
Gelsolin Facilitates Estrogen Receptor Beta Nuclear Translocation and Transcriptional Repression of Genes Associated with Alzheimer Disease


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Cell Treatments
2.3. Plasmid Constructs
2.4. Transient Transfections
2.5. Luciferase Assays
2.6. Western Blot
2.7. Immunofluorescence
2.8. Imaging
2.9. Statistics
3. Results
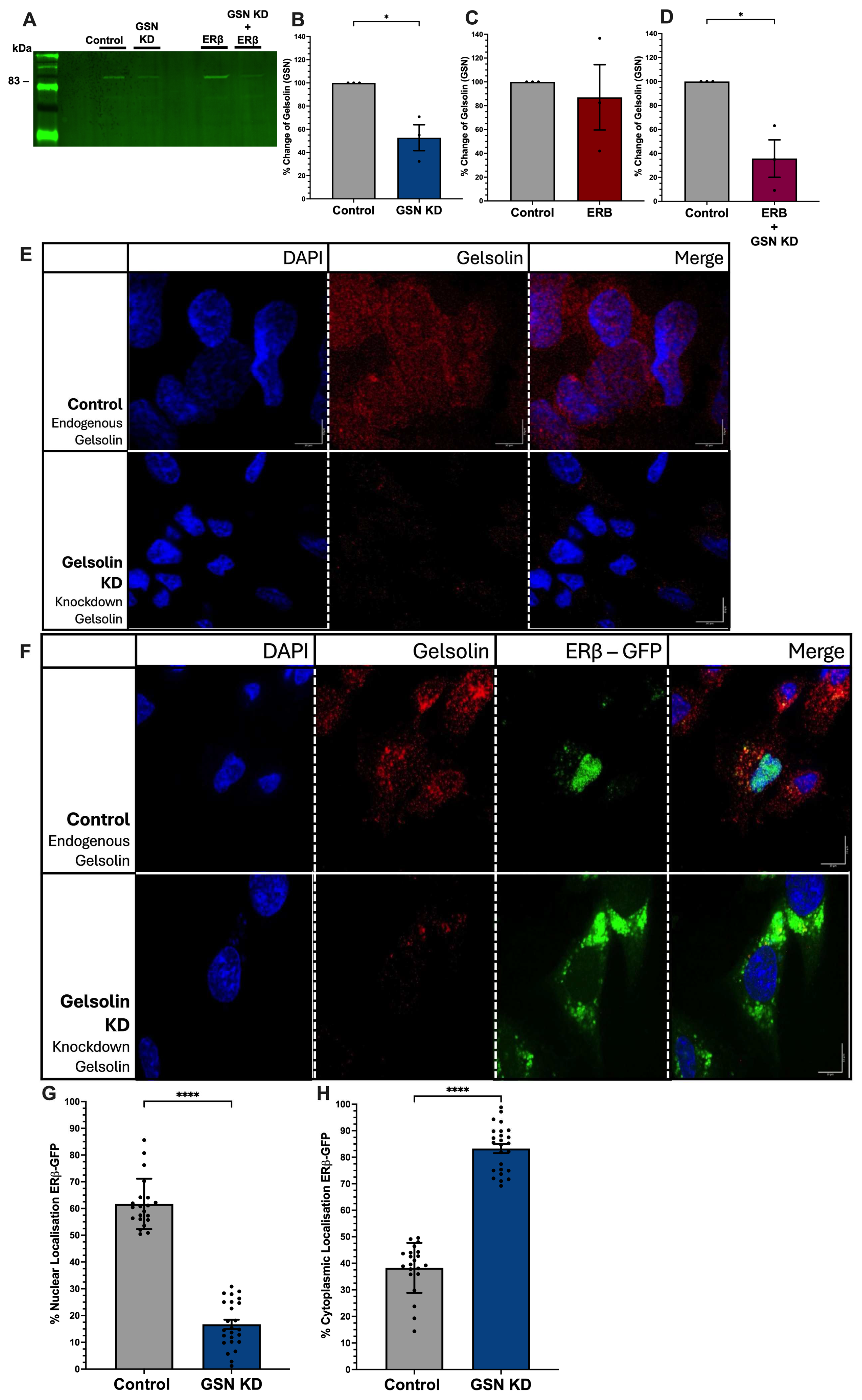
3.1. Gelsolin Is Required for Nuclear Translocation of ERβ1
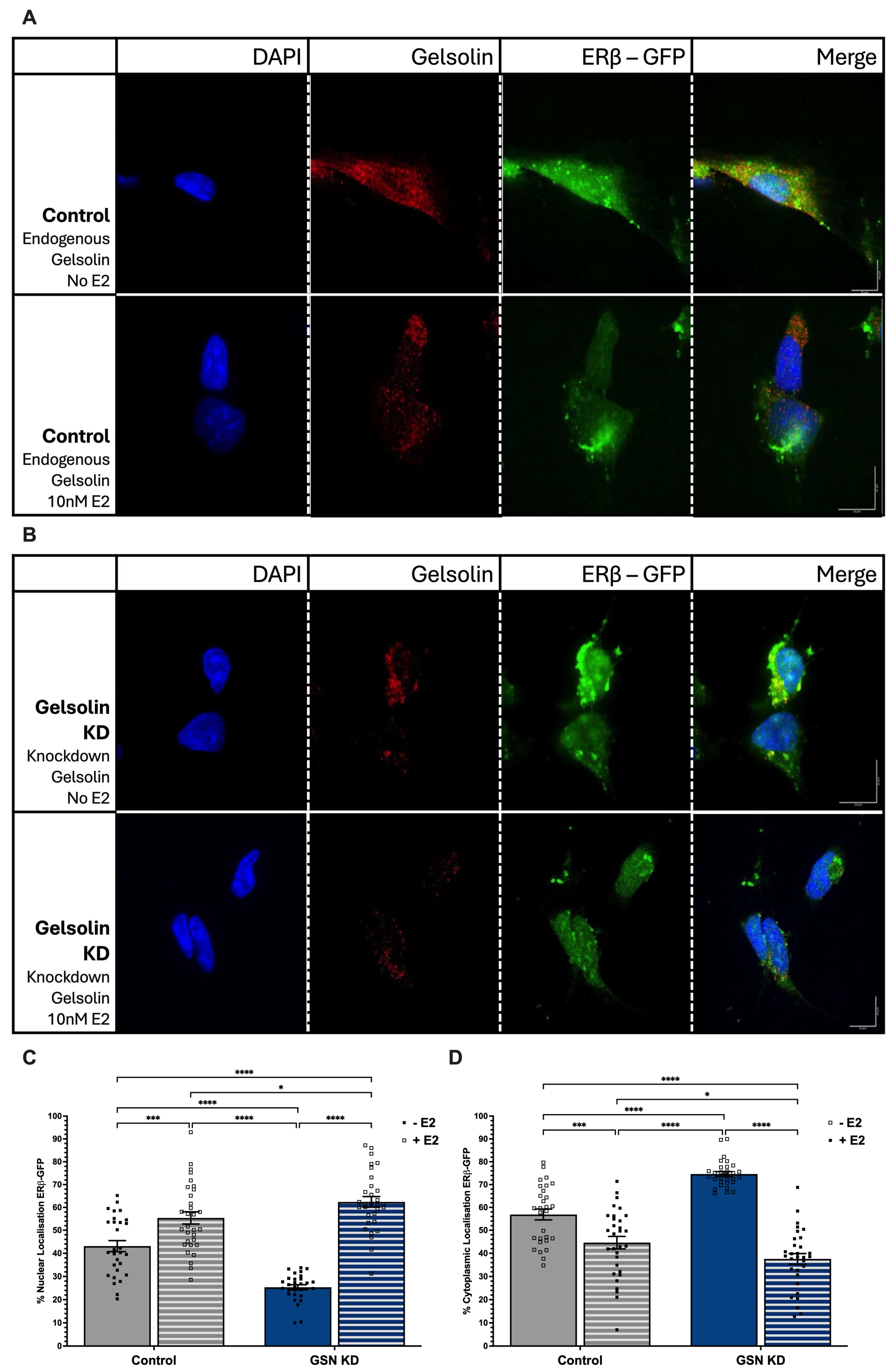
3.2. 17β-Estradiol Induces ERβ1 Nuclear Translocation in the Absence of GSN
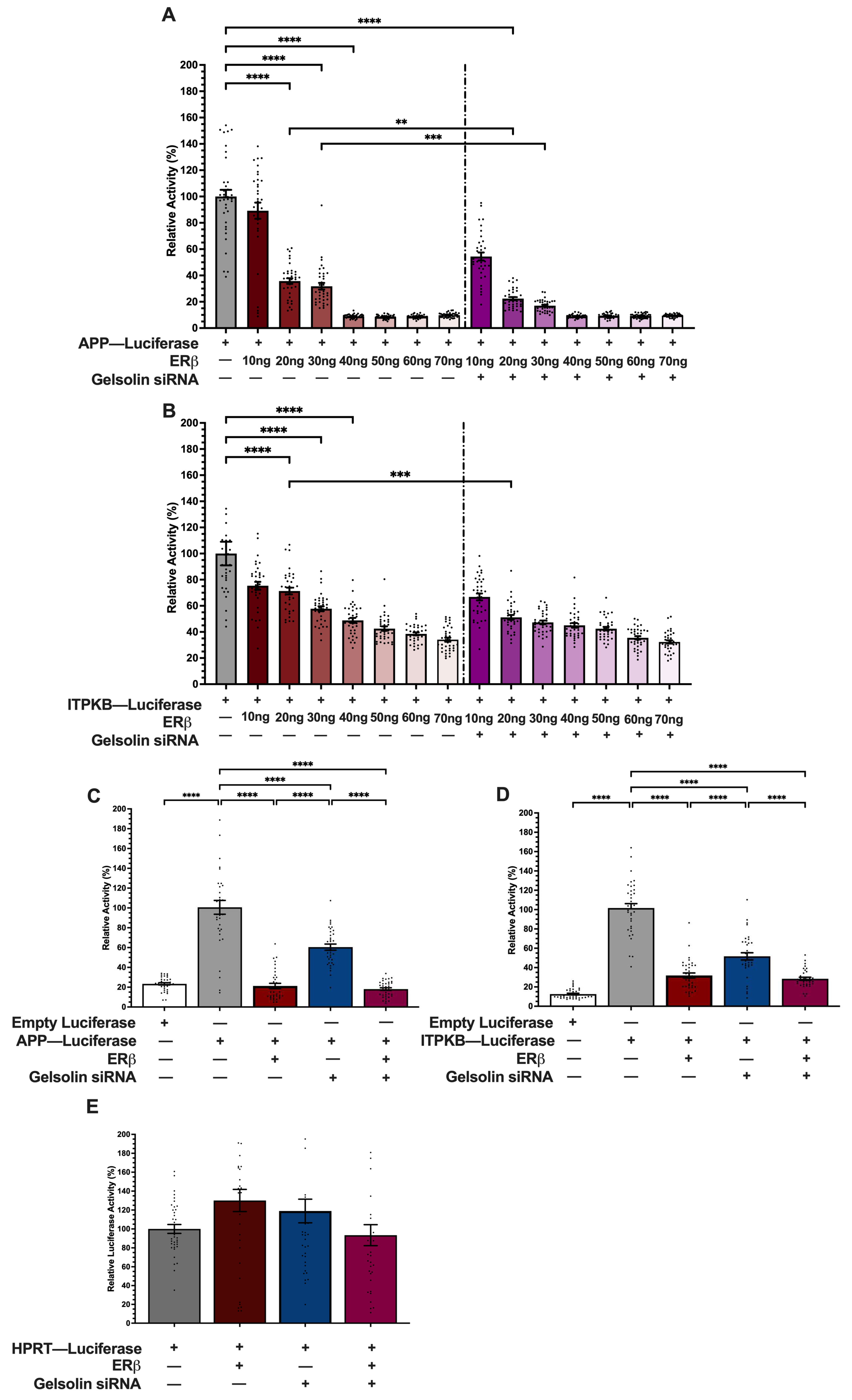
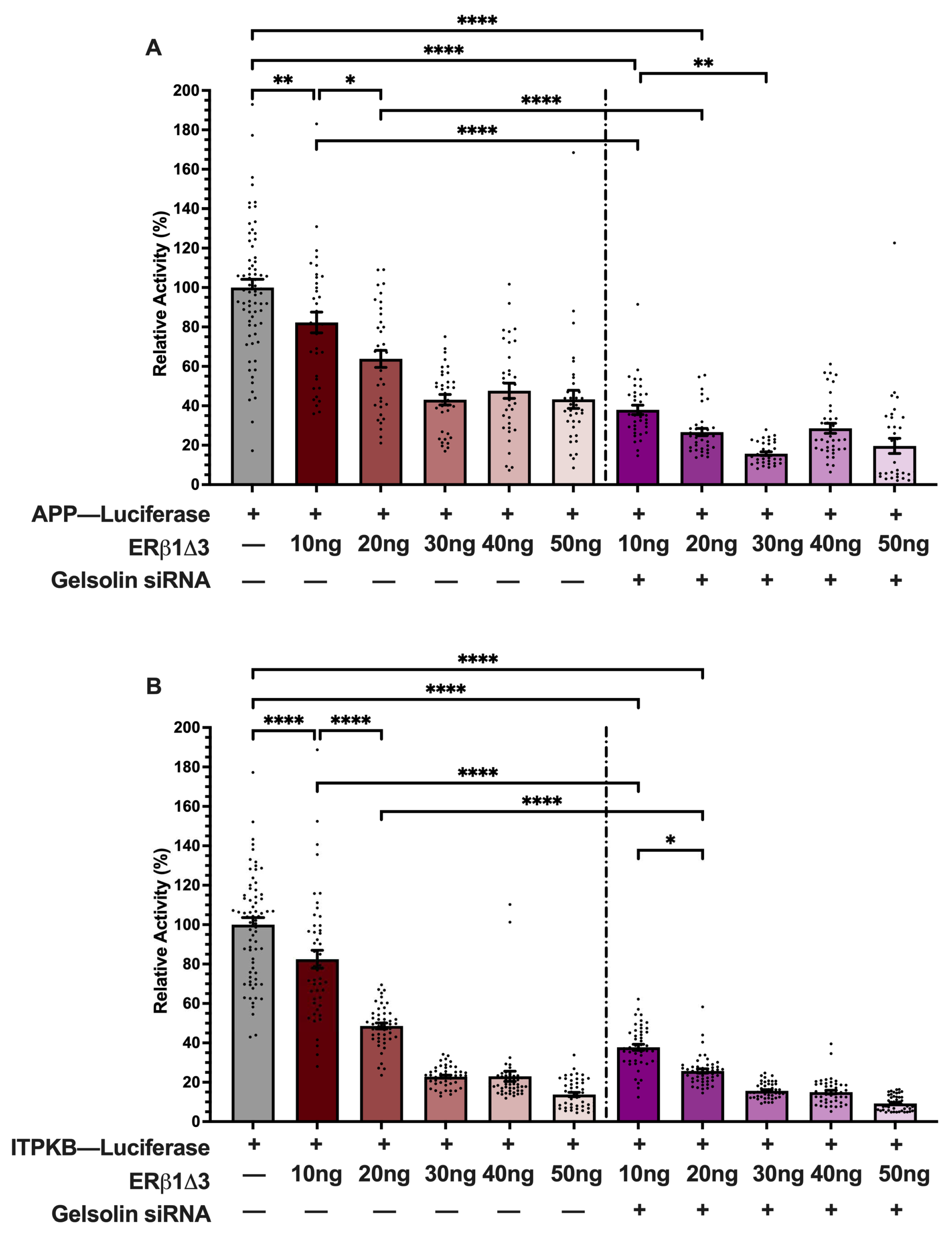
3.3. ERβ1 Represses APP and ITPKB Promoter Activity
3.4. GSN Enhances APP and ITPKB Promoter Activity
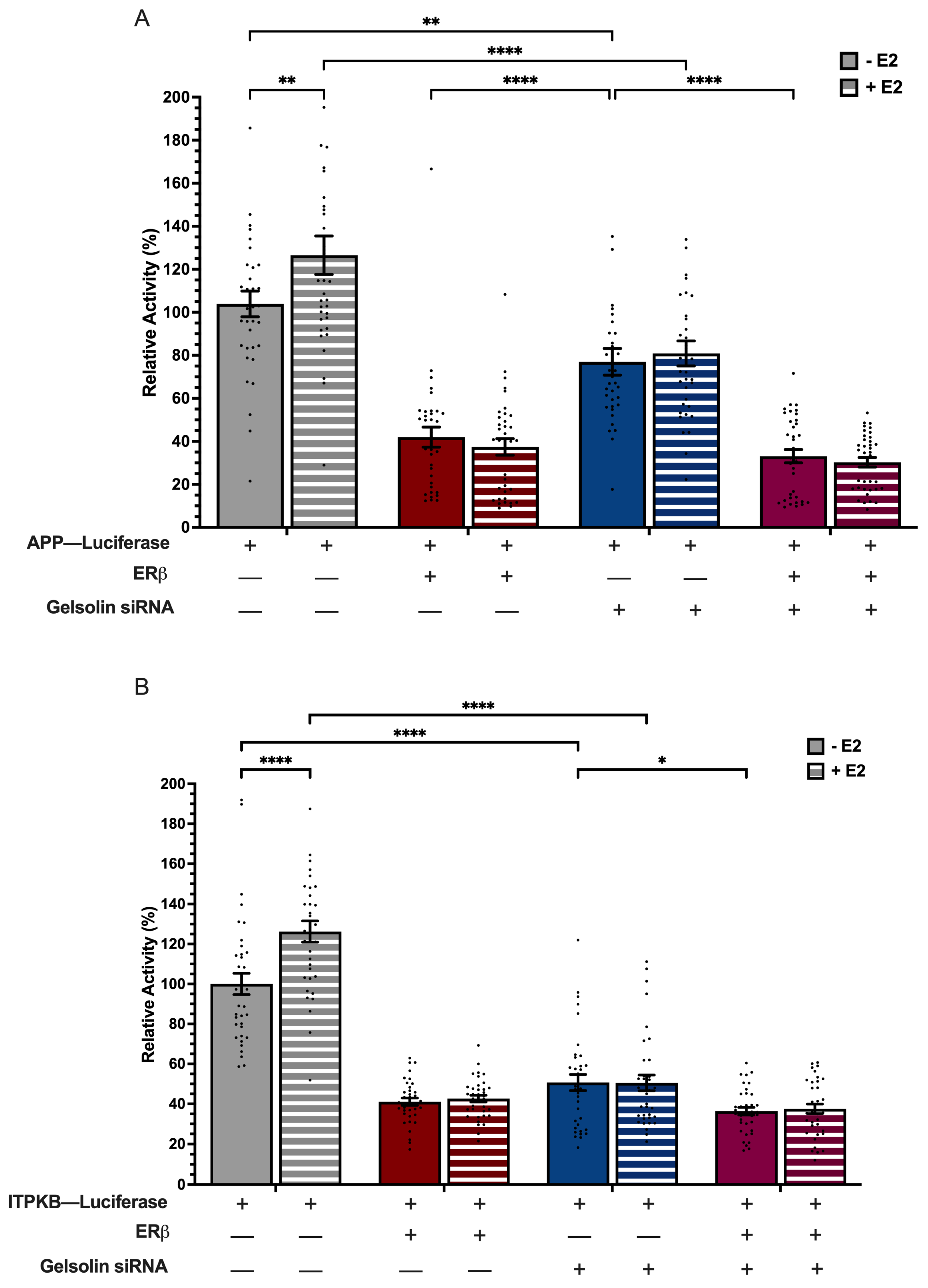
3.5. ERβ1 and GSN Regulation of APP and ITPKB Promoters Is Independent of E2
3.6. ERβ1-Induced Repression of APP and ITPKB Promoters Likely Occurs Through Indirect DNA Binding
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AD | Alzheimer Disease |
| AP-1 | Activator protein 1 |
| APP | Amyloid beta Precursor Protein |
| E2 | 17β-estradiol |
| ER, α, β1, β1Δ3 | Estrogen receptor, alpha, beta1, beta1delta3 |
| ERE | Estrogen response element |
| GSN | Gelsolin |
| HPRT | Hypoxanthine phosphoribosyltransferase 1 |
| ITPKB | Inositol–trisphosphate 3–kinase B |
| siRNA | Short interfering ribonucleic acid |
| SK-N-SH | Human neuroblastoma cell line |
References
- Kuiper, G.G.; Gustafsson, J.A. The novel estrogen receptor-beta subtype: Potential role in the cell- and promoter-specific actions of estrogens and anti-estrogens. FEBS Lett. 1997, 410, 87–90. [Google Scholar] [CrossRef] [PubMed]
- Paech, K.; Webb, P.; Kuiper, G.G.; Nilsson, S.; Gustafsson, J.; Kushner, P.J.; Scanlan, T.S. Differential ligand activation of estrogen receptors ERalpha and ERbeta at AP1 sites. Science 1997, 277, 1508–1510. [Google Scholar] [CrossRef]
- Umayahara, Y.; Kawamori, R.; Watada, H.; Imano, E.; Iwama, N.; Morishima, T.; Yamasaki, Y.; Kajimoto, Y.; Kamada, T. Estrogen regulation of the insulin-like growth factor I gene transcription involves an AP-1 enhancer. J. Biol. Chem. 1994, 269, 16433–16442. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Gao, H.; Liu, Y.; Papoutsi, Z.; Jaffrey, S.; Gustafsson, J.A.; Dahlman-Wright, K. Genome-wide mapping of estrogen receptor-beta-binding regions reveals extensive cross-talk with transcription factor activator protein-1. Cancer Res. 2010, 70, 5174–5183. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Steenbergen, C.; Murphy, E.; Sun, J. Estrogen receptor-beta activation results in S-nitrosylation of proteins involved in cardioprotection. Circulation 2009, 120, 245–254. [Google Scholar] [CrossRef]
- Luczak, E.D.; Leinwand, L.A. Sex-based cardiac physiology. Annu. Rev. Physiol. 2009, 71, 1–18. [Google Scholar] [CrossRef]
- Lund, T.D.; Rovis, T.; Chung, W.C.; Handa, R.J. Novel actions of estrogen receptor-beta on anxiety-related behaviors. Endocrinology 2005, 146, 797–807. [Google Scholar] [CrossRef]
- Nerattini, M.; Jett, S.; Andy, C.; Carlton, C.; Zarate, C.; Boneu, C.; Battista, M.; Pahlajani, S.; Loeb-Zeitlin, S.; Havryulik, Y.; et al. Systematic review and meta-analysis of the effects of menopause hormone therapy on risk of Alzheimer’s disease and dementia. Front. Aging Neurosci. 2023, 15, 1260427. [Google Scholar] [CrossRef]
- Simpkins, J.W.; Singh, M.; Brock, C.; Etgen, A.M. Neuroprotection and estrogen receptors. Neuroendocrinology 2012, 96, 119–130. [Google Scholar] [CrossRef]
- Xu, Y.; Arenas, I.A.; Armstrong, S.J.; Plahta, W.C.; Xu, H.; Davidge, S.T. Estrogen improves cardiac recovery after ischemia/reperfusion by decreasing tumor necrosis factor-alpha. Cardiovasc. Res. 2006, 69, 836–844. [Google Scholar] [CrossRef]
- Mott, N.N.; Pinceti, E.; Rao, Y.S.; Przybycien-Szymanska, M.M.; Prins, S.A.; Shults, C.L.; Yang, X.; Glucksman, M.J.; Roberts, J.L.; Pak, T.R. Age-dependent Effects of 17beta-estradiol on the dynamics of estrogen receptor beta (ERbeta) protein-protein interactions in the ventral hippocampus. Mol. Cell. Proteom. 2014, 13, 760–779. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, K.; Ting, H.J.; Harada, Y.; Tokizane, T.; Nonomura, N.; Kang, H.Y.; Chang, H.C.; Yeh, S.; Miyamoto, H.; Shin, M.; et al. Modulation of androgen receptor transactivation by gelsolin: A newly identified androgen receptor coregulator. Cancer Res. 2003, 63, 4888–4894. [Google Scholar] [PubMed]
- Feldt, J.; Schicht, M.; Garreis, F.; Welss, J.; Schneider, U.W.; Paulsen, F. Structure, regulation and related diseases of the actin-binding protein gelsolin. Expert Rev. Mol. Med. 2019, 20, e7. [Google Scholar] [CrossRef] [PubMed]
- Feng, B.; Hu, P.; Chen, J.; Liu, Q.; Li, X.; Du, Y. Analysis of Differentially Expressed Genes Associated With Alzheimer’s Disease Based on Bioinformatics Methods. Am. J. Alzheimer’s Dis. Other Dement. 2015, 30, 746–751. [Google Scholar] [CrossRef]
- Quitschke, W.W.; Goldgaber, D. The amyloid beta-protein precursor promoter. A region essential for transcriptional activity contains a nuclear factor binding domain. J. Biol. Chem. 1992, 267, 17362–17368. [Google Scholar] [CrossRef]
- Stygelbout, V.; Leroy, K.; Pouillon, V.; Ando, K.; D’Amico, E.; Jia, Y.; Luo, H.R.; Duyckaerts, C.; Erneux, C.; Schurmans, S.; et al. Inositol trisphosphate 3-kinase B is increased in human Alzheimer brain and exacerbates mouse Alzheimer pathology. Brain 2014, 137, 537–552. [Google Scholar] [CrossRef]
- Zhou, H.; Zarubin, T.; Ji, Z.; Min, Z.; Zhu, W.; Downey, J.S.; Lin, S.; Han, J. Frequency and distribution of AP-1 sites in the human genome. DNA Res. 2005, 12, 139–150. [Google Scholar] [CrossRef]
- Leung, Y.K.; Mak, P.; Hassan, S.; Ho, S.M. Estrogen receptor (ER)-beta isoforms: A key to understanding ER-beta signaling. Proc. Natl. Acad. Sci. USA 2006, 103, 13162–13167. [Google Scholar] [CrossRef]
- Price, R.H., Jr.; Butler, C.A.; Webb, P.; Uht, R.; Kushner, P.; Handa, R.J. A splice variant of estrogen receptor beta missing exon 3 displays altered subnuclear localization and capacity for transcriptional activation. Endocrinology 2001, 142, 2039–2049. [Google Scholar] [CrossRef]
- Castillo, A.B.; Triplett, J.W.; Pavalko, F.M.; Turner, C.H. Estrogen receptor-beta regulates mechanical signaling in primary osteoblasts. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E937–E944. [Google Scholar] [CrossRef]
- Pak, T.R.; Chung, W.C.; Roberts, J.L.; Handa, R.J. Ligand-independent effects of estrogen receptor beta on mouse gonadotropin-releasing hormone promoter activity. Endocrinology 2006, 147, 1924–1931. [Google Scholar] [CrossRef] [PubMed]
- Pinceti, E.; Shults, C.L.; Rao, Y.S.; Mott, N.N.; Pak, T.R. Phosphorylation Alters Oestrogen Receptor beta-Mediated Transcription in Neurones. J. Neuroendocr. 2015, 27, 861–871. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, A.; Tremblay, G.B.; Labrie, F.; Giguere, V. Ligand-independent recruitment of SRC-1 to estrogen receptor beta through phosphorylation of activation function AF-1. Mol. Cell 1999, 3, 513–519. [Google Scholar] [CrossRef]
- Zhang, G.; Yanamala, N.; Lathrop, K.L.; Zhang, L.; Klein-Seetharaman, J.; Srinivas, H. Ligand-independent antiapoptotic function of estrogen receptor-beta in lung cancer cells. Mol. Endocrinol. 2010, 24, 1737–1747. [Google Scholar] [CrossRef]
- Pemberton, L.F.; Paschal, B.M. Mechanisms of receptor-mediated nuclear import and nuclear export. Traffic 2005, 6, 187–198. [Google Scholar] [CrossRef]
- Schlegel, A.; Wang, C.; Katzenellenbogen, B.S.; Pestell, R.G.; Lisanti, M.P. Caveolin-1 potentiates estrogen receptor alpha (ERalpha) signaling. caveolin-1 drives ligand-independent nuclear translocation and activation of ERalpha. J. Biol. Chem. 1999, 274, 33551–33556. [Google Scholar] [CrossRef]
- Hager, G.L.; Lim, C.S.; Elbi, C.; Baumann, C.T. Trafficking of nuclear receptors in living cells. J. Steroid Biochem. Mol. Biol. 2000, 74, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Ishunina, T.A.; Swaab, D.F. Increased expression of estrogen receptor alpha and beta in the nucleus basalis of Meynert in Alzheimer’s disease. Neurobiol. Aging 2001, 22, 417–426. [Google Scholar] [CrossRef]
- Kocanova, S.; Mazaheri, M.; Caze-Subra, S.; Bystricky, K. Ligands specify estrogen receptor alpha nuclear localization and degradation. BMC Cell Biol. 2010, 11, 98. [Google Scholar] [CrossRef]
- Matsuda, K.; Ochiai, I.; Nishi, M.; Kawata, M. Colocalization and ligand-dependent discrete distribution of the estrogen receptor (ER)alpha and ERbeta. Mol. Endocrinol. 2002, 16, 2215–2230. [Google Scholar] [CrossRef]
- Yang, S.H.; Liu, R.; Perez, E.J.; Wen, Y.; Stevens, S.M., Jr.; Valencia, T.; Brun-Zinkernagel, A.M.; Prokai, L.; Will, Y.; Dykens, J.; et al. Mitochondrial localization of estrogen receptor beta. Proc. Natl. Acad. Sci. USA 2004, 101, 4130–4135. [Google Scholar] [CrossRef] [PubMed]
- Clarris, H.J.; Cappai, R.; Heffernan, D.; Beyreuther, K.; Masters, C.L.; Small, D.H. Identification of heparin-binding domains in the amyloid precursor protein of Alzheimer’s disease by deletion mutagenesis and peptide mapping. J. Neurochem. 1997, 68, 1164–1172. [Google Scholar] [CrossRef] [PubMed]
- Beher, D.; Hesse, L.; Masters, C.L.; Multhaup, G. Regulation of amyloid protein precursor (APP) binding to collagen and mapping of the binding sites on APP and collagen type I. J. Biol. Chem. 1996, 271, 1613–1620. [Google Scholar] [CrossRef]
- Kibbey, M.C.; Jucker, M.; Weeks, B.S.; Neve, R.L.; Van Nostrand, W.E.; Kleinman, H.K. beta-Amyloid precursor protein binds to the neurite-promoting IKVAV site of laminin. Proc. Natl. Acad. Sci. USA 1993, 90, 10150–10153. [Google Scholar] [CrossRef]
- Mok, S.S.; Sberna, G.; Heffernan, D.; Cappai, R.; Galatis, D.; Clarris, H.J.; Sawyer, W.H.; Beyreuther, K.; Masters, C.L.; Small, D.H. Expression and analysis of heparin-binding regions of the amyloid precursor protein of Alzheimer’s disease. FEBS Lett. 1997, 415, 303–307. [Google Scholar] [CrossRef] [PubMed]
- Storey, E.; Spurck, T.; Pickett-Heaps, J.; Beyreuther, K.; Masters, C.L. The amyloid precursor protein of Alzheimer’s disease is found on the surface of static but not activity motile portions of neurites. Brain Res. 1996, 735, 59–66. [Google Scholar] [CrossRef]
- Yamazaki, T.; Koo, E.H.; Selkoe, D.J. Cell surface amyloid beta-protein precursor colocalizes with beta 1 integrins at substrate contact sites in neural cells. J. Neurosci. 1997, 17, 1004–1010. [Google Scholar] [CrossRef]
- Young-Pearse, T.L.; Chen, A.C.; Chang, R.; Marquez, C.; Selkoe, D.J. Secreted APP regulates the function of full-length APP in neurite outgrowth through interaction with integrin beta1. Neural Dev. 2008, 3, 15. [Google Scholar] [CrossRef]
- Haass, C.; Selkoe, D.J. Soluble protein oligomers in neurodegeneration: Lessons from the Alzheimer’s amyloid beta-peptide. Nat. Rev. Mol. Cell. Biol. 2007, 8, 101–112. [Google Scholar] [CrossRef]
- Jaffe, A.B.; Toran-Allerand, C.D.; Greengard, P.; Gandy, S.E. Estrogen regulates metabolism of Alzheimer amyloid beta precursor protein. J. Biol. Chem. 1994, 269, 13065–13068. [Google Scholar] [CrossRef]
- Manthey, D.; Heck, S.; Engert, S.; Behl, C. Estrogen induces a rapid secretion of amyloid beta precursor protein via the mitogen-activated protein kinase pathway. Eur. J. Biochem. 2001, 268, 4285–4291. [Google Scholar] [CrossRef]
- Cui, J.; Ait-Ghezala, G.; Sambamurti, K.; Gao, F.; Shen, Y.; Li, R. Sex-Specific Regulation of beta-Secretase: A Novel Estrogen Response Element (ERE)-Dependent Mechanism in Alzheimer’s Disease. J. Neurosci. 2022, 42, 1154–1165. [Google Scholar] [CrossRef] [PubMed]
- Erneux, C.; Ghosh, S.; Koenig, S. Inositol(1,4,5)P3 3-kinase isoenzymes: Catalytic properties and importance of targeting to F-actin to understand function. Adv. Biol. Regul. 2016, 60, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Johnson, H.W.; Schell, M.J. Neuronal IP3 3-kinase is an F-actin-bundling protein: Role in dendritic targeting and regulation of spine morphology. Mol. Biol. Cell. 2009, 20, 5166–5180. [Google Scholar] [CrossRef] [PubMed]
- Koenig, S.; Moreau, C.; Dupont, G.; Scoumanne, A.; Erneux, C. Regulation of NGF-driven neurite outgrowth by Ins(1,4,5)P3 kinase is specifically associated with the two isoenzymes Itpka and Itpkb in a model of PC12 cells. FEBS J. 2015, 282, 2553–2569. [Google Scholar] [CrossRef]
- Rauscher, F.J., 3rd; Voulalas, P.J.; Franza, B.R., Jr.; Curran, T. Fos and Jun bind cooperatively to the AP-1 site: Reconstitution in vitro. Genes Dev. 1988, 2, 1687–1699. [Google Scholar] [CrossRef]
- Vivar, O.I.; Zhao, X.; Saunier, E.F.; Griffin, C.; Mayba, O.S.; Tagliaferri, M.; Cohen, I.; Speed, T.P.; Leitman, D.C. Estrogen receptor beta binds to and regulates three distinct classes of target genes. J. Biol. Chem. 2010, 285, 22059–22066. [Google Scholar] [CrossRef]
- Zhao, L.; Woody, S.K.; Chhibber, A. Estrogen receptor beta in Alzheimer’s disease: From mechanisms to therapeutics. Ageing Res. Rev. 2015, 24, 178–190. [Google Scholar] [CrossRef]
- Zhao, L.; Mao, Z.; Chen, S.; Schneider, L.S.; Brinton, R.D. Early intervention with an estrogen receptor beta-selective phytoestrogenic formulation prolongs survival, improves spatial recognition memory, and slows progression of amyloid pathology in a female mouse model of Alzheimer’s disease. J. Alzheimers Dis. 2013, 37, 403–419. [Google Scholar] [CrossRef]
- Said, S.A.; Isedowo, R.; Guerin, C.; Nar, N.N.; Lillie, L.; Bukovac, S.; Simone, J.J.; Green, M.R.; McCormick, C.M.; Stuart, J.A. Effects of long-term dietary administration of estrogen receptor-beta agonist diarylpropionitrile on ovariectomized female ICR (CD-1) mice. Geroscience 2018, 40, 393–403. [Google Scholar] [CrossRef]
- Sarvari, M.; Kallo, I.; Hrabovszky, E.; Solymosi, N.; Rodolosse, A.; Liposits, Z. Long-Term Estrogen Receptor Beta Agonist Treatment Modifies the Hippocampal Transcriptome in Middle-Aged Ovariectomized Rats. Front. Cell. Neurosci. 2016, 10, 149. [Google Scholar] [CrossRef] [PubMed]
- Vina, J.; Escudero, J.; Baquero, M.; Cebrian, M.; Carbonell-Asins, J.A.; Munoz, J.E.; Satorres, E.; Melendez, J.C.; Ferrer-Rebolleda, J.; Cozar-Santiago, M.D.P.; et al. Genistein effect on cognition in prodromal Alzheimer’s disease patients. The GENIAL clinical trial. Alzheimers Res. Ther. 2022, 14, 164. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yildiz, Y.; Fan, A.H.S.; Hartoun, A.A.; Flury, S.; Ngai, Y.; Pak, T.R. Gelsolin Facilitates Estrogen Receptor Beta Nuclear Translocation and Transcriptional Repression of Genes Associated with Alzheimer Disease. Receptors 2025, 4, 10. https://doi.org/10.3390/receptors4020010
Yildiz Y, Fan AHS, Hartoun AA, Flury S, Ngai Y, Pak TR. Gelsolin Facilitates Estrogen Receptor Beta Nuclear Translocation and Transcriptional Repression of Genes Associated with Alzheimer Disease. Receptors. 2025; 4(2):10. https://doi.org/10.3390/receptors4020010
Chicago/Turabian StyleYildiz, Yoldas, Angela H. S. Fan, Amanda A. Hartoun, Sarah Flury, Yan Ngai, and Toni R. Pak. 2025. "Gelsolin Facilitates Estrogen Receptor Beta Nuclear Translocation and Transcriptional Repression of Genes Associated with Alzheimer Disease" Receptors 4, no. 2: 10. https://doi.org/10.3390/receptors4020010
APA StyleYildiz, Y., Fan, A. H. S., Hartoun, A. A., Flury, S., Ngai, Y., & Pak, T. R. (2025). Gelsolin Facilitates Estrogen Receptor Beta Nuclear Translocation and Transcriptional Repression of Genes Associated with Alzheimer Disease. Receptors, 4(2), 10. https://doi.org/10.3390/receptors4020010