSc. Many of the current interventions primarily address symptoms or specific organ manifestations without altering the fundamental disease trajectory. The variability in clinical course and treatment response among patients underscores the urgent need for precision medicine approaches. Integrating molecular classifications, serum biomarkers, and tissue-based gene expression profiles into clinical decision-making will be essential for tailoring therapy and improving long-term outcomes.
Funding
SA was funded by “Köln Fortune“ [134/2023]. This research was funded by DFG [KR 558/17-1] and by Deutsche Forschungsgemeinschaft; Edith Busch Foundation.
Conflicts of Interest
P.M. reports lecture fees from Boehringer Ingelheim and Janssen, as well as advisory board fees of Almirall, unrelated to the present manuscript. T.K. and S.A.-G. declare no conflict of interest.
References
- Volkmann, E.R.; Andréasson, K.; Smith, V. Systemic sclerosis. Lancet 2023, 401, 304–318. [Google Scholar] [CrossRef]
- Gabrielli, A.; Avvedimento, E.V.; Krieg, T. Mechanisms of Disease Scleroderma. N. Engl. J. Med. 2009, 360, 1989–2003. [Google Scholar] [CrossRef]
- Distler, J.H.W.; Györfi, A.-H.; Ramanujam, M.; Whitfield, M.L.; Königshoff, M.; Lafyatis, R. Shared and distinct mechanisms of fibrosis. Nat. Rev. Rheumatol. 2019, 15, 705–730. [Google Scholar] [CrossRef] [PubMed]
- Ross, R.L.; Caballero-Ruiz, B.; Clarke, E.L.; Kakkar, V.; Wasson, C.W.; Mulipa, P.; De Lorenzis, E.; Merchant, W.; Di Donato, S.; Rindone, A.; et al. Biological hallmarks of systemic sclerosis are present in the skin and serum of patients with Very Early Diagnosis of Systemic Sclerosis (VEDOSS). Rheumatology 2024, keae698, ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Varga, J.; Abraham, D. Systemic sclerosis: A prototypic multisystem fibrotic disorder. J. Clin. Investigation. 2007, 117, 557–567. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A.; Ramalingam, T.R. Mechanisms of fibrosis: Therapeutic translation for fibrotic disease. Nat. Med. 2012, 18, 1028–1040. [Google Scholar] [CrossRef]
- Avouac, J.; Riemekasten, G.; Meune, C.; Ruiz, B.; Kahan, A.; Allanore, Y. Autoantibodies against endothelin 1 Type A receptor are strong predictors of digital ulcers in systemic sclerosis. J. Rheumatol. 2015, 42, 1801–1807. [Google Scholar] [CrossRef]
- Lescoat, A.; Varga, J.; Matucci-Cerinic, M.; Khanna, D. New promising drugs for the treatment of systemic sclerosis: Pathogenic considerations, enhanced classifications, and personalized medicine. Expert Opin. Investig. Drugs 2021, 30, 635–652. [Google Scholar] [CrossRef]
- Del Galdo, F.; Lescoat, A.; Conaghan, P.G.; Bertoldo, E.; Čolić, J.; Santiago, T.; Suliman, Y.A.; Matucci-Cerinic, M.; Gabrielli, A.; Distler, O.; et al. EULAR recommendations for the treatment of systemic sclerosis: 2023 update. Ann. Rheum. Dis. 2024, 84, 29–40. [Google Scholar] [CrossRef]
- Broen, J.C.A.; Radstake, T.R.D.J.; Rossato, M. The role of genetics and epigenetics in the pathogenesis of systemic sclerosis. Nat. Rev. Rheumatol. 2014, 10, 671–681. [Google Scholar] [CrossRef]
- López-Isac, E.; Acosta-Herrera, M.; Kerick, M.; Assassi, S.; Satpathy, A.T.; Granja, J.; Mumbach, M.R.; Beretta, L.; Simeón, C.P.; Carreira, P.; et al. GWAS for systemic sclerosis identifies multiple risk loci and highlights fibrotic and vasculopathy pathways. Nat. Commun. 2019, 10, 1–14. [Google Scholar] [CrossRef] [PubMed]
- De Martinis, M.; Ciccarelli, F.; Sirufo, M.M.; Ginaldi, L. An overview of environmental risk factors in systemic sclerosis. Expert Rev. Clin. Immunol. 2016, 12, 465–478. [Google Scholar] [CrossRef]
- Moroncini, G.; Svegliati, S.; Grieco, A.; Cuccioloni, M.; Mozzicafreddo, M.; Paolini, C.; Agarbati, S.; Spadoni, T.; Amoresano, A.; Pinto, G.; et al. Adeno-Associated Virus Type 5 Infection via PDGFRα Is Associated With Interstitial Lung Disease in Systemic Sclerosis and Generates Composite Peptides and Epitopes Recognized by the Agonistic Immunoglobulins Present in Patients With Systemic Sclerosis. Arthritis Rheumatol. 2024, 76, 620–630. [Google Scholar] [CrossRef]
- Jung, P.; Trautinger, F. Kapillarmikroskopie. JDDG-J. Ger. Soc. Dermatol. 2013, 11, 731–736. [Google Scholar] [CrossRef]
- Fieischmajer, R.; Perlish, J.S. Capillary Alterations in Scleroderma. J. Am. Acad. Dermatol. 1980, 2, 161–170. [Google Scholar] [CrossRef]
- Smith, V.; Herrick, A.L.; Ingegnoli, F.; Damjanov, N.; De Angelis, R.; Denton, C.P.; Distler, O.; Espejo, K.; Foeldvari, I.; Frech, T.; et al. Standardisation of nailfold capillaroscopy for the assessment of patients with Raynaud’s phenomenon and systemic sclerosis. Autoimmun. Rev. 2020, 19, 102458. [Google Scholar] [CrossRef] [PubMed]
- Rokni, M.; Shaker, M.S.; Kavosi, H.; Shokoofi, S.; Mahmoudi, M.; Farhadi, E. The role of endothelin and RAS/ERK signaling in immunopathogenesis-related fibrosis in patients with systemic sclerosis: An updated review with therapeutic implications. Arthritis Res. Ther. 2022, 24, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Allanore, Y.; Borderie, D.; Hilliquin, P.; Hernvann, A.; Levacher, M.; Lemaréchal, H.; Ekindjian, O.G.; Kahan, A. Low Levels of Nitric Oxide (NO) in Systemic Sclerosis: Inducible NO Synthase Production Is Decreased in Cultured Peripheral Blood Monocyteumacrophage Cells. Rheumatology 2001, 40, 1089–1096. [Google Scholar] [CrossRef]
- Hinz, B. The role of myofibroblasts in wound healing. Curr. Res. Transl. Med. 2016, 64, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Binda, M.; Moccaldi, B.; Civieri, G.; Cuberli, A.; Doria, A.; Tona, F.; Zanatta, E. Autoantibodies Targeting G-Protein-Coupled Receptors: Pathogenetic, Clinical and Therapeutic Implications in Systemic Sclerosis. Int. J. Mol. Sci. 2024, 25, 2299. [Google Scholar] [CrossRef]
- Rosendahl, A.; Schönborn, K.; Krieg, T. Pathophysiology of systemic sclerosis (scleroderma). Kaohsiung J. Med. Sci. 2022, 38, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Su, J.; An, X.-R.; Li, Q.; Li, X.-X.; Cong, X.-D.; Xu, M. Improvement of vascular dysfunction by argirein through inhibiting endothelial cell apoptosis associated with ET-1/Nox4 signal pathway in diabetic rats. Sci. Rep. 2018, 8, 12620. [Google Scholar] [CrossRef] [PubMed]
- Bruni, C.; Frech, T.; Manetti, M.; Rossi, F.W.; Furst, D.E.; De Paulis, A.; Rivellese, F.; Guiducci, S.; Matucci-Cerinic, M.; Bellando-Randone, S. Vascular leaking, a pivotal and early pathogenetic event in systemic sclerosis: Should the door be closed? Front. Immunol. 2018, 9, 2045. [Google Scholar] [CrossRef] [PubMed]
- De Ciuceis, C.; Amiri, F.; Brassard, P.; Endemann, D.H.; Touyz, R.M.; Schiffrin, E.L. Reduced vascular remodeling, endothelial dysfunction, and oxidative stress in resistance arteries of angiotensin II-infused macrophage colony-stimulating factor-deficient mice: Evidence for a role in inflammation in angiotensin-induced vascular injury. Arter. Thromb. Vasc. Biol. 2005, 25, 2106–2113. [Google Scholar] [CrossRef]
- Günther, J.; Rademacher, J.; van Laar, J.M.; Siegert, E.; Riemekasten, G. Functional autoantibodies in systemic sclerosis. Semin. Immunopathol. 2015, 37, 529–542. [Google Scholar] [CrossRef]
- Sartori-Valinotti, J.C.; Tollefson, M.M.; Reed, A.M. Updates on morphea: Role of vascular injury and advances in treatment. Autoimmune Dis. 2013, 2013, 1–8. [Google Scholar] [CrossRef]
- Talotta, R.; Atzeni, F.; Ditto, M.C.; Gerardi, M.C.; Batticciotto, A.; Bongiovanni, S.; Puttini, P.S. Certainties and uncertainties concerning the contribution of pericytes to the pathogenesis of systemic sclerosis. J. Scleroderma Relat. Disord. 2018, 3, 14–20. [Google Scholar] [CrossRef]
- Cozzani, E.; Javor, S.; Laborai, E.; Drosera, M.; Parodi, A. Endothelin-1 Levels in Scleroderma Patients: A Pilot Study. ISRN Dermatol. 2013, 2013, 125632. [Google Scholar] [CrossRef]
- Takagi, K.; Kawaguchi, Y.; Hara, M.; Sugiura, T.; Harigai, M.; Kamatani, N. Serum nitric oxide (NO) levels in systemic sclerosis patients: Correlation between NO levels and clinical features. Clin. Exp. Immunol. 2003, 134, 538–544. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Cantatore, F.P.; Maruotti, N.; Corrado, A.; Ribatti, D. Angiogenesis Dysregulation in the Pathogenesis of Systemic Sclerosis. Biomed Res. Int. 2017, 2017, 5345673. [Google Scholar] [CrossRef]
- Darby, I.A.; Laverdet, B.; Bonte, F.; Desmouliere, A. Fibroblasts and myofibroblasts in wound healing. Clin. Cosmet. Investig. Dermatol. 2014, 7, 301–311. [Google Scholar] [PubMed]
- Hunzelmann, N.; Krieg, T. Sklerodermie. In Braun-Falco’s Dermatologie, Venerologie Und Allergologie, 7th ed.; Plewig, G., Ruzicka, T., Kaufmann, R., Hertl, M., Eds.; Springer: Berlin/Heidelberg, Germany, 2018; pp. 919–936. [Google Scholar] [CrossRef]
- Pattanaik, D.; Brown, M.; Postlethwaite, A.E. Vascular involvement in systemic sclerosis (scleroderma). J. Inflamm. Res. 2011, 4, 105–125. [Google Scholar] [CrossRef]
- Roumm, A.D.; Whiteside, T.L.; Medsger, T.A.; Rodnan, G.P. Lymphocytes in the skin of patients with progressive systemic sclerosis. Arthritis Rheum. 1984, 27, 645–653. [Google Scholar] [CrossRef]
- Hasegawa, M.; Sato, S.; Ihn, H.; Takehara, K. Enhanced Production of Interleukin-6 (IL-6), Oncostatin M and Soluble IL-6 Receptor by Cultured Peripheral Blood Mononuclear Cells from Patients with Systemic Sclerosis. Rheumatology 1999, 38, 612–617. [Google Scholar] [CrossRef] [PubMed]
- Mori, R.; Shaw, T.J.; Martin, P. Molecular mechanisms linking wound inflammation and fibrosis: Knockdown of osteopontin leads to rapid repair and reduced scarring. J. Investig. Dermatol. 2008, 128, 1435–1447. [Google Scholar] [CrossRef] [PubMed]
- Massagué, J. TGFβ signalling in context. Nat. Rev. Mol. Cell Biol. 2012, 13, 616–630. [Google Scholar] [CrossRef] [PubMed]
- Agarbati, S.; Benfaremo, D.; Viola, N.; Paolini, C.; Baroni, S.S.; Funaro, A.; Moroncini, G.; Malavasi, F.; Gabrielli, A. Increased expression of the ectoenzyme CD38 in peripheral blood plasmablasts and plasma cells of patients with systemic sclerosis. Front. Immunol. 2022, 13, 1072462. [Google Scholar] [CrossRef]
- Yoshizaki, A.; Fukasawa, T.; Ebata, S.; Yoshizaki-Ogawa, A.; Sato, S. Involvement of B cells in the development of systemic sclerosis. Front. Immunol. 2022, 13, 938785. [Google Scholar] [CrossRef]
- Grassegger, A.; Pohla-Gubo, G.; Frauscher, M.; Hintner, H. Autoantibodies in systemic sclerosis (scleroderma): Clues for clinical evaluation, prognosis and pathogenesis. Wien. Med. Wochenschr. 2008, 158, 19–28. [Google Scholar] [CrossRef]
- Higashi-Kuwata, N.; Jinnin, M.; Makino, T.; Fukushima, S.; Inoue, Y.; Muchemwa, F.C.; Yonemura, Y.; Komohara, Y.; Takeya, M.; Mitsuya, H.; et al. Characterization of Monocyte/Macrophage Subsets in the Skin and Peripheral Blood Derived from Patients with Systemic Sclerosis. Arthritis Res. Ther. 2010, 12, R128. [Google Scholar] [CrossRef]
- Knipper, J.A.; Willenborg, S.; Brinckmann, J.; Bloch, W.; Maaß, T.; Wagener, R.; Krieg, T.; Sutherland, T.; Munitz, A.; Rothenberg, M.E.; et al. Interleukin-4 Receptor α Signaling in Myeloid Cells Controls Collagen Fibril Assembly in Skin Repair. Immunity 2015, 43, 803–816. [Google Scholar] [CrossRef] [PubMed]
- Khalil, N.; Bereznayj, I.O.; Sporn, M.; Greenberg, A.H. Macrophage Production of Transforming Growth Factor r and Fibroblast Collagen Synthesis in Chronic Pulmonary Inflammation. J. Exp. Med. 1989, 170, 727–737. [Google Scholar] [CrossRef]
- Vona, R.; Giovannetti, A.; Gambardella, L.; Malorni, W.; Pietraforte, D.; Straface, E. Oxidative stress in the pathogenesis of systemic scleroderma: An overview. J. Cell Mol. Med. 2018, 22, 3308–3314. [Google Scholar] [CrossRef]
- Tecchio, C.; Micheletti, A.; Cassatella, M.A. Neutrophil-derived cytokines: Facts beyond expression. Front. Immunol. 2014, 5, 508. [Google Scholar] [CrossRef]
- Thiam, H.R.; Wong, S.L.; Wagner, D.D.; Waterman, C.M. Cellular Mechanisms of NETosis. Annu. Rev. Cell Dev. Biol. 2020, 36, 191–218. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lande, R.; Lee, E.Y.; Palazzo, R.; Marinari, B.; Pietraforte, I.; Santos, G.S.; Mattenberger, Y.; Spadaro, F.; Stefanantoni, K.; Iannace, N.; et al. CXCL4 assembles DNA into liquid crystalline complexes to amplify TLR9-mediated interferon-α production in systemic sclerosis. Nat. Commun. 2019, 10, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Maugeri, N.; Capobianco, A.; Rovere-Querini, P.; Ramirez, G.A.; Tombetti, E.; Valle, P.D.; Monno, A.; D’Alberti, V.; Gasparri, A.M.; Franchini, S.; et al. Platelet Microparticles Sustain Autophagy-Associated Activation of Neutrophils in Systemic Sclerosis. Sci. Transl. Med. 2018, 10, 3089. [Google Scholar] [CrossRef] [PubMed]
- Conti, P.; Caraffa, A.; Mastrangelo, F.; Tettamanti, L.; Ronconi, G.; Frydas, I.; Kritas, S.K.; Theoharides, T.C. Critical role of inflammatory mast cell in fibrosis: Potential therapeutic effect of IL-37. Cell Prolif. 2018, 51, e12475. [Google Scholar] [CrossRef]
- Overed-Sayer, C.; Rapley, L.; Mustelin, T.; Clarke, D.L. Are mast cells instrumental for fibrotic diseases? Front. Pharmacol. Front. Pharmacol. 2014, 4, 174. [Google Scholar] [CrossRef]
- Gasparini, G.; Cozzani, E.; Parodi, A. Interleukin-4 and interleukin-13 as possible therapeutic targets in systemic sclerosis. Cytokine 2020, 125, 154799. [Google Scholar] [CrossRef]
- McGaha, T.; Saito, S.; Phelps, R.G.; Gordon, R.; Noben-Trauth, N.; Paul, W.E.; Bona, C. Lack of Skin Fibrosis in Tight Skin (TSK) Mice with Targeted Mutation in the Interleukin-4Ra and Transforming Growth Factor-b Genes. J. Investig. Dermatol. 2001, 116, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Foley, J.E.; Mariotti, J.; Ryan, K.; Eckhaus, M.; Fowler, D.H. Th2 Cell Therapy of Established Acute Graft-Versus-Host Disease Requires IL-4 and IL-10 and Is Abrogated by IL-2 or Host-Type Antigen-Presenting Cells. Biol. Blood Marrow Transplant. 2008, 14, 959–972. [Google Scholar] [CrossRef] [PubMed]
- Kaliterna, D.M.; Petrić, M. Biomarkers of skin and lung fibrosis in systemic sclerosis. Expert Rev. Clin. Immunol. 2019, 15, 1215–1223. [Google Scholar] [CrossRef]
- Lafyatis, R. Transforming growth factor β—At the centre of systemic sclerosis. Nat. Rev. Rheumatol. 2014, 10, 706–719. [Google Scholar] [CrossRef]
- Yamakage, A.; Kikuchi, K.; Smith, E.A.; Leroy, E.C.; Trojanowska, M. Selective Upregulation of Platelet-Derived Growth Factor C~Receptors by Transforming Growth Factor/~ in Scleroder Fibroblasts. Available online: http://rupress.org/jem/article-pdf/175/5/1227/1673399/1227.pdf (accessed on 18 May 2025).
- Lang, F.; Li, Y.; Yao, R.; Jiang, M. Osteopontin in Chronic Inflammatory Diseases: Mechanisms, Biomarker Potential, and Therapeutic Strategies. Biology 2025, 14, 428. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.; Misra, D.P.; Agarwal, V. Interleukin-17 pathways in systemic sclerosis-associated fibrosis. Rheumatol. Int. 2019, 39, 1135–1143. [Google Scholar] [CrossRef]
- Chizzolini, C.; Dufour, A.M.; Brembilla, N.C. Is there a role for IL-17 in the pathogenesis of systemic sclerosis? Immunol. Lett. 2018, 195, 61–67. [Google Scholar] [CrossRef]
- Tan, Y.; Mosallanejad, K.; Zhang, Q.; O’brien, S.; Clements, M.; Perper, S.; Wilson, S.; Chaulagain, S.; Wang, J.; Abdalla, M.; et al. IL11-mediated stromal cell activation may not be the master regulator of pro-fibrotic signaling downstream of TGFβ. Front. Immunol. 2024, 15, 1293883. [Google Scholar] [CrossRef]
- O’Reilly, S. Interleukin-11 and its eminent role in tissue fibrosis: A possible therapeutic target. Clin. Exp. Immunol. 2023, 214, 154–161. [Google Scholar] [CrossRef]
- Kuzumi, A.; Yoshizaki, A.; Matsuda, K.M.; Kotani, H.; Norimatsu, Y.; Fukayama, M.; Ebata, S.; Fukasawa, T.; Yoshizaki-Ogawa, A.; Asano, Y.; et al. Interleukin-31 promotes fibrosis and T helper 2 polarization in systemic sclerosis. Nat. Commun. 2021, 12, 1–18. [Google Scholar] [CrossRef]
- Yaseen, B.; Lopez, H.; Taki, Z.; Zafar, S.; Rosario, H.; Abdi, B.A.; Vigneswaran, S.; Xing, F.; Arumalla, N.; Black, S.; et al. Interleukin-31 promotes pathogenic mechanisms underlying skin and lung fibrosis in scleroderma. Rheumatology 2020, 59, 2625–2636. [Google Scholar] [CrossRef] [PubMed]
- Gabbiani, G.; Ryan, G.B.; Majno, G. Presence of modified fibroblasts in granulation tissue and their possible role in wound contraction. Experientia 1971, 27, 549–550. [Google Scholar] [CrossRef] [PubMed]
- Gabbiani, G. 50 Years of Myofibroblasts: How the Myofibroblast Concept Evolved. In Myofibroblasts: Methods and Protocols; Hinz, B., Lagares, D., Eds.; Springer: New York, NY, USA, 2021; pp. 1–5. [Google Scholar] [CrossRef]
- Hinz, B.; Phan, S.H.; Thannickal, V.J.; Galli, A.; Bochaton-Piallat, M.-L.; Gabbiani, G. The myofibroblast: One function, multiple origins. Am. J. Pathol. 2007, 170, 1807–1816. [Google Scholar] [CrossRef]
- Schuster, R.; Younesi, F.; Ezzo, M.; Hinz, B. The Role of Myofibroblasts in Physiological and Pathological Tissue Repair. Cold Spring Harb. Perspect. Biol. 2022, 15, a041231. [Google Scholar] [CrossRef]
- van Caam, A.; Vonk, M.; Hoogen, F.v.D.; van Lent, P.; van der Kraan, P. Unraveling SSc pathophysiology; The myofibroblast. Front. Immunol. 2018, 9, 2452. [Google Scholar] [CrossRef] [PubMed]
- Ihn, H. Scleroderma, Fibroblasts, Signaling, and Excessive Extracellular Matrix. Curr. Rheumatol. Rep. 2005, 7, 156–162. [Google Scholar] [CrossRef]
- Chadli, L.; Sotthewes, B.; Li, K.; Andersen, S.N.; Cahir-McFarland, E.; Cheung, M.; Cullen, P.; Dorjée, A.; de Vries-Bouwstra, J.K.; Huizinga, T.W.J.; et al. Identification of regulators of the myofibroblast phenotype of primary dermal fibroblasts from early diffuse systemic sclerosis patients. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef]
- Hinz, B.; Lagares, D. Evasion of apoptosis by myofibroblasts: A hallmark of fibrotic diseases. Nat. Rev. Rheumatol. 2020, 16, 11–31. [Google Scholar] [CrossRef] [PubMed]
- Santiago, B.; Galindo, M.; Rivero, M.; Pablos, J.L. Decreased susceptibility to Fas-induced apoptosis of systemic sclerosis dermal fibroblasts. Arthritis Rheum. 2001, 44, 1667–1676. [Google Scholar] [CrossRef] [PubMed]
- Jelaska, A.; Korn, J.H. Role of apoptosis and transforming growth factor beta1 in fibroblast selection and activation in systemic sclerosis. Arthritis Rheum. 2000, 43, 2230–2239. [Google Scholar] [CrossRef] [PubMed]
- Lagares, D.; Santos, A.; Grasberger, P.E.; Liu, F.; Probst, C.K.; Rahimi, R.A.; Sakai, N.; Kuehl, T.; Ryan, J.; Bhola, P.; et al. Targeted apoptosis of myofibroblasts with the BH3 mimetic ABT-263 reverses established fibrosis. Sci. Transl. Med. 2017, 9, eaal3765. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Samuel, G.H.; Lenna, S.; Bujor, A.M.; Lafyatis, R.; Trojanowska, M. Acid sphingomyelinase deficiency contributes to resistance of scleroderma fibroblasts to Fas-mediated apoptosis. J. Dermatol. Sci. 2012, 67, 166–172. [Google Scholar] [CrossRef]
- Jafarinejad-Farsangi, S.; Farazmand, A.; Gharibdoost, F.; Karimizadeh, E.; Noorbakhsh, F.; Faridani, H.; Mahmoudi, M.; Jamshidi, A.R. Inhibition of MicroRNA-21 induces apoptosis in dermal fibroblasts of patients with systemic sclerosis. Int. J. Dermatol. 2016, 55, 1259–1267. [Google Scholar] [CrossRef]
- Rajkumar, V.S.; Sundberg, C.; Abraham, D.J.; Rubin, K.; Black, C.M. Activation of microvascular pericytes in autoimmune Raynaud’s phenomenon and systemic sclerosis. Arthritis Rheum. 1999, 42, 930–941. [Google Scholar] [CrossRef]
- Sonnylal, S.; Shi-Wen, X.; Leoni, P.; Naff, K.; Van Pelt, C.S.; Nakamura, H.; Leask, A.; Abraham, D.; Bou-Gharios, G.; de Crombrugghe, B. Selective expression of connective tissue growth factor in fibroblasts in vivo promotes systemic tissue fibrosis. Arthritis Rheum. 2010, 62, 1523–1532. [Google Scholar] [CrossRef]
- Nikitorowicz-Buniak, J.; Denton, C.P.; Abraham, D.; Stratton, R. Partially evoked epithelial-mesenchymal transition (EMT) is associated with increased TGFβ signaling within lesional scleroderma skin. PLoS ONE 2015, 10, e0134092. [Google Scholar] [CrossRef] [PubMed]
- Blumbach, K.; Zweers, M.C.; Brunner, G.; Peters, A.S.; Schmitz, M.; Schulz, J.-N.; Schild, A.; Denton, C.P.; Sakai, T.; Fässler, R.; et al. Defective granulation tissue formation in mice with specific ablation of integrin-linked kinase in fibroblasts—Role of TGFβ1 levels and RhoA activity. J. Cell Sci. 2010, 123, 3872–3883. [Google Scholar] [CrossRef]
- Schulz, J.-N.; Zeltz, C.; Sørensen, I.W.; Barczyk, M.; Carracedo, S.; Hallinger, R.; Niehoff, A.; Eckes, B.; Gullberg, D. Reduced granulation tissue and wound strength in the absence of α11β1 integrin. J. Investig. Dermatol. 2015, 135, 1435–1444. [Google Scholar] [CrossRef] [PubMed]
- Schulz, J.-N.; Plomann, M.; Sengle, G.; Gullberg, D.; Krieg, T.; Eckes, B. New developments on skin fibrosis—Essential signals emanating from the extracellular matrix for the control of myofibroblasts. Matrix Biol. 2018, 68–69, 522–532. [Google Scholar] [CrossRef]
- Sawant, M.; Hinz, B.; Schönborn, K.; Zeinert, I.; Eckes, B.; Krieg, T.; Schuster, R. A story of fibers and stress: Matrix-embedded signals for fibroblast activation in the skin. Wound Repair Regen. 2021, 29, 515–530. [Google Scholar] [CrossRef]
- Lagares, D.; Busnadiego, O.; García-Fernández, R.A.; Kapoor, M.; Liu, S.; Carter, D.E.; Abraham, D.; Shi-Wen, X.; Carreira, P.; Fontaine, B.A.; et al. Inhibition of focal adhesion kinase prevents experimental lung fibrosis and myofibroblast formation. Arthritis Rheum. 2012, 64, 1653–1664. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Luo, H.; Skaug, B.; Tabib, T.; Li, Y.-N.; Tao, Y.; Matei, A.-E.; Lyons, M.A.; Schett, G.; Lafyatis, R.; et al. Fibroblast Subpopulations in Systemic Sclerosis: Functional Implications of Individual Subpopulations and Correlations with Clinical Features. J. Investig. Dermatol. 2024, 144, 1251–1261.e13. [Google Scholar] [CrossRef] [PubMed]
- Schönborn, K.; Willenborg, S.; Schulz, J.-N.; Imhof, T.; Eming, S.A.; Quondamatteo, F.; Brinckmann, J.; Niehoff, A.; Paulsson, M.; Koch, M.; et al. Role of collagen XII in skin homeostasis and repair. Matrix Biol. 2020, 94, 57–76. [Google Scholar] [CrossRef] [PubMed]
- Schulz, J.-N.; Nüchel, J.; Niehoff, A.; Bloch, W.; Schönborn, K.; Hayashi, S.; Kamper, M.; Brinckmann, J.; Plomann, M.; Paulsson, M.; et al. COMP-assisted collagen secretion—A novel intracellular function required for fibrosis. J. Cell Sci. 2016, 129, 706–716. [Google Scholar] [CrossRef]
- Pope, J.E.; Denton, C.P.; Johnson, S.R.; Fernandez-Codina, A.; Hudson, M.; Nevskaya, T. State-of-the-art evidence in the treatment of systemic sclerosis. Nat. Rev. Rheumatol. 2023, 19, 212–226. [Google Scholar] [CrossRef]
- Distler, O.; Highland, K.B.; Gahlemann, M.; Azuma, A.; Fischer, A.; Mayes, M.D.; Raghu, G.; Sauter, W.; Girard, M.; Alves, M.; et al. Nintedanib for Systemic Sclerosis–Associated Interstitial Lung Disease. N. Engl. J. Med. 2019, 380, 2518–2528. [Google Scholar] [CrossRef]
- Stratton, R.J.; Wilson, H.; Black, C.M. Pilot Study of Anti-Thymocyte Globulin plus Mycophenolate Mofetil in Recent-Onset Diffuse Scleroderma. Rheumatology 2001, 40, 84–88. [Google Scholar] [CrossRef]
- Franco-Fuquen, P.; Figueroa-Aguirre, J.; Martínez, D.A.; Moreno-Cortes, E.F.; Garcia-Robledo, J.E.; Vargas-Cely, F.; Castro-Martínez, D.A.; Almaini, M.; Castro, J.E. Cellular therapies in rheumatic and musculoskeletal diseases. J. Transl. Autoimmun. 2025, 10, 100264. [Google Scholar] [CrossRef]
- Kuzumi, A.; Ebata, S.; Baron, M.; Sato, S.; Yoshizaki, A. Usefulness of rituximab for the treatment of systemic sclerosis-associated interstitial lung disease: Further analysis of the DESIRES trial. J. Am. Acad. Dermatol. 2025, 92, 1402–1404. [Google Scholar] [CrossRef]
- Davis, J.S.; Ferreira, D.; Paige, E.; Gedye, C.; Boyle, M. Infectious complications of biological and small molecule targeted immunomodulatory therapies. Clin. Microbiol. Rev. 2020, 33, 1–117. [Google Scholar] [CrossRef]
- Colussi, L.; Dagri, A.; Pastore, S.; Tommasini, A.; Pin, A.; Taddio, A. Effect of the Janus kinase inhibitor tofacitinib in the treatment of juvenile scleroderma: A single-center experience. Int. J. Rheum. Dis. 2024, 27, 15295. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; Liu, C.; Wu, Y.; Liu, S.; Zheng, Y.; Hao, W.; Wang, D.; Sun, L. Efficacy and safety of mesenchymal stromal cell transplantation in the treatment of autoimmune and rheumatic immune diseases: A systematic review and meta-analysis of randomized controlled trials. Stem Cell Res. Ther. 2025, 16, 65. [Google Scholar] [CrossRef] [PubMed]
- Campochiaro, C.; De Luca, G.; Lazzaroni, M.-G.; Armentaro, G.; Spinella, A.; Vigone, B.; Ruaro, B.; Stanziola, A.; Benfaremo, D.; De Lorenzis, E.; et al. Real-life efficacy and safety of nintedanib in systemic sclerosis-interstitial lung disease: Data from an Italian multicentre study. RMD Open 2023, 9, e002850. [Google Scholar] [CrossRef] [PubMed]
- Humbert, M.; McLaughlin, V.; Gibbs, J.S.R.; Gomberg-Maitland, M.; Hoeper, M.M.; Preston, I.R.; Souza, R.; Waxman, A.; Subias, P.E.; Feldman, J.; et al. Sotatercept for the Treatment of Pulmonary Arterial Hypertension. N. Engl. J. Med. 2021, 384, 1204–1215. [Google Scholar] [CrossRef]
- Matucci-Cerinic, M.; Kahaleh, B.; Wigley, F.M. Review: Evidence that systemic sclerosis is a vascular disease. Arthritis Rheum. 2013, 65, 1953–1962. [Google Scholar] [CrossRef]
- Rigau, A.R.; Li, Y.-N.; Matei, A.-E.; Györfi, A.-H.; Bruch, P.-M.; Koziel, S.; Devakumar, V.; Gabrielli, A.; Kreuter, A.; Wang, J.; et al. Characterization of Vascular Niche in Systemic Sclerosis by Spatial Proteomics. Circ. Res. 2024, 134, 875–891. [Google Scholar] [CrossRef]
- Cutolo, M.; Sulli, A.; Smith, V. Assessing microvascular changes in systemic sclerosis diagnosis and management. Nat. Rev. Rheumatol. 2010, 6, 578–587. [Google Scholar] [CrossRef]
- Lemmers, J.; Velauthapillai, A.; van Herwaarden, N.; Vonk, M. Change of the microvascularization in systemic sclerosis, a matter of air. Best Pr. Res. Clin. Rheumatol. 2021, 35, 101683. [Google Scholar] [CrossRef]
- Allanore, Y.; Wung, P.; Soubrane, C.; Esperet, C.; Marrache, F.; Bejuit, R.; Lahmar, A.; Khanna, D.; Denton, C.P. A randomised, double-blind, placebo-controlled, 24-week, phase II, proof-of-concept study of romilkimab (SAR156597) in early diffuse cutaneous systemic sclerosis. Ann. Rheum. Dis. 2020, 79, 1600–1607. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).